")
Back to Journals » OncoTargets and Therapy » Volume 7
Targeting the epithelial to mesenchymal transition in glioblastoma: the emerging role of MET signaling
Authors Lee J, Joo KM, Lee J, Yoon Y, Nam D
Received 25 April 2014
Accepted for publication 9 July 2014
Published 20 October 2014 Volume 2014:7 Pages 1933—1944
DOI https://doi.org/10.2147/OTT.S36582
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jin-Ku Lee,1,2,* Kyeung Min Joo,3 Jeongwu Lee,4 Yeup Yoon,5,* Do-Hyun Nam2,5
1Samsung Biomedical Research Institute, Sungkyunkwan University School of Medicine, Seoul, Korea; 2Department of Neurosurgery, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea; 3Department of Anatomy and Cell Biology, Sungkyunkwan University School of Medicine, Seoul, Korea; 4Department of Stem Cell Biology and Regenerative Medicine, Lerner Research Institute, Cleveland Clinic, Cleveland, OH, USA; 5Samsung Advanced Institute for Health Sciences and Technology, Sungkyunkwan University School of Medicine, Seoul, Korea
*These authors contributed equally to this work
Abstract: Glioblastoma multiforme (GBM) is the most common human primary brain malignancy and has a dismal prognosis. Aggressive treatments using maximal surgical resection, radiotherapy, and temozolomide result in median survival of only 14.6 months in patients with GBM. Numerous clinical approaches using small molecule inhibitors have shown disappointing results because of the genetic heterogeneity of GBM. The epithelial to mesenchymal transition (EMT) is a crucial biological process occurring in the early development stages of many species. However, cancer cells often obtain the ability to invade and metastasize through the EMT, which triggers the scattering of cells. The hepatocyte growth factor (HGF)/MET signaling pathway is indicative of the EMT during both embryogenesis and the invasive growth of tumors, because HGF potently induces mesenchymal transition in epithelial-driven cells. Activation of MET signaling or co-overexpression of HGF and MET frequently represents aggressive growth and poor prognosis of various cancers, including GBM. Thus, efforts to treat cancers by inhibiting MET signaling using neutralizing antibodies or small molecule inhibitors have progressed during the last decade. In this review, we discuss HGF/MET signaling in the development of diseases, including cancers, as well as updates on MET inhibition therapy.
Keywords: glioblastoma multiforme, epithelial to mesenchymal transition, MET signaling
Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive form of adult brain tumor.1 Although its incidence is relatively low, it has attracted attention because of its dismal prognosis. Despite the aggressive treatment regimen, which includes surgery and radiotherapy plus temozolomide, median survival is only 12–15 months in patients with GBM.2,3
Improved understanding of the molecular pathogenesis of GBM has encouraged the adaptation of targeted molecular therapies to treat GBM.4 However, most clinical trials evaluating new drugs targeting cancer-specific signaling pathways have shown only minimal effects on the treatment of patients with GBM.4 A randomized controlled clinical trial assessing the antiangiogenic drug bevacizumab demonstrated that targeting angiogenesis increases tumor cell invasion in patients with GBM.5 Although molecular targeted agents potently inhibit receptor tyrosine kinases such as epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor, and vascular endothelial growth factor receptor, which are major survival and proliferation-promoting signaling molecules in GBM,4–7 treatment using a single agent has shown only minimal improvement, with response rates of 0%–15% of 6-monthprogression-free survival.8
The epithelial to mesenchymal transition (EMT) is a biological process that allows immobile epithelial cells to undergo biochemical changes and induces a mesenchymal cell phenotype, including enhanced migratory properties, invasiveness, and resistance to apoptosis.9,10 The EMT is also associated with tumor cell invasion that leads to metastatic dissemination.11 A study that investigated the association between mesenchymal markers and gliomas revealed a series of mesenchymal tissue-associated genes such as YKL-40, TNC, osteonectin, and CD105 to be overexpressed in GBM biopsies.12 A comprehensive gene expression analysis of 85 high-grade gliomas identified a subset of GBM tissues also overexpressing mesenchymal tissue-associated genes.13 TWIST, an EMT-associated molecule, also plays a key role in survival and invasion of human glioma cells.14
c-MET, a receptor tyrosine kinase, functions as a proto-oncogene by activating multiple cellular signal pathways that promote proliferation, migration, and invasion of cancer cells.15 Hepatocyte growth factor (HGF), a pleiotropic factor that promotes cell proliferation, survival, and motility, is a c-MET ligand.16,17 In addition, the MET tyrosine kinase induces glioma cell proliferation, survival, and migration.18 HGF/MET signaling also confers resistance to radiotherapy by promoting survival of glioma stem cells (GSCs).19 The MET oncogene is associated with the formation of neurospheres in mesenchymal and proneural subtypes of glioblastomas.20 HGF/MET signaling is also associated with invasive growth phenotype, which is a characteristic of EMT in GBM.21
In this review, we discuss issues related to identification of the MET signaling pathway as a therapeutic target via inhibition of the EMT in GBM.
EMT in development and disease
The EMT was originally defined to be a biological process that transforms mesenchymal cells from epithelial cells in different embryonic tissues.22 Both SNAI1 (Snail) and (SNAI2) Slug are critical factors in the delamination process of neuronal tissue development.23 Renal fibrosis is a characteristic kidney disease eventually leading to renal failure.24 Accumulating evidence has demonstrated that the majority of interstitial fibroblasts are derived from the kidney epithelium. The EMT is also a major issue in patients undergoing peritoneal dialysis, because long-term dialysis enhances injury of the mesothelial lining, which leads to the EMT, including loss of E-cadherin and increased Snail expression.25 In addition, the EMT is involved in anterior–subcapsular cataracts in humans.26 Eye lens epithelial cells undergo transdifferentiation into a myofibroblastic phenotype in combination with the production of type I and type III collagens, fibronectin, and tenascin.
