")
Back to Journals » OncoTargets and Therapy » Volume 14
Targeting the IDO-BCL2A1-Cytochrome c Pathway Promotes Apoptosis in Oral Squamous Cell Carcinoma
Authors Zheng Q, Gan G, Gao X, Luo Q , Chen F
Received 25 October 2020
Accepted for publication 4 February 2021
Published 4 March 2021 Volume 2021:14 Pages 1673—1687
DOI https://doi.org/10.2147/OTT.S288692
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Qiaoping Zheng,1,* Guifang Gan,1,* Xianfu Gao,2 Qingqiong Luo,1 Fuxiang Chen1
1Department of Clinical Immunology, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200011, People’s Republic of China; 2Shanghai Profleader Biotech Co., Ltd., Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fuxiang Chen
Department of Clinical Immunology, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200011, People’s Republic of China
Email [email protected]
Purpose: Indolamine 2,3-dioxygenase (IDO) is the rate limiting enzyme of tryptophan degradation and is a negative prognostic factor in oral squamous cell carcinoma (OSCC) patients, while the underlying molecular mechanism remains unclear. This research aimed to explore the IDO expression and its biological functions in OSCC.
Materials and Methods: IDO expression was analyzed by qPCR, Western blots, and immunohistochemistry (IHC) in OSCC cell lines and tissue specimens. Tryptophan and kynurenine content were determined by UPLC-MS/MS in serum samples of OSCC patients and healthy controls. Oncomine databases and Kaplan-Meier survival analyses were used to identify the IDO expression and its correlation with OSCC prognosis. Cell counting, CCK8 assay, flow cytometry, cell cycle, and EdU incorporation assays were used to assess the effect of IDO inhibition on OSCC growth either by shRNA or the IDO-specific inhibitor (epacadostat) in vitro. An OSCC xenograft mouse model was established to verify the predicted function of IDO inhibition in vivo. Mechanistically, an 84-gene apoptosis PCR array and rescue experiment were used to characterize the underlying mechanism involved in IDO-regulated apoptosis in OSCC.
Results: IDO expression was upregulated in OSCC cell lines and tissues and was negatively correlated with OSCC progression. Lentivirus-mediated IDO knockdown and epacadostat significantly reduced viability and promoted apoptosis of OSCC cells in vitro and in vivo. The apoptosis PCR array identified BCL2 related protein A1 (BCL2A1) as the most obviously changed gene at the transcriptional level. IDO inhibition downregulated BCL2A1 expression, increased the expression and translocation of cytochrome c, thus promoted apoptosis in OSCC. Overexpression of BCL2A1 reversed the pro-apoptotic effect of IDO inhibition.
Conclusion: The present results revealed that IDO directly affect the growth of OSCC cells by regulating BCL2A1 expression. IDO and the IDO-BCL2A1-cytochrome c axis may be potential therapeutic targets for OSCC.
Keywords: oral squamous cell carcinoma, indolamine 2, 3-dioxygenase, apoptosis, BCL2A1
Introduction
Head and neck cancer is the sixth most common cancer in the world, and oral squamous cell carcinoma (OSCC) accounts for 90% of the head and neck cancer.1,2 More than 300,000 patients are diagnosed with OSCC worldwide annually, and 25–50% of OSCC patients experience relapse regardless of treatment, location, or stage.3 Currently available therapies, including surgical resection, radiotherapy, and chemotherapy, have achieved modest success, but the 5-year overall survival rate of OSCC patients remains at approximately 50%.4 Therefore, seeking specific targets and designing new effective strategies for OSCC therapy is of great importance.
Tryptophan (Trp) is an essential amino acid that regulates protein synthesis and immune evasion during embryo implantation,5 thereby maintaining the stability of the internal environment.6 Indolamine 2,3-dioxygenase (IDO) catalyzes the rate limiting step of the degradation of Trp in the kynurenine (Kyn) pathway.7,8 In cancers, IDO activation boosts the formation of the immunosuppressive microenvironment, thereby inhibiting activation of anti-tumor immune responses. The mechanism underlying IDO-related immunosuppression relies on two processes: (1) overexpression of IDO in dendritic cells or tumor cells depletes the L-tryptophan in the tumor microenvironment, thus inhibiting cell cycle progression in lymphocytes through the general control nonderepressible 2 (GCN2) pathway;9,10 and (2) accumulation of toxic IDO metabolites (ie, Kyn) induces apoptosis of lymphocytes and differentiation of regulatory T cells.7,11 Treatment with IDO inhibitors decreases the tumor mass and stimulates anti-tumor immune responses.12 In addition to its immunosuppressive function, IDO has an effect on tumor cell growth, which remains to be explored. Increased expression of IDO promotes tumor growth and metastasis by activating the IDO-aryl hydrocarbon receptor (AHR) axis in primary breast cancer.13,14 IDO and its downstream metabolites promote colon cancer cell proliferation by activating β-catenin signaling.15 IDO is associated with poor clinical outcomes and chemotherapeutic responses in various malignant diseases.15–19 Elevated IDO expression is a significant negative prognostic factor in OSCC patients, especially in those undergoing adjuvant radiochemotherapy17 and nimotuzumab therapy.20 However, the underlying molecular mechanism remains to be explored.
In this study, we found that IDO was upregulated in OSCC cell lines and tissues and was negatively correlated with OSCC progression. IDO inhibition markedly reduced OSCC cell viability and promoted apoptosis of OSCC cells in vitro and in vivo. Mechanistically, IDO may promote OSCC by regulating BCL2A1 expression, and targeting the IDO-BCL2A1-cytochrome c pathway promoted apoptosis of OSCC cells. The present findings may reveal the mechanisms underlying the regulatory role of IDO in OSCC and suggest that IDO and the IDO-BCL2A1-cytochrome c axis are potential therapeutic targets for OSCC.
Materials and Methods
Animals
Four-week-old pathogen-free male BALB/c nude mice purchased from Shanghai Laboratory Animal Center (Shanghai, China) were reared in the Specific Pathogen Free Animal Center of the Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine.
Cell Lines
The human immortalized oral epithelial cells (HIOEC) and OSCC cell lines (WSU-HN4, WSU-HN6, CAL27, and WSU-HN30) used in the study were provided by the Oral Oncology Department, Ninth People’s Hospital. HIOEC cells were cultured in defined keratinocyte serum-free medium (Gibco, New York, NY, USA). OSCC cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. All cells were cultured in a humidified atmosphere containing 5% CO2 at 37°C.
Immunohistochemistry (IHC)
IHC was used to determine IDO expression in clinical samples. Before staining, paraffin-embedded OSCC tissue sections were placed in a 60°C incubator for 2 hours. Then, they were deparaffinized in xylene and soaked in the reducing concentrations of ethanol (90%, 80%, and 70%) to rehydrate. Sections were then boiled in citric acid buffer for 8 min to retrieve antigen, blocked in 5% goat serum for 20 min, and incubated in 3% H2O2 to suppress endogenous peroxidase activity. Sections were incubated in the 100-fold diluted anti-IDO (CST) antibody at 4°C overnight, rinsed with PBS the next day, and then incubated with the secondary antibody (peroxidase-conjugated goat anti-rabbit antibody) at room temperature for 1 hour. The sections were developed in DAB solution and the staining was observed under a microscope (Olympus, Japan). The staining was terminated with distilled water and counter-stained with hematoxylin. Light microscopy was used to observe the sections.
RNA Extraction, PCR Array and qPCR Validation
To determine the mRNA expression of IDO and analyze gene expression after IDO knockdown, a human apoptosis PCR assay (Wcgene Biotech, Shanghai, China) and qPCR were performed according to the manufacturer’s protocols. HIOEC and OSCC cells were collected using the TRIzol reagent (Invitrogen, USA) to obtain total RNA, and the PrimeScript RT Reagent kit (TaKaRa, Shiga, Japan) was used for reverse transcription. Genes that showed >2-fold change in expression level in the two lentivirus-mediated IDO knockdown OSCC cell lines were further analyzed by qPCR. qPCR was performed using the SYBR Premix Ex Taq II kit (TaKaRa, Shiga, Japan) according to the manufacturer’s protocols. Considering the difference of total RNA among different samples, β-actin was used as an endogenous control. The gene transcription levels were analyzed using 2−ΔΔCt. Primer sequences were as follows: IDO sense, 5ʹ-AGTTCTGGGATGCATCACCA-3ʹ, antisense, 5ʹ-ACTGCAGTCTCCATCACGAA-3ʹ; BCL2A1 sense, 5ʹ-GTCTCCATCACGAA-3ʹ, antisense, 5ʹ-CGCAACATTTTGTAGCACTCTG-3ʹ; Caspase-5 sense, 5ʹ-TCACCTGCCTGCAAGGAATG-3ʹ, antisense, 5′-TCTTTTCGTCAACCACAGTGTAG-3′; TNFRSF11B sense, 5′-GCGCTCGTGTTTCTGGACA-3′, antisense, 5′-AGTATAGACACTCGTCACTGGTG-3′; TNFRSF9 sense, 5′-TCCACCAGCAATGCAGAGTG-3′, antisense, 5′-CCAAAGCAACAGTCTTTACAACC-3′; CD27 sense, 5′-CAGAGAGGCACTACTGGGCT-3′, antisense 5′-CGGTATGCAAGGATCACACTG-3′; CD70 sense, 5′-GCTTTGGTCCCATTGGTCG-3′, antisense 5′-CGTCCCACCCAAGTGACTC-3′; DAPK sense, 5′-AATGGTGTTTACTACCTGCACTC-3′, antisense 5′-CTCAGGAGCGACAAACTCTGG-3′; β-actin sense, 5′-GCGGGAAATCGTGCGTGACAT-3′, antisense 5′-GATGGAGTTGAAGGTAGTTTCG-3′.
