")
Back to Journals » OncoTargets and Therapy » Volume 13
Targeting the MET-Signaling Pathway in Non-Small–Cell Lung Cancer: Evidence to Date
Authors Bylicki O , Paleiron N, Assié JB, Chouaïd C
Received 23 March 2020
Accepted for publication 30 May 2020
Published 17 June 2020 Volume 2020:13 Pages 5691—5706
DOI https://doi.org/10.2147/OTT.S219959
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr XuYu Yang
Olivier Bylicki,1,2 Nicolas Paleiron,1 Jean-Baptiste Assié,2– 4 Christos Chouaïd2,3
1Respiratory Disease Unit, HIA Sainte Anne, Toulon, France; 2University Paris–Est Créteil (UPEC), CEpiA (Clinical Epidemiology and Ageing), EA 7376- IMRB, UPEC, Créteil, France; 3Pneumology Department, Centre Hospitalier Intercommunal De Créteil, Créteil, France; 4Cordeliers Research Center, Inserm, Functional Genomics of Solid Tumors Laboratory, Sorbonne University, University of Paris, Paris, France
Correspondence: Olivier Bylicki
Respiratory Disease Unit, HIA Sainte Anne, BRCM Toulon, 2, Boulevard Saint-Anne, Toulon 83000, France
Tel +33(0)483162937
Fax +33(0)483162461
Email [email protected]
Abstract: The c-MET proto-oncogene (MET) plays an important role in lung oncogenesis, affecting cancer-cell survival, growth and invasiveness. The MET receptor in non-small–cell lung cancer (NSCLC) is a potential therapeutic target. The development of high-output next-generation sequencing techniques has enabled better identification of anomalies in the MET pathway, like the MET exon-14 (METex14) mutation. Moreover, analyses of epidermal growth factor-receptor (EGFR) and mechanisms of resistance to tyrosine-kinase inhibitors (TKIs) demonstrated the importance of MET amplification as an escape mechanism in patients with TKI-treated EGFR-mutated NSCLCs. This review summarizes the laboratory findings on MET and its anomalies, trial results on METex14 alterations and MET amplification in non-EGFR mutated NSCLCs, and acquired resistance to TKI in EGFR-mutated NSCLCs. The outcomes of the first trials with anti-MET agents on non-selected NSCLC patients or those selected for MET overexpression were disappointing. Two situations seem the most promising today for the use of anti-MET agents to treat these patients: tumors harboring METex14 and those EGFR-sensitive mutation mutated under TKI-EGFR with a MET-amplification mechanism of resistance or EGFR-resistance mutation.
Keywords: non-small–cell lung cancer, MET exon 14, MET amplification, MET pathway
Introduction
Targeted therapies have profoundly modified the prognoses of lung cancers with oncogenic mutations, achieving notably improved progression-free (PFS) and overall survival (OS) rates compared to reference chemotherapy regimens. That is particularly true for first- or second-line treatment of metastatic non-small–cell lung cancers (NSCLCs) harboring an epidermal growth factor-receptor (EGFR) mutation or anaplastic lymphoma kinase (ALK) translocation.1–5 Targeted therapies have also shown their efficacy in patients carrying the v-RAF murine sarcoma viral oncogene homolog B (BRAFV600E) mutation, tyrosine-protein kinase-1 protooncogene (ROS1) or rearranged-during-transfection (RET) translocation.6–8 More recently, the efficacy of targeting the neurotrophic tropomyosin receptor kinase (NTRK) in all patients whose cancers express it (making it a marker for tumor-agnostic therapy) was demonstrated.9 In other contexts, knowledge remains more fragmented, despite a potentially oncogenic target, with therapies having only modest activities. That is the case for NSCLCs expressing human epidermal growth factor receptor-2 (HER2), Kirsten rat-sarcoma viral oncogene (KRAS) or those with c-MET proto-oncogene (MET) protooncogene-pathway abnormalities.10–12
The MET pathway was identified in the 1980s and its carcinogenic role in lung cancer has been recognized since the 1990s.13,14 It is a complex pathway, poorly understood, with anomalies, ranging from MET overexpression, rare translocations, amplifications, de novo or acquired under tyrosine-kinase inhibitors of epidermal growth factor-receptor (EGFR) (EGFR-TKIs) and, finally, mutations, particularly of MET exon-14 (METex14) mutations. Numerous molecules targeting this pathway or its ligand, hepatocyte growth factor (HGF), are at various stages of development.
This review summarizes the data available on our understanding of the different molecular MET alterations, the results obtained with agents targeting this pathway and the contribution of immunotherapy to treating these patients.15,16
The MET Pathway and Its Alterations
The MET gene is located at 7q21–q31 on chromosome 7. It is comprised of ~125 kb and 21 exons.17,18 MET is a heterodimer tyrosine-kinase receptor with extracellular, transmembrane, juxtamembrane and kinase domains.19,20
MET binding to its exclusive ligand, HGF, leads to homodimerization and phosphorylation of the intracellular tyrosine residues.18 Receptor activation stimulates downstream signaling pathways, such as extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase-Akt (PI3K)/protein kinase B pathways and JAK/STAT (Janus kinase/signal transducer and activator of transcription).20 Those pathways are known to be involved in cell proliferation, migration, motility angiogenesis, survival and the epithelial-to-mesenchymal transition.21,22
During embryogenesis, MET and HGF favor the formation of trophoblasts and placental hepatocytes.23 In adults, the two proteins are strongly expressed in a wide variety of tissues and can be regulated positively in response to a tissue lesion.18
Deregulation of the MET pathway in oncology can be manifested in several ways: genetic mutation, amplification, rearrangement or overexpression of proteins. Other than NSCLC, breast, colon, kidney and stomach cancers overexpress MET. MET amplification is found in colon, esophageal and stomach cancers.24–28
Overexpression
Overexpression of MET or its ligand HGF, without amplification or mutation, is possible. This overexpression seems to induce activation independent of the MET ligand, phosphorylation and activation of downstream signaling pathways.29
Immunohistochemistry (IHC) is able to detect MET or HGF overexpression with several antibodies that are commercially available.
Rearrangement
The first MET rearrangement was described in the 1990s with the tryptophan (TRP) gene.30 Other rearrangements have since been found, notably in NSCLCs: kinesin family member 5B (KIF5B), F-actin–capping proteins bind in a Ca2+ (CAPZA2 (2)), cluster of differentiation 47 membrane protein (CD47 (2)), testin (TES), caveolin-1 (CAV1), integrin subunit alpha-9 (ITGA9), human leukocyte antigen (HLA-DRB1), transcription factor EC (TFEC), cortactin-binding protein-2 (CTTNBP2), ankyrin-1 (ANK1), steroidogenic acute regulatory-related lipid-transfer domain containing three N-terminal–like proteins (STARD3NL).31
Amplification
Amplification is an increased gene-copy number (GCN), linked to the focal duplication of a gene via breakage–fusion–bridge mechanisms.32 A higher GCN can also be secondary to polysomia of chromosome 7 (caused by chromosomal duplication, for example).33,34 MET amplification deregulates the MET signaling pathway by overexpression of the protein and constitutive activation of kinases.33 The number of MET copies can be evaluated by fluorescence in situ hybridization (FISH) or quantitative polymerase chain reaction. When using FISH, the MET/CEP7 (centromeric portion of chromosome 7) ratio remains unchanged, whereas, with amplification, the MET GCN increases at the expense of the number of centromeres, which results in a higher MET/CEP7 ratio.33
New techniques of hybridization capture-based next-generation sequencing (NGS) can analyze gene amplifications. The GCN modifications can be identified by comparing tumor sequences in targeted regions to a normal diploid sample.35 Unlike FISH, NGS and multiplex polymerase chain reaction are able to analyze in parallel other genes of interest to look for concomitant alterations having a clinical impact.36
No international consensus has been reached on the MET/CEP7 ratio threshold enabling characterization of a real amplification. Camidge et al proposed a classification scheme with several MET/CEP7-ratio categories (low, 1.8–2.2; intermediate, >2.2 and <5; and high, ≥5) but another classification (which changed the intermediate class to >2.2 and <4; and high to ≥4) has been applied in clinical settings when treating patients with MET inhibitors.37 Other scores exist: ≥5 MET signals per cell (Capuzzo scoring system) and a MET/CEP7 ratio ≥2 (PathVysion).38,39 Their harmonization seems essential to enable comparisons among studies and available data.
