")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
TGF-β1-Mediated Activation of SERPINE1 is Involved in Hemin-Induced Apoptotic and Inflammatory Injury in HT22 Cells
Authors Wang T, Lu H, Li D, Huang W
Received 24 November 2020
Accepted for publication 18 January 2021
Published 11 February 2021 Volume 2021:17 Pages 423—433
DOI https://doi.org/10.2147/NDT.S293772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yuping Ning
Tinggang Wang,1,* Haibin Lu,2,* Deqiang Li,2 Weichun Huang3
1Emergency Department, Affiliated Hospital of Zunyi Medical University, Zunyi, People’s Republic of China; 2Department of Critical Care Medicine, Daping Hospital, Army Medical University, Chongqing, People’s Republic of China; 3Radiology Department, First Affiliated Hospital of Army Medical University, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weichun Huang
Radiology Department, First Affiliated Hospital of Army Medical University, No. 30, Gao Tan Yan Main Street, Shapingba District, Chongqing, 400038, People’s Republic of China
Tel +86-023-68754425
Email [email protected]
Background: Intracerebral hemorrhage (ICH) is a severe subtype of stroke with high mortality and morbidity. Serpin Family E Member 1 (SERPINE1) has been documented to be upregulated following ICH, however, the participation of SERPINE1 in the development of ICH has never been studied.
Methods: Hemin was utilized to develop an in vitro model of ICH. Gene levels were evaluated by the use of quantitative reverse transcription polymerase chain reaction, Western blot, as well as enzyme-linked immunoassay assay. The activity of caspase-3 was determined using a commercial kit. Cell viability and apoptosis were assessed using 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay and Terminal deoxynucleotidyl transferase (TdT) d UTP Nick-End Labeling assay.
Results: SERPINE1 was upregulated in hemin-treated HT22 cells. Silencing of SERPINE1 attenuated hemin-induced inhibition of cell viability. Moreover, knockdown of SERPINE1 repressed hemin-induced apoptosis in HT22 cells, as evidenced by the decrease in the number of TUNEL positive cells, caspase-3 activity, and Bax expression, and the increase in Bcl-2 expression. Meanwhile, knockdown of SERPINE1 repressed hemin-induced inflammation in HT22 cells, as indicated by reduced levels of tumor necrosis factor-α, interleukin-6 (IL-6), IL-1β, and inducible nitric oxide synthase. We also found that transforming growth factor-beta 1 (TGF-β 1) induced SERPINE1 expression in a dose-dependent manner. Besides, SERPINE1 knockdown attenuated the effects of TGF-β 1 on hemin-induced neuronal damage.
Conclusion: TGF-β 1-induced SERPINE1 activation exacerbated hemin-induced apoptosis and inflammation in HT22 cells, manifesting a novel mechanism for ICH progression.
Keywords: intracerebral hemorrhage, serpin family E member 1, transforming growth factor-beta 1
Introduction
Intracerebral hemorrhage (ICH) is a severe form of stroke, accounting for 10% of the stroke patients.1 ICH is a common cerebrovascular disorder with high mortality, which has become a heavy burden for families and countries.2 ICH refers to the extravasation of blood caused by the rupture of blood vessels within the brain parenchyma, resulting in neuronal death.3 In the last years, the morbidity of ICH is increasing, however, it is still devoid of effective treatment.4 Evidences during the past decades prove that the pathological process of ICH includes the primary brain damage resulting from mass effect owing to hematoma formation, and the secondary brain injury triggered by the extension of hematoma into the ventricles.5 Hence, a depth understanding of the mechanism underlying ICH-induced secondary brain damage has considerable significance that will help to develop efficient therapeutic strategies for ICH.
Hemin is a breakdown product of hemoglobin and is the main cause of neuronal death during ICH.6 Following ICH, hemolysis of erythrocytes leads to hematoma resolution and the release of hemoglobin into the brain tissues.7 Subsequently, hemin liberated from hemoglobin may intercalate into cell membranes directly or enter brain cells via the heme carrier protein 1/proton-coupled folate transporter.8 When hemin internalizes to brain cells, hemin can trigger DNA fragmentation, oxidative stress and inflammatory response, resulting in cell death.9 Besides, hemin is catabolized by heme oxygenases into biliverdin, CO, and iron, which in turn causes lipid peroxidation.10 Increasing evidence suggests that hemin-induced apoptosis and inflammation have emerged as vital contributors to ICH-induced brain damage.11 Nevertheless, the mechanism of hemin-induced apoptosis and inflammation remains elusive. Understanding the mechanism underlying hemin-induced apoptosis and inflammation, therefore, is of significance for ICH therapy.
Serpin Family E Member 1 (SERPINE1), also known as plasminogen activator inhibitor 1 (PAI-1), belongs to the serine protease inhibitor superfamily and is a multifunctional glycoprotein that plays a crucial role in various cellular processes.12 A recent study by Liu’s group found that SERPINE1 was upregulated in the perihematomal brain tissues of ICH patients as well as the brain tissues of ICH rats, indicating the involvement of SERPINE1 in the progression of ICH.13 However, few studies have studied the contribution of SERPINE1 to the development of ICH. In this study, we sought to determine the contribution of SERPINE1 to the development of ICH. We found that SERPINE2 was upregulated following hemin treatment, and its silencing mitigated hemin-induced apoptosis and inflammatory response. Our findings represented a novel mechanism for ICH progression.
Materials and Methods
Cell Culture and Treatment
Mouse HT22 hippocampal neuronal cells were purchased from Procell (Wuhan, China) and grown in DMEM medium enriched with 10% fetal bovine serum at 37°C in a humidified incubator maintained at 5% CO2 and 5% O2. To establish an in vitro model of ICH, HT22 cells were stimulated with different doses (0, 10, 20, 40, 60, 80, 100, and 120 μM) of hemin for 24 h.