Western Blot Analysis
Total protein determinations used the RIPA lysis buffer (Beyotime Biotechnology, Nanjing, China) supplemented with 1 mM PMSF on ice for 30 min; proteins were quantified by the Nanodrop assay (Thermo) and denatured using a 100°C water bath. Equal amounts of total protein calculated according to the concentration of each sample were separated on 12.5% SDS PAGE. The protein bands were transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA). The non-specific bands were blocked by 5% nonfat milk powder dissolved in Tris-buffered saline (TBS)/Tween20 (TBS/T) at a final concentration of 5% used to block the non-specific sites. The blots were detected using specific antibodies overnight at 4°C and washed three times by TBST followed by incubated in HRP-conjugated secondary antibody for 1 hour at room temperature. Finally, all immunoreactive signals were analyzed with the Allcam detection system (Tanon, Canton, MA, USA).
Serum Sample Collection
Serum was collected from OSCC patients (n=34) and healthy controls (n=35) during January 2018 and December 2018. None of the patients had received previous chemotherapy or radiotherapy, nor did they have a history of prior malignancies. Healthy controls were volunteers without oral diseases, autoimmune disorders, or immunodeficiency. The TNM staging was according to the standards of the American Joint Committee on Cancer (AJCC). All subjects provided written informed consents. The clinical information of the OSCC patients and healthy controls is summarized in Table 1. There was no statistically significant difference in average age between patients and healthy controls. Each blood sample was clotted for 45 min at room temperature prior to centrifugation at 3000 × g for 10 min. The serum was collected into a 1.5 mL centrifuge tube and frozen at −80°C before analysis.
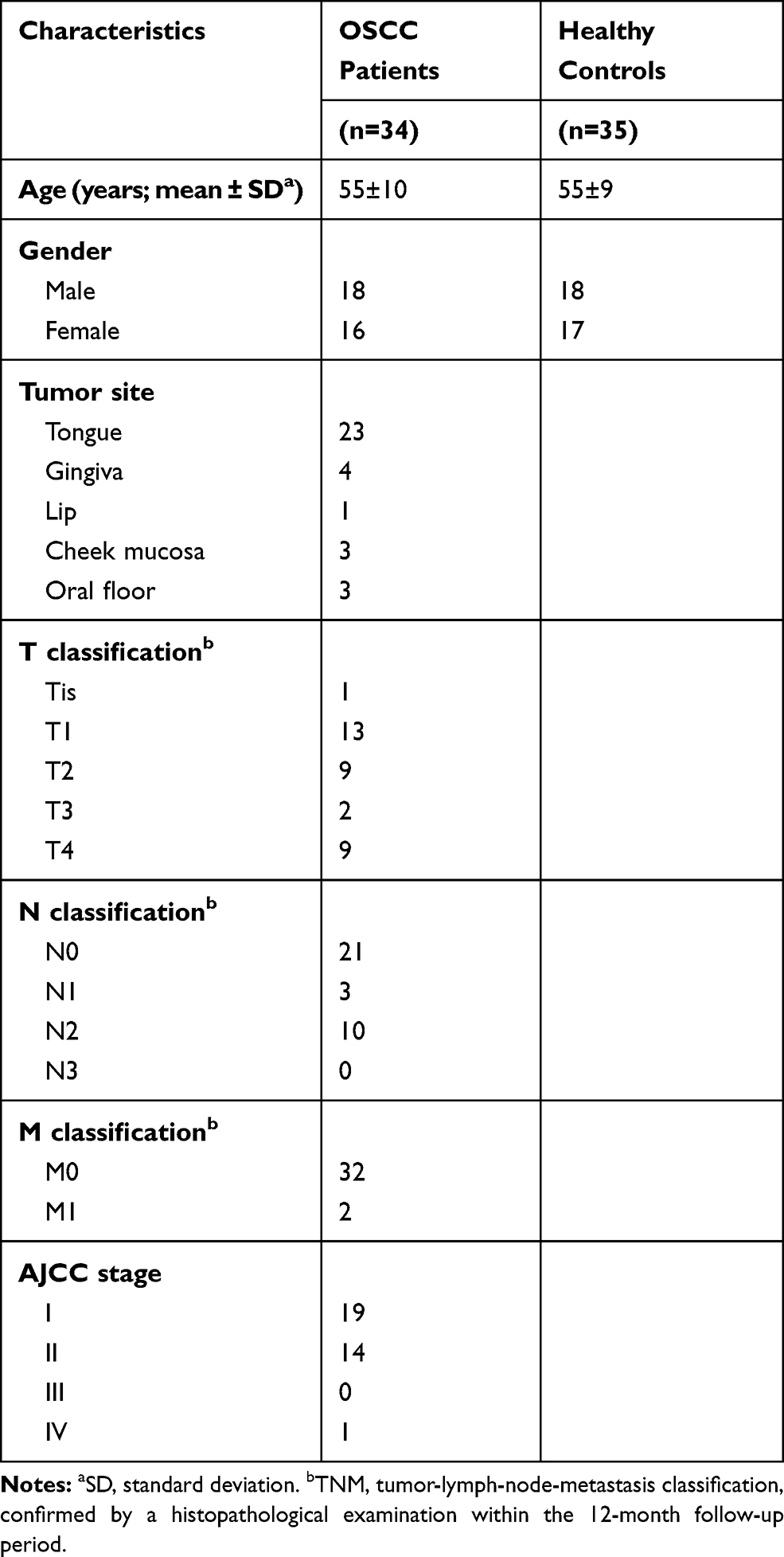 |
Table 1 Clinical Characteristics of the Patients with OSCC and Healthy Controls |
Gene Knockdown
Short hairpin RNAs (shRNAs) (Sangon Biotech, Shanghai, China) was used to knockdown the expression of IDO in OSCC cells. The sequences of the shRNA duplex targeting IDO (IDO-SH1) were as follows: sense, 5ʹ-TGGATGTTCATTGCTAAAC-3ʹ, antisense, 5ʹ-TAAAGCCTGAGGAGGCCCTGGG-3′. Those targeting IDO-SH2 were as follows: sense, 5′-ACGATCATGTGAACCCAAA-3′, antisense, 5′-TTGTTGGCAATTTCCATCCAAG-3′. The sequence of the negative control (mock) vector was as follows: 5′-GAAGCAGCACGACTTCTTC-3′. After 12-hour lentivirus particle transfection, stable transfected cells were selected with puromycin for another 7 days. The efficiency of gene knockdown was confirmed by qPCR and Western blotting.
Cell Proliferation
Cells were seeded into 96-well plates at a density of 5 × 103/well. After 1, 2, or 4 days, the culture medium was removed and the cell counting kit-8 (CCK8) solution (Dojindo, Japan) (10%/well) was added into each well to measure cell proliferation. After incubation for 2 hours, absorbance was measured at a wavelength of 450 nm on a microplate reader (Bio-Rad).
Cell Counting
Cells were seeded into 6-well plates at a density of 4 × 105/well). After 1, 2, or 4 days, cells were digested by trypsinization and suspended in DMEM. Cell number was counted under light microscopy using a Neubauer improved chamber.
Apoptosis and Cell Cycle Analysis
IDO knockdown (IDO-SH1 and IDO-SH2) and control CAL27 cells (mock) were seeded in 6-well plates, and after 24 hours, when reaching 70% confluence, they were harvested by trypsinization for apoptosis and cell cycle analysis. OSCC cells (2 × 105 cells/well) were seeded in 6-well plates. After 24 hours, the medium was replaced with fresh medium containing 0, 20, or 40 μmol/L (μM) epacadostat for another 24 hours. Cells were prepared for apoptosis and cell cycle analysis. For apoptosis analysis, cells were stained with Annexin V-APC and 7-AAD. For cell cycle analysis, cells were fixed with cold 70% ethanol overnight followed by staining with PI/RNase staining buffer according to the manufacturer’s procedures. Apoptosis rate and cell cycle distribution were determined by a flow cytometry (BD FACSCanto II, San Francisco, CA, USA).
