")
Back to Journals » The Application of Clinical Genetics » Volume 11
Tetrasomy 18p: case report and review of literature
Authors Bawazeer S, Alshalan M, Alkhaldi A, AlAtwi N, AlBalwi M, Alswaid A , Alfadhel M
Received 7 October 2017
Accepted for publication 27 November 2017
Published 8 February 2018 Volume 2018:11 Pages 9—14
DOI https://doi.org/10.2147/TACG.S153469
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Shahad Bawazeer,1 Maha Alshalan,2 Aziza Alkhaldi,3 Nasser AlAtwi,3 Mohammed AlBalwi,1,3,4 Abdulrahman Alswaid,2 Majid Alfadhel,1,2,4
1Developmental Medicine Department, King Abdullah International Medical Research Center, King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Saudi Arabia; 2Department of Pediatrics, Genetic Division, King Abdullah Specialized Children Hospital, King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Saudi Arabia; 3Department of Pathology and Laboratory Medicine, King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Saudi Arabia; 4King Saud Bin Abdulaziz University for Health Sciences, King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Saudi Arabia
Abstract: Tetrasomy 18p syndrome (Online Mendelian Inheritance in Man 614290) is a very rare chromosomal disorder that is caused by the presence of isochromosome 18p, which is a supernumerary marker composed of two copies of the p arm of chromosome 18. Most tetrasomy 18p cases are de novo cases; however, familial cases have also been reported. It is characterized mainly by developmental delays, cognitive impairment, hypotonia, typical dysmorphic features, and other anomalies. Herein, we report de novo tetrasomy 18p in a 9-month-old boy with dysmorphic features, microcephaly, growth delay, hypotonia, and cerebellar and renal malformations. We compared our case with previously reported ones in the literature. Clinicians should consider tetrasomy 18p in any individual with dysmorphic features and cardiac, skeletal, and renal abnormalities. To the best of our knowledge, we report for the first time an association of this syndrome with partial agenesis of cerebellar vermis.
Keywords: tetrasomy 18p, chromosomal, isochromosome, 18p, dysmorphic, CGH microarray, chromosome, syndrome
Background
Tetrasomy 18p syndrome (Online Mendelian Inheritance in Man 614290) is a very rare chromosomal disorder that is caused by the presence of isochromosome 18p, which is a supernumerary marker, composed of two copies of the p arm of chromosome 18.1 While most tetrasomy 18 cases are de novo, familial inheritance has also been described, mostly of maternal origin.2–4 Takeda et al4 described a phenotypically normal mother with two 18p tetrasomic daughters, one of which was a stillbirth. It was believed that the daughters inherited one normal chromosome 18 from the father and one normal chromosome 18 and one isochromosome 18 from the mother. Moreover, Abeliovich et al2 reported a case wherein the mother was a mosaic for isochromosome 18p(i18p) with mild manifestation of the syndrome, and her daughter, who had i18p in all her cells, showed full manifestation of the syndrome with a striking resemblance to her mother. Boyle et al3 also presented data on inheritance through maternal gonadal mosaicism in a case of two maternal half-sisters with isochromosome 18p. The mechanism of isochromosome 18p has been postulated to be linked to maternal meiosis II nondisjunction and centromeric misdivision or U-shaped exchange.5 Clinically, this condition is characterized mainly by developmental delays, microcephaly, abnormalities in muscle tone, and feeding problems, in addition to dysmorphic features.6 Diagnosis can be achieved through routine karyotyping or fluorescence in situ hybridization (FISH) by centromere-specific probes or comparative genomic hybridization microarray.6,7 Currently, there is no cure for this disorder and management is supportive, involving a multidisciplinary team approach, including detailed genetic counseling and evaluation; periodic ophthalmology evaluation; ear, nose, and throat referral for management of otitis media; cardiology evaluation with electrocardiography and echocardiography; renal ultrasound; orthopedic evaluation for management of foot abnormalities; kyphosis and scoliosis monitoring; neurological evaluation for seizures; gastroenterological evaluation for failure to thrive, constipation, and gastroesophageal reflux; endocrinological evaluation for short stature and growth hormone deficiency; physiotherapy; and occupational therapy.