To evaluate the role of SERPINE1 in ICH, si-SERPINE1 and si-negative control (NC) synthesized by GenePharma (Shanghai, China) were respectively transfected into HT22 cells using Lipofectamine 2000 (Invitrogen, Darmstadt, Germany) as instructed by the manufacturer. Following this, HT22 cells were exposed to 100 μM hemin for 24 h.
To evaluate the impact of transforming growth factor-beta 1 (TGF-β1) on ICH progression in vitro, HT22 cells were incubated in RPMI-1640 medium containing 100 μM hemin and 10 ng/mL of recombinant TGF-β1.
Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA from HT22 cells was extracted using Tri®-Reagent (Sigma, St. Louis, USA) per manufacturer’s instructions. A NanoDrop One spectrophotometer from Thermo Fisher Scientific (Waltham, MA, USA) was applied to assess the quality and concentration of the extracted RNA. The conversion of extracted RNA into cDNA was undertaken by the use of PrimeScript™ RT reagent Kit with gDNA Eraser from Takara (Dalian, China). qRT-PCR reaction was conducted using TB Green Premix Ex Taq from Takara. The experiment was performed for 3 times and the relative expression of SERPINE1, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1β, and inducible nitric oxide synthase (iNOS) was normalized to that of β-actin by the 2−ΔΔCT method.
Western Blot
After treatment, HT22 cells were collected and lysed to extract the total proteins. The extracted proteins were separated by 14% sodium dodecyl sulfate-polyacrylamide gel and then transferred onto a polyvinylidene fluoride membrane. After blocking in 5% skim milk, the membranes were probed overnight at 4°C with the following antibodies from Novus (Littleton, Colorado, USA) or Abcam (Cambridge, MA, USA): anti-SERPINE1 antibody, anti-TGF-β1 antibody, anti-Bax antibody, anti-Bcl-2 antibody and anti-β-actin antibody, followed by immunoblot for 1 h at room temperature with the horseradish peroxidase-conjugated secondary antibodies from Affinity Biosciences (Cincinnati, OH, USA). Signals were visualized using chemiluminescence from Solarbio (Beijing, China) and quantified using Image J software (National Institutes of Health, NY, USA).
3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (MTT) Assay
After treatment, HT22 cells were harvested and cultured in RPMI-1640 medium plus MTT solution from Solarbio. After 4 h of incubation, HT22 cells were treated for 10 min with formazan solution and then assayed for the absorbance at 490 nm.
Terminal Deoxynucleotidyl Transferase (TdT) dUTP Nick-End Labeling (TUNEL) Assay
Cell apoptosis was evaluated using TUNEL apoptosis assay kit from Beyotime (Shanghai, China), by following the manufacturer’s instruction. HT22 cells were harvested after treatment. Thereafter, we removed cell media and treated HT22 cells with TUNEL working solution for 1 h. Following this, we washed HT22 cells with PBS and then added reaction buffer (Component B) to each sample. Afterward, HT22 cells were treated with formaldehyde fixative buffer for 20 min at room temperature. Then, the fixative was removed and HT22 cells were washed and stained with 4ʹ 6-diamidino-2-phenylindole (DAPI). The number of TUNEL positive cells was analyzed with a fluorescence microplate reader.
Determination of Caspase-3 Activity
The activity of caspase-3 was measured using Caspase 3 Activity Apoptosis Assay Kit (BBI life sciences corporation, Shanghai, China), under the guidance of the manual. After treatment, HT22 cells were harvested and treated with caspase-3 detection buffer at room temperature for 1 h away from the light. After centrifugation, the sample was tested for fluorescence intensity using a microplate reader.
Enzyme-Linked Immunoassay (ELISA) Assay
The levels of TNF-α, IL-6, IL-1β, and iNOS in HT22 cell supernatants were examined according to the manufacturer’s instructions for TNF-α, IL-6, IL-1β, and iNOS ELISA kits (BBI life sciences corporation).
Statistical Analysis
All the data were represented as the mean ± standard deviation, and statistical analyses were done using SPSS 20.0 software. Comparisons between groups were analyzed using student’s t-test or one-way analysis of variance, and results were thought to be significantly different when a P-value<0.05 was obtained.
Results
SERPINE1 is Upregulated in Hemin-Treated HT22 Cells
First, we established an in vitro model of ICH by treatment of HT22 cells with hemin. As shown in Figure 1A, hemin administration reduced the viability of HT22 cells in a dose-dependent manner. Thus, a dose of 100 μM of hemin was chosen to develop an in vitro model of ICH. As determined by qRT-PCR assay, hemin treatment markedly increased the expression of SERPINE1 (Figure 1B). Consistent with this, the elevation of SERPINE1 protein expression was also discovered in HT22 cells stimulated with hemin, compared with the control group (Figure 1C).
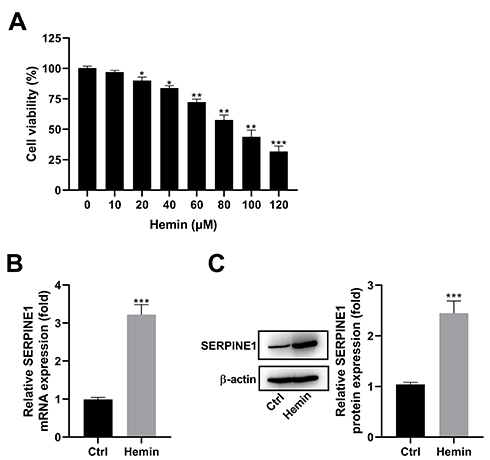 |
Figure 1 SERPINE1 is upregulated in hemin-treated HT22 cells. (A) HT22 cells were treated with different doses (0, 10, 20, 40, 60, 80, 100, and 120 μM) of hemin for 24 h and then subjected to MTT assay. (B) HT22 cells were treated with 100 μM of hemin for 24 h and then assayed for SERPINE1 expression using qRT-PCR assay. (C) Representative image of Western blot analysis and quantification of SERPINE1 expression in HT22 cells stimulated with hemin. *p < 0.05, **p <0.01, and ***p <0.001. |
Silencing of SERPINE1 Attenuates Hemin-Induced Neuronal Apoptosis
To evaluate the contribution of SERPINE1 to the development of ICH, we knockdown SERPINE1 in HT22 cells using si-SERPINE1. As shown by qRT-PCR and Western blot assay, the downregulation of SERPINE1 was noted in HT22 cells transfected with si-SERPINE1 relative to the si-NC group (Figure 2A and B). An MTT assay indicated that hemin administration dramatically decreased the viability of HT22 cells, which was blocked following si-SERPINE1 transfection (Figure 2C). In parallel, treatment of HT22 cells with hemin led to a remarkable increase in the number of TUNEL positive cells, but this increase was conversed by SERPINE1 knockdown (Figure 2D). Furthermore, we found that the activity of caspase-3 was increased in HT22 cells treated with hemin compared with the control group, however, silencing of SERPINE1 could block hemin-induced elevation of caspase-3 activity (Figure 2E). Similarly, Bax was upregulated, while Bcl-2 was downregulated in HT22 cells treated with hemin in comparison to the control group. And these changes caused by hemin administration were prominently abrogated following si-SERPINE1 transfection (Figure 2F).
