")
Back to Journals » Journal of Inflammation Research » Volume 8
Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives
Authors Ozaki E, Campbell M, Doyle S
Received 30 September 2014
Accepted for publication 14 November 2014
Published 16 January 2015 Volume 2015:8 Pages 15—27
DOI https://doi.org/10.2147/JIR.S51250
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Ema Ozaki,1 Matthew Campbell,1 Sarah L Doyle2,3
1Department of Genetics, 2Department of Clinical Medicine, School of Medicine, Trinity College Dublin, 3National Children's Research Centre, Our Lady's Children's Hospital Crumlin, Dublin, Ireland
Abstract: The inflammasome is a molecular platform formed by activation of an innate immune pattern recognition receptor seed, such as NLRP3. Once activated, NLRP3 recruits the adapter ASC (apoptosis-related speck-like protein containing a caspase recruitment domain), which in turn recruits procaspase-1. Procaspase-1 autocatalyzes its cleavage and activation, resulting in maturation of the precursor forms of interleukin (IL)-1β and IL-18 into active proinflammatory cytokines and initiation of pyroptotic cell death. The NLRP3 inflammasome has been implicated in the pathogenesis of a wide variety of diseases, including genetically inherited autoinflammatory conditions as well as chronic diseases in which NLRP3 is abnormally activated. The NLRP3 inflammasome has been linked to diseases such as Alzheimer's disease, atherosclerosis, metabolic syndrome, and age-related macular degeneration. In this review, we describe the NLRP3 inflammasome complex and its activation in disease, and detail the current therapies that modulate either the NLRP3 inflammasome complex itself or the two cytokines it is responsible for activating, ie, IL-1β and IL-18.
Keywords: NLRP3, interleukin-1, interleukin-18, caspase-1, therapeutics, inflammasome
Introduction
The inflammasome was first described in 2002 by Tschopp as a large molecular platform that triggers the activation of inflammatory caspases and processing of prointerleukin-1β.1 It is now known that these inflammasomes act as signaling platforms that can respond to a plethora of microbial products, as well as endogenous host products associated with cellular stress and damage, and that they play a vital role in innate immunity. These multiprotein complexes contain a pattern recognition receptor seed, typically a nucleotide-binding oligomerization domain-like receptor (NLR) or an absent in melanoma 2 (AIM2)-like receptor, which upon activation by pathogen-associated molecular patterns or damage-associated molecular patterns, oligomerize and recruit the adaptor protein ASC (apoptosis-related speck-like protein containing a caspase recruitment domain [CARD]) and the cysteine protease procaspase-1, leading to the autocatalysis and activation of caspase-1. Active caspase-1 ultimately cleaves the precursor proinflammatory cytokines pro-IL-1β and pro-IL-18 into their mature secreted forms. Activated caspase-1 also triggers a form of cell death known as pyroptosis.2
The NLRs are an evolutionarily conserved family of cytosolic receptors with a tripartite structure that share a common central nucleotide-binding and oligomerization (NACHT) domain that is usually flanked by a C-terminal leucine-rich repeat (LRR) domain and a N-terminal effector pyrin domain (PYD) or CARD. The LRR domain is important for ligand sensing and autoregulation, the NACHT domain is vital for forming oligomeric structures, while the N-terminal effector domain mediates downstream signal transduction.3 So far, 23 human and 34 mouse NLR genes have been identified in humans, and have phylogenetically been grouped into three subfamilies, ie, the nucleotide-binding oligomerization domain-containing proteins (NOD1-5 and CIITA), the NACHT, LRR, and PYD domain-containing proteins (NLRP1–14), and the IPAF family (IPAF/NLRC4 and NAIPs). The AIM2-like receptor family consists of four members in humans (IFI16, IFIX, MNDA, and AIM2) that contain an N-terminal PYD domain and one or two C-terminal hematopoietic interferon-inducible nuclear antigens with 200 amino acid repeat (HIN-200) DNA-binding domains.4
To date, five different inflammasomes have been clearly identified, ie, NLRP1, NLRP2, NLRP3, AIM2, and IPAF/NLRC4 (see Table 1).3,5 Each inflammasome is activated in response to different stimuli. The NLRP1 inflammasome is activated by directly binding to bacterial ligands, such as anthrax lethal toxin and muramyl dipeptide.6 The IPAF inflammasome senses Gram-negative bacteria possessing type III or IV secretion systems, such as Salmonella typhimurium and Shigella flexneri.7,8 The HIN-200 domain of AIM2 recognizes foreign cytoplasmic double-stranded DNA, and the AIM2 inflammasome, consisting of AIM2, ASC, and caspase-1, is activated in response to both viruses and bacteria.9 Other members of the NLR family, namely NLRP6, NLRP7 and NLRP12, have also been described to form inflammasomes with ASC leading to caspase-1 activation, although their specific ligands are still unknown.10,11 Herein we focus on the NLRP3 inflammasome as the best characterized inflammasome, which has been shown to have major implications in the development of chronic diseases.
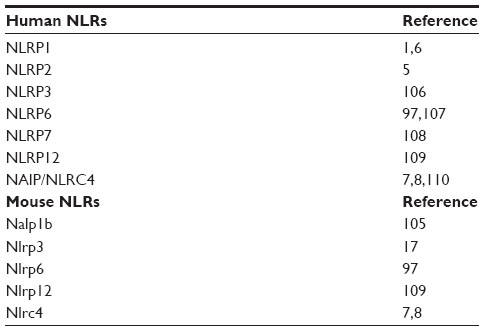 | Table 1 Inflammasome-forming NLRs in humans and mice |
NLRP3 inflammasome
The NLRP3 inflammasome is by far the most studied inflammasome. It recognizes a wide variety of microbes, including Staphylococcus aureus, Escherichia coli,12 Influenza A virus,13,14 and Candida albicans,15 as well as host-derived factors associated with damage, such as extracellular adenosine triphosphate (ATP),16 uric acid crystals,17 β-amyloid plaques,18 islet amyloid polypeptide,19 and drusen (Table 2).20 NLRP3 is expressed in the cytosol of monocytes, dendritic cells, neutrophils, lymphocytes, epithelial cells, and osteoblasts.21 The inflammasome platform is composed of NLRP3, ASC, and procaspase-1. The LRR domain of NLRP3 is likely to be involved in sensing the danger signal, which results in oligomerization of NLRP3 monomers via their NACHT domains. The PYD effector domain of NLRP3 then interacts with the PYD domain of ASC. ASC serves as an adaptor protein, recruiting procaspase-1 via its CARD domain. The NACHT domain of NLRP3 has also been shown to bind to and hydrolyze ATP. This ATPase activity of NLRP3 has been shown to be essential for NLRP3-mediated functions, and mutations in the NACHT domain reduce inflammasome oligomerization, caspase-1 activation, IL-1β and IL-18 secretion, and pyroptosis. Additionally, disruption of the NACHT domain in diseases associated with NLRP3 gain-of-function mutations greatly reduces the disease phenotype.22
 | Table 2 Sterile inflammatory NLRP3 activators and associated diseases |
Activation of the NLRP3 inflammasome is tightly regulated and requires two independent signals (Figure 1). Basal expression of the precursor proform of IL-1β as well as the NLRP3 protein is barely detectable, so a priming step or “signal 1” is required to drive their transcription.23 This is usually facilitated through activation of Toll-like receptors or NOD2 to initiate the nuclear factor kappa B (NF-κB) signaling pathway. Pattern recognition receptor signaling through Toll-like receptor-4 and MyD88 was also shown to rapidly prime free cytosolic NLRP3 maintained in an inactive ubiquitinated state, through deubiquitination of its LRR domain, which was dependent on generation of reactive oxygen species (ROS).24 A role for deubiquitination in ASC oligomerization has also been described.25 Once primed, subsequent activation of the NLRP3 inflammasome, referred to as “signal 2”, results in its oligomerization and inflammasome assembly, eventually leading to caspase-1-dependent cleavage and secretion of pro-IL-1β and pro-IL-18.