Since its discovery a long time ago, the description of this syndrome at the phenotypic level has been scarce in the literature. In this report, we present de novo tetrasomy 18p in a male Saudi baby featuring cerebellar and renal malformations, in addition to the typical dysmorphic features. We reviewed the literature regarding the clinical features of tetrasomy 18p and compared it with our patient.
Case report
Clinical features
A 41-year-old woman – gravida 9, para 7, abortus 1 – was referred for genetic testing (chromosomal analysis and FISH), as ultrasound at the second- and third-trimester scans revealed multiple anomalies in the fetus, including dilated posterior fossa, absence of vermis, mild skin edema in the back of the skull, abnormal heart (regurgitation and thick-walled right ventricle), and bilateral hydronephrosis. The fetus position was cephalic. The proband was a product of full-term normal spontaneous vaginal delivery, diagnosed antenatally as trisomy 18, and admitted to the neonatal intensive care unit for 2 weeks for further investigation. The birth weight of the male infant was 3.2 g (25th–50th percentile), height, 46 cm (below 3rd percentile), and head circumference 33.5 cm (10th–25th percentile). He had the following dysmorphic features: low-set ears, intact high-arched palate, depressed nasal bridge, and micrognathia (Figure 1). He had feeding difficulties and neonatal jaundice, which resolved after a few days of life. The baby was discharged at 1 week of age in good condition. At the age of 9 months, the growth parameters were as follows: length 67.5 cm (below 3rd percentile), weight 5 kg (below 3rd percentile), and head circumference 42 cm (below 3rd percentile). He displayed dysmorphic features, including microcephaly, low-set posteriorly rotated ears, synophrys, pinched nose, long philtrum, strabismus, depressed nasal bridge, epicanthal fold, micrognathia, and low anterior hairline (Figure 1). The proband showed feeding problems and global developmental delay in the form of gross and fine-motor delay (he could not sit or support his head). Developmentally, he showed functions and characteristics of a 3-month-old baby. He had central hypotonia and kyphosis. At 3 years of age, he showed severe, expected developmental delay in terms of gross- and fine-motor skills, in that he could sit with support and roll over, but could not crawl or walk and had no speech. Developmentally, he showed functions and characteristics of a 6-month-old baby. He has the same dysmorphic, neurological, and skeletal features with all growth parameters below the 3rd percentile.