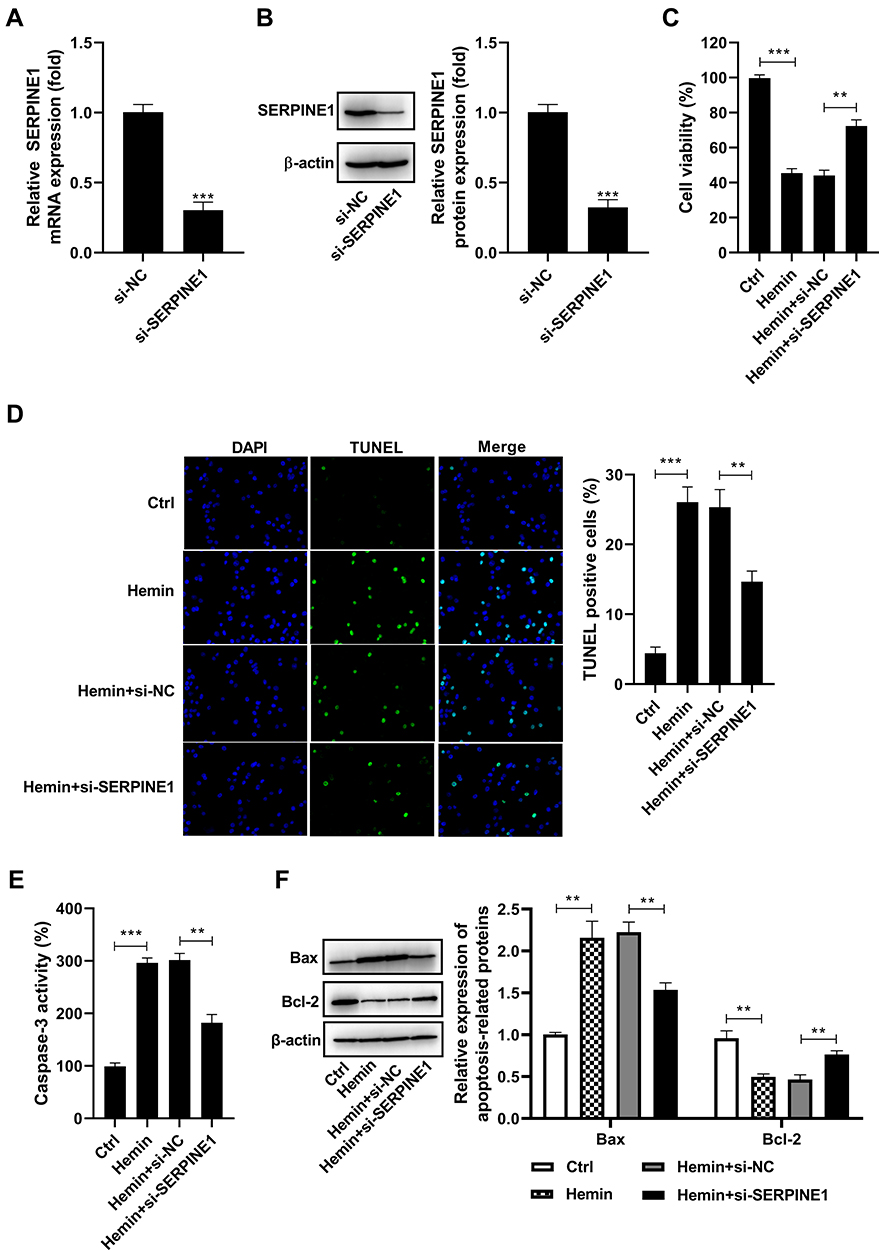 |
Figure 2 Silencing of SERPINE1 attenuates hemin-induced neuronal apoptosis. (A and B) HT22 cells were transfected with si-NC or si-SERPINE1 and the silencing efficiency was identified by qRT-PCR assay and Western blot. HT22 cells were transfected with si-NC or si-SERPINE1, followed by treatment with 100 μM hemin. (C) MTT assay was conducted to measure the viability of HT22 cells. (D) The apoptosis of HT22 cells was examined using TUNEL assay. (E) The activity of caspase-3 was measured using a commercial kit. (F) Western blot analysis of Bax and Bcl-2 expression in HT22 cells. **p <0.01 and ***p <0.001. |
Silencing of SERPINE1 Attenuates Hemin-Induced Neuroinflammation
To study the impact of SERPINE1 on hemin-induced neuroinflammation, HT22 cells were treated with si-NC or si-SERPINE1 and 100 μM hemin, and then the levels of IL-6, TNF-α, iNOS and IL-1β were detected using ELISA assay and qRT-PCR assay. As a result, administration of hemin resulted in a marked elevation of IL-6, TNF-α, iNOS, and IL-1β levels in the supernatant of HT22 cells, as determined by ELISA assay. However, silencing of SERPINE1 counteracted hemin-induced elevations of IL-6, TNF-α, iNOS, and IL-1β levels (Figure 3A–D). Consistently, qRT-PCR assay showed that knockdown of SERPINE1 abolished hemin-induced elevations of IL-6, TNF-α, iNOS, and IL-1β mRNA expression in HT22 cells (Figure 3E–H).