Mutations
METex14 mutations provoke the suppression of the juxtamembrane domain or abnormal splicing leading to the suppression of the juxtamembrane domain that prevents the degradation of the MET receptor, which leads to increased MET-receptor activity. There can be punctual mutations at the Y1003 catalytic site (Sema-3C, encoded by exon 2) and the juxtamembrane (encoded by exons 14 and 15) domains. In NSCLCs, punctual MET mutations are often situated in the extracellular or juxtamembrane domains (exon 14).40 The first NSCLC patients with METex14 mutations were described in 2005.41
In the absence of mutation, the introns adjacent to METex14 in the premessenger RNA (pre-mRNA) are spliced, which gives rise to an mRNA containing METex14 that becomes the functional MET receptor. METex14 codes for a part of the of the juxtamembrane domain containing Y1003, the binding site of E3 ubiquitin ligase c-Cbl (protooncogene Casitas B-lineage lymphoma). Ubiquitination marks the MET receptor for degradation.42 These mutations lead to METex14 skipping, which yields a truncated MET receptor lacking a Y1003 c-Cbl–binding site. The loss of that site leads to less ubiquitination of the MET protein and its degradation, and prolonged MET activation that favors the tumor oncogenicity.43 MET overexpression detectable by IHC for it may detect the degradation of the protein.
METex14 alterations are highly variable and represent a diagnostic challenge. Substitutions or insertions of bases at splice sites in introns 13 and 14, respectively at 3′ and 5′ termini, for example.42–45 METex14 mutations are mutually exclusive from other mutations, suggesting its role as a true oncogenic driver. Based on an analysis of 933 non-squamous NSCLCs, no patient with a METex14 mutation had any other associated oncogenic abnormality.45,46
Epidemiology of the MET Pathway in NSCLC
MET Overexpression
Its frequency ranges between 22% and 75%, depending on the series.47–52 MET overexpression is considered a poor-prognosis factor.48,52 A meta-analysis including 18 studies (5516 NSCLC patients) showed that MET overexpression was associated with a significantly increased risk of death (hazard ratio (HR): 1.52 [95% confidence interval (CI): 1.08–2.15]).52 Another meta-analysis of 4454 NSCLC patients (based on 22 studies) confirmed that IHC MET-positivity was significantly associated with worse OS (HR: 1.55 [95% CI: 1.10–2.18]).53
Rearrangements
The prevalence of MET rearrangement is unknown. Based on a series of 2410 NSCLC patients, that rate was 0.04% (one patient with MET–ATXN7L1 (ataxin-7-like protein-1) fusion).54
Amplification
De Novo Amplification
The reported frequency of de novo MET amplification in NSCLCs ranges from 1%–5%, depending on the level of preselection, the assay and the positivity threshold applied.4,38,46,55,56 No consensus has yet been reached on the definition of MET positivity based on GCN. Different classification thresholds among studies has complicated comparisons of reported MET-amplification/GCN gain relative to the underlying frequency.47–49,57 These amplifications are more frequent in poorly differentiated adenocarcinomas with a poor prognosis.
A few meta-analyses on the prognostic role of MET amplification in NSCLC have been published.52,58,59 A meta-analysis of 21 studies that had enrolled 7647 patients showed that MET amplification was associated with shorter OS (HR: 1.45 [95% CI: 1.16–1.80]). Subgroup analyses based on histology and ethnicity indicated that MET amplification was significantly associated with shorter survival, especially for patients with adenocarcinomas (HR: 1.41 [95% CI: 1.11–1.79]) and of Asian ethnicity (HR: 1.58 [95% CI: 1.32–1.88]).58
Amplification as a Resistance Mechanism in Tumors Becoming EGFR-Mutated Under TKIs
MET amplification represents a mechanism of acquired resistance in 5–20% of patients, whose NSCLCs harbor EGFR mutations and were treated with EGFR-inhibitors, particularly after first-line third-generation therapy.60–65 In the AURA3 trial of 83 patients who cancers progressed on second-line osimertinib, 19% exhibited MET amplification.66 When osimertinib was given as a first-line therapy, MET amplification was the most common resistance mechanism, found in 15% of patients by NGS of circulating-tumor DNA analysis. Moreover, that percentage is expected to be higher in tissues, because of the underestimation of gene amplification in plasma.67 Consistent with those findings, the results of several preclinical and clinical studies demonstrated that the combined use of MET inhibitors, osimertinib and other EGFR-TKIs can potentially overcome the resistance in osimertinib-resistant EGFR-mutant NSCLC lines with MET-gene amplification.68–70
Mutations
The frequencies of METex14 mutations was 1.7–4.3% in metastatic lung adenocarcinomas, according to NGS analyses.46–48,55,58 METex14-skipping mutations tend to be more frequent in relatively elderly populations and mutually exclusive of other lung cancer-driver mutations.71,72 METex14-skipping mutations have been identified across different major histological subtypes of lung cancers, eg adenosquamous (8.2%) or sarcomatoid subtypes (7.7%), adenocarcinomas (2.9%) and squamous-cell carcinomas (2.1%).72,73
Anti-MET Therapies
The MET pathway can be targeted via several mechanisms. Anti-MET therapies are divided among selective TKIs, non-selective (also known as multitarget) TKIs and antibodies directed against MET or its ligand HGF.74 Table 1 summarizes the molecules being evaluated as NSCLC treatments. TKIs can be separated into three types according to their binding mechanisms and their conformations.75,76 TKI types I and II are ATP-competitive MET inhibitors but with different selectivities, conformations and binding sites. Those two groups include the majority of TKIs currently used or being developed, such as crizotinib, capmatinib and savolitinib (type I) or cabozantinib, merestinib and glesatinib (type II). Tivantinib is an exception because its activity is only partially linked to MET inhibition (with non-ATP–competitive binding; other mechanisms are involved, eg, microtubule rupture and blocked assembly).77 Type III TKIs bind to allosteric sites distinct from the ATP-binding site. At present, no type III inhibitor has been developed for use in oncology.75
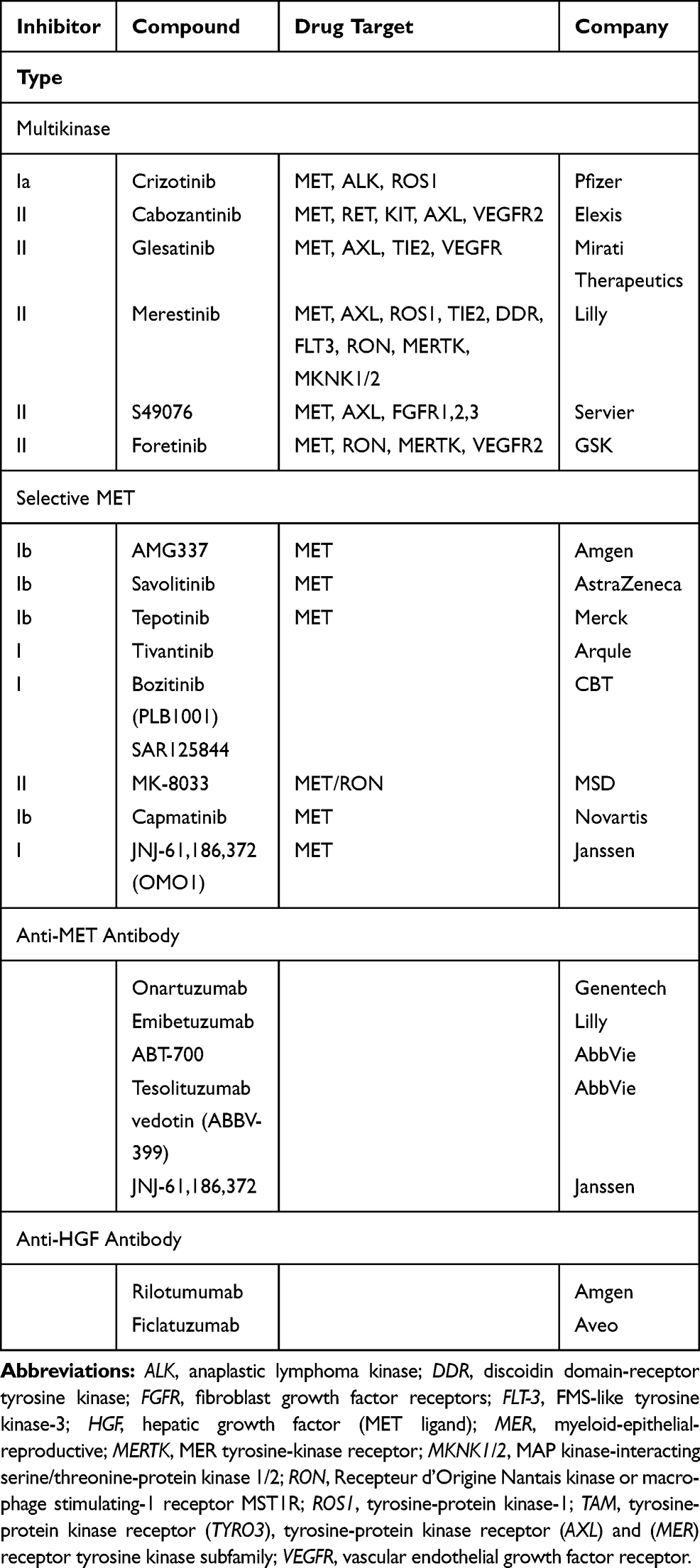 |
Table 1 Agents Being Evaluated as Treatments for Non-Small–Cell Lung Cancers |
Therapies Developed for Non-Selected or Selected MET-Overexpression Patients
Anti-MET results obtained for NSCLC patients not selected for a MET pathway anomaly have been disappointing, even when they were analyzed as a function of their IHC-detected MET expression.