EdU Incorporation Assay
The cell preparation was as described for apoptosis and cell cycle analysis. To test DNA replication, duplicate DNA was marked using an EdU (5-ethynyl-2ʹ-deoxyuridine) Detection Kit (Beyotime Biotechnology) according to the manufacturer’s instructions. The EdU incorporation rate was measured using immunofluorescence (Olympus fluorescent microscope, Japan).
Transient Plasmid Transfection
IDO knockdown (IDO-SH1 and IDO-SH2) and control CAL27 cells were transfected with the vectors pTSB (empty vector) and pTSB-BCL2A1 (BCL2A1 overexpression plasmid), which were purchased from TranSheepBio (Shanghai, China). The BCL2A1 sequence was as follows: gaattcgccaccatgacagactgtgaatttggatatatttacaggctggctcaggactatctgcagtgcgtcctacagataccacaacctggatcaggtccaagcaaaacgtccagagtgctacaaaatgttgcgttctcagtccaaaaagaagtggaaaagaatctgaagtcatgcttggacaatgttaatgttgtgtccgtagacactgccagaacactattcaaccaagtgatggaaaaggagtttgaagacggcatcattaactggggaagaattgtaaccatatttgcatttgaaggtattctcatcaagaaacttctacgacagcaaattgccccggatgtggatacctataaggagatttcatattttgttgcggagttcataatgaataacacaggagaatggataaggcaaaacggaggctgggggaaatggcacaatcacacacctatgctggtagagtcagtggcccacaagaagaggaaaatggctttgggatcc. Aliquots containing 4 μg of plasmid/vector and 10 μL PEI were used per well (6-well plates). After 6 hours, the medium was replaced with antibiotics-free culture medium. The transfection efficiency was estimated to be 70% after using a GFP (green fluorescent protein) expressing plasmid. After 48 hours, cells were collected and lysed to obtain total protein and prepared for Western blot analysis.
Cellular Distribution of Cytochrome c
Confocal microscopy was used to show the distribution of cytochrome c. Cells were incubated with MitoTracker dye (MitoTracker Deep Red, Molecular Probes, Invitrogen) for 45 min in an incubator in the dark. Then, the slides were fixed with 4% formaldehyde at room temperature for 30 min and blocked with 5% normal goat serum in PBS containing 0.2% Triton X-100 (TPBS) for 1 hour at room temperature to reduce nonspecific labeling. The fixed slides were stained with a primary anti-cytochrome c antibody (12963S; Cell Signaling Technology, Danvers, MA, USA) at 4°C for 12–16 hours, then with an AF-561-labeled secondary antibody at 37°C for 60 min. The cells were also subjected to 4′,6-diamidino-2-phenylindole staining to visualize the nuclei. Images were obtained using different excitation filters and merged.
Mouse Xenograft Models
To assess the effect of IDO knockdown on OSCC tumor xenograft progression, 5 × 106 control cells and IDO-SH1 CAL27 cells were injected into the right side and left side, respectively, on the mouse inguen and allowed to grow until visible (five mice total). Tumors were measured every 7 days for 49 days with Vernier digital calipers.
To verify the effect of the IDO inhibitor in vivo, 5 × 106 CAL27 cells were injected into the left side and right side of the mouse inguen. When the tumor volume reached approximately 80 mm3, mice were randomly distributed to control and treated groups (five mice in each group), and treatment was initiated. The treated and control groups received intraperitoneal injection of epacadostat (100 mg/kg) and DMSO, respectively, every 3 days for a total of five injections and tumors were measured with Vernier digital calipers on the same day.
Mice were sacrificed at the end of the experiments. Tumors obtained were fixed with 4% phosphate-buffered paraformaldehyde and embedded in paraffin for IHC.
Statistical Analysis
All results were verified by at least three independent experiments and are presented as the mean ± SD. Statistical analyses between two groups were performed with t-tests using SPSS version 23.0 software.
Results
The Expression Level of IDO is Correlated with OSCC Progression
IDO expression in HIOEC and OSCC cells was analyzed by qPCR and Western blot analysis. The study included four OSCC cell lines: CAL27, WSU-HN4, WSU-HN6, and WSU-HN30. The qPCR results indicated that IDO mRNA expression of the OSCC cells was remarkably higher than that of HIOEC cells (Figure 1A, p < 0.01). The overexpression of IDO in OSCC cells was confirmed by Western blot analysis (Figure 1B). Higher expression at the transcriptional and translational levels suggested that IDO played a functional role in OSCC. IDO expression in tissues was assessed by collecting four pairs of OSCC tissue specimens and normal oral epithelial tissue specimens. Samples were fixed and embedded in paraffin, and tissue sections were processed as described in Materials and methods. IDO expression in human tissues was determined by IHC staining, which verified that IDO expression was higher in OSCC tissues than that in peritumoral tissues (Figure 1D), and this was confirmed by the Oncomine database (Figure 1E and S1). Trp and Kyn were analyzed by UPLC-MS/MS in serum samples from 34 patients with OSCC (55.31 ± 10.43 years old) and 35 healthy controls (55.46 ± 9.13 years old) (Table 1). As shown in Figure 1C, the Kyn/Trp ratio was higher in OSCC patients than that in healthy controls (9.87 ± 2.57% and 8.73 ± 1.48%, respectively, p < 0.05), indicating that IDO was activated in OSCC. Kaplan-Meier survival analysis showed that OSCC patients with higher IDO expression had a worse prognosis than those with lower IDO expression (Figure 1F, p < 0.05), suggesting that IDO promoted OSCC progression.