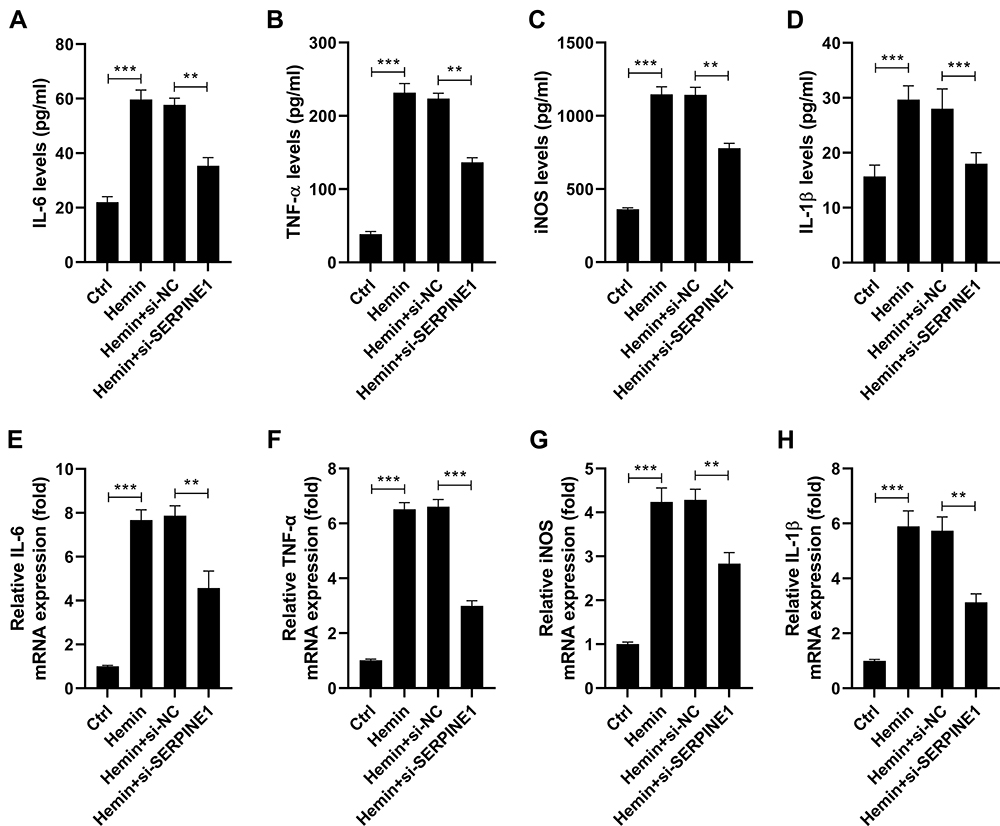 |
Figure 3 Silencing of SERPINE1 attenuates hemin-induced neuroinflammation. HT22 cells were transfected with si-NC or si-SERPINE1, followed by treatment with 100 μM hemin. ELISA assay was performed to detect the levels of IL-6 (A), TNF-α (B), iNOS (C), and IL-1β (D) in the supernatant of HT22 cells. qRT-PCR analysis of IL-6 (E), TNF-α (F), iNOS (G), and IL-1β (H) mRNA levels in HT22 cells. **p <0.01 and ***p <0.001. |
Upregulation of SERPINE1 Expression is Dependent on the Activation of TGF-β1
TGF-β1 mRNA and protein expression were upregulated in HT22 cells exposed to hemin (Figure 4A and B). It was reported that TGF-β1 participates in the regulation of SERPINE1.14 To study whether TGF-β1 takes part in the regulation of SERPINE1 in HT22 cells, HT22 cells were stimulated with different doses (0, 2, 5, and 10 ng/mL) of recombinant TGF-β1. As determined by qRT-PCR and Western blot assay, TGF-β1 increased the expression of SERPINE1 in a dose-dependent manner (Figure 4C and D). In addition, we also discovered that the expression levels of SERPINE1 mRNA and protein were significantly decreased in cells in which TGF-β1 levels were reduced by siRNA (Figure 4E and F).