The GO27820 study evaluated onartuzumab (Genentech, Inc, South San Francisco, CA), a recombinant, fully humanized, monovalent monoclonal antibody that binds to the extracellular domain of MET, in combination with first-line platinum-based doublet chemotherapy, in patients with squamous cell NSCLCs. Its results were considered negative, with median PFS at 4.9 months in both treatment arms. For patients whose cancers expressed IHC-detected MET, median PFS lasted 5.0 and 5.2 months, respectively, in the onartuzumab or placebo arms.12 In another Phase II trial, onartuzumab in combination with chemotherapy comprised of platinum salt–pemetrexed–bevacizumab in patients with non-squamous NSCLC (GO27281) did not reach its principle objective, with median PFS at 5.0 months vs 6.8 months for the placebo arm. In patients with IHC MET-positive expression, median PFS was 4.8 (95% CI: 3.7–6.2) months for onartuzumab recipients vs 6.9 (95% CI: 4.9–10.9) months for the placebo arm, with an unstratified HR of 1.71.78
The combination of erlotinib and onartuzumab, tested in two studies, did not prolong PFS or OS in the general NSCLC population or those with MET overexpression.79,80
Crizotinib (PF-02341066, Xalkori, developed by Pfizer; 200 mg twice daily) was evaluated in combination with dacomitinib (NCT01121575; maximum tolerated dose: 30 mg once daily) in a Phase I study on 70 patients were treated during the dose-escalation (n=33) and expansion phases (n=37). Grade-3 or −4 treatment-related adverse events occurred in 43% of patients.81 The crizotinib–dacomitinib combination had limited antitumor activity against advanced NSCLC and was associated with substantial toxicity. Further assessment of that combination was not pursued.
Tivantinib (formerly ARQ 197; ArQule, Woburn, MA; Daiichi Sankyo, Tokyo, Japan) a non-ATP–competitive small-molecule MET inhibitor (TKI) was evaluated in three trials (NCT00777309, MARQUEE, ATTENTION) in combination with erlotinib, as second- or third-line therapy for advanced NSCLC.82–84 None of the three trials obtained positive results. The ATTENTION study was stopped, after 307 patients had been randomized, as recommended by the Safety Review Committee because of the very different between-group frequencies and impacts of interstitial lung disease: 14 (three deaths) tivantinib recipients and six (0 deaths) placebo-group patients.84