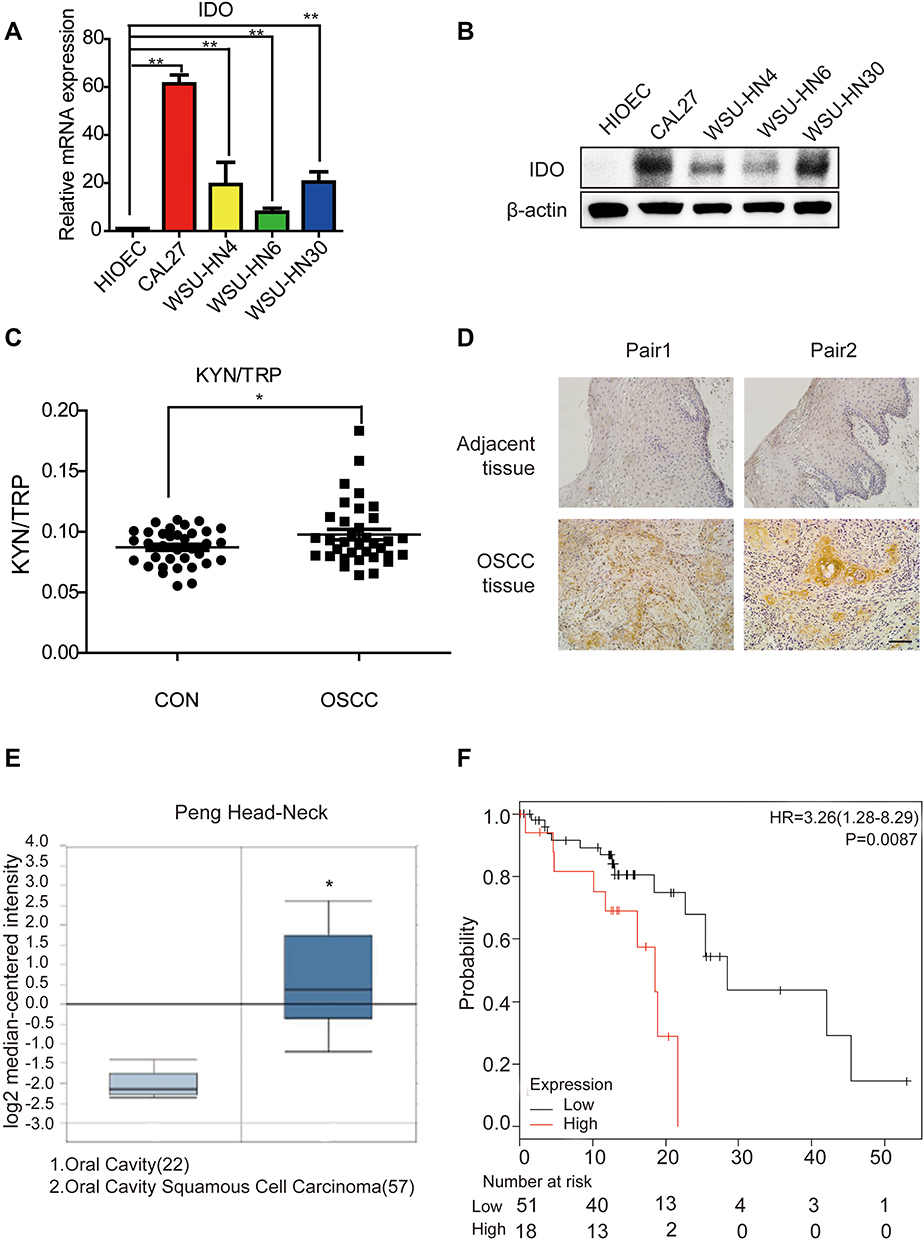 |
Figure 1 The expression level of IDO is correlated with oral squamous cell carcinoma progression. (A) Expression analysis of IDO by qPCR in oral squamous cell carcinoma cell lines. Analysis of IDO expression in normal (HIOEC) and OSCC (CAL27, WSU-HN4, WSU-HN6 and WSU-HN30) cell lines. (B) Expression analysis of IDO by Western blot in OSCC cell lines. Analysis of IDO expression in normal (HIOEC) and OSCC (CAL27, WSU-HN4, WSU-HN6 and WSU-HN30) cell lines. The Western blot shown here is a representative of three independent experiments. (C) The ratio of KYN (Kynuenine)/TRP (Tryptophan) in OSCC patients. TRP and KYN were analyzed by UPLC-MS/MS. (D) IDO immunohistochemistry analysis of OSCC or paratumor tissues (original magnification 200×, scale bar, 200 μm). (E) IDO gene analysis in OSCC (Oncomine database). Box plots derived from gene expression data in Oncomine comparing expression of IDO gene in oral cavity (left plot) and OSCC (right plot). (F) Kaplan-Meier analysis of OSCC patients with low level (112 cases) vs high level (33 cases) of IDO expression. P-values were calculated by Log rank test. (*P < 0.05, **P < 0.01). |
IDO Inhibition Decreases the Viability of OSCC Cells
As the qPCR results indicated that higher IDO expression was found in CAL27 cells, we chose CAL27 cells to investigate whether IDO potentiates OSCC progression. IDO shRNAs were constructed and transfected into CAL27 cells (Figure 1A and B). The knockdown efficacy was determined by qPCR (Figure 2A) and Western blotting (Figure 2B), which showed that IDO was markedly downregulated by shRNA transfection in OSCC cells. Cell counting and CCK8 assays were used to explore the effect of ectopic IDO expression on the malignant phenotype of OSCC cells. IDO knockdown significantly decreased the growth (Figure 2C) and viability (Figure 2D) of OSCC cells, indicating that IDO might play an important role in OSCC cell growth. To further confirm this, we used an IDO inhibitor—epacadostat, which competes with the Kyn pathway substrate Trp to bind to the catalytic domain of IDO1. Dose-dependent and time-dependent effects of epacadostat on OSCC cells were determined by the CCK-8 assay. Results indicated that epacadostat significantly reduced cell viabilities at the concentration of 2.5 μM, and the inhibiting effect was much more obvious at the concentrations of 20 μM and 40 μM (Figure 2E and F). Thus, we finally chose the higher concentration for further experiment, which was similar in a study of colorectal cancer with the concentration of 50 μM.16 Epacadostat also inhibited cell growth in a time-dependent manner, as shown in Figure 2G and H. Therefore, inhibiting IDO by shRNA or a specific inhibitor decreased the viability of OSCC cells.
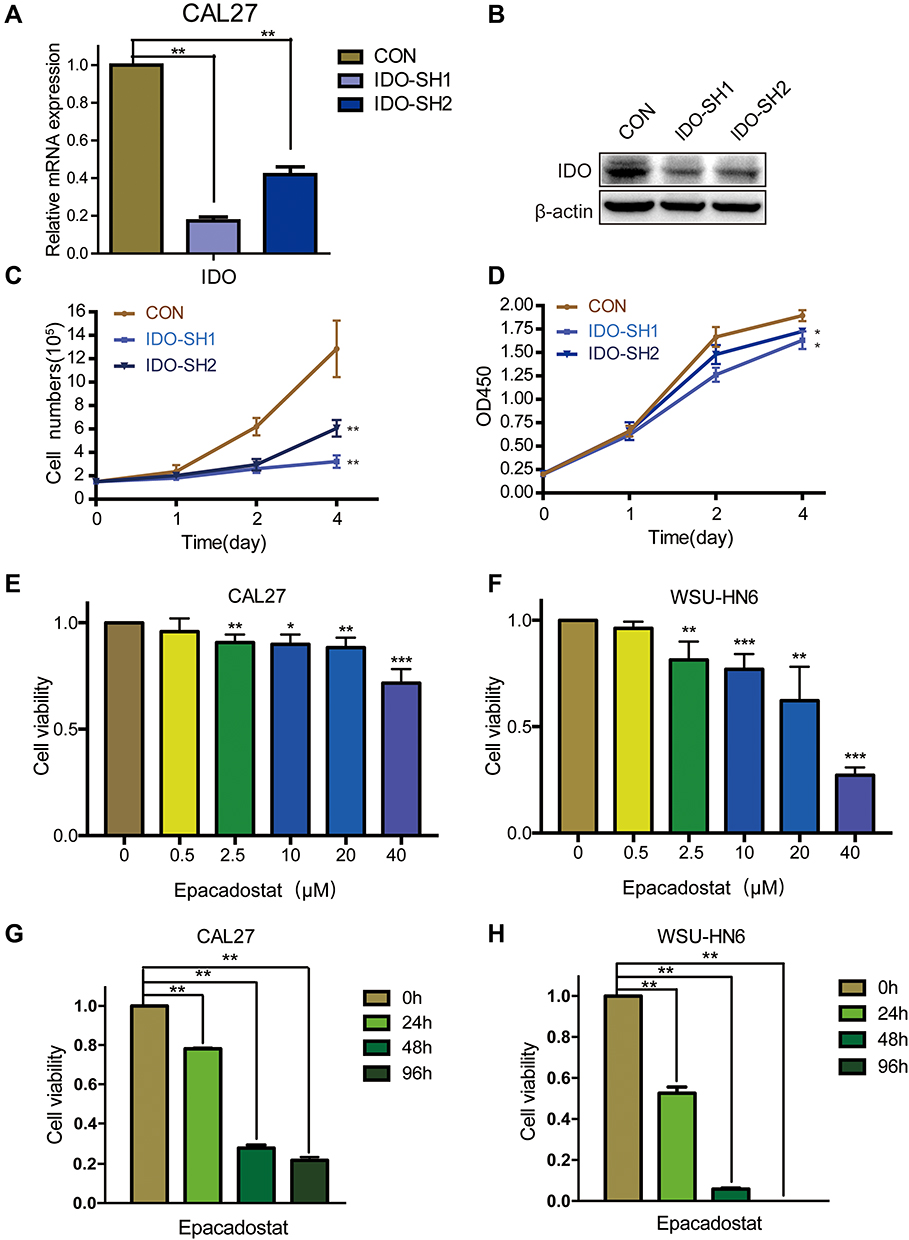 |
Figure 2 IDO inhibition decreased the cell viabilities of OSCC cells. (A and B) Knockdown efficiency detection of IDO in CAL27 cells. Expression analysis of IDO by (A) qPCR and (B) Western blot in CAL27 cells. IDO was stably knocked down in CAL27 cells using lentivirus-generated shRNA. (C) Effects of IDO knockdown on growth of OSCC cells. Cytometry analysis of IDO knockdown and control CAL27 cells at the indicated time points. (D) Effects of IDO knockdown on cell viability of OSCC cells. CCK8 analysis of IDO knockdown and control CAL27 cells at the indicated time points. (E and F) OSCC cells were treated without or with epacadostat (0.5, 2.5, 10, 20, or 40 μM) for 24 hours. Cell viability was analyzed using the CCK8 assay. (G and H) IDO inhibitor epacadostat induced a significant decrease of cell viabilities to CAL27 and WSU-HN6 cell lines by CCK8. CAL27 and WSU-HN6 cells were treated with 40 μM of epacadostat for 0, 24, 48 and 96 h. (*P < 0.05, **P < 0.01, ***P<0.001). |
IDO Inhibition Promotes Apoptosis of OSCC Cells
Flow cytometry analysis was used to explore whether blocking IDO expression caused apoptosis in OSCC cells. The results showed that knockdown of IDO in CAL27 cells increased the percentage of apoptotic cells from 3.85 ± 0.47% to 12.39 ± 2.89% or 9.59 ± 0.57% (Figure 3A). Using the Western blot assay, we found that IDO knockdown markedly increased the expression of cleaved caspase-3 in OSCC cells (Figure 3C), which identified the pro-apoptotic effect of IDO knockdown. Similar results were observed in CAL27 and WSU-HN6 cells treated with the IDO inhibitor epacadostat (Figure 3B and D, S2). CAL27 and WSU-HN6 cells were treated with 0, 20, and 40 μM of epacadostat for 24 hours and analyzed by flow cytometry. Results showed that epacadostat significantly increased the percentage of apoptosis in OSCC cells. For CAL27 cells, the percentage of apoptosis was 8.56 ± 0.54% in cells treated with 20 μM epacadostat and 11.84 ± 1.75% in those treated with 40 μM, compared with 5.82 ± 0.40% of apoptosis in the control group. The same tendency was observed for WSU-HN6 cells. These data indicated that blocking IDO expression by IDO-shRNA or IDO inhibitor suppressed OSCC growth by promoting apoptosis.