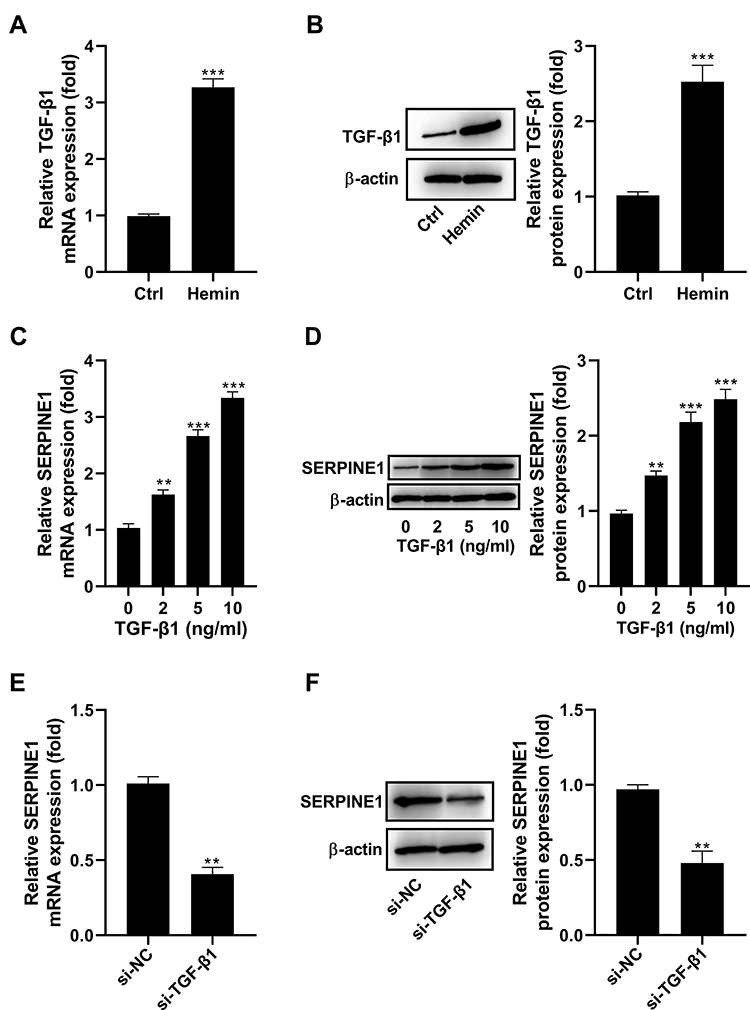 |
Figure 4 Regulatory effect of TGF-β1 on the expression of SERPINE1 in HT22 cells. (A and B) TGF-β1 mRNA and protein levels were increased in HT22 cells in the presence of hemin. (C and D) HT22 cells were stimulated with different doses (0, 2, 5, and 10 ng/mL) of recombinant TGF-β1, and then assayed for SERPINE1 expression using qRT-PCR assay and Western blot. (E and F) HT22 cells were transfected with TGF-β1 siRNA, and then assayed for SERPINE1 expression using qRT-PCR assay and Western blot. **p <0.01 and ***p <0.001. |
SERPINE1 Knockdown Attenuates the Effect of TGF-β1 on Hemin-Induced Neuronal Damage
To further confirm whether TGF-β1 controls the effect of SERPINE1 knockdown on hemin-induced neuronal damage, we transfected HT22 cells with si-SERPINE1 or si-NC and then treated them with hemin or recombinant TGF-β1. As a result, TGF-β1 treatment enhanced hemin-mediated inhibition of cell viability, and this action was blocked by silencing of SERPINE1 (Figure 5A). Meanwhile, TGF-β1 promoted the apoptosis of HT22 cells in the presence of hemin, which was abrogated following si-SERPINE1 transfection (Figure 5B). In line with this, hemin-induced elevation of caspase-3 activity was enhanced by TGF-β1 administration. However, the effect of TGF-β1 on hemin-induced elevation of caspase-3 activity was blocked by downregulation of SERPINE1 (Figure 5C). Additionally, TGF-β1 increased the expression of Bax and reduced the expression of Bcl-2 in hemin-treated HT22 cells, which were counteracted after si-SERPINE1 transfection (Figure 5D). Besides, TGF-β1 augments hemin-induced inflammation in HT22 cells, as evidenced by the increased expression of IL-6, TNF-α, iNOS, and IL-1β. Notably, downregulation of SERPINE1 attenuated the promotion effect of TGF-β1 on hemin-induced inflammation (Figure 6).
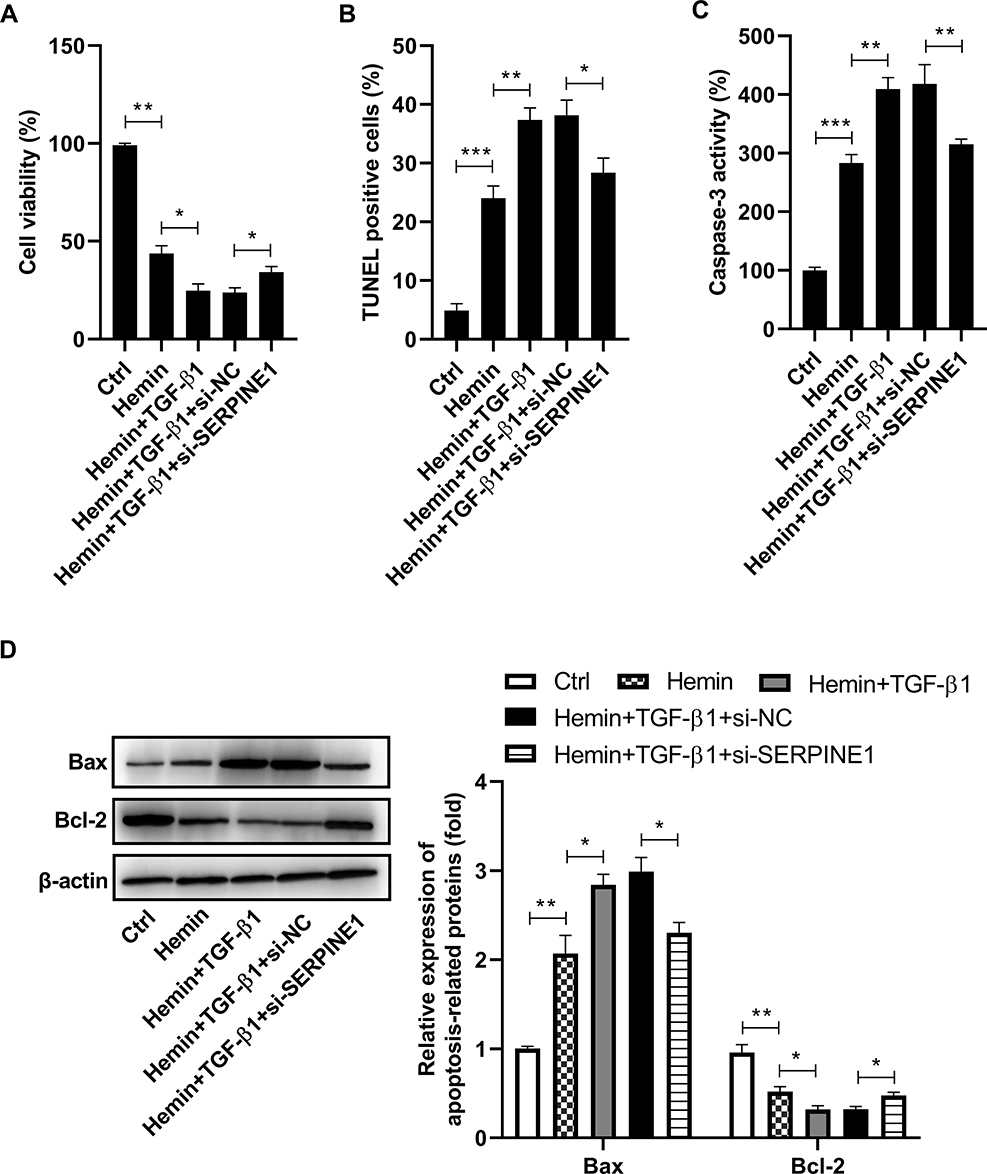 |