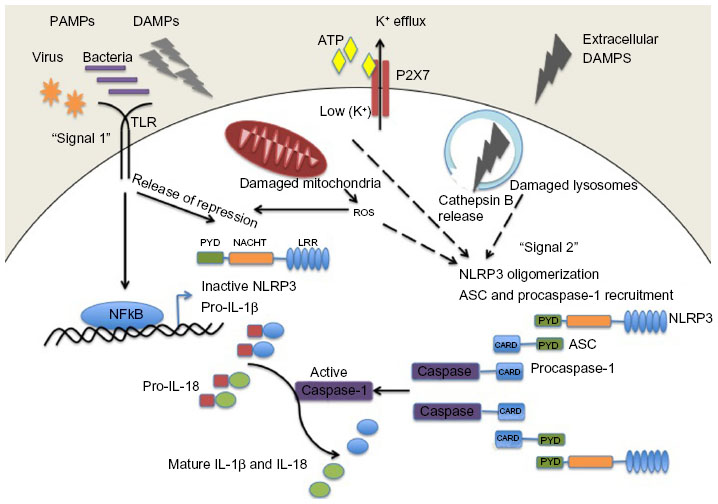 | Figure 1 NLRP3 inflammasome activation. Pathogenic PAMPs from virus or bacteria, or sterile DAMPs “prime” the inflammasome by activating a TLR inducing NFκB activation and the expression of NLRP3 and pro-IL-1β (signal 1). NLRP3 oligomerizes and recruits ASC and procaspase-1 in response to an activation signal (signal 2). NLRP3 can be activated in response to potassium ion efflux through the ATP-gated P2X7 channel, in response to reactive oxygen species released from damaged mitochondria, or in response to cathepsin B release from damaged lysosomes. Once activated the NLRP3 inflammasome causes the activation of caspase-1 which cleaves the precursor proforms of IL-1β and IL-18 into their mature forms. |
NLRP3 inflammasome activation models
The NLRP3 inflammasome is activated by a range of molecules that differ both chemically and structurally, and the exact mechanisms of NLRP3 activation remain unclear. It seems unlikely that NLRP3 binds directly with any of its agonists, and indeed no studies have yet shown a direct interaction between LRR and any NLRP3 activators. Three different models of NLRP3 activation have often been suggested, ie, potassium efflux, phagolysosomal destabilization, and mitochondrial ROS generation (Figure 1). The first model proposes that activation of P2X purinergic receptor 7 (P2X7) ATP-gated ion channels by extracellular ATP results in potassium ion efflux through the cell membrane. Supporting this model, it has been shown that potassium ion concentrations below 90 mM result in spontaneous NLRP3 inflammasome assembly, whereas this is prevented at higher potassium ion concentrations.26 ATP-induced activation of P2X7 receptors has also been linked to a hemichannel protein, pannexin-1. Pannexin-1 associates with P2X7 post-ATP stimulation and stimulates the formation of a large nonselective pore.27,28 These pores would allow NLRP3 agonists to gain access into the cytosol and may activate a host cellular factor that activates NLRP3. The second model proposes a mechanism by which large particulate matter, such as alum, silica, and amyloid-β, may activate the NLRP3 inflammasome. Engulfment of these particles by phagocytes and fusion with lysosomes leads to swelling and damage of the newly formed phagolysosome. As a result, lysosomal matter, such as the lysosomal protease cathepsin B, is released into the cytosol. Cathepsin B is thought to be sensed by NLRP3, leading to its activation.29 The third model predicts involvement of ROS, as many NLRP3 agonists have been shown to promote ROS formation. Production of ROS likely activates the inflammasome via an intermediate, and it has previously been demonstrated that an increase in ROS leads to activation of thioredoxin-interacting protein, which then binds to NLRP3, resulting in inflammasome activation.30 The source of ROS is believed to be the mitochondria,31 and mitochondrial DNA release by damaged mitochondria has also been shown to activate the NLRP3 inflammasome.32 However, a recent study has suggested that ROS are only required for priming the inflammasome and not for its activation.23
More recently, osmotic pressure and cell volume regulation have also been implicated in activation of NLRP3.33 In this study, macrophages subjected to hypotonic solutions underwent cellular swelling with a decrease in intracellular potassium and chloride ion concentrations. This led to a regulatory volume decrease response controlled by the transient receptor potential cation channels transient receptor potential (TRP) and transient receptor potential cation channel subfamily M member 7 (TRPM7) and the subsequent mobilization of intracellular calcium. Both low intracellular calcium concentrations and a regulatory volume decrease response controlled by TRP channels were shown to be required for activation of caspase-1. Ultimately, it is unlikely that the above mechanisms are independent, and integration of these models may explain how NLRP3 becomes activated by a diverse range of agonists.