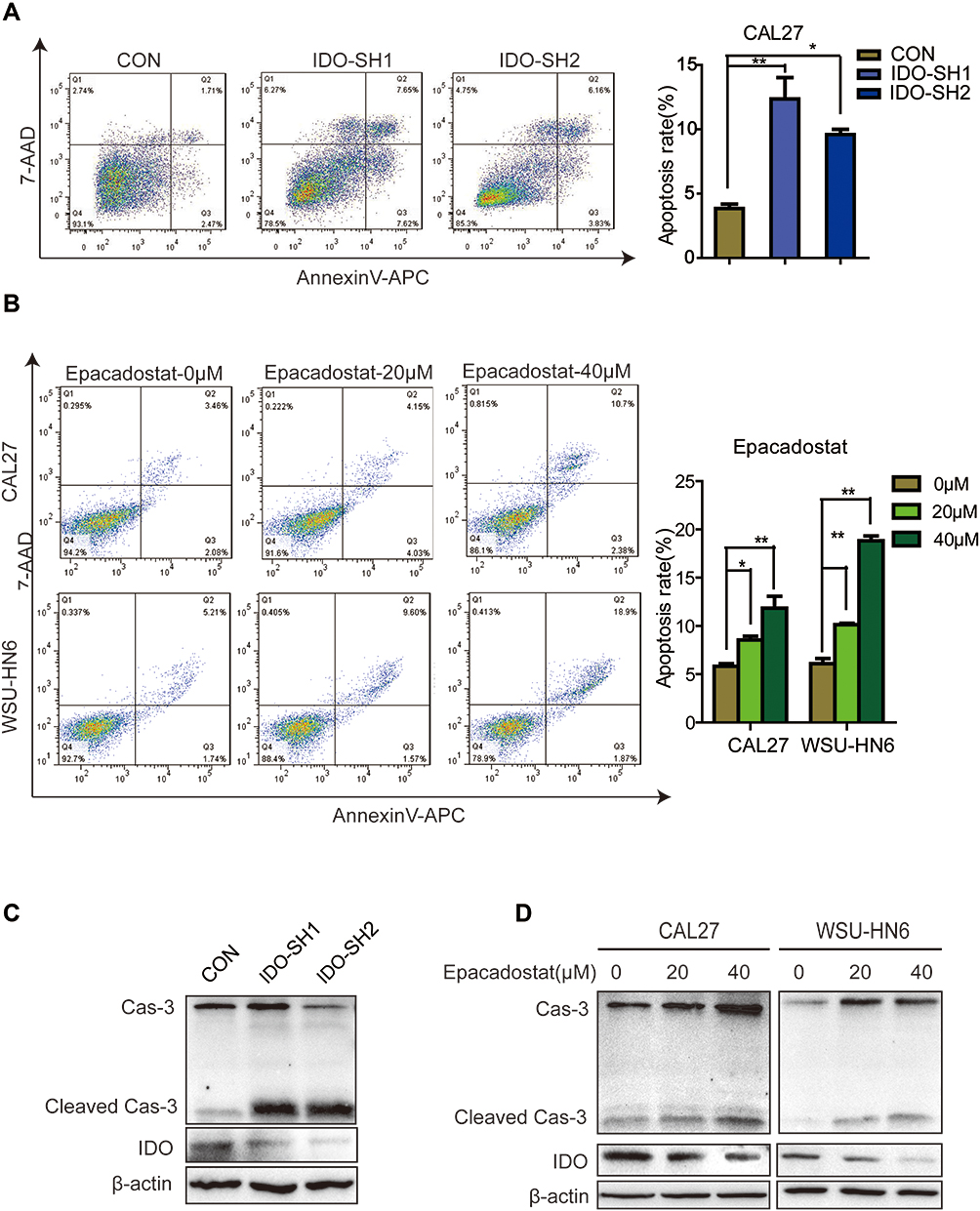 |
Figure 3 IDO inhibition promoted apoptosis of OSCC cells. (A) Effects of IDO knockdown on apoptosis of OSCC cells. Flow cytometry analysis of Annexin V expression in IDO knockdown and control CAL27 cells. (B) Effects of epacadostat on apoptosis of OSCC cells. CAL27 and WSU-HN6 cells were treated with 0, 20, and 40 μM of epacadostat for 24 hours, stained for Annexin V and analyzed by flow cytometry. (C) Representative Western blots show the expression of caspase-3 and IDO in IDO knockdown and control CAL27 cells. (D) Representative Western blots show the expression of caspase-3 and IDO in epacadostat-treated CAL27 and WSU-HN6 cells. (*P < 0.05, **P < 0.01). |
IDO Inhibition Induces Apoptosis by Repressing BCL2A1 Expression in OSCC Cells
To explore the signaling pathways involved in IDO-regulated apoptosis in OSCC, an 84-gene qPCR-based apoptosis array was performed. The apoptosis array identified 15 and 13 genes with a more than two-fold change in transcription level in IDO-SH1 and IDO-SH2 (Figure S4A), respectively. Seven apoptosis-related genes—BCL2A1, caspase-5, TNFRSF11B, TNFRSF9, CD27, CD70, and DAPK—showed similar changes in two knockdown OSCC cell lines (Figure 4A). As shown in Figure 4B, IDO knockdown significantly decreased BCL2A1 expression by 23.78-fold and 3.15-fold in IDO-SH1 and IDO-SH2, respectively, compared with the controls (p < 0.05). Similar results were observed in CAL27 and WSU-HN6 cells treated with the IDO inhibitor epacadostat (Figure S4B). BCL2A1 is a member of the BCL-2 protein family. The protein encoded by the BCL2A1 gene decreases the release of pro-apoptotic cytochrome c from mitochondria and blocks caspase activation, thus acting as an anti-apoptotic regulator involved in a wide variety of cellular activities such as embryonic development, homeostasis, and tumorigenesis.18 To examine the role of the IDO-BCL2A1-cytochrome c axis in regulating apoptosis in OSCC cells, the expression of molecules involved and the localization of cytochrome c relative to mitochondria was determined by Western blotting and immunofluorescence staining, respectively. The results showed that IDO knockdown and epacadostat treatment downregulated BCL2A1 and upregulated cytochrome c, thereby promoting apoptosis (Figure 4C). Immunofluorescence staining results revealed that cytochrome c co-localized with the mitochondrial marker (Mito Tracker) in the control WSU-HN6 and CAL27 cells, while cytosolic cytochrome c was increased in the IDO knockdown and epacadostat-treated cells (Figure 4D). Overexpression of BCL2A1 decreased the release of cytochrome c and restored apoptosis induction in IDO knockdown OSCC cells (Figure 4E). These data indicated that IDO inhibition induced apoptosis in OSCC by regulating BCL2A1 and targeting the IDO-BCL2A1-cytochrome c pathway promoted OSCC cell apoptosis.