Figure 5 SERPINE1 knockdown attenuates the effect of TGF-β1 on hemin-induced neuronal damage. HT22 cells were transfected with si-SERPINE1 or si-NC, and then treated them with hemin or recombinant TGF-β1. (A) Cell viability was assessed using MTT assay. (B) The apoptosis of HT22 cells was examined using TUNEL assay. (C) The activity of caspase-3 was measured using a commercial kit. (D) Western blot analysis of Bax and Bcl-2 expression in HT22 cells. *p < 0.05, **p <0.01, and ***p <0.001. |
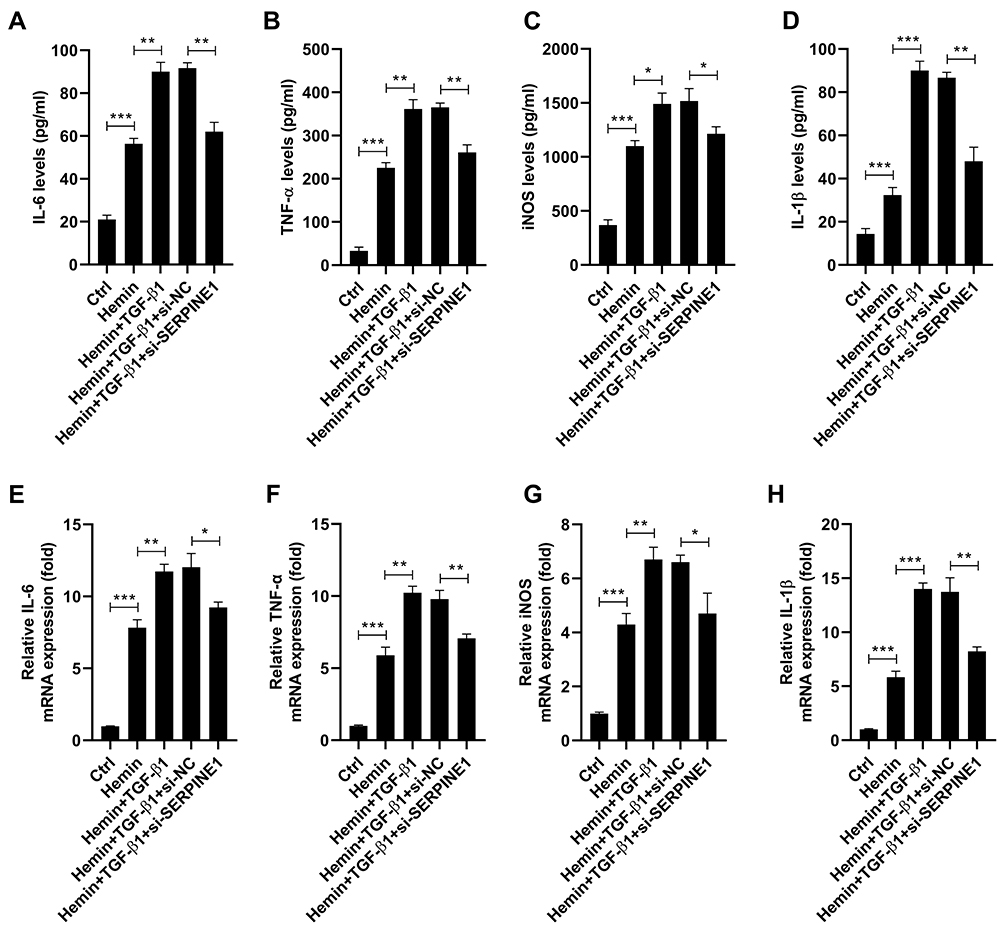 |
Figure 6 Downregulation of SERPINE1 attenuates the promotion effect of TGF-β1 on hemin-induced inflammation. HT22 cells were transfected with si-SERPINE1or si-NC, and then treated with hemin or recombinant TGF-β1. (A–D) ELISA assay was conducted to evaluate the levels of IL-6, TNF-α, iNOS, and IL-1β in the supernatant of HT22 cells. (E–H) qRT-PCR assay was conducted to detect mRNA levels in HT22 cells. *p < 0.05, **p <0.01, and ***p <0.001. |
Discussion
SERPINE1 is widely recognized as an inhibitor of fibrinolysis by inhibiting the activation of intrinsic plasminogen.12 Increased levels of SERPINE1 have been demonstrated to be tightly related to the prognosis of patients with stroke.15 Recently, Hendrix et al suggested that SERPINE1 gene polymorphisms were closely related to the functional outcome of patients with aneurysmal subarachnoid hemorrhage, indicating the participation of SERPINE1 in aneurysmal subarachnoid hemorrhage.16 Similarly, Lin et al found that G5GGGT homozygotes for SERPINE1 gene increased the risk of delayed cerebral ischemia and clinical vasospasm in patients with aneurysmal subarachnoid hemorrhage, which further supports the important regulatory role of SERPINE1 in the progression of aneurysmal subarachnoid hemorrhage.17 To date, although the functional role of SERPINE1 in ICH progression was not explicitly studied, a putative role in the development of ICH is supported by an increased expression of SERPINE1 in the perihematomal brain tissues of ICH patients, and the brain tissues of ICH rats. Herein, the upregulation of SERPINE1 was found in hemin-stimulated HT22 cells, and silencing of SERPINE1 could restrain hemin-mediated inhibition of cell viability in HT22 cells, indicating the participation of SERPINE1 in the progression of ICH.