NLRP3 inflammasome in disease
Inflammasome dysregulation has been implicated in almost every age-related condition that afflicts man. Age is one of the greatest “risk” factors for some of the most common chronic inflammatory diseases, eg, atherosclerosis, metabolic syndrome, age-related macular degeneration (AMD), Alzheimer’s disease, and gout. Indeed, the term “inflamm-aging” was coined by Franceschi et al to illustrate the concept that the aging process is associated with a progressive increase in proinflammatory status due to the body’s inability to manage the continuous antigenic load and stress.35 The inflammatory response observed during aging is different from the more traditional acute inflammation; it is a low-grade, controlled, chronic systemic inflammatory state and most often asymptomatic. The NLRP3 inflammasome has been shown to be triggered by many of the metabolic byproducts associated with these diseases, eg, monosodium urate crystals associated with gout,17 Beta-amyloid plaques associated with Alzheimer’s disease,18 drusen deposits associated with AMD,20 islet amyloid polypeptide involved in type 2 diabetes,19 and cholesterol crystals involved in atherosclerosis (Table 2).36
IL-1β is known to play a causative role in many of the NLRP3 inflammasome-related diseases. For example, in mice suffering from atherosclerosis, IL-1β plays a role in stimulating lipid plaque development and destabilizing plaques in mice, and cholesterol crystals which accumulate in this disease can activate the NLRP3 inflammasome causing increased IL-1β production. Moreover, when hypercholesterolemic mice that are deficient in the low-density lipoprotein receptor are reconstituted with bone marrow from mice deficient in various inflammasome components (NLRP3, ASC, IL-1β), fewer atherosclerotic plaques are seen to develop than in those reconstituted with wild-type bone marrow.37 Similarly, in patients suffering from gout, monosodium urate crystals have been shown to activate the NLRP3 inflammasome, and the resulting IL-1β released can trigger the recruitment of neutrophils and peritoneal inflammation. Interestingly, in a mouse model of wet AMD, production of IL-18 has been shown to have a protective role against progression to the end-stage neovascular form of the disease.20 IL-1 is an important mediator of cartilage destruction in rheumatic diseases, and evidence of NLRP3 inflammasome activation in rheumatoid arthritis (RA) exists due to the analysis of genetic variants in various population cohorts.38 However, until recently, the mechanism underlying the role of the NLRP3 inflammasome in the pathogenesis of RA was unknown. Myeloid cell-specific deletion of the RA susceptibility gene A20/Tnfaip3 in mice (A20[myel-KO] mice) triggers a spontaneous erosive polyarthritis that resembles RA in patients. Deletion of Nlrp3, caspase-1, and interleukin-1 receptor markedly protects against RA-associated inflammation and cartilage destruction in A20 (myel-KO) mice,39 indicating that increased NLRP3 inflammasome activation contributes to the pathology of RA in vivo, at least in mice.
Inflammatory bowel disease (IBD) has also been linked with uncontrolled inflammasome activation. A complex balance exists between the microbiota residing in the human gut and the systemic immune system of the host. Alterations in the composition of the microbiota, the gut epithelial barrier function, or inappropriate immune responses and genetic predispositions can lead to IBD or intestinal cancer. A possible role of the inflammasome in IBD arose from early studies demonstrating the association of variants in NOD2 (a pattern recognition receptor with structural similarities to NLRP3) with susceptibility to Crohn’s disease.40,41 Other NLR family members, as well as caspase-1, IL-1β, IL-18, and IL-18R, have been associated with IBD.42 However, there has been debate as to whether inflammasome activation, and in particular production of IL-18, is beneficial or harmful in IBD.43–45 Recent studies have demonstrated that the location of IL-18 activation may be crucial in this regard: IL-18 activation in intestinal epithelial cells may initiate a compensatory proliferative response and preservation of the intestinal barrier, whereas activation in the lamina propria may result in a harmful proinflammatory phenotype.46
The inflammasome has also been implicated in various cancers, including gastrointestinal cancers, melanoma, breast cancer, and hepatitis C virus-associated hepatocellular carcinoma, with both procarcinogenic and anticarcinogenic functions attributed.47 Anticarcinogenic functions originate from the ability of the inflammasome to promote pyroptotic cell death pathways. This enables maintenance of epithelial barrier function and wound repair processes as seen for IBD, and by stimulating anticancer immune responses during chemotherapy.48–50 Increased cytotoxicity of natural killer cells has been observed after IL-18 stimulation, which also contributes to the inflammasome’s antitumor effects.51–53 In contrast, the inflammasome and its products, IL-1β and IL-18, have also been seen to suppress natural killer-mediated and T-cell-mediated antimetastatic actions and immunosurveillance.54 Trophic factors of IL-1β have also been implicated, whereby IL-1β derived from myeloma was shown to induce production of IL-6 in stromal cells, which can act as a growth factor for these cells.55
The above conditions can loosely be grouped together and considered NLRP3 “activation” disorders. NLRP3 “genetic” disorders also exist and were previously known as periodic fever syndromes, but are now collectively referred to as cryopyrin-associated periodic syndromes (CAPS).56 CAPS have a distinct disease phenotype compared with the more chronic NLRP3 activation disorders and result in autoinflammatory diseases. CAPS disorders are due to gain-of-function mutations in the NLRP3 gene, which leads to increased secretion of IL-1β and associated manifestation of disease (Table 3). The clinical severity of CAPS varies, with familial cold autoinflammatory syndrome (FCAS) being the mildest form, neonatal-onset multisystem inflammatory disease, or chronic infantile neurological cutaneous and articular syndrome being of intermediate severity, and Muckle-Wells syndrome (MWS) being the most severe.57 Unlike autoimmune diseases, these autoinflammatory syndromes do not present typical characteristics of adaptive immunity, such as high-titer autoantibodies and antigen-specific T-cells.58 Instead, they are disorders of the innate immune system and are characterized by recurrent fever, urticaria-like rashes, and systemic inflammation.59
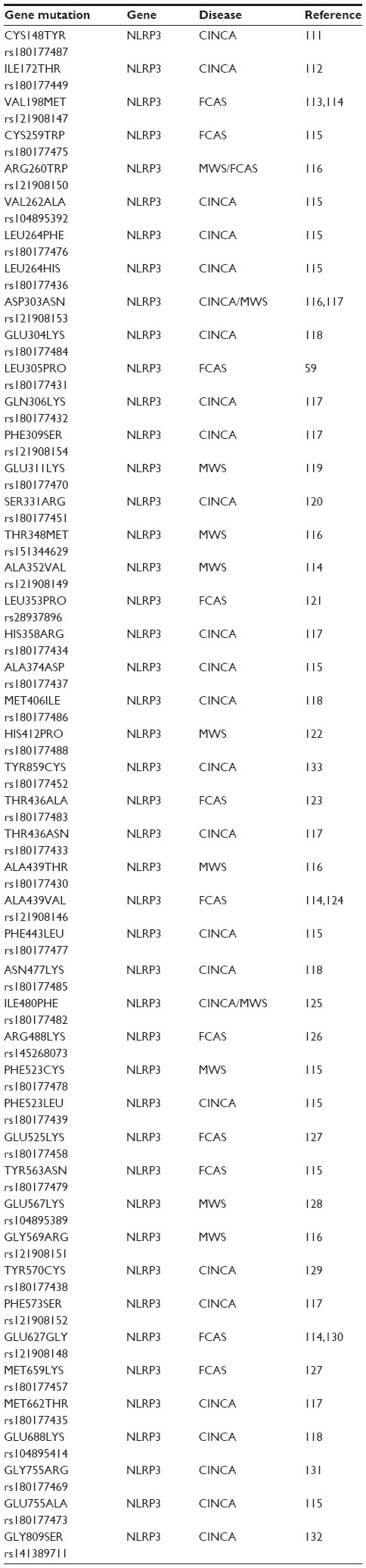 | Table 3 Some common NLRP3 mutations identified with associated diseases |
Remarkable clinical outcomes have been observed for these disorders by treatment with the IL-1-blocking agents, rilonacept (Arcalyst®; Regeneron Pharmaceuticals, Tarrytown, NY, USA), canakinumab (Ilaris®; Novartis, Basel, Switzerland), and anakinra (Kineret®; Swedish Orphan Biovitrum Ltd, Waltham, MA, USA).60–63 Given the evidence that NLRP3 is involved in a diverse range of diseases, there is considerable interest in the discovery of effective therapeutics that selectively inhibit the NLRP3 inflammasome pathway. We discuss the current therapies available and those under clinical trial and in the early stages of characterization in detail below.