EMT in human cancers
EMT in cancers
Epithelial cell plasticity is a hallmark of invasive and/or metastatic malignancies. Evidence indicates that EMT occurs at certain sites in primary tumors.27 E-cadherin-negative cells from colon cancer are found at sites of tumor invasion and bud into the stroma, which contributes to local dissemination and metastasis of primary tumors. One study demonstrated that fibroblast-specific protein-1, together with a conversion signal for local formation of fibroblasts by the EMT, provokes acquisition of a metastatic phenotype in genetically engineered mice with breast cancer.28 The EMT induced by ectopic TWIST expression promotes invasiveness, suppressing E-cadherin expression in hepatoma cell lines.29 Irradiation-induced EMT confers invasive properties in endometrial cancer cells.30
EMT in therapy resistance
The EMT also confers resistance to both radiotherapy and chemotherapy. Kajiyama et al31 discovered that paclitaxel-resistant cells, which develop following chronic exposure to paclitaxel, demonstrate cellular and molecular characteristics of the EMT. Snail and Slug, transcriptional repressors and inducers of the EMT, contribute to radio- and chemoresistance by inhibiting p53-mediated apoptosis in ovarian cancer cells.32 The endothelin-1/endothelin-A receptor axis is involved in chemoresistance by promoting the EMT in ovarian cancer cells in vitro and in vivo.33 Stable expression of SNAI1, an EMT inducer, contributes to drug resistance and acquisition of stem-like characteristics in MCF10A cells.34 Suda et al35 determined that the EMT plays a part in the acquired resistance to EGFR tyrosine kinase inhibitors such as erlotinib in non-small-cell lung cancer (NSCLC) cells.36
EMT in cancer stem cells
Reports have demonstrated that the EMT also helps maintain cancer stem cells. The EMT process promotes the emergence of cancer cells with mesenchymal traits essential for tumor invasion/metastasis and the self-renewal properties needed for colonization of a secondary tumor.37 Stem cells derived from malignant breast tissue have a basal-like phenotype and highly upregulated expression of EMT-associated genes.38,39 Mani et al40 found that EMT induction in immortalized human mammary epithelial cells promotes the acquisition of stem cell-like properties, including formation of mammospheres and stem cell marker expression. Morel et al41 discovered that CD44+CD24-/low mammary epithelial cells demonstrate an EMT phenotype, including loss of E-cadherin and gain of vimentin expression. They also demonstrated that treatment with transforming growth factor (TGF)-β, a potent EMT-inducing factor, induced the switch of CD24+ cells into stem-like CD24–cells.
EMT molecular mechanisms
The EMT is characterized by the loss of polarity and downregulation of epithelial cell-associated proteins, including E-cadherin, γ-catenin/plakoglobin, and Zo-1, along with induced expression of mesenchymal proteins, including smooth muscle actin, fibronectin, N-cadherin, and vimentin.42,43 Expression of these molecules enables epithelial cells to acquire a more flexible and migratory mesenchymal cell-like phenotype.27
EMT-inducing signals in many cancers, notably HGF, EGF, platelet-derived growth factor, and TGF-β, are derived from the tumor-associated stroma (Figure 1). These ligands stimulate cells and activate a series of EMT-promoting transcription factors such as Snail, Slug, zinc finger E-box binding homeobox 1, and TWIST.9,27 A series of intracellular signaling networks, including extracellular regulated kinase, mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), Akt, SMADs, β-catenin, and Ras pathways, is involved in the EMT process.44 Cell surface molecules, including β4 integrins and α5β1 and αVβ6 integrins, are also associated with transmitting EMT-promoting signals.45
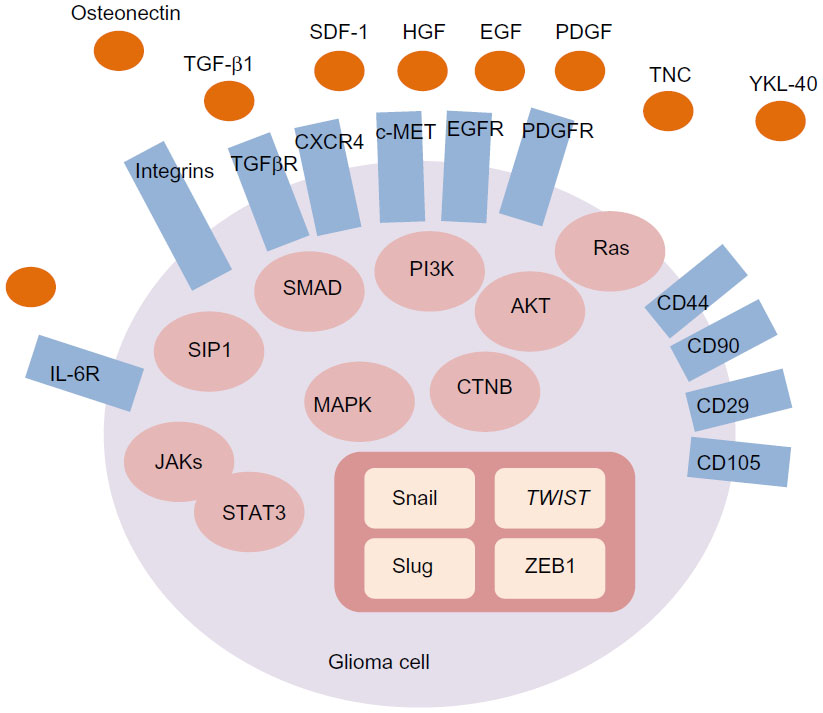 | Figure 1 EMT-associated signaling molecules in glioma cell. Glioma-associated stromal molecules, including TNC, YKL-40, osteonectin, SDF-1, TGF-β1, HGF, EGF, PDGF, and IL-6, trigger the EMT via stimulating corresponding receptors in glioma cells. Cell surface molecules, including IL-6R, TGF-βR, CXCR4, c-MET, EGFR, and PDGF receptor, initiate the oncogenic signaling cascade when they are activated by their specific ligands. β4 integrins and α5β1 and αVβ6 integrins are also associated with transmitting the EMT-promoting signals. In addition, glioma cells express the mesenchymal stem cell-associated surface markers such as CD29, CD44, CD90, and CD105. Intracellular signaling networks, including ERK, MAPK, PI3K, Akt, SMADs, β-catenin, and the Ras pathway, are involved in the EMT. These signaling networks activate a series of EMT-promoting transcription factors such as Snail, Slug, ZEB1, and TWIST. |