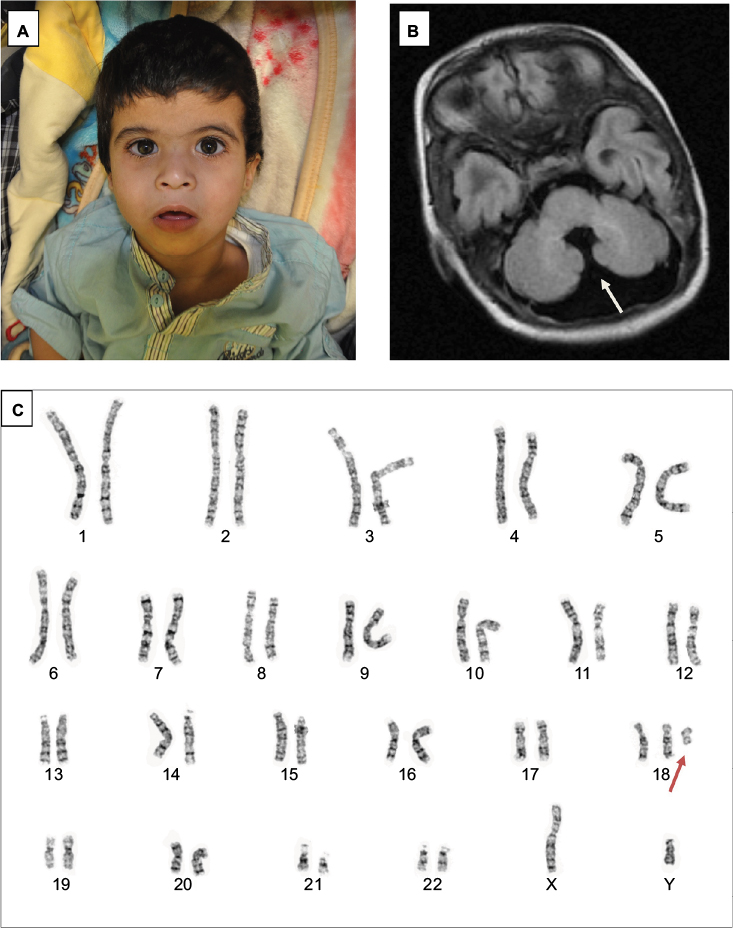 | Figure 1 (A) Dysmorphic features of the patient, including pinched nose, long philtrum, and micrognathia; (B) brain magnetic resonance imaging showing partial agenesis of cerebellar vermis; (C) G-banding chromosomal analysis showing isochromosome 18p (arrow). |
Echocardiography showed a large high-secundum atrial septal defect (6 mm) with left–right shunt and a small patent ductus arteriosus with left–right shunt. Brain magnetic resonance imaging (MRI) revealed partial agenesis of cerebellar vermis (Figure 1), and renal ultrasound showed bilateral hydroureter and hydronephrosis. Moreover, the patient also displayed bilateral vesicoureteral reflux and bilateral undescended testes.
Chromosomal and molecular analysis
Chromosomal analysis from cultured cordocentesis was done at 32 weeks of pregnancy and revealed 47,XY,+mar karyotype with an extra free marker chromosome in all examined cells (Figure 1). Further investigation using FISH signal patterns revealed the presence of three copies of chromosome 18 centromere in all examined cells with the possibility of isochromosome 18p (Figure 1). Parental chromosomal analysis was normal. Postnatal analysis of peripheral blood revealed a karyotype of 47,XY,+i(18)(p10). Further investigation by comparative genomic hybridization microarray confirmed the diagnosis of tetrasomy 18p, as it revealed amplification of at least 15 Mb extending from cytogenetic band 18p11.32 to 18p11.21 (Arr[hg19]18p11.32p11.21[163,323-15,276,360]x4).
Ethics approval and consent to participate
The study was approved by the research committee of King Abdullah International Medical Research Centre in Riyadh, Saudi Arabia. The parents of the patient provided written informed consent for publication of the case details and the accompanying images.
Discussion
A thorough review of the literature identified a range of characteristics for tetrasomy 18p cases (Table 1, Box 1). We compared our case with previously reported cases, and the constant clinical features of tetrasomy 18p that have been presented in all cases are developmental delays, cognitive impairments, and dysmorphic features. The typical dysmorphic features found in most patients are long philtrum, low-set ears, palatal abnormalities, clinodactyly, microcephaly, small mouth, and micrognathia.8 Our patient showed the same dysmorphic features, in addition to mild synophrys (Figure 1), which was not reported as a common facial feature for this syndrome.