Inhibiting effectors of the inflammasome
It is logical to conceive that inhibiting the final products of the inflammasome, ie, mature IL-1β and IL-18, could hold therapeutic potential. Indeed, both of these cytokines are potent immune modulators, so their expression at high levels in distinct cellular beds could have disastrous consequences if left uncontrolled. For example, prolonged expression of these cytokines in the brain calvarium could add to the malignancies observed in Alzheimer’s disease and other forms of dementia.64
Strategies for IL-1β blockade
Inhibition of IL-1β is currently a therapeutic strategy for multiple diseases. Antibodies directed against IL-1β have therapeutic potential for the treatment of MWS and FCAS, with multiple clinical trials having been undertaken in these patients using a fully humanized IL-1β antibody, canakinumab. Canakinumab is a human immunoglobulin Gκ monoclonal antibody that specifically targets IL-1β.65 It has been developed by Novartis as a treatment for a range of immune disorders, but predominant among these are FCAS and MWS. A fusion protein developed by Regeneron Pharmaceuticals has also been investigated in the treatment of CAPS. Their drug (rilonacept) comprises the extracellular portion of the IL-1 receptor and the Fc domain of human immunoglobulin G1; however, as rilonacept can bind to both IL-1α and IL-1β, it may have therapeutic potential over and above that of canakinumab, and its uses could extend to conditions such as diabetes and gout.66 In addition, the IL-1 receptor antagonist, anakinra, has found utility in numerous conditions, including RA. Anakinra is a recombinant human IL-1 receptor antagonist protein that is now a commonly used subcutaneously administered drug for the treatment of RA and has been shown to prevent the cartilage degeneration associated with this condition.67 A vaccine designed to inhibit IL-1β is also currently in development by Cytos Biopharmaceuticals (Durham, NC, USA) for the treatment of diabetes and RA;67 however, data pertaining to its safety/efficacy are not currently available. An indepth review of strategies for IL-1 blockade is discussed in Dinarello et al.68
Strategies for IL-18 blockade
With regard to inhibiting IL-18 directly, numerous clinical trials have been undertaken using an antibody directed against IL-18 (termed GSK1070806) which is a clinical asset of GlaxoSmithKline (Brentford, UK). This molecule has been used for conditions as diverse as B-cell non-Hodgkin’s lymphoma and IBD. Data pertaining to the safety/efficacy of using systemic IL-18 antibodies are currently limited to information obtained from http://www.clinicaltrials.gov, but it is prudent to consider that numerous trials have been listed as “completed”, which would suggest that systemic inhibition of IL-18 is at the very least tolerated in human subjects (http://www.clinicaltrials.gov).
Targeting inflammasome constituents
Currently, the best therapies for treating conditions associated with an overtly active inflammasome target the main pyogenic product of inflammasome activity, ie, IL-1β. However, alternative modes of inhibiting inflammasome activation are being actively pursued (Figure 2,Table 4). Manipulating the inflammasome therapeutically prior to activation would have the benefit of blocking the release of IL-1β and IL-18, rather than “sponging” them up, in conditions where these cytokines are deleterious. In some cases, it has been discovered that previously established pharmacological agents, in use for treatment of a variety of diseases, in fact target components of the inflammasome. The development of small-molecule inhibitors that directly target the building blocks of the inflammasome would benefit from being cheaper and less invasive modes of therapy compared with the larger biologics currently in use and aimed at sequestering IL-1β after it has been secreted.
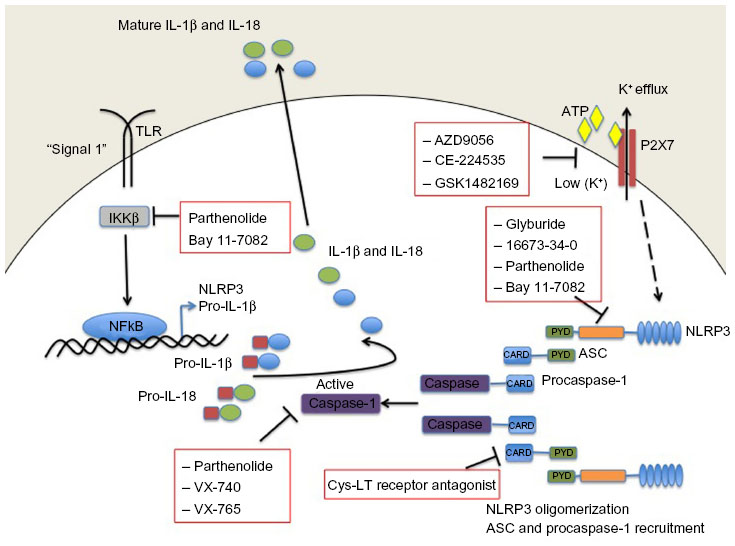 | Figure 2 Small-molecule blockade of the NLRP3 inflammasome. Cartoon depicting the mode of action of the various small molecule inhibitors described in detail in the text. |
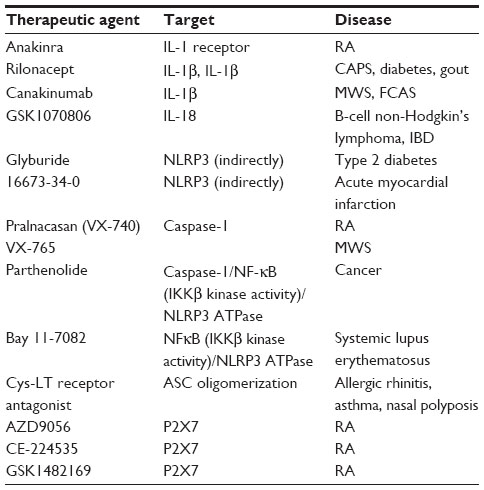 | Table 4 Therapeutic agents that inhibit inflammasome components and their targeted diseases |
Indirect NLRP3 inhibitors
Glyburide is a sulfonylurea drug commonly used in the treatment of type 2 diabetes. It acts by inhibiting ATP-sensitive potassium channels in pancreatic β-cells.69 However, in addition to inhibiting IL-1β in response to ATP, glyburide can also inhibit production of IL-1β in response to multiple other NLRP3 stimuli. In fact, the inhibitory activity of glyburide is not dependent on potassium channel blockade or on the ATPase activity of NLRP3 (required for caspase-1 activity);22 however, it does appear to be a specific inhibitor of NLRP3 because it does not block the production of IL-1β following NLRC4 or NLRP1 activation. Furthermore, although effective at inhibiting release of IL-1β upon activation of NLRP3 by pathogen-associated molecular patterns, damage-associated molecular patterns, and crystals, glyburide does not inhibit temperature-induced IL-1β release from monocytes of FCAS patients, suggesting that while it acts downstream of the P2X7 receptor, it acts upstream of NLRP3 activation.70 These properties suggest that glyburide may be a treatment option for conditions where excessive NLRP3 activation is considered a major contributor to pathology. However, while glyburide has clear NLRP3 inhibitory activity in vitro, it requires very high doses in vivo, which are associated with hypoglycemia, limiting its use outside of type 2 diabetes. A recent publication has described 16673-34-0, an intermediate substrate free of the cyclohexylurea moiety of glyburide, as an NLRP3 inhibitor effective in a model of acute myocardial infarction that does not affect glucose metabolism.71 It is likely that this intermediate in the glyburide synthesis pathway will prove useful as a tool to discover exactly how these sulfonyl compounds inhibit NLRP3 inflammasome activation. Further investigations will determine if it is a useful therapeutic in chronic inflammatory conditions.