TGF-β is an important inducer of the EMT in various cancers. SMAD proteins mediate signaling induced by TGF-β via the anaplastic lymphoid kinase (ALK)-5 receptor.46,47 p38 MAPK and RhoA also mediate TGF-β-induced EMT in mouse mammary epithelial cells.48,49 Active β-catenin, bound to the cytoplasmic tail of E-cadherin, accumulates in the nucleus, where it forms part of the Tcf/LEF complex. This β-catenin-mediated signaling pathway correlates with acquisition of the EMT and tumor invasiveness.50 Loss of E-cadherin is induced by several EMT-promoting transcription factors such as Snail, Slug, SIP1, and E12.51 SIP1 in human epithelial cells induces morphological changes from the epithelial to mesenchymal phenotype.52 The transcription repressor SIP1, also known as ZEB2, suppresses E-cadherin expression by binding to the E-cadherin promoter along with Snail.53 The major regulators of embryogenesis, TWIST 1 and TWIST 2, cooperate with Ras to transform embryonic fibroblasts. The basic helix–loop–helix regulatory factor TWIST 1 induces the EMT and metastatic dissemination of cancer cells by promoting Snail expression.54,55
Xie et al56 reported that interleukin (IL)-6 is capable of generating stem-like CD44+ cells by inducing the EMT in the T47D luminal breast cancer cell line. IL-6 also promotes EMT-related phenotypic changes and mesenchymal cell-specific gene expression by activating the Jak/Stat3/Snail signaling pathway in head and neck squamous cells and immortalized oral epithelial cells.57 Recent evidence indicates that the IL-6/casein kinase 2 signaling pathway promotes EMT and cancer cell migration by stabilizing TWIST at the post-translational level.58
EMT in malignant glioma
Overexpression of TWIST, an EMT-promoting factor, significantly enhances tumor cell invasion in a human glioma cell line (Table 1).14 One study demonstrated that primary glioblastoma and its stem cell lines express cellular and molecular characteristics of mesenchymal stem cells (MSCs).12 Additionally, GBM cell lines largely express MSC-associated surface markers, including CD29, CD44, CD90, and CD105. Genes associated with mesenchymal cells, such as YKL-40, tenascin C (TNC), osteonectin, and CD105, were highly expressed in primary GBM tumors and glioma cell lines. Knockdown of signal transducer and activator of transcription 3 (STAT3), a master regulator of the EMT and cell migration, inhibits glioma cell infiltration and tumor growth in vivo.59 EMT signature genes discovered in a study using mammary cancer tissue, including TAGLN2, insulin-like growth factor-binding protein 2 (IGFBP2), IGFBP3, periostin (POSTN), TNC, and TGF-β1, are predictors for survival in patients with malignant glioma.60 These genes are also upregulated in each of the GBM subtypes: mesenchymal, classical, neural, and proneural. Among the four GBM subtypes, the mesenchymal subtype demonstrates the highest correlation with the EMT-inducing genes.
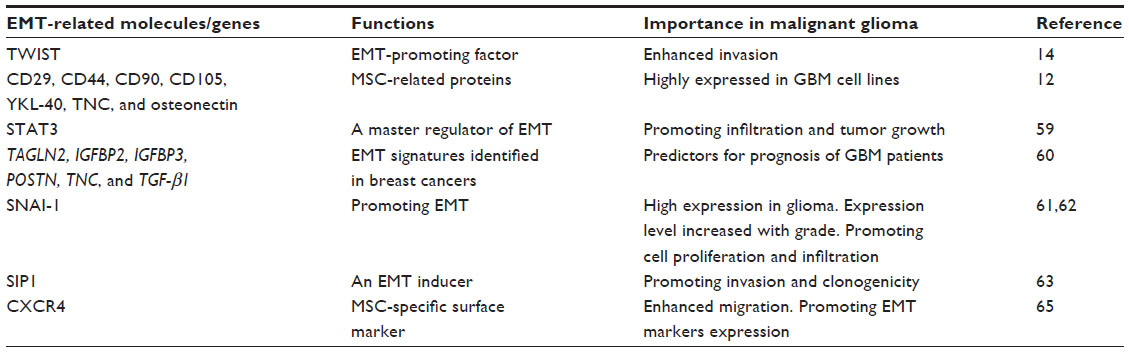 | Table 1 Importance of EMT-related molecules in malignant glioma |
Snail homology 1 (SNAI-1), an EMT-promoting factor, is upregulated in glioma specimens when compared with the normal brain tissue, and SNAI-1 expression is higher in high-grade than in low-grade gliomas.61 Interference of SNAI-1 inhibits the proliferation and migration of glioma cell lines, which confirms a critical role of the EMT in the migration and invasion of glioma cells.62 Another EMT inducer, SMAD-interacting protein-1, promotes invasion, migration, and clonogenecity of glioma cells.63 The chemokine receptor CXCR4 has been regarded to mediate MSC-specific migration.64 Silencing CXCR4 inhibits invasion of the U87 human glioma cell line by suppressing the EMT, and it also upregulates E-cadherin and decreases N-cadherin and vimentin expression.65
MET signaling and EMT
c-MET is a receptor tyrosine kinase involved in a variety of cellular signaling pathways, including those associated with proliferation, invasion, and self-renewal.15 This cell surface receptor is located in epithelial cells of various organs, including the liver, prostate, kidney, muscle, and bone marrow.66 The extracellular portion of c-MET is composed of three domains: the Sema; plexins, semasphorins, and integrins; and the immunoglobulin–plexin–transcription domain.67 The intracellular portion of the c-MET contains a tyrosine kinase catalytic domain with a flanked juxtamembrane and a carboxy-terminal sequence.15 This intracellular portion of the c-MET receptor consists of catalytic tyrosines Y1234 and Y1235, which positively regulate the enzymatic activity of c-MET, whereas the juxtamedullary portion contains Y1003, which negatively regulates c-MET signaling (Figure 2).