Cabozantinib, an available oral TKI active against MET and vascular endothelial growth-factor–receptor-2 (VEGFR2), RET, ROS1, tyrosine-protein kinase receptor (AXL), tyrosine-protein kinase KIT (KIT), and tyrosine kinase with immunoglobulin and EGF homology domains (TIE2/TEK), was tested alone and combined with erlotinib, as second- or third-line therapy for NSCLCs. That study included 125 patients: 42 assigned to receive erlotinib, 40 cabozantinib and 43 the combination. PFS was significantly longer for the cabozantinib (4.3 months, HR: 0.39 [80% CI: 0.27–0.55]; P=0.0003) and erlotinib plus cabozantinib arms (4.7 months, HR: 0.37 [80% CI: 0.25–0.53]; P=0.0003) than erlotinib alone (median: 1.8 months). For the 74/125 patients with IHC-detected MET-positive expression, median PFS lasted 1.8 months for patients randomized to erlotinib vs 5.0 months for patients given cabozantinib alone or in combination. This agent is not currently being evaluated in any study.85
An ongoing phase II study (NCT03539536) is evaluating telisotuzumab vedotin (ABBV-399), an anti-MET antibody, as second-line therapy for NSCLCs, especially IHC MET-positive NSCLCs, as assessed by an AbbVie-designated IHC laboratory or known documented MET-gene amplification.
The results with anti-MET agents have been disappointing in patients with tumors overexpressing MET. Notably, onartuzumab, tivantinib and cabozantinib yielded negative findings (Table 2).
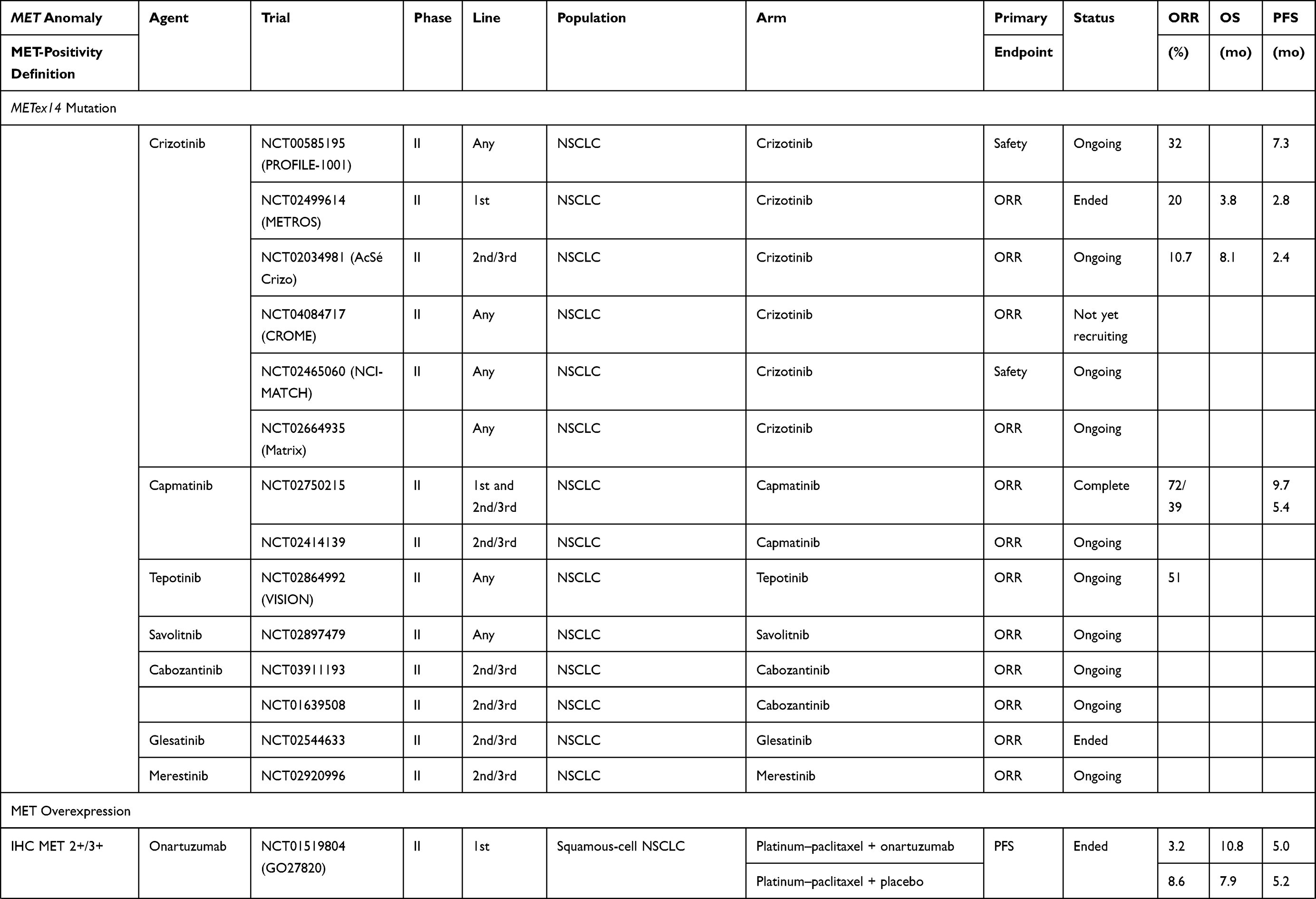 | 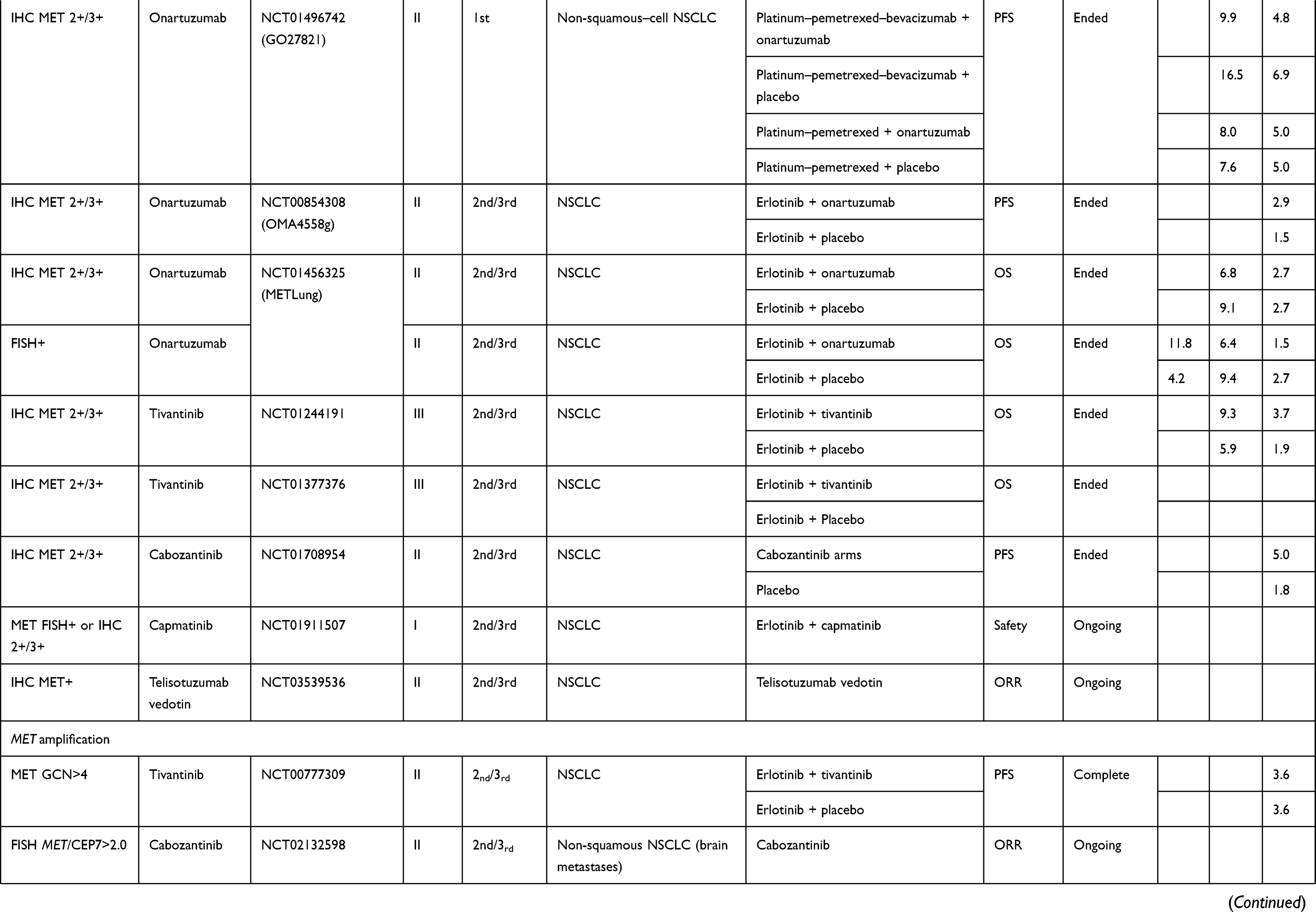 | 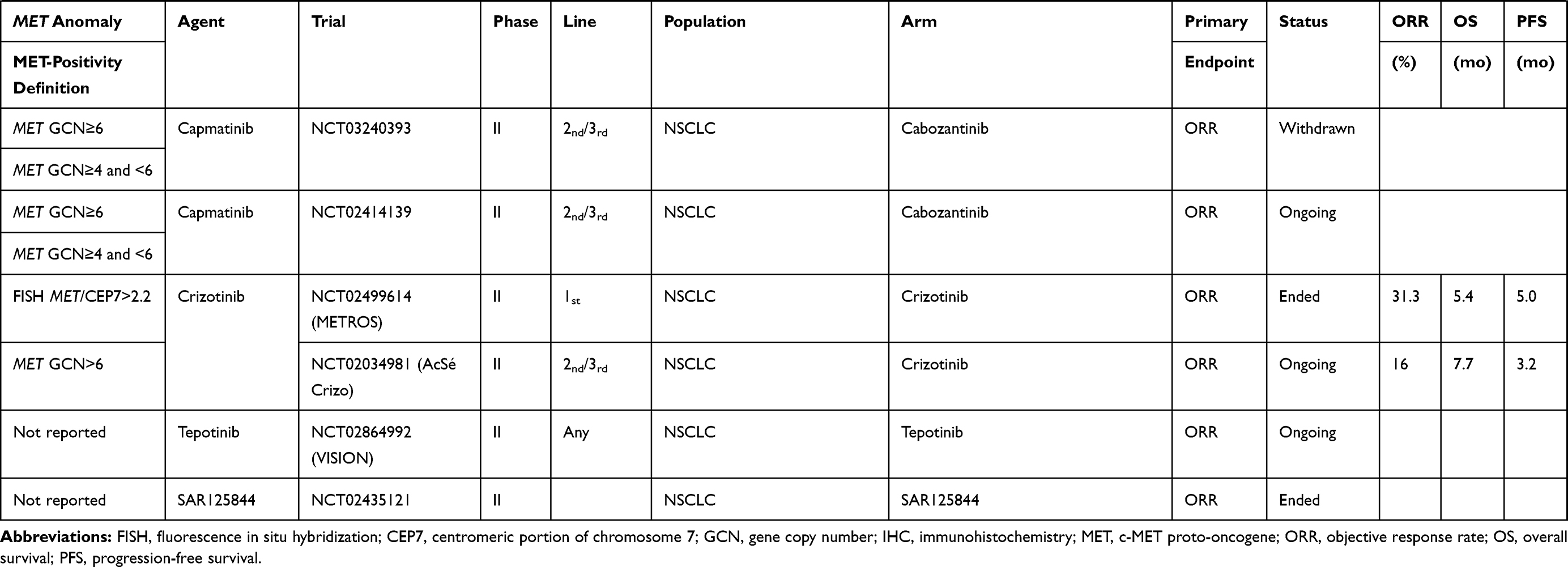 |
Table 2 Clinical Trials on NSCLC Patients Without EGFR-Mutation(s) |
MET Rearrangement
At this time, no molecule is being developed to overcome this anomaly. However, published case reports have described crizotinib efficacy against NSCLCs harboring a KIF5B–MET rearrangement.86
MET-Amplified NSCLCs
De Novo Amplification
Results obtained with agents tested in patients with this genetic abnormality are summarized in Table 2. In the two arm, non-comparative phase II METROS trial, among the 16 patients with MET amplification (Camidge-classification intermediate for 14 patients or high for 2) treated with oral crizotinib (250 mg twice daily), the objective response rate (ORR) was 31.3% (95% CI: 5.2–71.4), with respective median PFS and OS at 5.0 (95% CI: 2.7–7.3) and 5.4 months (95% CI: 3.4–7.4).87 The AcSé phase II trials on 25 crizotinib-treated patients with MET amplification (GCN>6), the ORR was 16%, and the respective median PFS and OS were 3.2 (95% CI: 1.9–3.7) months and 7.7 (95% CI: 4.6–15.7) months.88
Tivantinib also yielded disappointing results for patients with MET amplification (defined as GCN>4): median PFS last 3.6 months for those given the erlotinib–tivantinib combination, as for those taking erlotinib alone.83 Other molecules, like tepotinib or capmatinib, are being tested to treat this anomaly.