The pathomechanism of ICH is complicated, in which neuronal apoptosis serves as a major player.18 Neuronal apoptosis is considered as a principal event that depends on the dynamic modulation of apoptosis-related genes during ICH. Bax is a pro-apoptosis factor that can induce the release of cytochrome c from the mitochondria to the cytoplasm, conversely, Bcl-2 is an anti-apoptosis protein that restrains the release of cytochrome c. The release of cytochrome c, in turn, triggers the activation of caspase cascade, resulting in cell apoptosis. Caspase-3 is regarded as a key executioner of cell apoptosis. It follows that reducing neuronal apoptosis is a vital step toward the development of effective treatment for ICH. It is worth noting that SERPINE1 serves as a regulatory player in the progression of neuronal apoptosis.19 PAI-039 treatment restrained neuronal apoptosis and attenuated the brain damage improved the neurofunctional outcome in the mouse controlled cortical impact model of traumatic brain injury.20 Moreover, middle cerebral artery occlusion mice with SERPINE1 deficiency exerted a reduced infarct volume, suggesting that SERPINE1 plays a crucial role in acute ischemic stroke-induced brain damage.21 However, the impact of SERPINE1 on hemin-induced neuronal apoptosis is still obscure. In this work, we found that knockdown of SERPINE1 could inhibit hemin-induced neuronal apoptosis, as evidenced by the decrease in the number of TUNEL positive cells, caspase-3 activity, and Bax expression, and the increase in Bcl-2 expression. This finding reveals that SERPINE1 may be a therapeutic target for ICH.
Apart from neuronal apoptosis, neuronal inflammation is also recognized as a crucial contributor to ICH-induced brain damage.22 Various stimuli produced by ICH can induce an inflammatory response, including the activation of microglia and the release of pro-inflammatory cytokines and chemokines.23 Among them, the levels of pro-inflammatory factors, IL-1β, TNF-α, IL-6 and iNOS, are widely used to reflect the severity of inflammatory response. There is a growing body of evidence that SERPINE1 plays a vital role in the respect of neuronal inflammation. As an example, SERPINE1 antagonist TM5484 reduced the expression of TNF-α, IL-6, chemokine Ligand-2 and Cd68, and increased the expression of IL-10 in the spinal cord and spleen of mice.24 In addition, SERPINE1 reportedly promoted leukocyte infiltration, and tissue damage following ischemia-reperfusion injury, revealing a role of SERPINE1 as an inflammatory mediator.25 In the present research, we found that knockdown of SERPINE1 repressed hemin-induced inflammation in HT22 cells, as indicated by reduced levels of TNF-α, IL-1β, IL-6 and iNOS, indicating the regulatory role of SERPINE1 in hemin-induced inflammation.
TGF-β1 is a member of the TGF-β superfamily that participates in the regulation of cellular growth fibrosis and differentiation.26 Several studies provide evidence that TGF-β1 act as a vital player in the progression of ICH. The polymorphism of TGF-β1 is markedly correlated with the risk of ICH in north Indian population.27 Furthermore, it was reported that TGF-β1 partly mediated SR144528 (cannabinoid receptor 2 antagonist)-induced inhibition of the fibrosis of the ventricular system and reduction of hydrocephalus in the rat model of intraventricular hemorrhage.28 Besides, inhibition of TGF-β mitigated the brain damage and improved neurological deficits following germinal matrix hemorrhage.29 Activation of TGF-β1 exacerbated inflammation and apoptosis in in vitro and in vivo models of mild traumatic brain injury.30 More importantly, SERPINE1 is a long-established downstream molecular of TGF-β1.31 However, whether TGF-β1 takes part in the impact of SERPINE1 silencing on hemin-induced neuronal apoptosis and inflammation remains in doubt. In our study, TGF-β1 administration dose-dependently increased the expression of SERPINE1 in HT22 cells. Notably, SERPINE1 knockdown attenuated the effect of TGF-β1 on hemin-induced neuronal damage, supporting the notion that TGF-β1-induced SERPINE1 activation is implicated in hemin-induced neuronal apoptosis and inflammation in HT22 cells.
In summary, our work suggested that TGF-β1-induced SERPINE1 activation contributed to hemin-induced neuronal apoptosis and inflammation, providing novel evidence for the linkage between TGF-β1-induced SERPINE1 activation and ICH progression. Further exploration in the functional role of SERPINE1 in ICH is needed to elucidate its possible mechanism. Our data manifested that targeting SERPINE1 might be a potential strategy for ICH therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart disease and stroke statistics-2017 update: a report from the American heart association. Circulation. 2017;135(10):e146–e603. doi:10.1161/CIR.0000000000000485
2. Morotti A, Goldstein JN. Diagnosis and management of acute intracerebral hemorrhage. Emerg Med Clin North Am. 2016;34(4):883–899. doi:10.1016/j.emc.2016.06.010
3. Sansing LH. Intracerebral Hemorrhage. Semin Neurol. 2016;36(3):223–224. doi:10.1055/s-0036-1583296
4. Sembill JA, Huttner HB, Kuramatsu JB. Impact of recent studies for the treatment of intracerebral hemorrhage. Curr Neurol Neurosci Rep. 2018;18(10):71. doi:10.1007/s11910-018-0872-0
5. Mittal MK, LacKamp A. Intracerebral hemorrhage: perihemorrhagic edema and secondary hematoma expansion: from bench work to ongoing controversies. Front Neurol. 2016;7:210. doi:10.3389/fneur.2016.00210
6. Chu X, Wu X, Feng H, et al. Coupling between interleukin-1R1 and necrosome complex involves in hemin-induced neuronal necroptosis after intracranial hemorrhage. Stroke. 2018;49(10):2473–2482. doi:10.1161/STROKEAHA.117.019253
7. Gram M, Sveinsdottir S, Ruscher K, et al. Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J Neuroinflammation. 2013;10:100. doi:10.1186/1742-2094-10-100
8. Babu R, Bagley JH, Di C, Friedman AH, Adamson C. Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurg Focus. 2012;32(4):E8. doi:10.3171/2012.1.FOCUS11366
9. Tan Y, Tan SW, Fan BY, Li L, Zhou YG. Hemin induces the activation of NLRP3 inflammasome in N9 microglial cells. Iran J Immunol. 2018;15(2):122–132.