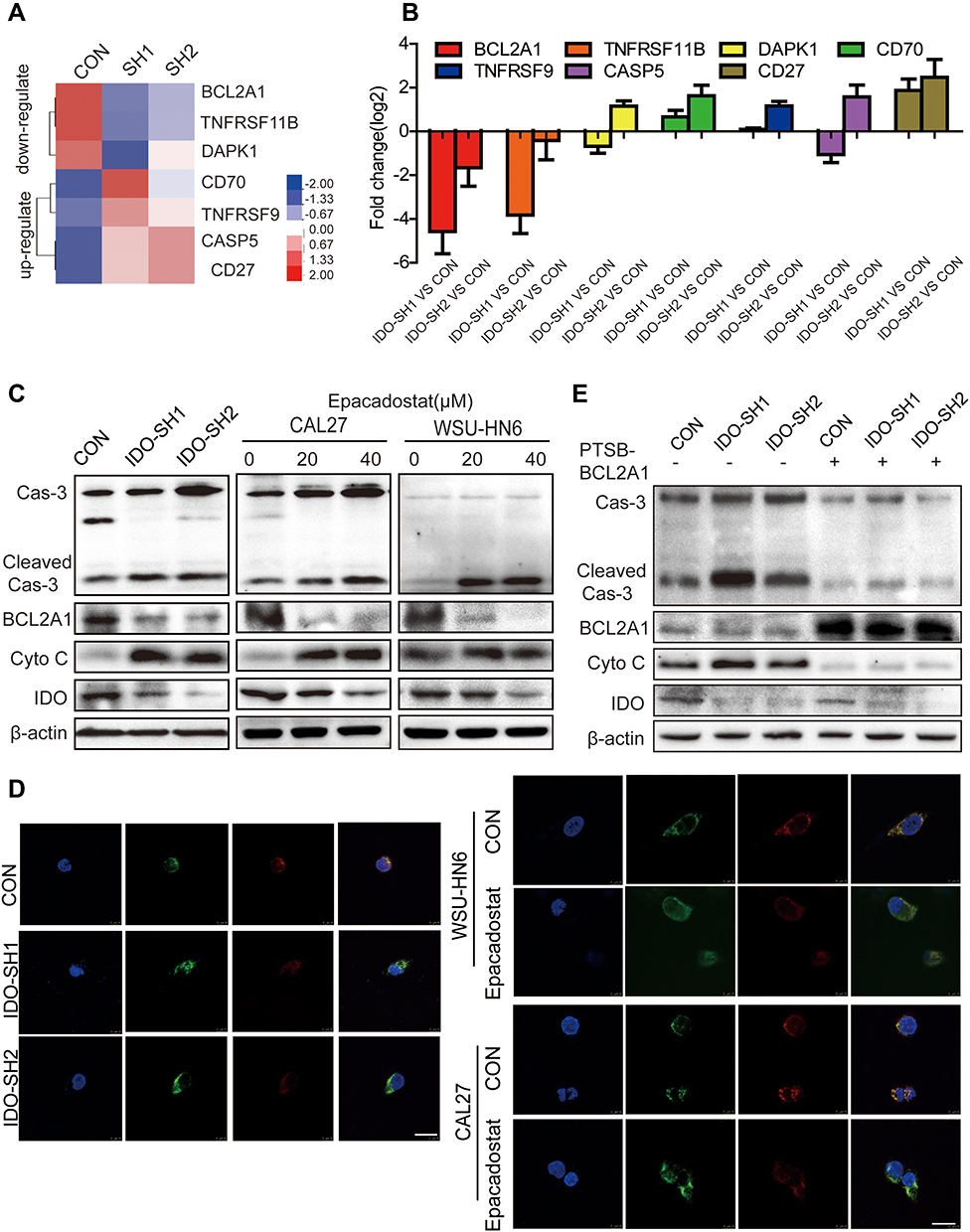 |
Figure 4 IDO inhibition induced apoptosis of OSCC cells through repressing BCL2A1 expression. (A) Heatmap of the consistently down-regulated and up-regulated genes of IDO knockdown and control CAL27 cells. (B) Top consistently down-regulated and up-regulated genes were individually verified using qPCR. (C) The expression of IDO, BCL2A1, cytochrome c and caspase-3 in IDO knockdown and OSCC cells (CAL27 and WSU-HN6) treated with 0, 20, and 40 μM of epacadostat for 24 hours was detected by Western blots. (D) The co-localization of cytochrome c with mitochondria in IDO knockdown and OSCC cells (treated with 20 μM epacadostat for 24 h) is presented in cells labeled using Mito tracker dye and observed using a confocal microscope. Merged images are shown, and the yellow color represents co-localization of cytochrome c (green) and mitochondria (red) (scale bar, 20 μm). (E) Effects of BCL2A1 overexpression on apoptosis of IDO knockdown and control CAL27 cells. IDO knockdown CAL27 cells and control CAL27 cells were treated with BCL2A1 activator for 24 hours, and the expression of IDO, BCL2A1, cytochrome c and caspase-3 were detected by Western blots. |
Inhibition of IDO Suppresses OSCC Xenograft Tumor Growth
The animal model of CAL27 transplanted in nude was established to observe the effect of IDO inhibition on OSCC progression in vivo. When the tumor volume reached 80 mm3, mice were randomly assigned into control and treated groups. As epacadostat was used at the concentration of 100mg/kg in vivo in a previous study of Lewis lung cancer,21 we chose the same dose in our study. Mice received 100 mg/kg of epacadostat intraperitoneally every 3 days. Tumor volumes were calculated and tumor weights were recorded. No obvious side effects were found in epacadostat-treated mice (Figure S5A). Consistent with the in vitro results, epacadostat markedly inhibited tumor growth by 69.35% (Figure 5A). Compared with the control group, epacadostat-treated mice had a significantly lower tumor burden, as indicated by the smaller size and less weight of tumors (Figure 5B and C). The mean weight of the tumors from mice treated with epacadostat was approximately 65.58% lower than that from control mice. Moreover, to confirm the impact of IDO knockdown on the growth of OSCC xenograft tumors, mock vector-transfected cells and shRNA- transfected cells (IDO-SH1) were subcutaneously implanted into each side of the inguen of mice, respectively. The results showed that tumors in mice receiving IDO knockdown cells had smaller size and less weight than those of control mice (Figure 5D–F and S5B). Furthermore, to assess the effect of IDO inhibition on BCL2A1 protein levels and apoptosis of tumor cells in vivo, BCL2A1 and cleaved caspase-3 expression were analyzed by IHC in xenograft tumors. In line with the results in vitro, IDO-SH1 and epacadostat significantly reduced BCL2A1 expression and promoted the cleavage of caspase-3 compared with those in control cells (Figure 5G–H). These data indicated that IDO knockdown suppressed the expression of BCL2A1 and promoted apoptosis, thereby suppressing the growth of OSCC cells in vivo.
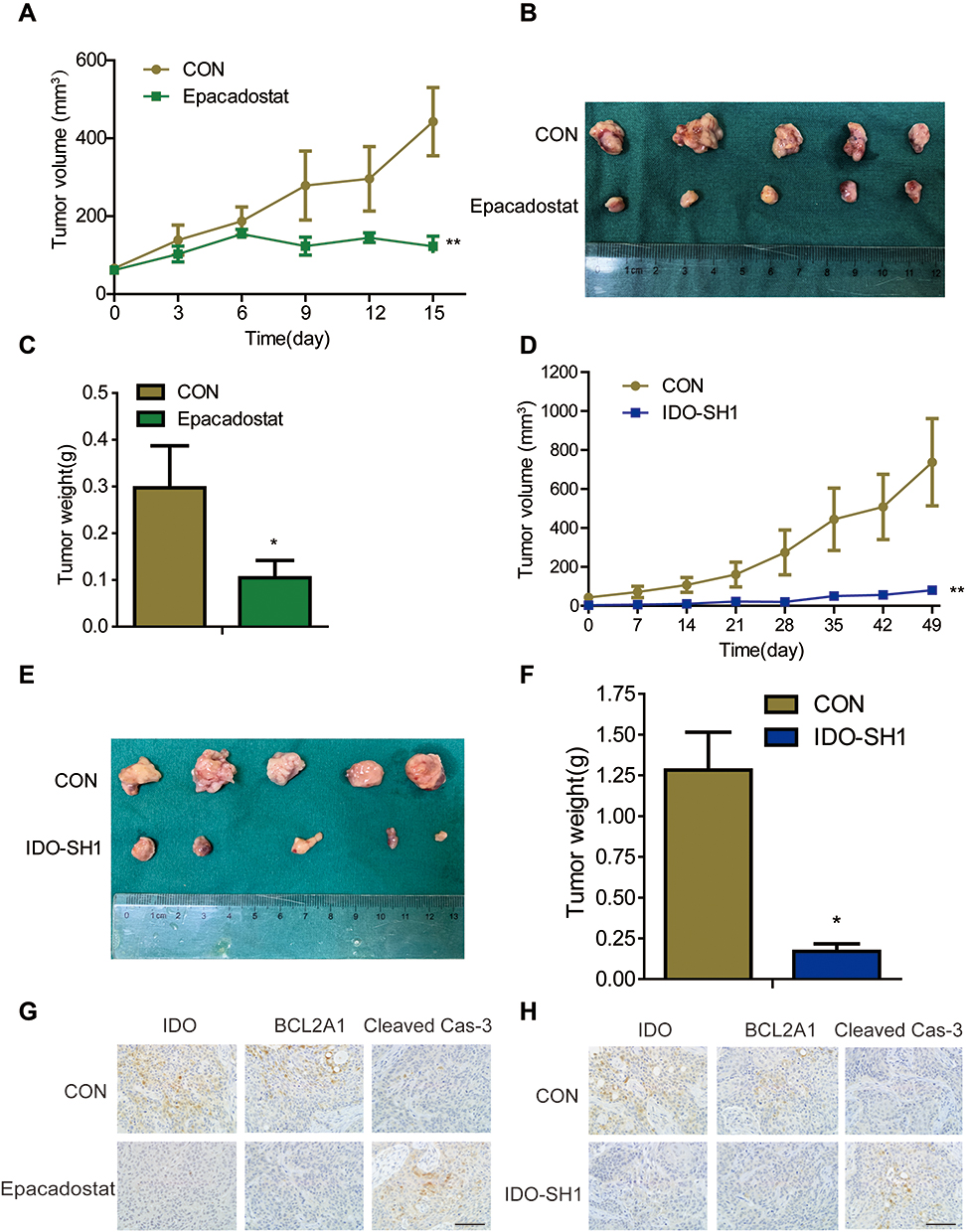 |
Figure 5 IDO inhibition suppressed OSCC xenograft growth in vivo. (A–C) Epacadostat reduced tumor formation of OSCC. The OSCC xenograft mouse model was established with CAL27 cells, and mice were grouped and treated with 100 mg/kg epacadostat or DMSO. Graphs showed the (A) tumor volumes and (C) tumor weight from control and epacadostat-treated groups. Results are presented as the mean ± SD. (B) Representative image of tumors in each group. (D–F) IDO knockdown attenuated tumor growth of OSCC. Mock vector-transfected cells and shRNA- transfected cells (IDO-SH1) were implanted into each side of the inguen of mice. Graphs showed the (D) tumor volumes and (F) tumor weight of IDO-SH1 and control CAL27 cells groups. The results are presented as the mean ± SD. (E) Representative image of tumors in each group. (G and H) The expression of IDO, BCL2A1, and caspase-3 in tumors from epacadostat-treated (G) and IDO knockdown (H) mice were indicated by immunohistochemical staining (original magnification 400×; scale bar, 500 μm). Tumor volume (mm3) = a × b2/2, where a is the longest diameter and b is the shortest diameter. Results are representative of three independent experiments. (*P < 0.05, **P < 0.01). |
Discussion
OSCC accounts for the majority of head and neck cancer, and it can arise in different locations including the tongue, gingiva, floor of the mouth, and buccal area.2,4 The complex carcinogenesis of the disease from precancerous dysplastic lesions to carcinoma is still poorly understood. In the present study, we found that IDO expression was upregulated in OSCC and was negatively correlated with OSCC progression. IDO knockdown or IDO inhibitor induced apoptosis of OSCC cells in vitro. Silencing IDO inhibited the expression of the anti-apoptotic regulator BCL2A1, thereby increased the release of cytochrome c and promoted OSCC cell apoptosis. BCL2A1 overexpression reversed the induction of apoptosis caused by IDO knockdown. Silencing IDO expression significantly inhibited OSCC tumor growth in vivo. The present results indicated that IDO may promote OSCC growth by regulating BCL2A1 expression and targeting the IDO-BCL2A1-cytochrome c pathway may help to the therapy of OSCC.