Amplification as a Resistance Mechanism
Several TKIs with anti-MET activity have been evaluated in this context (Table 3). According to a phase I trial combining crizotinib and erlotinib, respective maximum tolerated doses were 150 mg twice daily and 100 mg/day.89 However, no ongoing clinical trial is testing this combination therapy. In a phase II study that combined cabozantinib (40 mg/day) and erlotinib (150 mg/day) for patients with EGFR-mutated tumors that progressed under EGFR-TKI, ORR was 10.8% for the 37 analyzable patients, none of whom had MET amplification.90
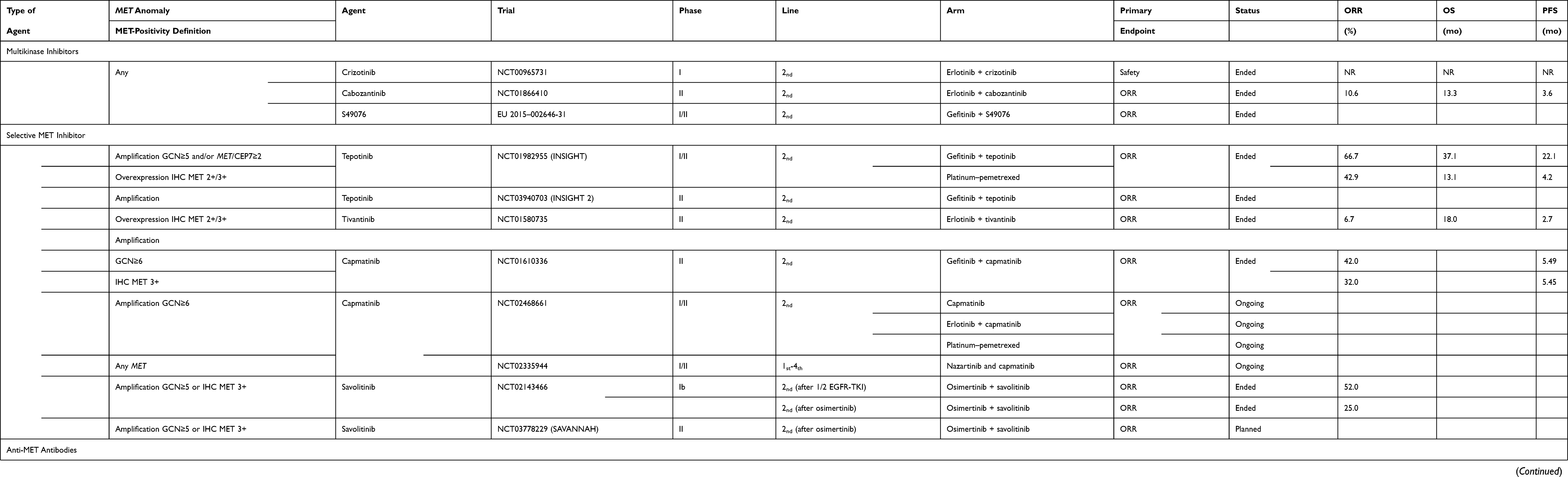 |
Table 3 Clinical Trials on Patients with EGFR-Mutated NSCLC |
The combination of emibetuzumab and erlotinib versus erlotinib alone as first-line therapy for EGFR-mutated metastatic NSCLC, without selection according to MET status, yielded respective negative outcomes for its principal criterion (PFS) of 9.3 vs 9.5 months. Exploratory analysis of patients with MET-high expressing tumors (IHC MET 3+) showed that PFS was prolonged by 15.3 months (combination: 20.7 months vs 5.4 months, HR: 0.39 [90% CI: 0.17–0.91]).91
The combination of tepotinib plus gefitinib versus platinum–pemetrexed chemotherapy in patients with EGFR-mutated but EGFRT790M-negative, IHC MET 2+/3+ or with MET amplification (GCN≥5 and/or MET/CEP7 ratio ≥2) that progressed under TKI, respective median PFS lasted 21.2 vs 4.2 months (HR: 0.13 [90% CI: 0.04–0.43]), and median OS of 37.3 vs 13.1 months (HR: 0.08 [90% CI: 0.01–0.51]).92 ORR was also higher for the combination, respectively: 66.7% vs 42.9%. Patients with MET-amplification experienced ≥15% grade ≥3 treatment-related adverse events (increased amylase or lipase) in both arms. A new phase II study is now underway.
The tivantinib plus erlotinib combination had an ORR of 6.7% in a Japanese phase II study that had enrolled 45 patients with advanced EGFR-mutated NSCLC with acquired resistance to gefitinib or erlotinib and MET expression.93 Half the patients enrolled in that study were EGFRT790M-positive and 48.9% had high MET expression (IHC MET 2+/3+), including the three responders with both genetic anomalies.
In a phase Ib/II study on EGFR-TKI–pretreated patients, with EGFRT790M-negative and MET amplification-positive (GCN≥6) NSCLCs, the gefitinib–capmatinib (400 mg twice per day) plus gefitinib (250 mg/day) achieved an ORR of 47%.68 No significant drug–drug interactions were observed in that study. Other ongoing studies are combining capmatinib and aunazartinib or erlotinib.
The TATTON (phase Ib) study tested the combination of osimertinib (80 mg/day) and savolitinib (600 mg/day) on two cohorts of patients with EGFR-mutated MET-amplified NSCLCs.69,70 In the first cohort of first- and second-generation EGFR-TKI–pretreated patients with EGFRT790M-negative/MET-positive (GCN≥5 or IHC MET 3+) disease, the ORR was 52%. In the other cohort that included third-generation EGFR-TKI–pretreated patients, the combination therapy obtained an ORR of 25%.
The ongoing phase II SAVANNAH study (NCT03778229) will further evaluate the osimertinib–savolitinib combination in first-generation EGFR-TKI–pretreated patients with EGFR-mutant, MET-amplified NSCLCs that progressed on prior osimertinib.
Emibetuzumab (LY2875358) is a humanized IgG4 bivalent monoclonal anti-MET antibody-blocking ligand-dependent and -independent HGF/MET signaling. In a study examining whether acquired resistance to erlotinib in MET-positive (expression) NSCLC patients, with a predominance of EGFR-mutated tumors, that resistance could be overcome by emibetuzumab or emibetuzumab + erlotinib; the ORRs for patients with MET overexpression (≥60%) were 3.8% and 4.8% in the combination and monotherapy arms, respectively.94 In a phase Ib study combining tesolituzumab vedotin (ABBV-399) and erlotinib for patients with IHC MET-positive (H-score >150 or MET-amplification) NSCLCs, the ORR was 34.5% for the 29 EGFR-TKI–pretreated patients.95
JNJ-61,186,372, an antibody bispecific to EGFR and MET, binds the two proteins, thereby blocking their ligand binding, promoting receptor degradation and triggering antibody-dependent cellular cytotoxicity in models of EGFR-mutated NSCLC. Results of the phase I study were reported at ASCO 2019 (NCT02609776).96 Response-assessable patients’ ORR was 28% and their best timepoint response was partial. Among 47 patients with prior third-generation TKI therapy, 10 had best timepoint response of partial response (six confirmed), including four with EGFRC797S mutation, one with MET amplification and five with no identifiable EGFR/MET-dependent resistance. Enrollment in that trial’s expansion phase is ongoing. It also evaluated another cohort with the combination of JNJ-61,186,372 and lazertinib (third-generation EGFR-TKI).
Evaluation of Anti-MET Agents in Patients with NSCLCs Harboring METex14
The METex14 mutation clearly appears to be an oncogenic driver. According to a multicenter series of patients carrying the METex14 mutation, 61/148 patients not exposed to anti-MET and for whom survival data was available, median OS lasted 8.1 [95% CI: 5.3–not reached] vs 24.6 [95% CI 12.1–not reached] months for those who had received at least one TKI anti-MET (crizotinib, glesatinib or capmatinib) during management of their NSCLCs.97 Several MET inhibitors are under development for this indication.
Crizotinib efficacy was addressed in several case reports and Phase I–II trials (Table 2).73,98,99 Updated results from the PROFILE-1001 study, in which 69 treatment-naïve or chemotherapy-refractory, METex14+ NSCLC patients participated, showed three complete responses and 18 partial responses (ORR, 32% [95% CI: 21–45]) with median PFS at 7.3 [95% CI: 5.4–9.1] months.100
In the METRO study, which included 26 crizotinib-treated patients (16 with MET amplification, nine harboring the METex14 mutation and one with concurrent abnormalities), the ORR was 20% for patients with METex14 mutations, with median PFS at 2.6 [95% CI: 2.2–3.0] months and median OS at 3.8 months [95% CI: 1.7–5.8]. No difference between MET-amplified and METex14-mutated patients was found for any clinical endpoint.87
In the AcSé crizotinib study,88 among 28 patients with MET anomalies, 25 had the METex14 mutation; the ORR at 2 cycles was 10.7% [95% CI 2.3–28.2%]; median PFS was 2.4 [95% CI 1.6–5.9] months and median OS was 8.1 months [95% CI 4.1–12.7].
In light of the outcomes of the Profile-1001 study, in 2018, the FDA granted, crizotinib (Xalkori) a breakthrough-therapy designation for the treatment of patients with NSCLC harboring METex14 alterations that progressed after receiving platinum-based chemotherapy.
Capmatinib (INC280; Novartis), an oral, ATP-competitive, type Ib MET inhibitor has also been developed for this indication. In the phase I study, the four METex14-mutated–NSCLC patients enrolled achieved significant tumor-volume reductions (>45%).101 In the phase II GEOMETRY mono-1 study, among the 94 METex14-mutated NSCLC patients included (69 receiving second- or third-line therapy and 25 treatment-naïve), respective ORRs were 39.1% (95% CI: 27.6–51.6) and 72.0% [95% CI: 50.6–87.9]. The median first-line PFS was 9.7 months and 5.4 months for the subsequent lines.102 In September 2019, the FDA designated capmatinib (INC280) a breakthrough therapy as first-line treatment for patients with METex14-mutated NSCLC.
Tepotinib (EMD1214063, MSC2156119J; Merck), an oral, ATP-competitive, and highly selective MET inhibitor, was evaluated in a phase II study on patients with METex14-mutated NSCLCs. The intermediate results for 35/90 patients included and assessable showed the ORR at 51.4% [95% CI: 34.0–68.6], with median treatment duration of 9.8 [95% CI:1.1–18.0] months.103 The FDA accorded this investigational targeted therapy breakthrough-therapy designation for patients with METex14-mutated NSCLCs that progressed after platinum-based chemotherapy.