10. Chen-Roetling J, Regan RF. Targeting the Nrf2-heme oxygenase-1 axis after intracerebral hemorrhage. Curr Pharm Des. 2017;23(15):2226–2237. doi:10.2174/1381612822666161027150616
11. Shao Z, Tu S, Shao A. Pathophysiological mechanisms and potential therapeutic targets in intracerebral hemorrhage. Front Pharmacol. 2019;10:1079. doi:10.3389/fphar.2019.01079
12. Flevaris P, Vaughan D. The role of plasminogen activator inhibitor type-1 in fibrosis. Semin Thromb Hemost. 2017;43(2):169–177. doi:10.1055/s-0036-1586228
13. Liu Z, Zhang R, Chen X, et al. Identification of hub genes and small-molecule compounds related to intracerebral hemorrhage with bioinformatics analysis. PeerJ. 2019;7:e7782. doi:10.7717/peerj.7782
14. Rawson R, Yang T, Newbury RO, et al. TGF-β1-induced PAI-1 contributes to a profibrotic network in patients with eosinophilic esophagitis. J Allergy Clin Immunol. 2016;138(3):791–800.e794. doi:10.1016/j.jaci.2016.02.028
15. Hu X, Zan X, Xie Z, et al. Association between plasminogen activator inhibitor-1 genetic polymorphisms and stroke susceptibility. Mol Neurobiol. 2017;54(1):328–341. doi:10.1007/s12035-015-9549-8
16. Hendrix P, Foreman PM, Harrigan MR, et al. Association of plasminogen activator inhibitor 1 (SERPINE1) polymorphisms and aneurysmal subarachnoid hemorrhage. World Neurosurg. 2017;105:672–677. doi:10.1016/j.wneu.2017.05.175
17. Lin M, Griessenauer CJ, Starke RM, et al. Haplotype analysis of SERPINE1 gene: risk for aneurysmal subarachnoid hemorrhage and clinical outcomes. Mol Genet Genomic Med. 2019;7(8):e737. doi:10.1002/mgg3.737
18. Shi E, Shi K, Qiu S, Sheth KN, Lawton MT, Ducruet AF. Chronic inflammation, cognitive impairment, and distal brain region alteration following intracerebral hemorrhage. FASEB J. 2019;33(8):9616–9626. doi:10.1096/fj.201900257R
19. Gerenu G, Martisova E, Ferrero H, et al. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer’s neuropathology and cognitive deficits. Biochim Biophys Acta Mol Basis Dis. 2017;1863(4):991–1001. doi:10.1016/j.bbadis.2017.01.023
20. Griemert EV, Schwarzmaier SM, Hummel R, et al. Plasminogen activator inhibitor-1 augments damage by impairing fibrinolysis after traumatic brain injury. Ann Neurol. 2019;85(5):667–680. doi:10.1002/ana.25458
21. Griemert EV, Recarte Pelz K, Engelhard K, Schafer MK, Thal SC. PAI-1 but not PAI-2 gene deficiency attenuates ischemic brain injury after experimental stroke. Transl Stroke Res. 2019;10(4):372–380. doi:10.1007/s12975-018-0644-9
22. Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25–44. doi:10.1016/j.pneurobio.2013.11.003
23. Zhang Z, Zhang Z, Lu H, Yang Q, Wu H, Wang J. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol Neurobiol. 2017;54(3):1874–1886. doi:10.1007/s12035-016-9785-6
24. Pelisch N, Dan T, Ichimura A, et al. Plasminogen activator inhibitor-1 antagonist TM5484 attenuates demyelination and axonal degeneration in a mice model of multiple sclerosis. PLoS One. 2015;10(4):e0124510. doi:10.1371/journal.pone.0124510
25. Praetner M, Zuchtriegel G, Holzer M, et al. Plasminogen activator inhibitor-1 promotes neutrophil infiltration and tissue injury on ischemia-reperfusion. Arterioscler Thromb Vasc Biol. 2018;38(4):829–842. doi:10.1161/ATVBAHA.117.309760
26. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–338. doi:10.1038/nrneph.2016.48
27. Kumar P, Kumar A, Misra S, et al. Association of transforming growth factor-beta1 gene C509T, G800A and T869C polymorphisms with intracerebral hemorrhage in North Indian population: a case-control study. Neurol Sci. 2016;37(3):353–359. doi:10.1007/s10072-015-2426-4
28. Tan Q, Chen Q, Feng Z, et al. Cannabinoid receptor 2 activation restricts fibrosis and alleviates hydrocephalus after intraventricular hemorrhage. Brain Res. 2017;1654(Pt A):24–33. doi:10.1016/j.brainres.2016.10.016
29. Manaenko A, Lekic T, Barnhart M, Hartman R, Zhang JH. Inhibition of transforming growth factor-beta attenuates brain injury and neurological deficits in a rat model of germinal matrix hemorrhage. Stroke. 2014;45(3):828–834. doi:10.1161/STROKEAHA.113.003754
30. Patel RK, Prasad N, Kuwar R, Haldar D, Abdul-Muneer PM. Transforming growth factor-beta 1 signaling regulates neuroinflammation and apoptosis in mild traumatic brain injury. Brain Behav Immun. 2017;64:244–258. doi:10.1016/j.bbi.2017.04.012
31. Wang X, Liu C, Wang J, Fan Y, Wang Z, Wang Y. Oxymatrine inhibits the migration of human colorectal carcinoma RKO cells via inhibition of PAI-1 and the TGF-beta1/Smad signaling pathway. Oncol Rep. 2017;37(2):747–753. doi:10.3892/or.2016.5292
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.