Trp is an essential amino acid that plays crucial roles in immune suppression and tumor progression.6,7 IDO, the rate-limiting enzyme in the Trp degradation pathway, is present in two isoforms, IDO1 and IDO2. Both isoforms regulate the degradation of Trp to Kyn, although IDO1 has a 20–30-fold higher enzymatic activity than IDO2. IDO1 is expressed in cancer cells and surrounding cells, whereas IDO2 is mainly expressed in the liver and kidney,22,23 and both isoforms have different expression patterns in different tumors. In this study, qPCR results showed that IDO1, but not IDO2 or tryptophan-2,3-dioxygenase, was upregulated in OSCC cell lines, which is consistent with the results reported by Laimer et al.17 In cancers, increased IDO activity promotes the development of an immunosuppressive microenvironment that supports tumor growth. IDO is associated with poor clinical outcome and chemotherapeutic response in various malignant diseases including OSCC.15–19 High expression of IDO is a significant negative prognostic factor in patients with OSCC, especially in those undergoing adjuvant radiochemotherapy17 and nimotuzumab therapy.20 Thus, inhibition of IDO alone or in combination with classical chemotherapeutic agents has become a hot topic in the field of cancer treatment.24,25 Chronic inflammation increases the rates of necrotic and apoptotic cell death in IDO-KO mice,26 which may promote immune escape and tumor cell proliferation in OSCC.27 In addition to its immunosuppressive function, IDO overexpression promotes tumor growth and metastasis by activating IDO-AHR or β-catenin signaling in primary breast cancer and colon cancer.14,28 Limited research has reported that overexpression of IDO contributed to the differentiation of Treg, leading to immunosuppression responses in chronic periodontitis, which is a dysbiotic disease linked to increased risk of OSCC.29 However, the clinical significance of IDO expression in OSCC is unclear, and the underlying molecular mechanism remains to be explored.
In the present study, we found that IDO expression was upregulated in OSCC and was negatively correlated with OSCC progression. By the CCK8 assay, we found that IDO inhibition reduced OSCC cell viabilities. As cell viability can be affected by cell death and proliferation, we later determined cell apoptosis, cell cycle and cell proliferation with the flow cytometry and EdU incorporation assay (Figure S3). Results indicated that IDO inhibition induced OSCC cell apoptosis but not cell proliferation decrease. Although there was report that IDO inhibition induced G2/M cycle arrest in colorectal cancer cells,16 we guess this could be attributed to the difference of cancer cell type. Kyn is a natural ligand of AHR and their interaction, which leads to the nuclear translocation of AHR, is related to oncogene expression, angiogenesis, and cell survival, as well as effects on immune cell function.10,30,31 Here, exogenous Kyn failed to rescue the growth retardation and apoptosis caused by IDO knockdown (data not shown), and IDO knockdown did not affect the expression of CYP1A1, a target gene of AHR, in OSCC, suggesting that the promotion of apoptosis by IDO inhibition was independent of the Kyn-AHR interaction.
Deregulation of apoptosis is a hallmark of cancer cells, and the agents that activate apoptosis in cancer cells could be valuable as anticancer therapeutics.32 In the present study, the results of flow cytometry indicated that knocking down IDO or IDO inhibitor treatment induced apoptosis in OSCC cells.33 The BCL2 family of proteins constitutes a critical checkpoint in apoptotic programmed cell death by regulating apoptotic molecules, including the release of cytochrome c from mitochondria. An apoptosis PCR array identified BCL2A1 as a markedly downregulated protein in response to IDO knockdown. BCL2A1, which has not been studied extensively compared with other BCL2-family proteins, is processed by the ubiquitin-proteasome system.34 BCL2A1 inhibits apoptosis by interacting with several proapoptotic proteins, including BAX, BID, and PUMA, and decreases the release of pro-apoptotic cytochrome c from mitochondria, as well as blocking caspase activation.35,36 BCL2A1 is overexpressed in various cancer cells,28,37–39 and it is overexpressed in 88.9% of OSCC samples.37 BCL2A1 increased 5-fold in OSCC stem cell-like cancer cells (side population),40 indicating that it may play a facilitative role in OSCC progression. Mandal et al reported that overexpression of BCL2A1 contributes to T cell leukemia by inhibiting caspase-3 activation.34 Hu et al showed that the microRNA-573/apoM/BCL2A1 pathway is essential for hepatocyte apoptosis and hepatocarcinogenesis.41 In addition, BCL2A1 contributes to anti-cancer drug resistance.42,43 Therefore, the development of BCL2A1 inhibitors may be a promising approach to sensitize tumor cells to apoptosis and thus improve the efficiency of anti-cancer therapy.35 In this study, knockdown of IDO by lentivirus-mediated transfection or treatment with an IDO inhibitor downregulated BCL2A1, increased the release of cytochrome c, and promoted caspase-3 cleavage, thereby promoting apoptosis in OSCC. Overexpression of BCL2A1 reversed apoptosis induced by IDO knockdown. These results indicated that IDO may play a facilitative role in OSCC by regulating BCL2A1 expression.
Conclusion
This study showed that IDO promoted tumor progression in OSCC by modulating BCL2A1 expression and targeting the IDO-BCL2A1-cytochrome c pathway promoted apoptosis of OSCC. These findings provide new insight into the mechanisms underlying OSCC progression and identify IDO as a potential prognostic biomarker and therapeutic target for OSCC.
Abbreviations
IDO, indolamine 2.3-dioxygenase; OSCC, oral squamous cell carcinoma; IHC, immunohistochemistry; BCL2A1, BCL2 related protein A1; Trp, Tryptophan; Kyn, kynurenine; AHR, aryl hydrocarbon receptor.
Ethics and Consent Statement
The animal experiments were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, USA) and the related animal ethical rules of our hospital. We did our best to minimize the number and pain of animals used. The use of HIOEC and OSCC cell lines and all experiments acquired ethical approval by the Ethics Committee of Shanghai Ninth People’s Hospital (SH9H-2020-A332-1). All patients gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Shanghai Ninth People’s Hospital (SH9H-2020-TK148-1).
Acknowledgments
The authors thank the Shanghai Key Laboratory of Stomatology for kindly providing HIOEC and OSCC cell lines. We thank International Science Editing for editing this manuscript. Qiaoping Zheng and Guifang Gan are co-first authors for this study.
Funding
This work was supported by the National Natural Science Foundation of China (81870762) and the Fundamental Research Program Funding of the Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (JYZZ059, JYZZ043).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Mignogna MD, Fedele S, Lo Russo L. The world cancer report and the burden of oral cancer. Eur J Cancer Prev. 2004;13(2):139–142. doi:10.1097/00008469-200404000-00008
3. Chi AC, Day TA, Neville BW. Oral cavity and oropharyngeal squamous cell carcinoma–an update. CA Cancer J Clin. 2015;65(5):401–421. doi:10.3322/caac.21293
4. Blatt S, Kruger M, Ziebart T, et al. Biomarkers in diagnosis and therapy of oral squamous cell carcinoma: a review of the literature. J Cranio Maxillofac Surg. 2017;45(5):722–730. doi:10.1016/j.jcms.2017.01.033
5. Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–1193. doi:10.1126/science.281.5380.1191
6. Platten M, Nollen EAA, Röhrig UF, Fallarino F, Ca O. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. 2019. [Epub ahead of print].
7. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435–5440. doi:10.1158/0008-5472.CAN-12-0569
8. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37(3):193–207. doi:10.1016/j.it.2016.01.002
9. Munn DH, Sharma MD, Baban B, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–642. doi:10.1016/j.immuni.2005.03.013
10. Nguyen NT, Kimura A, Nakahama T, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107(46):19961–19966. doi:10.1073/pnas.1014465107
11. Hayashi T, Mo JH, Gong X, et al. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. P Natl Acad Sci USA. 2007;104(47):18619–18624. doi:10.1073/pnas.0709261104
12. Rohrig UF, Majjigapu SR, Chambon M, et al. Detailed analysis and follow-up studies of a high-throughput screening for indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. Eur J Med Chem. 2014;84:284–301. doi:10.1016/j.ejmech.2014.06.078
13. Cheong JE, Sun LJ. Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy - Challenges and opportunities. Trends Pharmacol Sci. 2018;39(3):307–325. doi:10.1016/j.tips.2017.11.007
14. Yu J, Sun J, Wang SE, et al. Upregulated expression of indoleamine 2, 3-dioxygenase in primary breast cancer correlates with increase of infiltrated regulatory T cells in situ and lymph node metastasis. Clin Dev Immunol. 2011;2011:469135. doi:10.1155/2011/469135
15. Thaker AI, Rao MS, Bishnupuri KS, et al. IDO1 metabolites activate β-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology. 2013;145(2):416–425. doi:10.1053/j.gastro.2013.05.002
16. Liu X, Zhou W, Zhang X, Ding Y, Du Q, Hu R. 1-L-MT, an IDO inhibitor, prevented colitis-associated cancer by inducing CDC20 inhibition-mediated mitotic death of colon cancer cells. Int J Cancer. 2018;143(6):1516–1529. doi:10.1002/ijc.31417
17. Laimer K, Troester B, Kloss F, et al. Expression and prognostic impact of indoleamine 2,3-dioxygenase in oral squamous cell carcinomas. Oral Oncol. 2011;47(5):352–357. doi:10.1016/j.oraloncology.2011.03.007
18. Vogler M. BCL2A1: the underdog in the BCL2 family. Cell Death Differ. 2012;19(1):67–74. doi:10.1038/cdd.2011.158
19. Prendergast GC, Metz R, Muller AJ, Merlo LM, Mandik-Nayak L. IDO2 in immunomodulation and autoimmune disease. Front Immunol. 2014;5:585. doi:10.3389/fimmu.2014.00585
20. Chen W. IDO: more than an enzyme. Nat Immunol. 2011;12(9):809–811. doi:10.1038/ni.2088
21. Liu C, Nan Y, Xia Z, et al. Discovery of novel hydroxyamidine derivatives as indoleamine 2,3-dioxygenase 1 inhibitors with in vivo anti-tumor efficacy. Bioorg Med Chem Lett. 2020;30(8):127038. doi:10.1016/j.bmcl.2020.127038
22. Opitz CA, Litzenburger UM, Sahm F, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203. doi:10.1038/nature10491
23. Mitchell TC, Hamid O, Smith DC, et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: Phase I results from a multicenter, open-label Phase I/II trial (ECHO-202/KEYNOTE-037). J Clin Oncol. 2018;36(32):3223–3230. doi:10.1200/JCO.2018.78.9602
24. Lionnard L, Duc P, Brennan MS, et al. TRIM17 and TRIM28 antagonistically regulate the ubiquitination and anti-apoptotic activity of BCL2A1. Cell Death Differ. 2019;26(5):902–917. doi:10.1038/s41418-018-0169-5
25. Vogler M, Butterworth M, Majid A, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113(18):4403–4413. doi:10.1182/blood-2008-08-173310
26. Qin X, Liu JY, Wang T, et al. Role of indoleamine 2,3-dioxygenase in an inflammatory model of murine gingiva. J Periodontal Res. 2017;52(1):107–113. doi:10.1111/jre.12374
27. You G, Feng L, Yan W, et al. BCL2A1 is a potential biomarker for postoperative seizure control in patients with low-grade gliomas. CNS Neurosci Ther. 2013;19(11):882–888. doi:10.1111/cns.12148
28. Fougeray S, Mami I, Bertho G, Beaune P, Thervet E, Pallet N. Tryptophan depletion and the kinase GCN2 mediate IFN-gamma-induced autophagy. J Immunol. 2012;189(6):2954–2964. doi:10.4049/jimmunol.1201214
29. Arjunan P, Meghil MM, Pi W, et al. Oral pathobiont activates anti-apoptotic pathway, promoting both immune suppression and oncogenic cell proliferation. Sci Rep. 2018;8(1):16607. doi:10.1038/s41598-018-35126-8
30. Champa D, Russo MA, Liao XH, Refetoff S, Ghossein RA, Di Cristofano A. Obatoclax overcomes resistance to cell death in aggressive thyroid carcinomas by countering Bcl2a1 and Mcl1 overexpression. Endocr Relat Cancer. 2014;21(5):755–767. doi:10.1530/ERC-14-0268
31. Rasooly R, Schuster GU, Gregg JP, Xiao JH, Chandraratna RA, Stephensen CB. Retinoid x receptor agonists increase bcl2a1 expression and decrease apoptosis of naive T lymphocytes. J Immunol. 2005;175(12):7916–7929. doi:10.4049/jimmunol.175.12.7916
32. Yajima T, Ochiai H, Uchiyama T, Takano N, Shibahara T, Azuma T. Resistance to cytotoxic chemotherapy-induced apoptosis in side population cells of human oral squamous cell carcinoma cell line Ho-1-N-1. Int J Oncol. 2009;35(2):273–280.
33. Ball HJ, Sanchez-Perez A, Weiser S, et al. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene. 2007;396(1):203–213. doi:10.1016/j.gene.2007.04.010
34. Mandal M, Borowski C, Palomero T, et al. The BCL2A1 gene as a pre-T cell receptor-induced regulator of thymocyte survival. J Exp Med. 2005;201(4):603–614. doi:10.1084/jem.20041924
35. Wang W, Huang L, Jin JY, et al. IDO immune status after chemoradiation may predict survival in lung cancer patients. Cancer Res. 2018;78(3):809–816. doi:10.1158/0008-5472.CAN-17-2995
36. Corm S, Berthon C, Imbenotte M, et al. Indoleamine 2,3-dioxygenase activity of acute myeloid leukemia cells can be measured from patients’ sera by HPLC and is inducible by IFN-gamma. Leuk Res. 2009;33(3):490–494. doi:10.1016/j.leukres.2008.06.014
37. Brandacher G, Perathoner A, Ladurner R, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. 2006;12(4):1144–1151. doi:10.1158/1078-0432.CCR-05-1966
38. Eleftheriadis T, Pissas G, Antoniadi G, Liakopoulos V, Stefanidis I. Indoleamine 2,3-dioxygenase depletes tryptophan, activates general control non-derepressible 2 kinase and down-regulates key enzymes involved in fatty acid synthesis in primary human CD4+ T cells. Immunology. 2015;146(2):292–300. doi:10.1111/imm.12502
39. Reyes-Ocampo J, Ramírez-Ortega D, Cervantes GI, et al. Mitochondrial dysfunction related to cell damage induced by 3-hydroxykynurenine and 3-hydroxyanthranilic acid: non-dependent-effect of early reactive oxygen species production. Neurotoxicology. 2015;50:81–91. doi:10.1016/j.neuro.2015.08.003
40. Lee SM, Lee YS, Choi JH, et al. Tryptophan metabolite 3-hydroxyanthranilic acid selectively induces activated T cell death via intracellular GSH depletion. Immunol Lett. 2010;132(1–2):53–60. doi:10.1016/j.imlet.2010.05.008
41. Hu YW, Chen ZP, Hu XM, et al. The miR-573/apoM/Bcl2A1-dependent signal transduction pathway is essential for hepatocyte apoptosis and hepatocarcinogenesis. Apoptosis. 2015;20(10):1321–1337. doi:10.1007/s10495-015-1153-x
42. Haq R, Yokoyama S, Hawryluk EB, et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc Natl Acad Sci U S A. 2013;110(11):4321–4326. doi:10.1073/pnas.1205575110
43. Hiraki M, Maeda T, Mehrotra N, et al. Targeting MUC1-C suppresses BCL2A1 in triple-negative breast cancer. Signal Transduct Target Ther. 2018;3(1):13. doi:10.1038/s41392-018-0013-x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.