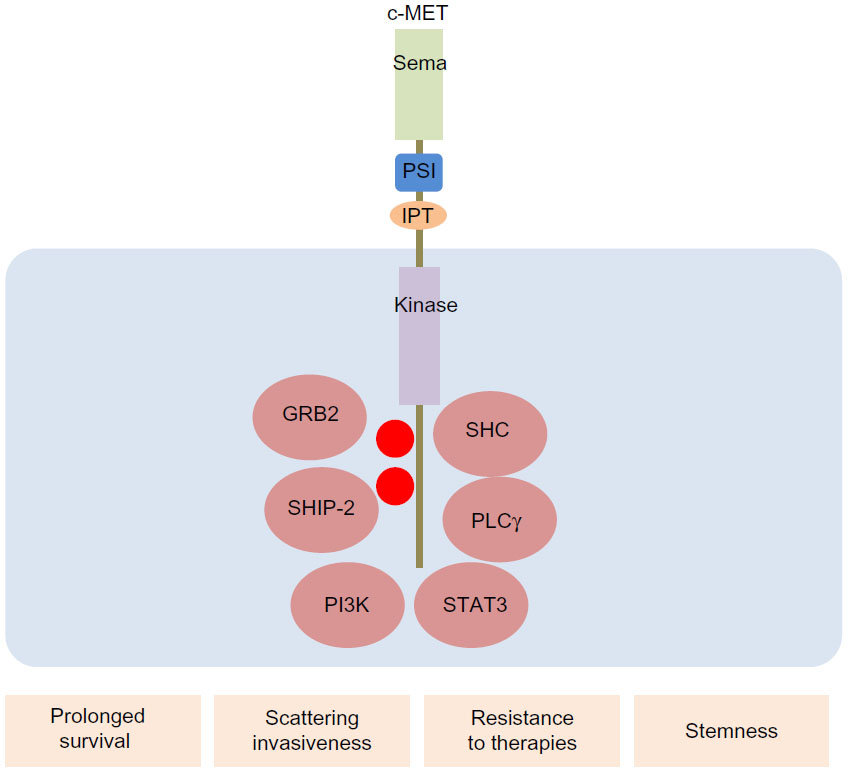 | Figure 2 c-MET mediates EMT-promoting signals in cancers. The extracellular portion of c-MET is composed of three domains: the Sema, PSI, and IPT. The intracellular portion of c-MET contains a tyrosine kinase catalytic domain with flanked juxtamembrane and carboxy-terminal sequence. HGF binding to c-MET induces receptor homodimerization and phosphorylation of tyrosine residues (Y1234 and Y1235) within the catalytic loop, followed by phosphorylation of Y1349 and Y1356 in the carboxyl-terminal tail. When these tyrosine residues are phosphorylated, various adaptor and effecter molecules, including growth factor receptor bound protein 2, Src homology-2-containing phosphatidylionositol 3-kinase, phospholipase Cγ, Src homology domain-containing 5′ inositol phosphatase, and STAT3, are recruited. These intracellular signaling molecules mediate EMT-related cellular responses that promote prolonged survival, scattering/invasiveness, resistance to conventional and molecular targeted therapies, and self-renewal/stemness in cancer cells. |
HGF, also known as scatter factor (SF), is the ligand for c-MET that is considered a motility factor and an SF.16,68 HGF acts as a pleiotropic factor that promotes proliferation, motility, scattering, and differentiation. HGF binding to c-MET induces receptor homodimerization and phosphorylation of tyrosine residues (Y1234 and Y1235) within the catalytic loop, followed by phosphorylation of Y1349 and Y1356 in the carboxyl-terminal tail.69,70 When these tyrosine residues are phosphorylated, various adaptor and effector molecules are recruited, including growth factor receptor bound protein 2, Src homology-2-containing PI3K, phospholipase Cγ, Src homology domain-containing 5′ inositol phosphatase, and STAT3 (Figure 2).70–73 The c-MET receptor interacts with multiple signaling adaptors and cell surface receptors to mediate a variety of biological responses. Recent large-scale proteomic studies have demonstrated notable intricacies in the HGF/c-MET signaling axis.74,75
In certain types of cell models, the HGF/c-MET signaling pathway is equivalent to the EMT.76 The EMT or scattering is a crucial event in the morphogenic processes of embryogenesis.77 HGF and c-MET are coexpressed in progenitor cells during the early stage of mouse embryogenesis and are involved in the generation of endoderm and mesoderm during development of Hensen’s node.78 HGF promotes the EMT during somitogenesis and development of the endocardial cushion through the c-MET/Crk adaptor protein.79 Paracrine HGF derived from mesenchymal cells stimulates epithelial and myoblast progenitors during organogenesis.78 Axonal guidance of motor neurons by HGF results in innervations of striatal muscles, triggering coordinated development.80
Canadas et al81 demonstrated that inhibition of MET signaling results in the reversal of biomarkers associated with the EMT and subsequently increases chemosensitivity in SCLC models. EMT-like phenotypes, including downregulated E-cadherin and increased expression of Snail and N-cadherin, were observed in HGF-treated hepatocellular carcinoma cells. Sorafenib potently reverses the EMT process by inhibiting HGF-mediated MET and downstream kinases such as MAPK.82 Lu et al83 demonstrated that inhibiting MET blocks the EMT and invasive growth in a GBM mouse model provoked by VEGF ablation (Figure 2).