Caspase-1 inhibitors
Parthenolide is a naturally occurring plant sesquiterpene lactone with multiple anti-inflammatory properties, and has been used extensively as a herbal remedy for a variety of inflammatory diseases with few and mild side effects.72 It was thought to be an NFκB inhibitor through selective inhibition of κB kinase β (IKKβ) kinase activity; however, in addition to its effects on NFκB activation, parthenolide has now been shown to inhibit activation of caspase-1 in response to NLRP3, NLRP1, and NLRC4 stimulation. The effect of parthenolide on the spectrum of inflammasomes implies that it acts on a common component and, accordingly, it has been shown that parthenolide is a direct inhibitor of caspase-1, causing alkylation of caspase-1 on a number of cysteine residues.73
Two newly developed caspase 1 inhibitors, ie, pralnacasan (VX-740) and VX-765 have also been described. Pralnacasan was effective in vitro and in several animal models, and has been tested in a clinical trial in RA; however, the outcome of the trial has not been reported. VX-765 has been administered to six patients with MWS, resulting in a 40%–70% decrease in markers of inflammation as well as a significant reduction in recurrent fevers and arthritis.68
NLRP3 ATPase inhibitors
Interestingly, in addition to its role in inhibiting caspase-1, parthenolide also directly inhibits NLRP3 by inhibiting its ATPase activity, which is required for activation of caspase-1. Another NF-κB inhibitor, Bay 11-7082, which, like parthenolide, inhibits NFκB by blocking IKKβ kinase activity, has also been shown to inhibit the ATPase activity of NLRP3, and it appears likely that the mechanism of inhibition, at the level of NLRP3 or upstream, involves NLRP3 alkylation of nucleophilic residues.73
ASC inhibitors
More recently, a cysteinyl leukotriene receptor antagonist developed by Bayer Pharmaceuticals (Bayer AG, Leverkusen, Germany) (Härter et al, US Patent 7,498,460, 200974) was described and found to inhibit both NLRP3 and AIM2 inflammasome induced IL-1β processing, by preventing ASC oligomerization. ASC oligomerization is essential for both NLRP3 and AIM2 inflammasome formation. In contrast, ASC appears to be dispensable for NLRC4 inflammasome formation as the CARD domain of NLRC4 can interact with caspase-1 directly. This cysteinyl leukotriene receptor antagonist does not inhibit IL-1β release following activation of the NLRC4 inflammasome. Interestingly, ASC appears to have further roles in innate immune responses separate from its role as an adaptor for inflammasome formation. Hence, this small-molecule inhibitor of ASC may hold therapeutic promise as a dual-purpose therapy in some inflammatory conditions.74
P2X7 antagonists
Due to the requirement for a reduction in potassium levels inside the cell prior to NLRP3-induced caspase-1 activation, regulating potassium levels presents a target for therapeutic intervention. This process is controlled by the ATP receptor P2X7 and therefore small-molecule inhibitors of P2X7 have been developed and tested in humans. AZD9056 (a P2X7 inhibitor) resulted in a significant clinical improvement in joint inflammation in patients with RA, but there was no concurrent reduction in C-reactive protein levels.75 In fact, other P2X7 inhibitors have likewise had disappointing outcomes in relation to RA trials; CE-224535, for example, was ineffective in a Phase IIa trial in RA (ClinicalTrials.gov identifier NCT00628095).76 Similarly, GSK1482169 disappointed in its inability to suppress IL-1β release (Phase I trial, ClinicalTrials.gov identifier NCT00849134).77 Overall, oral inhibitors of P2X7 do not appear to be effective in diminishing RA disease symptoms.78
Immunotherapies
There is no doubt that the use of drugs that target IL-1β in order to block its effects has been a real success story over the last decade in tackling both autoinflammatory and chronic inflammatory disease. However, there are some conditions in which IL-1β and/or IL-18 appear to play a protective role, and support the notion that, in some cases, the use of immunotherapy in the form of administration of recombinant cytokines may be therapeutically beneficial.
Therapeutic use of IL-1β
IL-1 is a multifunctional cytokine.79 Alongside its well known proinflammatory and fever-inducing properties, there is also an established protective effect of IL-1β in mice after irradiation or treatment with cytotoxic drugs, most likely due to the ability of IL-1β to induce levels of colony stimulating growth factors.80–83 Administration of IL-lβ to mice has also resulted in tumor regression by either nonspecific or tumor-specific immune-mediated mechanisms. Because of these properties, IL-1β has been administered to patients in an effort to improve bone marrow function84 and to treat a variety of solid tumors.85–87 While the benefits of IL-1β were inconsistent in the cancer trials, levels of peripheral white blood cells did increase significantly even with administration of low doses (0.027–0.1 μg/kg) of IL-1β.84,85 Despite the benefits associated with IL-1β therapy, at least for the improvement of bone marrow function, the acute toxicities observed in these patients were unacceptable and incompatible with use of IL-1β as a drug in practical terms. Patients experienced fever and significant hypotension, loss of appetite, generalized muscle and joint aches and pains, and headache, nausea, and fatigue, as well as gastrointestinal and sleep disturbances.85 In fact, the signs and symptoms exhibited post-IL-1 injection into humans are practically indistinguishable from those seen with low doses of endotoxin.