Other anti-MET TKIs are currently being tested, like savolitinib (AZD6094, volitinib, HMPL-504; AstraZeneca) or glesatinib (MGCD265; Mirati Therapeutics) in phase II trials, but no information is available at this time (Table 2).
Immunotherapy for Patients with a MET-Pathway–Signaling Abnormality
In pathophysiological terms, the presence of a MET anomaly seems to induce programed cell-death protein-1–ligand-1 (PD-L1) expression.104–106 An analysis of 622 surgical NSCLC samples showed that PD-L1 expression was significantly higher in patients with MET amplifications than those without. In addition, peritumoral lymphocyte infiltration was more abundant in patients with MET amplification.105 In that paper, six patients with MET anomalies were treated with immunotherapy, which yielded three partial responses, one disease stabilization and two progressions. In an analysis of 148 patients harboring the METex14 mutation, 63% of the cohort’s NSCLCs expressed PD-L1: 1–49% for 22% and >50% for 41%.100 Their median tumor mutation burden was 3.8 mutations/megabase, lower than that of a control historical cohort, whose tumors did not carry the METex14 mutation (5.7 mutation/megabase: P<0.001).
Retrospective analysis of registries provided information on the inefficacy of immune-checkpoint inhibitors (ICIs) for patients with oncogenic driver alterations. The Immunotarget Registry included 34 patients, whose NSCLCs harbored the METex14 mutation, 30% expressing PD-L1 and their ORR was 16%, with median PFS at 4.7 months.15 In another analysis of 30 patients with MET mutations, 43% expressing PD-L1, ORR was 35.7% and median PFS lasted 4.9 months.16 ICIs do not seem to have any remarkable efficacy against MET anomalies but it appeared better than for other oncogenic anomalies, eg ALK or RET translocations. Phase I–II trials combining ICIs with anti-MET TKIs, like glesatinib, are ongoing but no information is available at this time.105
Conclusion
Inspired by the major breakthrough of targeted therapies in treating lung cancers, the identification of new pertinent targets remains a high priority. After the discoveries of the EGFR or BRAF mutations, or ALK or ROS1 rearrangements, new, less frequent mutations have been identified, such as RET or NTRK. MET pathway anomalies also have major clinical impact, especially for patients with the METex14 mutation, for which promising therapeutics have been developed, eg first-line capmatinib or second-line crizotinib and tepotinib after chemotherapy failure. Other agents are being investigated as any treatment line in patients with metastatic METexon14-mutation–positive NSCLCs.
It is necessary to distinguish between de novo amplifications and amplifications as a resistance mechanism to EGFR-TKIs. Among the latter, capmatinib, tepotinib or savolitinib have yielded promising results in combination with an EGFR-TKI, like gefitinib or osimertinib. For this indication, anti-MET or -HGF antibodies can also represent a therapeutic option.
Clinical findings about other MET anomalies, like overexpression or rearrangement, have been disappointing and do not represent an avenue for clinical research at this time.
In light of the promising results obtained for patients whose NSCLCs harbor the METex14 mutation or MET amplification as a mechanism of resistance to EGFR-TKI, inclusion of such patients in clinical trials should be strongly encouraged.
Disclosure
Dr Olivier Bylicki reports personal fees from ROCHE, personal fees from MSD, personal fees from ASTRA-ZENECA, during the conduct of the study. Professor Christos Chouaid reports grants, personal fees from Roche, grants, personal fees from AZ, grants, personal fees from Amgen, grants, personal fees from BMS, grants, personal fees from MSD, grants, personal fees from Bayer, grants, personal fees from Janssen, grants, personal fees from Pierre Fabre, grants, personal fees from Mundi Pharma, grants, personal fees from Takeda, grants, personal fees from Pfizer, personnal feesfrom Novartis, outside the submitted work. The other authors report no other conflicts of interest in this work.
References
1. Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small–cell lung cancer. N Engl J Med. 2017;377(9):829–838. doi:10.1056/NEJMoa1704795
2. Sorai JC, Tan DSW, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small–cell lung cancer (ASCEND-4): a randomised, open-label, Phase 3 study. Lancet. 2017;389(10072):917–929. doi:10.1016/S0140-6736(17)30123-X
3. Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382(1):41–50. doi:10.1056/NEJMoa1913662
4. Wu YL, Cheng Y, Zhou X, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small–cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18(11):1454–1466. doi:10.1016/S1470-2045(17)30608-3
5. Park K, Tan EH, O’Byrne K, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016;17(5):577–589. doi:10.1016/S1470-2045(16)30033-X
6. Planchard D, Smit EF, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–1316. doi:10.1016/S1470-2045(17)30679-4
7. Wu YL, Yang JC, Kim DW. Phase II study of crizotinib in East Asian patients with ROS1-positive advanced non-small-cell lung cancer. J Clin Oncol. 2018;36(14):1405–1411. doi:10.1200/JCO.2017.75.5587
8. Drilon A, Oxnard G, Wirth L, et al. Registrational results of libretto-001: a phase 1/2 trial of loxo-292 in patients with ret fusion-positive lung cancers. J Thorac Oncol. 2019:14(10);S6–S7. doi:10.1016/j.jtho.2019.08.059
9. Farago AF, Le LP, Zheng Z, et al. Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J Thorac Oncol. 2015;10(12):1670–1674. doi:10.1097/01.JTO.0000473485.38553.f0
10. Mazières J, Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: results from the European EUHER2 cohort. Ann Oncol. 2016;27(2):281–286. doi:10.1093/annonc/mdv573
11. Jänne PA, van den Heuvel MM, Barlesi F, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical trial. JAMA. 2017;317(18):1844–1853. doi:10.1001/jama.2017.3438
12. Hirsch FR, Govindan R, Zvirbule Z, et al. Efficacy and safety results from a phase II, placebo-controlled study of onartuzumab plus first-line platinum-doublet chemotherapy for advanced squamous cell non-small-cell lung cancer. Clin Lung Cancer. 2017;18(1):43–49. doi:10.1016/j.cllc.2016.05.011
13. Ichimura E, Maeshima A, Nakajima T, et al. Expression of c-met/HGF receptor in human non-small cell lung carcinomas in vitro and in vivo and its prognostic significance. Jpn J Cancer Res. 1996;87(10):1063–1069. doi:10.1111/j.1349-7006.1996.tb03111.x
14. Siegfried JM, Weissfeld LA, Luketich JD, et al. The clinical significance of hepatocyte growth factor for non-small cell lung cancer. Ann Thorac Surg. 1998;66:1915–1918. doi:10.1016/S0003-4975(98)01165-5
15. Mazières J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30(8):1321–1328. doi:10.1093/annonc/mdz167
16. Guisier F, Dubos-Arvis C, Viñas F, et al. Efficacy and safety of anti-PD-1 immunotherapy in patients with advanced non small cell lung cancer with BRAF, HER2 or MET mutation or RET-translocation. GFPC 01-2018. J Thorac Oncol. 2020;pii:
17. Liu Y. The human hepatocyte growth factor receptor gene: complete structural organization and promoter characterization. Gene. 1998;215(1):159–169. doi:10.1016/S0378-1119(98)00264-9
18. Skead G, Govender D. Gene of the month: MET. J Clin Pathol. 2015;68(6):405–409. doi:10.1136/jclinpath-2015-203050
19. Giordano S, Ponzetto C, Di Renzo MF, et al. Tyrosine kinase receptor indistinguishable from the c-MET protein. Nature. 1989;339(6220):155–156. doi:10.1038/339155a0
20. Birchmeir C, Birchmeir W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915–925. doi:10.1038/nrm1261
21. Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3(1_suppl):S7–S19. doi:10.1177/1758834011422556
22. Finocchiaro G, Toschi L, Gianoncelli L, et al. Prognostic and predictive value of MET deregulation in non-small cell lung cancer. Ann Transl Med. 2015;3(6):83.
23. Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7(6):504–516. doi:10.1038/nrd2530
24. Edakuni G, Sasatomi E, Satoh T, et al. Expression of the hepatocyte growth factor/c-MET pathway is increased at the cancer front in breast carcinoma. Pathol Int. 2001;51(3):172–178. doi:10.1046/j.1440-1827.2001.01182.x
25. Otte JM, Schmitz F, Kiehne K, et al. Functional expression of HGF and its receptor in human colorectal cancer. Digestion. 2000;61(4):237–246. doi:10.1159/000007764
26. Sweeney P, El-Naggar AK, Lin SH, Pisters LL. Biological significance of c-MET over expression in papillary renal cell carcinoma. J Urol. 2002;168(1):51–55. doi:10.1016/S0022-5347(05)64830-6
27. Wu JG, Yu JW, Wu HB, et al. Expressions and clinical significances of c-MET, p-MET and E2f-1 in human gastric carcinoma. BMC Res Notes. 2014;7(6):6. doi:10.1186/1756-0500-7-6
28. Yano S, Nakagawa T. The current state of molecularly targeted drugs targeting HGF/MET. Jpn J Clin Oncol. 2014;44(1):9–12. doi:10.1093/jjco/hyt188
29. Qiao H, Hung W, Tremblay E, et al. Constitutive activation of MET kinase in non-small-cell lung carcinomas correlates with anchorage-independent cell survival. J Cell Biochem. 2002;86(4):665–677. doi:10.1002/jcb.10239
30. Soman NR, Correa P, Ruiz BA, Wogan GN. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc Natl Acad Sci U S A. 1991;88(11):4892–4896. doi:10.1073/pnas.88.11.4892
31. Zhao J, Chen J, Ma H, et al. MET kinase domain rearrangements across 10 cancer types. J Clin Oncol. 2019;37(15_suppl):Abstr3078. doi:10.1200/JCO.2019.37.15_suppl.3078
32. Hellman A, Zlotorynski E, Scherer SW, et al. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 2002;1(1):89–97. doi:10.1016/S1535-6108(02)00017-X
33. Kawakami H, Okamoto I, Okamoto W, et al. Targeting MET amplification as a new oncogenic driver. Cancers (Basel). 2014;6(3):1540–1552. doi:10.3390/cancers6031540
34. Albertson DG, Collins C, McCormick F, et al. Chromosome aberrations in solid tumors. Nat Genet. 2003;34(4):369–376. doi:10.1038/ng1215
35. Cheng DT, Mitchell T, Zehir A, et al. MSK-IMPACT: a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264. doi:10.1016/j.jmoldx.2014.12.006
36. Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20(12):1479–1484. doi:10.1038/nm.3729
37. Camidge DR, Otterson GA, Clark JW, et al. Crizotinib in patients (pts) with MET-amplified non-small cell lung cancer (NSCLC): updated safety and efficacy findings from a phase 1 trial. J Clin Oncol. 2018;36(15_suppl):Abstr 9062. doi:10.1200/JCO.2018.36.15_suppl.9062
38. Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27(10):1667–1674. doi:10.1200/JCO.2008.19.1635
39. Tanaka A, Sueoka-Aragane N, Nakamura T, et al. Co-existence of positive MET FISH status with EGFR mutations signifies poor prognosis in lung adenocarcinoma patients. Lung Cancer. 2012;75(1):89–94. doi:10.1016/j.lungcan.2011.06.004
40. Krishnaswamy S, Kanteti R, Duke-Cohan JS, et al. Ethnic differences and functional analysis of MET mutations in lung cancer. Clin Cancer Res. 2009;15(18):5714–5723. doi:10.1158/1078-0432.CCR-09-0070
41. Ma P, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-MET and its therapeutic inhibition with SU11274 and small interfering RNA in non–small cell lung cancer. Cancer Res. 2005;65(4):1479–1488. doi:10.1158/0008-5472.CAN-04-2650
42. Onozato R, Kosaka T, Kuwano H, et al. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. 2009;4(1):5–11. doi:10.1097/JTO.0b013e3181913e0e
43. Kong-Beltran M, Seshagiri S, Zha J, et al. Somatic mutations lead to an oncogenic deletion of MET in lung cancer. Cancer Res. 2006;66(1):283–289. doi:10.1158/0008-5472.CAN-05-2749
44. The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi:10.1038/nature13385
45. Awad MM, Oxnard GR, Jackman DM, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-MET overexpression. J Clin Oncol. 2016;34(7):721–730. doi:10.1200/JCO.2015.63.4600
46. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–859. doi:10.1158/2159-8290.CD-15-0285
47. Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5(8):842–849. doi:10.1158/2159-8290.CD-14-1467
48. Song Z, Wang X, Zheng Y, Su H, Zhang Y. MET gene amplification and overexpression in Chinese non-small-cell lung cancer patients without EGFR mutations. Clin Lung Cancer. 2017;18(2):213–219. doi:10.1016/j.cllc.2016.09.011
49. Sterlacci W, Fiegl M, Gugger M, Bubendorf L, Savic S, Tzankov A. MET overexpression and gene amplification: prevalence, clinico-pathological characteristics and prognostic significance in a large cohort of patients with surgically resected NSCLC. Virchows Arch. 2017;471(1):49–55. doi:10.1007/s00428-017-2131-1
50. Dziadziuszko R, Wynes MW, Singh S, et al. Correlation between MET gene copy number by silver in situ hybridization and protein expression by immuno-histochemistry in non-small cell lung cancer. J Thorac Oncol. 2012;7(2):340–347. doi:10.1097/JTO.0b013e318240ca0d
51. Watermann I, Schmitt B, Stellmacher F, et al. Improved diagnostics targeting c-MET in non-small cell lung cancer: expression, amplification and activation? Diagn Pathol. 2015;10(1):130. doi:10.1186/s13000-015-0362-5
52. Guo B, Cen H, Tan X, et al. Prognostic value of MET gene copy number and protein expression in patients with surgically resected non-small cell lung cancer: a meta-analysis of published literatures. PLoS One. 2014;9(6):e99399. doi:10.1371/journal.pone.0099399
53. Pyo J-S, Kang G, Cho WJ, Choi SB. Clinicopathological significance and concordance analysis of c-MET immunohistochemistry in non-small cell lung cancers: a meta-analysis. Pathol Res Pract. 2016;212(8):710–716. doi:10.1016/j.prp.2016.05.006
54. Wang WX, Xu C, Chen Y, et al. MET gene fusions in non-small cell lung cancer (NSCLC) in the Chinese population: a multicenter study. J Clin Oncol. 2018;15(Suppl):e13539. doi:10.1200/JCO.2018.36.15_suppl.e13539
55. Okuda K, Sasaki H, Yukiue H, et al. MET gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008;99(11):2280–2285. doi:10.1111/j.1349-7006.2008.00916.x
56. Frampton GM, Ali SM, Rosenzweig M, et al. Comprehensive genomic profiling (CGP) of advanced cancers to identify MET exon 14 alterations that confer sensitivity to MET inhibitors. J Clin Oncol. 2015;33(15_suppl):Abstr11007. doi:10.1200/jco.2015.33.15_suppl.11007
57. Sacco JJ, Clague MJ. Dysregulation of the MET pathway in non-small cell lung cancer: implications for drug targeting and resistance. Transl Lung Cancer Res. 2015;4(3):242–252. doi:10.3978/j.issn.2218-6751.2015.03.05
58. Kim JH, Kim HS, Kim BJ. Prognostic value of MET copy number gain in non-small-cell lung cancer: an updated meta-analysis. J Cancer. 2018;9(10):1836–1845. doi:10.7150/jca.24980
59. Dimou A, Non L, Chae YK, Tester WJ, Syrigos KN. MET gene copy number predicts worse overall survival in patients with non-small cell lung cancer (NSCLC); a systematic review and meta-analysis. PLoS One. 2014;9(9):e107677. doi:10.1371/journal.pone.0107677
60. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi:10.1126/science.1141478
61. Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–1400. doi:10.1038/nm.3388
62. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi:10.1126/scitranslmed.3002003
63. Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. doi:10.1158/1078-0432.CCR-12-2246
64. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725–737. doi:10.1038/s41416-019-0573-8
65. Le X, Puri S, Negrao MV, et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin Cancer Res. 2018;24(24):6195–6203. doi:10.1158/1078-0432.CCR-18-1542
66. Papadimitrakopoulou V, Wu YL, Han JY, et al. LBA51 analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. 2018;29:viii741. doi:10.1093/annonc/mdy424.064
67. Ramalingam SS, Cheng Y, Zhou C, et al. LBA50 mechanisms of acquired resistance to first-line osimertinib: preliminary data from the Phase III FLAURA study. Ann Oncol. 2018;29:viii740. doi:10.1093/annonc/mdy424.063
68. Wu Y-L, Zhang L, Kim D-W, et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor- dysregulated non-small-cell lung cancer. J Clin Oncol. 2018;36(31):3101–3109. doi:10.1200/JCO.2018.77.7326
69. Sequist LV, Lee JS, Han JY, et al. Abstract CT033: TATTON phase Ib expansion cohort: osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-amplified NSCLC after progression on prior third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). Atlanta, GA:
70. Oxnard GR, Yang JC, Yu H, et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol. 2020;31(4):507–516. doi:10.1016/j.annonc.2020.01.013
71. Tong JH, Yeung SF, Chan AW, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22(12):3048–3056. doi:10.1158/1078-0432.CCR-15-2061
72. Gow CH, Hsieh MS, Wu SG, Shih JY. A comprehensive analysis of clinical outcomes in lung cancer patients harboring a MET exon 14 skipping mutation compared to other driver mutations in an East Asian population. Lung Cancer. 2017;103:82–89. doi:10.1016/j.lungcan.2016.12.001
73. Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol. 2017;12(1):15–26. doi:10.1016/j.jtho.2016.10.014
74. Cui JJ. Targeting receptor tyrosine kinase MET in cancer: small molecule inhibitors and clinical progress. J Med Chem. 2014;57(11):4427–4453. doi:10.1021/jm401427c
75. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103. doi:10.1038/nrc3205
76. Backes A, Zech B, Felber B, Klebl B, Müller G. Small-molecule inhibitors binding to protein kinases. Part I: exceptions from the traditional pharmacophore approach of type I inhibition. Expert Opin Drug Discov. 2008;3(12):1409–1425. doi:10.1517/17460440802579975
77. Katayama R, Aoyama A, Yamori T, et al. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013;73(10):3087–3096. doi:10.1158/0008-5472.CAN-12-3256
78. Wakelee H, Zvirbule Z, De Braud F, et al. Efficacy and safety of onartuzumab in combination with first-line bevacizumab- or pemetrexed-based chemotherapy regimens in advanced non-squamous non-small-cell lung cancer. Clin Lung Cancer. 2017;18(1):50–59. doi:10.1016/j.cllc.2016.09.013
79. Spigel DR, Edelman MJ, O’Byrne K, et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: METLung. J Clin Oncol. 2017;35(4):412–420. doi:10.1200/JCO.2016.69.2160
80. Spigel DR, Ervin TJ, Ramlau RA, et al. Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non- small-cell lung cancer. J Clin Oncol. 2013;31(32):4105–4114. doi:10.1200/JCO.2012.47.4189
81. Janne PA, Shaw AT, Camidge DR, et al. Combined pan-HER and ALK/ROS1/MET inhibition with dacomitinib and crizotinib in advanced non- small cell lung cancer: results of a phase I study. J Thorac Oncol. 2016;11(5):737–747. doi:10.1016/j.jtho.2016.01.022
82. Sequist LV, von Pawel J, Garmey EG, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29(24):3307–3315. doi:10.1200/JCO.2010.34.0570
83. Scagliotti G, von Pawel J, Novello S, et al. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol. 2015;33(24):2667–2674. doi:10.1200/JCO.2014.60.7317
84. Yoshioka H, Azuma K, Yamamoto N, et al. A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-MET inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study). Ann Oncol. 2015;26(10):2066–2072. doi:10.1093/annonc/mdv288
85. Neal JW, Dahlberg SE, Wakelee HA, et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): a randomised, controlled, open-label, multicentre, phase 2 trial. Lancet Oncol. 2016;17(12):1661–1671. doi:10.1016/S1470-2045(16)30561-7
86. Cho JH, Ku BM, Sun JM, et al. KIF5B-MET gene rearrangement with robust antitumor activity in response to crizotinib in lung adenocarcinoma. J Thorac Oncol. 2018;13(3):e29–e31. doi:10.1016/j.jtho.2017.10.014
87. Landi L, Chiari R, Tiseo M, et al. Crizotinib in MET deregulated or ROS1 rearranged pretreated non-small-cell lung cancer (METROS): a phase II, prospective, multicentre, two-arms trial. Clin Cancer Res. 2019;25(24):7312–7319. doi:10.1158/1078-0432.CCR-19-0994
88. Moro-Sibilot D, Cozic N, Pérol M, et al. Crizotinib in c-MET- or ROS1-positive NSCLC: results of the AcSé phase II trial. Ann Oncol. 2019;30(12):1985–1991. doi:10.1093/annonc/mdz407
89. Ou SI, Govindan R, Eaton KD, et al. Phase I results from a study of crizotinib in combination with erlotinib in patients with advanced nonsquamous non-small cell lung cancer. J Thorac Oncol. 2017;12(1):145–151. doi:10.1016/j.jtho.2016.09.131
90. Reckamp KL, Frankel PH, Ruel N, et al. Phase II trial of cabozantinib plus erlotinib in patients with advanced epidermal growth factor receptor (EGFR)-mutant non-small cell lung cancer with progressive disease on epidermal growth factor receptor tyrosine kinase inhibitor therapy: a California Cancer Consortium phase II trial (NCI 9303). Front Oncol. 2019;9(11):132.
91. Scaglotti GV, Moro-Sibilot D, Kollmeier J, et al. A randomized, controlled, open label phase II study of erlotinib (E) with or without the MET antibody emibetuzumab (Emi) as first-line treatment for EGFR mt non-small cell lung cancer (NSCLC) patients who have disease control after an 8-week lead-in treatment with erlotinib. J Clin Oncol. 2017;15(37 Suppl):Abstr9019.
92. Wu Y, Zhou J, Lu S, et al. Phase 2 study of tepotinib + gefitinib (TEP+GEF) in MET-positive (MET+)/epidermal growth factor receptor (EGFR)-mutant (MT) non-small cell lung cancer (NSCLC). Ann Oncol. 2018;29:ix159. doi:10.1093/annonc/mdy425.026
93. Azuma K, Hirashima T, Yamamoto N, et al. Phase II study of erlotinib plus tivantinib (ARQ 197) in patients with locally advanced or metastatic EGFR mutation-positive non-small-cell lung cancer just after progression on EGFR-TKI, gefitinib or erlotinib. ESMO Open. 2016;1(4):e000063. doi:10.1136/esmoopen-2016-000063
94. Camidge DR, Moron T, Demedts I, et al. A randomized, open label, phase 2 study of emibetuzumab plus erlotinib (LY+E) and emibetuzumab monotherapy (LY) in patients with acquired resistance to erlotinib and MET diagnostic positive (MET DX+) metastatic NSCLC. J Clin Oncol. 2016;34(15_suppl):Abstr9070. doi:10.1200/JCO.2016.34.15_suppl.9070
95. Camidge DR, Barlesi F, Goldman JW. Results of the phase 1b study of ABBV-399 (telisotuzumab vedotin; teliso-v) in combination with erlotinib in patients with c-Met+ non-small cell lung cancer by EGFR mutation status. J Clin Oncol. 2019;37(15_suppl):Abstr3011. doi:10.1200/JCO.2019.37.15_suppl.3011
96. Haura EB, Cho BC, Lee JS, et al. JNJ-61186372 (JNJ-372), an EGFR-cMet bispecific antibody, in EGFR-driven advanced non-small cell lung cancer (NSCLC). J Clin Oncol. 2019;37(15_suppl):Abstr9009. doi:10.1200/JCO.2019.37.15_suppl.9009
97. Awad MM, Leonardi GC, Kravets S, et al. Impact of MET inhibitors on survival among patients with non-small cell lung cancer harboring MET exon 14 mutations: a retrospective analysis. Lung Cancer. 2019;133:96–102. doi:10.1016/j.lungcan.2019.05.011
98. Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol. 2016;11(9):1493–1502. doi:10.1016/j.jtho.2016.06.004
99. Katakura S, Kobayashi N, Somekawa K, Masumoto N, Kudo M, Kaneko T. Non-small cell lung cancer with mesenchymal-epithelial transition gene exon 14 skipping mutation treated with crizotinib. Respirol Case Rep. 2019;7(7):e00453.
100. Drilon A, Clark JW, Weiss J, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Met. 2020;26(1):47–51.
101. Esaki T, Hirai F, Makiyama A, et al. Phase I dose-escalation study of capmatinib (INC280) in Japanese patients with advanced solid tumors. Cancer Sci. 2019;110(4):1340–1351. doi:10.1111/cas.13956
102. Wolf J, Seto T, Han JH, et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): efficacy data from the phase II GEOMETRY mono-1 study. J Clin Oncol. 2019;37(15_suppl):Abstr9004. doi:10.1200/JCO.2019.37.15_suppl.9004
103. Paik PK, Veillon R, Cortot AB, et al. Phase II study of tepotinib in NSCLC patients with MET ex14 mutations. J Clin Oncol. 2019;37(15_suppl):Abstr9005. doi:10.1200/JCO.2019.37.15_suppl.9005
104. Yoshimura K, Inoue Y, Tsuchiya K, et al. Elucidation of the relationships of MET protein expression and gene copy number status with PD-L1 expression and the immune microenvironment in non-small cell lung cancer. Lung Cancer. 2020;141:21–31. doi:10.1016/j.lungcan.2020.01.005
105. Sabari JK, Leonardi GC, Shu CA, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. 2018;29(10):2085–2091. doi:10.1093/annonc/mdy334
106. Neumunaitis J, Borghaei H, Akerley W, et al. P2.06-014 phase 2 study of glesatinib or sitravatinib with nivolumab in non-small cell lung cancer (NSCLC) after checkpoint inhibitor therapy. J Thorac Oncol. 2016;12(1):S1078. doi:10.1016/j.jtho.2016.11.1507
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.