MET signaling in cancers and GBM
MET signaling pathway in general cancers
Somatic mutations in MET are associated with tumor aggressiveness and metastasis in various human malignancies.84 Cancer cells harboring MET mutations are observed particularly in the metastatic lymph node region in certain cases of head and neck cancer.85 In addition, NIH 3T3 cells, which endogenously express HGF, become tumorigenic in nude mice when MET is overexpressed ectopically. Furthermore, cells co-overexpressing human MET and HGF metastasize to the lung and produce invasive tumors in the heart, diaphragm, and retroperitoneum.86 The c-MET autocrine loop has been identified in cancer cells and is indicative of tumor aggressiveness and poor prognosis in NSCLC and breast cancer.87,88 MET amplification was detected in approximately 22% of lung cancer specimens that are resistant to EGFR inhibitors such as gefitinib and erlotinib. Ectopic expression of MET induces resistance to gefitinib in gefitinib-sensitive lung cancer cells. Moreover, MET knockdown restores sensitivity to gefitinib.89
MET signaling pathway in GBM
The high expression level of HGF/SF and MET is significantly correlated with the tumor grade in human primary brain tumors. Moreover, HGF/MET signaling induces cell proliferation and invasive growth in GBM cell lines but does not affect proliferation in normal human astrocytes.90 Kong et al91 reported that MET overexpression is associated with shorter overall survival and poor treatment responses in GBM. They also found that MET expression is significantly associated with matrix metalloproteinase (MMP)-2 and -9, which are indicative of the EMT in cancer. Liu et al92 demonstrated that recurrent GBM expresses a higher level of MET than do primary tumors, and that MET overexpression is associated with shorter progression-free survival in patients with GBM. Activation of the HGF/MET axis has been detected in some human glioblastoma cell lines such as U-373 MG and T98G, and it promotes activation of the Ras, MAPK, AP-1, PI3K, and protein kinase C signaling pathways.93 Also, MET signaling activation was detected in a GSC-rich fraction, and MET expression correlates with stem cell marker expression in GBM. Activating MET also inhibits the forced differentiation of GBM neurospheres.94 Joo et al19 found that a distinct fraction of MET overexpressing cells from patient-derived GSCs were highly clonogenic, tumorigenic, and resistant to radiation. Furthermore, inhibiting MET signaling resulted in disrupted tumorigenecity and invasive growth in GSCs derived from patient tumor specimens. One study reported that HGF/MET signaling induces invasive tumor growth through direct activation of Wnt/β-catenin signaling in GSCs.95
Targeting MET signaling in cancers and GBM
Targeting the MET signaling pathway in general cancers
NK4, a four-kringle domain containing intracellular fragment of HGF, was identified as a competitive inhibitor of HGF/MET signaling via suppression of the specific binding of HGF to its receptor (Figure 3 and Table 2).96 Administrating NK4 by gene therapy suppresses tumor invasion and metastasis in colon cancer cells by inactivating the HGF/MET signaling pathway.97 An engineered soluble MET receptor, decoy MET, interferes with HGF binding to MET and MET homodimerization.98 The decoy MET inhibits cell proliferation, angiogenesis, and spontaneous metastasis in various cancer cells.
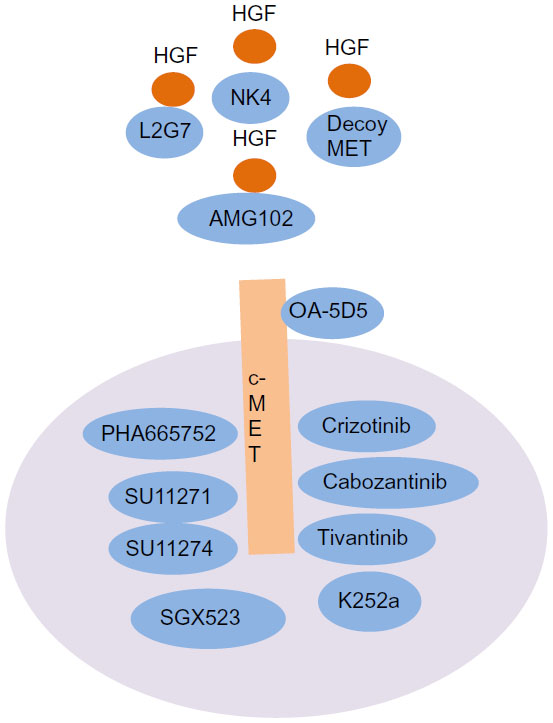 | Figure 3 Current therapeutics targeting the HGF/MET signaling pathway. NK4, a four-kringles-containing intracellular fragment of HGF, was identified as a competitive inhibitor of HGF/MET signaling through suppression of specific binding of HGF to the receptor. An engineered soluble MET receptor, decoy MET, interferes with HGF binding to MET. L2G7 is an anti-HGF monoclonal antibody that suppresses tumor growth by inhibition of the MET signaling pathway. A neutralizing, fully human monoclonal antibody against human and nonhuman primate HGF, AMG 102, inhibited HGF-induced c-MET autophosphorylation. A novel OA-5D5 anti-c-MET antibody suppresses the growth of U87MG cells. An adenosine triphosphateanalog, K252a, potently inhibits HGF-mediated morphology changes and suppresses MET-mediated proliferation. A selective small molecular inhibitor, PHA-665752, inhibits the catalytic activity of c-MET and downstream kinases. PHA-665752 also suppresses the HGF-driven cell growth, motility, and morphology changes. SU11271, a potent and selective pyrrole-indolinone inhibitor of MET tyrosine kinase, suppresses HGF-mediated cell proliferation, survival, and invasion in various cancer cells. SU11274 reduces the activity of constitutively active TPR-MET fusion protein, followed by inducing apoptosis in cells transformed by the oncogenic TPR-MET. Crizotinib, a dual inhibitor of MET and ALK tyrosine kinases, inhibits cells dependent upon c-MET or ALK activity. Cabozantinib, a multityrosine kinase inhibitor, suppresses the activity of MET and vascular endothelial growth factor receptor 2. Tivantinib, a potent small molecular inhibitor of c-MET, induces cell-cycle arrest and inhibits the invasive growth of various cancer cells. |