Therapeutic use of IL-18
IL-18 and IL-1β are cleaved in a similar manner, ie, by caspase-1, downstream of inflammasome activation. They also share common signaling pathways and are both considered to be proinflammatory cytokines. However, there are several important differences between IL-18 and IL-1β, which caution that assumptions made for one cannot be made for the other. One striking difference is in the expression pattern of IL-18 and IL-1β. Whereas IL-1β gene expression is absent basally in all cell types tested, pro-IL-18 is constitutively present in nearly all cells, including blood monocytes, keratinocytes, endothelial cells, and epithelial cells.88 The fact that IL-18 is constitutively expressed is consistent with a role for IL-18 in homeostasis. Furthermore, IL-18-deficient animals start to overeat at a young age, become obese, and exhibit symptoms reminiscent of the metabolic syndrome,89 further implicating IL-18 in the regulation of homeostasis. In contrast, IL-1-deficient animals are relatively normal under specific pathogen-free conditions. As discussed above, the use of IL-1β as a treatment is not practical due to its toxicity and induction of fever. Conversely, the induction of a fever is not a significant result of IL-18 treatment, so at least in that regard the use of IL-18 as an immune stimulant is feasible.90
In fact, IL-18 (SB-485232, GlaxoSmithKline) has been administered to humans for the treatment of various cancers in Phase I and II trials.91–93 IL-18 induces interferon-gamma together with either IL-12 or IL-15, promoting the differentiation of CD4+ T lymphocytes into Type 1 helper cells and inducing the generation of memory cytotoxic CD8+ T lymphocytes and natural killer cells, resulting in enhanced cellular immunity.94 The rationale for investigating the use of recombinant IL-18 in the treatment of cancer is to harness these immune stimulatory effects of IL-18, and to increase the activity, expansion, and tumor infiltration of the immune cells. The initial trials where IL-18 was evaluated as monotherapy in patients with advanced solid tumors served the purpose to indicate that IL-18 was safe and well tolerated up to a dose of 1,000 μg/kg and has immunomodulatory activity.95 A follow-up Phase I study has investigated the feasibility of use of IL-18 as an adjunct therapy with PEGylated liposomal doxorubicin and reported no positive drug interactions.92 Common drug-related adverse events were grade 1 or 2 chills and nausea. However, it remains to be seen whether IL-18 administration is in fact efficacious for the treatment of cancer.
Interestingly, one of the less common symptoms experienced by patients receiving IL-18 (with PEGylated liposomal doxorubicin) was mucosal inflammation. The role of IL-18 in inflammation of the bowel appears to be a complicated one. There are a number of studies that indicate a protective role for IL-18 in models of IBD.96,97 These studies indicate that mice deficient for NLRP396 or NLRP697,98 are more vulnerable to dextran sodium sulfate-induced colitis and that this is due to a lack of IL-18 not IL-1β. Similarly, mice deficient in caspase-1 are also more susceptible to dextran sodium sulfate-induced colitis, and administration of IL-18 initiates mucosal healing in these mice.99 On the other hand, blocking IL-18 with a neutralizing antibody or with IL-18 binding protein, a natural inhibitor of IL-18, reduces colitis in wild-type mice. The most likely explanation is that IL-18, which is constitutively expressed in intestinal epithelial cells, has a role in maintaining the intestinal barrier under homeostatic conditions. However, once the integrity of the barrier is lost, production of IL-18 by infiltrating macrophages causes inflammation.46 Therefore, despite the protective role of IL-18 in barrier maintenance, there appears to be a rationale for blocking IL-18 in Crohn’s disease and IBD, when the intestinal barrier is lost and chronic inflammation ensues.
AMD is the leading cause of central vision loss in industrialized nations. It is widely considered that the inflammatory response plays a role in the development of AMD, although the mechanisms involved remain unknown. A number of studies have indicated that the NLRP3 inflammasome is activated in AMD.20,100–102 Further studies, including our own, have indicated that IL-18 acts in an antiangiogenic capacity in the context of both AMD and diabetic retinopathy,20,103,104 suggesting that immunotherapy, in the form of recombinant IL-18, may provide some therapeutic protection in ocular conditions where neovascularization is a symptom. This hypothesis is currently being tested in preclinical trials.
Conclusion
The discoveries of IL-18 and IL-1β predate the discovery of the inflammasome by 1 and 2 decades, respectively. However, the time between the first description of the NLRP3 inflammasome and the first attempts to target this inflammasome therapeutically has been very short. There is no doubt that great leaps forward have been made in the treatment of genetically inherited autoinflammatory diseases through IL-1β blockade, which has brought untold relief to those who suffer with CAPS. It is clear that abnormal activation of the NLRP3 inflammasome is involved in many chronic inflammatory conditions, as detailed above. However, it remains to be seen whether targeting the NLRP3 inflammasome will provide therapeutic benefit in these more chronic disease states. At worst, it is possible that simply blocking IL-1β or IL-18 in these conditions may prove ineffective if the cytokines themselves are not causative of the pathology but rather resulting from it. In these cases, targeting the NLRP3 inflammasome itself, as early as possible in disease progression, may prove more beneficial, blocking not only cytokine release but also pyroptotic cell death and the ensuing tissue destruction; however, this would require reliable methods for early prediction of onset of disease. Still, even with this caveat in mind, identification of small-molecule inhibitors of the NLRP3 inflammasome offers considerable therapeutic promise, and continued research in this area is an important step in the process of developing effective medicines for the treatment of chronic diseases in which inflammation is a known component.
Disclosure
The authors report no conflicts of interest in this work.