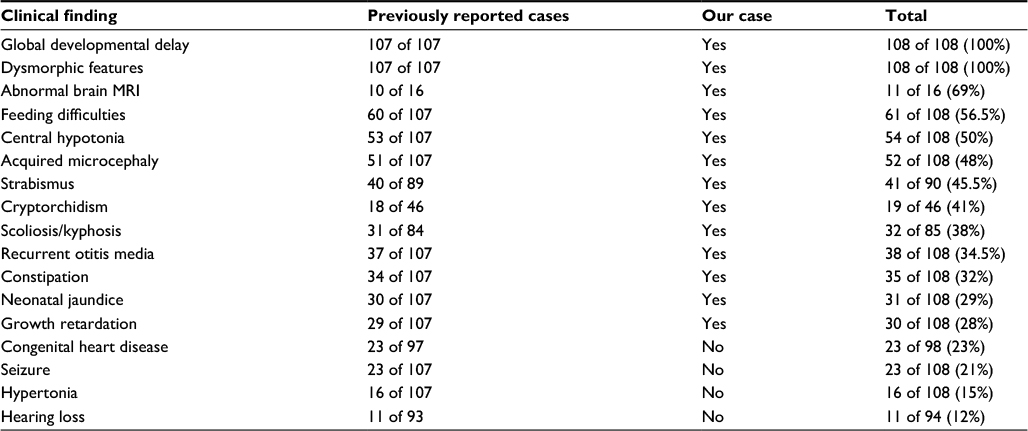 | Table 1 Summary of phenotypic characteristics of cases with tetrasomy 18p compared to our case Notes: Data from previous studies.2–5,7,8,10–35 See Figure 1 for more information on dysmorphic features. |
 | Box 1 Method of literature review |
Interestingly, although the neurological manifestations constitute the majority of the clinical features of tetrasomy 18p, brain MRI findings are not frequently reported. Only 16 of 108 patients underwent brain MRI. Approximately 70% of them showed abnormal findings. The reported abnormalities included thin corpus callosum, brain atrophy, and lateral ventricle enlargement.6 The current patient had partial agenesis of the cerebellar vermis, which has not been reported previously.
Cardiac manifestations are not uncommon in this disorder, and account for ~23% of all complications (Table 1). The patient presented had a large high-secundum atrial septal defect and small patent ductus arteriosus with left–right shunt. Other reported congenital heart diseases include ventricular septal defect, pulmonary stenosis, and valvular abnormalities.6,7,9,10 Additionally, the proband had bilateral vesicoureteral reflux and bilateral undescended testes. Genitourinary abnormalities account for 41% of all complications and include a small kidney, cryptorchidism, micropenis, and hypospadias.3,4,6 Other common features include microcephaly, strabismus, feeding difficulties, neonatal jaundice, hypotonia, kyphosis, scoliosis, recurrent otitis media, hearing loss, constipation, and growth retardation (Table 1). Abnormalities in laboratory parameters include thyroid abnormalities, growth hormone deficiency, and IgA deficiency.4,6
The implications for care of tetrasomy 18p should ideally be tailored to meet the specific needs of affected individuals. Children with this chromosomal abnormality commonly have feeding disorders and swallowing difficulties that in many cases place them at risk of aspiration with oral feeding, with potential pulmonary complications. They also commonly show failure to thrive, malnutrition, and prolonged stressful mealtimes. Therefore, regular and frequent dietitian follow-up and swallowing assessments are mandatory to prevent these children from the aforementioned complications. In several patients, a feeding tube like a gastrostomy tube would be an efficient solution for children with significant eating, drinking, and swallowing difficulties to ensure they receive enough calories. Additionally, hearing and vision monitoring is essential. Furthermore, frequent orthopedic, psychological, and neurological evaluation is mandatory for affected children.6 The prognosis of these patients is uncertain and insufficiently discussed in the literature. If the child is monitored appropriately through multidisciplinary care, there is no reason for premature mortality. However, several patients died, mainly due to cardiac complications or recurrent infections. The survivors had global developmental delays and behavioral abnormalities, including aggression, tendency to self-injure, and destructive behaviour.8,9
Finally, we emphasize the need for longitudinal data, as such information will provide a profile encompassing care recommendations and better inform clinical teams about proper management of tetrasomy 18p patients. In conclusion, we alert clinicians to consider tetrasomy 18p in any individual with dysmorphic features (microcephaly, low-set posteriorly rotated ears, pinched nose, long philtrum, strabismus, depressed nasal bridge, epicanthal fold, micrognathia, and low anterior hairline) and cardiac, skeletal, and renal abnormalities. Future research is needed in order to elucidate the long-term outcome of these patients.