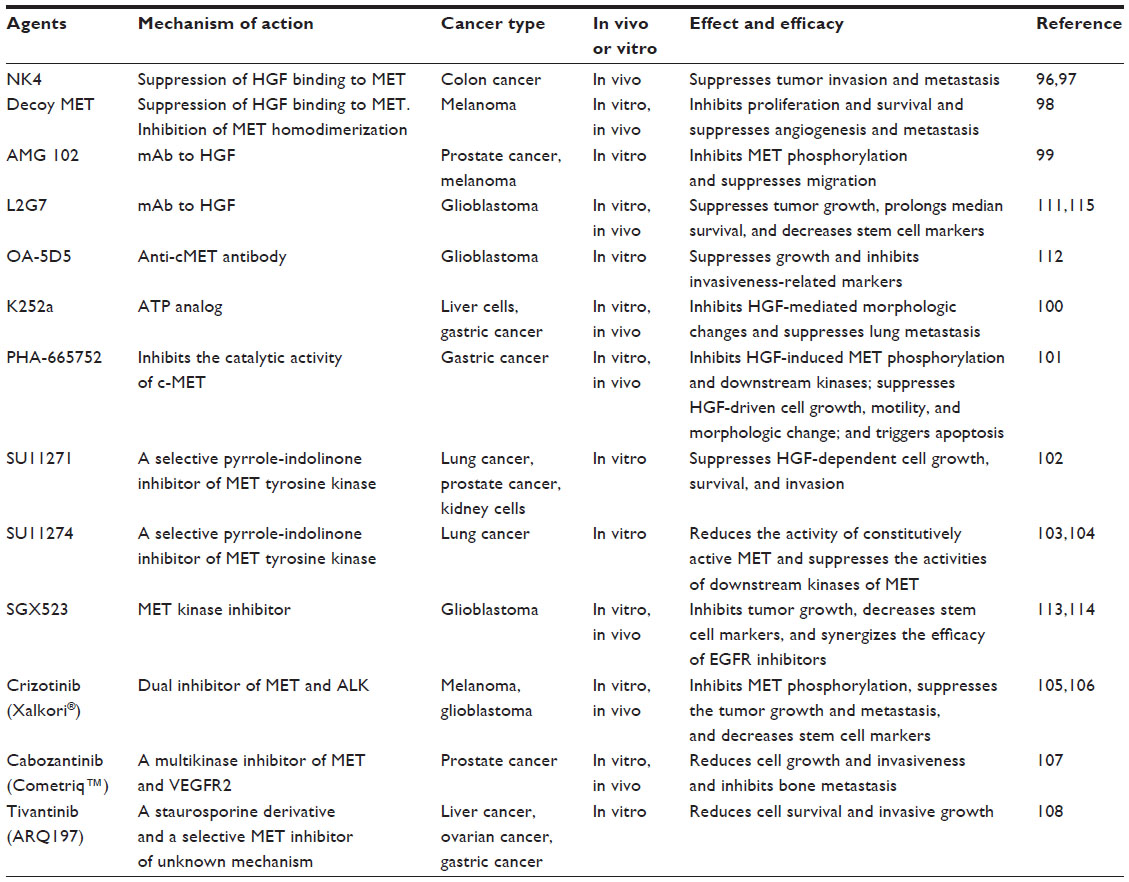 | Table 2 Summary of targeted therapies for MET signaling |
AMG 102, a neutralizing, fully human monoclonal antibody against human and nonhuman primate HGF, inhibited HGF-induced c-MET autophosphorylation in PC3 cells and suppressed HGF-induced migration of human MDA-MB-435 cells (Figure 3).99
The adenosine triphosphateanalog K252a potently inhibited HGF-mediated morphological changes in MLP-29 cells and suppressed MET-mediated proliferation in GTL-16 gastric carcinoma cells. Moreover, administrating K252a inhibited metastasis to the lungs following injection of GTL-16 cells into the caudal vein of a mouse metastasis model.100 The selective small molecular inhibitor PHA-665752, which inhibits c-MET catalytic activity, potently inhibits HGF-induced c-MET phosphorylation and downstream kinases, including extracellular regulated kinase, Akt, and STAT3. PHA-665752 also suppressed HGF-driven cell growth, motility, and morphological changes.101 PHA-665752 specifically triggered apoptosis in gastric cancer cells with MET amplification. SU11271, a potent and selective pyrrole–indolinone inhibitor of MET tyrosine kinase, suppresses HGF-mediated cell proliferation, survival, and invasion in various cancer cells.102 SU11274, another inhibitor of MET kinase activity, reduced the activity of the constitutively active TPR-MET fusion protein, followed by induction of apoptosis in cells transformed with the oncogenic TPR-MET.103 SU11274 also inhibits phosphorylation of c-MET and downstream kinases, including Akt, in NSCLC cells (Figure 3).104
Crizotinib, a dual inhibitor of MET and ALK tyrosine kinases, inhibits cells dependent upon c-MET or ALK activity.105 In addition, crizotinib inhibits the growth and metastasis of uveal melanoma cells by inactivating MET in a metastatic melanoma mouse model.106 Cabozantinib, a multityrosine kinase inhibitor of MET and vascular endothelial growth factor receptor 2, reduces cell growth and invasiveness in vitro, as well as bone metastasis of prostate cancer cells in vivo, by inhibiting the MET signaling pathway.107 A potent small molecule inhibitor of c-MET, tivantinib, induces cell-cycle arrest and reduces cell viability and invasive growth in various cancer cells (Figure 3).108
Targeting the MET signaling pathway in GBM
Neutralizing monoclonal antibodies potently inhibit growth of xenograft tumors from human GBM cells expressing MET and HGF.109 Additionally, the anti-HGF L2G7 monoclonal antibody is capable of suppressing tumor growth by inhibiting the MET signaling pathway. Systemic administration of L2G7 significantly prolongs the median survival of mice with intracranial tumors.110 An anti-HGF antibody manufactured using human–mouse chimeric HGF proteins inhibits HGF-dependent tumor growth and suppresses tumor growth in a xenograft model of the U87MG glioma cell line.111 A novel one-armed 5D5 (OA-5D5) anti-c-MET antibody suppresses growth of U87MG cells by >95%. In addition, treatment with OA-5D5 downregulates the expression of urokinase-type plasminogen activator and MMP-16 in GBM cells.112
The MET kinase inhibitor SGX523 inhibits in vivo tumor growth dramatically in GBM cell lines such as U87M2, U118, and SF295SQ1.113 Xie et al113 demonstrated that activation of HGF autocrine mechanism and serum HGF levels in a xenograft model, rather than MET amplification or HGF paracrine effects, serve as biomarkers for predicting sensitivity to MET inhibition in GBM cell lines. A combination of SGX523 and erlotinib, an EGFR inhibitor, synergistically decreased tumor growth in U87M2 cells.114
Inhibiting the MET pathway by neutralizing the anti-HGF monoclonal antibody L2G7 or crizotinib potently decreased tumor growth and the expression of stem cell markers such as CD133, Sox2, Nanog, and Musashi in a pre-established GBM xenograft model.115 Furthermore, serial transplantation of xenograft-derived cells from mice administrated c-MET inhibition therapy resulted in depleted tumor formation ability and smaller tumor size, compared with control mice.115 L2G7 also synergized the antitumor efficacy of erlotinib against a PTEN-null/EGFRvIII+ GBM xenograft model.116
These results support that inhibiting MET signaling alone may diminish tumor growth in certain GBM cell types, and also that MET inhibitors may synergize the effect of EGFR inhibitors in GBM cells with EGFR vIII espression and PTEN deletion.
Conclusion
The EMT has emerged as a critical event in tumor invasion and metastasis. The EMT also plays a pivotal role in tumor aggressiveness and the development of therapeutic resistance in GBM patients. Activating the HGF/MET signaling pathway is indicative of the EMT because HGF potently induces cell scattering.