References
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. | |
Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol. 2011;11(3):213–220. | |
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. | |
Cridland JA, Curley EZ, Wykes MN, et al. The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol Biol. 2012;12:140. | |
Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013;61(7):1113–1121. | |
Faustin B, Lartigue L, Bruey JM, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25(5):713–724. | |
Miao EA, Alpuche-Aranda CM, Dors M, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7(6):569–575. | |
Franchi L, Amer A, Body-Malapel M, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7(6):576–582. | |
Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–518. | |
Grenier JM, Wang L, Manji GA, et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett. 2002;530(1–3):73–78. | |
Wang L, Manji GA, Grenier JM, et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem. 2002;277(33):29874–29880. | |
Rathinam VA, Vanaja SK, Waggoner L, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150(3):606–619. | |
Allen IC, Scull MA, Moore CB, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30(4):556–565. | |
Thomas PG, Dash P, Aldridge JR Jr, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30(4):566–575. | |
Gross O, Poeck H, Bscheider M, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459(7245):433–436. | |
Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. | |
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. | |
Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9(8):857–865. | |
Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010;11(10):897–904. | |
Doyle SL, Campbell M, Ozaki E, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. 2012;18(5):791–798. | |
Kummer JA, Broekhuizen R, Everett H, et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. 2007;55(5):443–452. | |
Duncan JA, Bergstralh DT, Wang Y, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A. 2007;104(19):8041–8046. | |
Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187(2):613–617. | |
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287(43):36617–36622. | |
Lopez-Castejon G, Luheshi NM, Compan V, et al. Deubiquitinases regulate the activity of caspase-1 and interleukin-1beta secretion via assembly of the inflammasome. J Biol Chem. 2013;288(4):2721–2733. | |
Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14(9):1583–1589. | |
Kanneganti TD, Lamkanfi M, Kim YG, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26(4):433–443. | |
Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25(21):5071–5082. | |
Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9(8):847–856. | |
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–140. | |
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. | |
Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. | |
Compan V, Baroja-Mazo A, Lopez-Castejon G, et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity. 2012;37(3):487–500. | |
Gong YN, Wang X, Wang J, et al. Chemical probing reveals insights into the signaling mechanism of inflammasome activation. Cell Res. 2010;20(12):1289–1305. | |
Franceschi C, Bonafe M, Valensin S, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. | |
Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. | |
Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. | |
Mathews RJ, Robinson JI, Battellino M, et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis. 2014;73(6):1202–1210. | |
Vande Walle L, Van Opdenbosch N, Jacques P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512(7512):69–73. | |
Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603. | |
Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–606. | |
Aguilera M, Darby T, Melgar S. The complex role of inflammasomes in the pathogenesis of inflammatory bowel diseases – lessons learned from experimental models. Cytokine Growth Factor Rev. April 18, 2014. [Epub ahead of print.] | |
Kanai T, Watanabe M, Okazawa A, et al. Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn’s disease. Gastroenterology. 2001;121(4):875–888. | |
Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32(3):379–391. | |
Villani AC, Lemire M, Fortin G, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet. 2009;41(1):71–76. | |
Siegmund B. Interleukin-18 in intestinal inflammation: friend and foe? Immunity. 2010;32(3):300–302. | |
Kolb R, Liu GH, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell. 2014;5(1):12–20. | |
Winter RN, Rhee JG, Kyprianou N. Caspase-1 enhances the apoptotic response of prostate cancer cells to ionizing radiation. Anticancer Res. 2004;24(3a):1377–1386. | |
Winter RN, Kramer A, Borkowski A, Kyprianou N. Loss of caspase-1 and caspase-3 protein expression in human prostate cancer. Cancer Res. 2001;61(3):1227–1232. | |
Allen IC, TeKippe EM, Woodford RM, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207(5):1045–1056. | |
Dao T, Mehal WZ, Crispe IN. IL-18 augments perforin-dependent cytotoxicity of liver NK-T cells. J Immunol. 1998;161(5):2217–2222. | |
Micallef MJ, Yoshida K, Kawai S, et al. In vivo antitumor effects of murine interferon-gamma-inducing factor/interleukin-18 in mice bearing syngeneic Meth A sarcoma malignant ascites. Cancer Immunol Immunother. 1997;43(6):361–367. | |
Osaki T, Peron JM, Cai Q, et al. IFN-gamma-inducing factor/IL-18 administration mediates IFN-gamma- and IL-12-independent antitumor effects. J Immunol. 1998;160(4):1742–1749. | |
Terme M, Ullrich E, Aymeric L, et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 2011;71(16):5393–5399. | |
Lust JA, Lacy MQ, Zeldenrust SR, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009;84(2):114–122. | |
Ozkurede VU, Franchi L. Immunology in clinic review series; focus on autoinflammatory diseases: role of inflammasomes in autoinflammatory syndromes. Clin Exp Immunol. 2012;167(3):382–390. | |
Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14(1):10–22. | |
Federici S, Caorsi R, Gattorno M. The autoinflammatory diseases. Swiss Med Wkly. 2012;142:w13602. | |
Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 2002;46(9):2445–2452. | |
Hoffman HM, Rosengren S, Boyle DL, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364(9447):1779–1785. | |
Neven B, Marvillet I, Terrada C, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010;62(1):258–267. | |
Lepore L, Paloni G, Caorsi R, et al. Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with anakinra. J Pediatr. 2010;157(2):310–315. e311. | |
Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. A 2006;355(6):581–592. | |
Tan MS, Yu JT, Jiang T, Zhu XC, Tan L. The NLRP3 inflammasome in Alzheimer’s disease. Mol Neurobiol. 2013;48(3):875–882. | |
Moll M, Kuemmerle-Deschner JB. Inflammasome and cytokine blocking strategies in autoinflammatory disorders. Clin Immunol. 2013;147(3):242–275. | |
Zhang H. Anti-IL-1beta therapies. Recent Pat DNA Gene Seq. 2011;5(2):126–135. | |
Kone-Paut I, Galeotti C. Anakinra for cryopyrin-associated periodic syndrome. Expert Rev Clin Immunol. 2014;10(1):7–18. | |
Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–652. | |
Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115(8):2047–2058. | |
Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187(1):61–70. | |
Marchetti C, Chojnacki J, Toldo S, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. 2014;63(4):316–322. | |
Heinrich M, Robles M, West JE, Ortiz de Montellano BR, Rodriguez E. Ethnopharmacology of Mexican asteraceae (Compositae). Annu Rev Pharmacol Toxicol. 1998;38:539–565. | |
Juliana C, Fernandes-Alnemri T, Wu J, et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285(13):9792–9802. | |
Coll RC, O’Neill LA. The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS One. 2011;6(12):e29539. | |
Keystone EC, Wang MM, Layton M, Hollis S, McInnes IB. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann Rheum Dis. 2012;71(10):1630–1635. | |
Stock TC, Bloom BJ, Wei N, et al. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J Rheumatol. 2012;39(4):720–727. | |
Ali Z, Laurijssens B, Ostenfeld T, et al. Pharmacokinetic and pharmacodynamic profiling of a P2X7 receptor allosteric modulator GSK1482160 in healthy human subjects. Br J Clin Pharmacol. 2013;75(1):197–207. | |