Acknowledgments
We are grateful to the patient and his family for their genuine support.
Author contributions
Shahad Bawazeer and Majid Alfadhel performed the majority of work associated with preparing, writing, and submitting the manuscript, and contributed to the clinical diagnosis and management of the patient. Maha Alshalan summarized the clinical data. Aziza Alkhaldi, Nasser AlAtwi, and Mohammed AlBalwi performed and interpreted the chromosomal analysis, molecular analysis, and edited the manuscript. Abdulrahman Alswaid edited the manuscript and contributed to the clinical diagnosis and management of the patient. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Plaiasu V, Ochiana D, Motei G, Georgescu A. A rare chromosomal disorder: isochromosome 18p syndrome. Maedica (Buchar). 2011;6:132–136. | ||
Abeliovich D, Dagan J, Levy A, Steinberg A, Zlotogora J. Isochromosome 18p in a mother and her child. Am J Med Genet. 1993;46:392–393. | ||
Boyle J, Sangha K, Dill F, Robinson WP, Yong SL. Grandmaternal origin of an isochromosome 18p present in two maternal half-sisters. Am J Med Genet. 2001;101:65–69. | ||
Takeda K, Okamura T, Hasegawa T. Sibs with tetrasomy 18p born to a mother with trisomy 18p. J Med Genet. 1989;26:195–197. | ||
Kotzot D, Bundscherer G, Bernasconi F, et al. Isochromosome 18p results from maternal meiosis II nondisjunction. Eur J Hum Genet. 1996;4:168–174. | ||
Sebold C, Roeder E, Zimmerman M, et al. Tetrasomy 18p: report of the molecular and clinical findings of 43 individuals. Am J Med Genet A. 2010;152A:2164–2172. | ||
Blennow E, Nielsen KB. Molecular identification of a small supernumerary marker chromosome by in situ hybridization: diagnosis of an isochromosome 18p with probe L1.84. Clin Genet. 1991;39:429–433. | ||
Swingle HM, Ringdahl J, Mraz R, Patil S, Keppler-Noreuil K. Behavioral management of a long-term survivor with tetrasomy 18p. Am J Med Genet A. 2006;140:276–280. | ||
Park VM, Gustashaw KM, Bilenker RM, Golden WL. Diagnosis of tetrasomy 18p using in situ hybridization of a DNA probe to metaphase chromosomes. Am J Med Genet. 1991;41:180–183. | ||
Back E, Toder R, Voiculescu I, Wildberg A, Schempp W. De novo isochromosome 18p in two patients: cytogenetic diagnosis and confirmation by chromosome painting. Clin Genet. 1994;45:301–304. | ||
Bakshi S, Brahmbhatt M, Trivedi P, Chudoba I. Constitutional tetrasomy 18p. Indian Pediatr. 2006;43:357–360. | ||
Balíĉek P, Zižka J, Lichý J. An isochromosome of the short arms of the no. 18 chromosome in a mentally retarded girl. Clin Genet. 1976;9(2):192–196. | ||
Balkan M, Duran H, Budak T. Tetrasomy 18p in a male dysmorphic child in southeast Turkey. J Genet. 2009;88:337–340. | ||
Batista D, Vianna-Morgante A, Richieri-Costa A. Tetrasomy 18p: tentative delineation of a syndrome. J Med Genet. 1983;20:144–147. | ||
Blennow E, Nielsen KB, Telenius H, et al. Fifty probands with extra structurally abnormal chromosomes characterized by fluorescence in situ hybridization. Am J Med Genet. 1995;55:85–94. | ||
Bugge M, Blennow E, Friedrich U, et al. Tetrasomy 18p de novo: parental origin and different mechanisms of formation. Eur J Hum Genet. 1996;4:160–167. | ||