Preclinical and clinical trials to inhibit MET signaling by neutralizing human HGF, antagonizing the MET receptor, and inhibiting MET enzymatic activity have progressed. Several monoclonal antibodies and receptor tyrosine kinase inhibitors have been tested clinically for treatment of advanced/metastatic cancer.
Several issues should be considered to maximize the effect of MET inhibition therapy. First, selecting the target population selection is crucial because MET inhibition is often insufficient to suppress the invasive growth of tumors with normal MET expression. Tumors harboring amplified MET and activating MET mutations could be an initial selection strategy for establishing a target population for MET inhibition therapy. Another target population of interest could be those patients administered antiangiogenic therapy with VEGF ablation. Because VEGF ablation may induce invasive growth by rebound activation of the MET signaling pathway,83 MET inhibition could suppress the emerging outgrowth of cells induced by antiangiogenic therapy. Second, considering a combination of chemotherapeutics or small molecule inhibitors of other major growth pathways would be beneficial, since inhibition of MET signaling alone may be insufficient to induce massive apoptosis or reduce tumor volume. Inhibiting other major growth pathways, including EGFR or FGFR, in combination with HGF/MET, may trigger effective reductions in tumor burden as well as invasive growth in certain cancer cases.
Acknowledgments
This work was supported by a Samsung Medical Center grant (GFO1140011) and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI09C1552).
Disclosure
The authors report no conflicts of interest in this work.
References
Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. | |
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | |
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | |
Ohka F, Natsume A, Wakabayashi T. Current trends in targeted therapies for glioblastoma multiforme. Neurol Res Int. 2012;2012:878425. | |
Keunen O, Johansson M, Oudin A, et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A. 2011;108(9):3749–3754. | |
Uhm JH, Ballman KV, Wu W, et al. Phase II evaluation of gefitinib in patients with newly diagnosed grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int J Radiat Oncol Biol Phys. 2011;80(2):347–353. | |
Lu-Emerson C, Norden AD, Drappatz J, et al. Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. J Neurooncol. 2011;104(1):287–291. | |
Chi AS, Wen PY. Inhibiting kinases in malignant gliomas. Expert Opin Ther Targets. 2007;11(4):473–496. | |
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. | |
Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776–1784. | |
Arrieta O, Saavedra-Perez D, Kuri R, et al. Brain metastasis development and poor survival associated with carcinoembryonic antigen (CEA) level in advanced non-small cell lung cancer: a prospective analysis. BMC Cancer. 2009;9:119. | |
Tso CL, Shintaku P, Chen J, et al. Primary glioblastomas express mesenchymal stem-like properties. Mol Cancer Res. 2006;4(9):607–619. | |
Freije WA, Castro-Vargas FE, Fang Z, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64(18):6503–6510. | |
Elias MC, Tozer KR, Silber JR, et al. TWIST is expressed in human gliomas and promotes invasion. Neoplasia. 2005;7(9):824–837. | |
Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3(Suppl 1):S7–S19. | |
Nakamura T, Nishizawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342(6248):440–443. | |
Basilico C, Arnesano A, Galluzzo M, Comoglio PM, Michieli P. A high affinity hepatocyte growth factor-binding site in the immunoglobulin-like region of MET. J Biol Chem. 2008;283(30):21267–21277. | |
Abounader R, Laterra J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol. 2005;7(4):436–451. | |
Joo KM, Jin J, Kim E, et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012;72(15):3828–3838. | |
De Bacco F, Casanova E, Medico E, et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012;72(17):4537–4550. | |
Boccaccio C, Comoglio PM. The MET oncogene in glioblastoma stem cells: implications as a diagnostic marker and a therapeutic target. Cancer Res. 2013;73(11):3193–3199. | |
Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15(6):740–746. | |
Aybar MJ, Nieto MA, Mayor R. Snail precedes Slug in the genetic cascade required for the specification and migration of the Xenopus neural crest. Development. 2003;130(3):483–494. | |
Eitner F, Floege J. Novel insights into renal fibrosis. Curr Opin Nephrol Hypertens. 2003;12(3):227–232. | |
Yanez-Mo M, Lara-Pezzi E, Selgas R, et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med. 2003;348(5):403–413. | |
Saika S, Miyamoto T, Tanaka S, et al. Response of lens epithelial cells to injury: role of lumican in epithelial-mesenchymal transition. Invest Ophthalmol Vis Sci. 2003;44(5):2094–2102. | |
Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. | |
Xue C, Plieth D, Venkov C, Xu C, Neilson EG. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res. 2003;63(12):3386–3394. | |
Lee TK, Poon RT, Yuen AP, et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin Cancer Res. 2006;12(18):5369–5376. | |