Arulkumaran N, Unwin RJ, Tam FW. A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin Investig Drugs. 2011;20(7):897–915. | |
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. | |
Neta R, Douches S, Oppenheim JJ. Interleukin 1 is a radioprotector. J Immunol. 1986;136(7):2483–2485. | |
Hancock SL, Chung RT, Cox RS, Kallman RF. Interleukin 1 beta initially sensitizes and subsequently protects murine intestinal stem cells exposed to photon radiation. Cancer Res. 1991;51(9):2280–2285. | |
Bergmann M, Barnes PJ, Newton R. Molecular regulation of granulocyte macrophage colony-stimulating factor in human lung epithelial cells by interleukin (IL)-1beta, IL-4, and IL-13 involves both transcriptional and post-transcriptional mechanisms. Am J Respir Cell Mol Biol. 2000;22(5):582–589. | |
Bagby GC Jr. Interleukin-1 and hematopoiesis. Blood Rev. 1989;3(3):152–161. | |
Crown J, Jakubowski A, Gabrilove J. Interleukin-1: biological effects in human hematopoiesis. Leuk Lymphoma. 1993;9(6):433–440. | |
Crown J, Jakubowski A, Kemeny N, et al. A phase I trial of recombinant human interleukin-1 beta alone and in combination with myelosuppressive doses of 5-fluorouracil in patients with gastrointestinal cancer. Blood. 1991;78(6):1420–1427. | |
Nemunaitis J, Appelbaum FR, Lilleby K, et al. Phase I study of recombinant interleukin-1 beta in patients undergoing autologous bone marrow transplant for acute myelogenous leukemia. Blood. 1994;83(12):3473–3479. | |
Kitamura K, Takahashi T, Yamaguchi T, et al. Immunochemical characterization of the antigen recognized by the murine monoclonal antibody A7 against human colorectal cancer. Tohoku J Exp Med. 1989;157(2):83–93. | |
Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci U S A. 1999;96(5):2256–2261. | |
Netea MG, Joosten LA, Lewis E, et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med. 2006;12(6):650–656. | |
Gatti S, Beck J, Fantuzzi G, Bartfai T, Dinarello CA. Effect of interleukin-18 on mouse core body temperature. Am J Physiol Regul Integr Comp Physiol. 2002;282(3):R702–R709. | |
Robertson MJ, Kirkwood JM, Logan TF, et al. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res. 2008;14(11):3462–3469. | |
Simpkins F, Flores A, Chu C, et al. Chemoimmunotherapy using PEGylated liposomal dDoxorubicin and interleukin-18 in recurrent ovarian cancer: a pphase I dose-escalation study. Cancer Immunol Res. 2013;1(3):168–178. | |
Tarhini AA, Millward M, Mainwaring P, et al. A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer. 2009;115(4):859–868. | |
Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289. | |
Robertson MJ, Mier JW, Logan T, et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin Cancer Res. 2006;12(14 Pt 1):4265–4273. | |
Hirota SA, Ng J, Lueng A, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17(6):1359–1372. | |
Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145(5):745–757. | |
Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13(7):851–856. | |
Dupaul-Chicoine J, Yeretssian G, Doiron K, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010;32(3):367–378. | |
Tarallo V, Hirano Y, Gelfand BD, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149(4):847–859. | |
Tseng WA, Thein T, Kinnunen K, et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2013;54(1):110–120. | |
Marneros AG. NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration. Cell Rep. 2013;4(5):945–958. | |
Doyle SL, Ozaki E, Brennan K, et al. IL-18 attenuates experimental choroidal neovascularization as a potential therapy for wet age-related macular degeneration. Sci Transl Med. 2014;6(230):230ra244. | |
Shen J, Choy DF, Yoshida T, et al. Interleukin-18 has antipermeablity and antiangiogenic activities in the eye; reciprocal suppression with VEGF. J Cell Physiol. 2014;229(8):974–983. | |
Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38(2):240–244. | |
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20(3):319–325. | |
Kempster SL, Belteki G, Forhead AJ, et al. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am J Physiol Gastrointest Liver Physiol. 2011;300(2):G253–G263. | |
Khare S, Dorfleutner A, Bryan NB, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity. 2012;36(3):464–476. | |
Vladimer GI, Weng D, Paquette SW, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37(1):96–107. | |
Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem. 2001;276(30):28309–28313. | |
Jesus AA, Silva CA, Segundo GR, et al. Phenotype-genotype analysis of cryopyrin-associated periodic syndromes (CAPS):description of a rare non-exon 3 and a novel CIAS1 missense mutation. J Clin Immunol. 2008;28(2):134–138. | |
Stojanov S, Weiss M, Lohse P, Belohradsky BH. A novel CIAS1 mutation and plasma/cerebrospinal fluid cytokine profile in a German patient with neonatal-onset multisystem inflammatory disease responsive to methotrexate therapy. Pediatrics. 2004;114(1):e124–e127. | |
Vlagopoulos T, Townley R, Villacorte G. Familial cold urticaria. Ann Allergy. 1975;34(6):366–369. | |
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29(3):301–305. | |
Aksentijevich I, D Putnam C, Remmers EF, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56(4):1273–1285. | |
Dodé C, Le Dû N, Cuisset L, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet. 2002; 70(6):1498–1506. | |
Feldmann J, Prieur AM, Quartier P, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71(1):198–203. | |
Caroli F, Pontillo A, D’Osualdo A, et al. Clinical and genetic characterization of Italian patients affected by CINCA syndrome. Rheumatology (Oxford). 2007;46(3):473–478. | |
Mirault T, Launay D, Cuisset L, et al. Recovery from deafness in a patient with Muckle-Wells syndrome treated with anakinra. Arthritis Rheum. 2006;54(5):1697–1700. | |
Boschan C, Witt O, Lohse P, Foeldvari I, Zappel H, Schweigerer L. Neonatal-onset multisystem inflammatory disease (NOMID) due to a novel S331R mutation of the CIAS1 gene and response to interleukin-1 receptor antagonist treatment. Am J Med Genet A. 2006;140(8):883–886. | |
Hoffman HM, Gregory SG, Mueller JL, et al. Fine structure mapping of CIAS1: identification of an ancestral haplotype and a common FCAS mutation, L353P. Hum Genet. 2003;112(2):209–216. | |
Koike R, Kubota T, Hara Y, et al. A case of Muckle-Wells syndrome caused by a novel H312P mutation in NALP3 (cryopyrin). Mod Rheumatol. 2007;17(6):496–499. | |
Zeft A, Bohnsack JF. Cryopyrin-associated autoinflammatory syndrome: a new mutation. Ann Rheum Dis. 2007;66(6):843–844. | |
Shepard MK. Cold hypersensitivity. Birth Defects Orig Artic Ser. 1971;7(8):352. | |
Dalgic B, Egritas O, Sari S, Cuisset L. A variant Muckle-Wells syndrome with a novel mutation in CIAS1 gene responding to anakinra. Pediatr Nephrol. 2007;22(9):1391–1394. | |
Arostegui JI, Aldea A, Modesto C, et al. Clinical and genetic heterogeneity among Spanish patients with recurrent autoinflammatory syndromes associated with the CIAS1/PYPAF1/NALP3 gene. Arthritis Rheum. 2004;50(12):4045–4050. | |
Stack JH, Beaumont K, Larsen PD, et al. IL-converting enzyme/caspase-1 inhibitor VX-765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J Immunol. 2005;175(4):2630–2634. | |
Saito M, Nishikomori R, Kambe N, et al. Disease-associated CIAS1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood. 2008;111(4):2132–2141. | |
Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID):a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46(12):3340–3348. | |
Wanderer AA. An ‘allergy’ to cold. Hosp Pract. 1979;14(6):136–137. | |
Matsubayashi T, Sugiura H, Arai T, Oh-Ishi T, Inamo Y. Anakinra therapy for CINCA syndrome with a novel mutation in exon 4 of the CIAS1 gene. Acta Paediatr. 2006;95(2):246–249. | |
Ohnishi H, Teramoto T, Iwata H, et al. Characterization of NLRP3 variants in Japanese cryopyrin-associated periodic syndrome patients. J Clin Immunol. 2012;32(2):221–229. | |
Frenkel J, van Kempen MJ, Kuis W, van Amstel HK. Variant chronic infantile neurologic, cutaneous, articular syndrome due to a mutation within the leucine-rich repeat domain of CIAS1. Arthritis Rheum. 2004;50(8):2719–2720. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.