Callen D, Freemantle C, Ringenbergs M, et al. The isochromosome 18p syndrome: confirmation of cytogenetic diagnosis in nine cases by in situ hybridization. Am J Hum Genet. 1990;47:493–498. | ||
Côté G, Petmezaki S, Bastakis N. A gene for hypospadias in a child with presumed tetrasomy 18p. Am J Med Genet. 1979;4:141–146. | ||
Condron CJ, Cantwell R, Kaufman RL, Brown SB, Warren RJ. The supernumerary isochromosome 18 syndrome (+ 18pu). Birth Defects Orig Artic Ser. 1974;10:36–42. | ||
DeBerardinis R, Medne L, Spinner N, Zackai E. DiGeorge anomaly in a patient with isochromosome 18p born to a diabetic mother. Am J Med Genet A. 2005;138A:155–159. | ||
Eggermann T, Engels H, Apacik C, et al. Tetrasomy 18p caused by paternal meiotic nondisjunction. Eur J Hum Genet. 1997,5:175–177. | ||
Esmer CM, Frias S, Gomez L, Carnevale A. Tetrasomy 18p in two cases: confirmation by in situ hybridization. Ann Genet. 1994;37:156–159. | ||
Fryns JP, Kleczkowska A, Marien P, van den Berghe H. 18p Tetrasomy: further evidence for a distinctive clinical syndrome. Ann Genet. 1985;28:111–112. | ||
Kleckzkowska A, Fryns JP, Buttiens M, de Bisschop F, Emmery L, van den Berghe H. Trisomy (18q) and tetrasomy (18p) resulting from isochromosome formation. Clin Genet. 1986;30:503–508. | ||
Mewar R, Harrison W, Overhauser J. Confirmation of isochromosome 18p using whole chromosome arm-specific fluorescence in situ hybridization. Cytogen Cell Genet. 1993;64:1–4. | ||
Nielsen K, Dyggve H, Friedrich U, Hobolth N, Lyngbye T, Mikkelsen M. Small metacentric nonsatellited extra chromosome: report of five mentally retarded individuals and review of literature – contribution to further delineation of a new syndrome. Hum Genet. 1978;44:59–69. | ||
Ogata K, Iinuma K, Kammura K, Morinaga R, Kato J. A case report of a presumptive +i(18p) associated with serum IgA deficiency. Clin Genet. 1977;11:184–188. | ||
Park V, Gustashaw K, Bilenker R, Golden W. Diagnosis of tetrasomy 18p using in situ hybridization of a DNA probe to metaphase chromosomes. Am J Med Genet. 1991;41:180–183. | ||
Ramegowda S, Gawde H, Hyderi A, et al. De novo isochromosome 18p in a female dysmorphic child. J Appl Genet. 2006;47:397–401. | ||
Rauch A, Pfeiffer RA, Trautmann U, Liehr T, Rott HD, Ulmer R. A study of ten small supernumerary (marker) chromosomes identified by fluorescence in situ hybridization (FISH). Clin Genet. 1992;42:84–90. | ||
Rivera H, Möller M, Hernández A, Enríque-Guerra MA, Arreola R, Cantú JM. Tetrasomy 18p: a distinctive syndrome. Ann Genet. 1984;27:187–189. | ||
Rocchi M, Stormi M, Archidiacono N, Filippi G. Extra small metacentric chromosome identified as i(18p). J Med Genet. 1979;16:69–73. | ||
Singer TS, Kohn G, Yatziv S. Tetrasomy 18p in a child with trisomy 18 phenotype. Am J Med Genet. 1990;36:144–147. | ||
Tangheroni W, Cao A, Furbetta M. Multiple anomalies associated with an extra small metacentric chromosome: modified Giemsa stain results. Humangenetik. 1973;18:291–295. | ||
Taylor KM, Wolfinger HL, Brown MG, Chadwick DL. Origin of a small metacentric chromosome: familial and cytogenetic evidence. Clin Genet. 1975;8:364–369. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.