Tsukamoto H, Shibata K, Kajiyama H, Terauchi M, Nawa A, Kikkawa F. Irradiation-induced epithelial-mesenchymal transition (EMT) related to invasive potential in endometrial carcinoma cells. Gynecol Oncol. 2007;107(3):500–504. | |
Kajiyama H, Shibata K, Terauchi M, et al. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31(2):277–283. | |
Kurrey NK, Jalgaonkar SP, Joglekar AV, et al. Snail and Slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. 2009;27(9):2059–2068. | |
Rosano L, Cianfrocca R, Spinella F, et al. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin Cancer Res. 2011;17(8):2350–2360. | |
Lim S, Becker A, Zimmer A, Lu J, Buettner R, Kirfel J. SNAI1-mediated epithelial-mesenchymal transition confers chemoresistance and cellular plasticity by regulating genes involved in cell death and stem cell maintenance. PloS One. 2013;8(6):e66558. | |
Suda K, Tomizawa K, Fujii M, et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011;6(7):1152–1161. | |
Krysan K, Lee JM, Dohadwala M, et al. Inflammation, epithelial to mesenchymal transition, and epidermal growth factor receptor tyrosine kinase inhibitor resistance. J Thorac Oncol. 2008;3(2):107–110. | |
Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69(10):4116–4124. | |
Stingl J, Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer. 2007;7(10):791–799. | |
Liao MJ, Zhang CC, Zhou B, et al. Enrichment of a population of mammary gland cells that form mammospheres and have in vivo repopulating activity. Cancer Res. 2007;67(17):8131–8138. | |
Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. | |
Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PloS One. 2008;3(8):e2888. | |
Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4(8):657–665. | |
Jechlinger M, Grunert S, Tamir IH, et al. Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003;22(46):7155–7169. | |
Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–829. | |
Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem. 2007;101(4):816–829. | |
Miyazono K. TGF-beta signaling by Smad proteins. Cytokine Growth Factor Rev. 2000;11(1–2):15–22. | |
Miyazono K. TGF-beta/SMAD signaling and its involvement in tumor progression. Biol Pharm Bull. 2000;23(10):1125–1130. | |
Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem. 2001;276(50):46707–46713. | |
Bhowmick NA, Ghiassi M, Bakin A, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12(1):27–36. | |
Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26(5):463–476. | |
Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell. 2008;19(11):4875–4887. | |
Vandewalle C, Comijn J, De Craene B, et al. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 2005;33(20):6566–6578. | |
Comijn J, Berx G, Vermassen P, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7(6):1267–1278. | |
Ansieau S, Bastid J, Doreau A, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14(1):79–89. | |
Smit MA, Geiger TR, Song JY, Gitelman I, Peeper DS. A Twist-Snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol Cell Biol. 2009;29(13):3722–3737. | |
Xie G, Yao Q, Liu Y, et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int J Oncol. 2012;40(4):1171–1179. | |
Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011;9(12):1658–1667. | |
Su YW, Xie TX, Sano D, Myers JN. IL-6 stabilizes Twist and enhances tumor cell motility in head and neck cancer cells through activation of casein kinase 2. PloS One. 2011;6(4):e19412. | |
Priester M, Copanaki E, Vafaizadeh V, et al. STAT3 silencing inhibits glioma single cell infiltration and tumor growth. Neuro Oncol. 2013;15(7):840–852. | |
Zarkoob H, Taube JH, Singh SK, Mani SA, Kohandel M. Investigating the link between molecular subtypes of glioblastoma, epithelial-mesenchymal transition, and CD133 cell surface protein. PloS One. 2013;8(5):e64169. | |
Motta FJ, Valera ET, Lucio-Eterovic AK, et al. Differential expression of E-cadherin gene in human neuroepithelial tumors. Genet Mol Res. 2008;7(2):295–304. | |
Han SP, Kim JH, Han ME, et al. SNAI1 is involved in the proliferation and migration of glioblastoma cells. Cell Mol Neurobiol. 2011;31(3):489–496. | |
Xia M, Hu M, Wang J, et al. Identification of the role of Smad interacting protein 1 (SIP1) in glioma. J Neurooncol. 2010;97(2):225–232. | |
Honczarenko M, Le Y, Swierkowski M, Ghiran I, Glodek AM, Silberstein LE. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells. 2006;24(4):1030–1041. | |
Zhu Y, Yang P, Wang Q, et al. The effect of CXCR4 silencing on epithelial-mesenchymal transition related genes in glioma U87 cells. Anat Rec. 2013;296(12):1850–1856. | |
Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7(6):504–516. | |
Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat RevCancer. 2002;2(4):289–300. | |
Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327(6119):239–242. | |
Rodrigues GA, Park M. Autophosphorylation modulates the kinase activity and oncogenic potential of the MET receptor tyrosine kinase. Oncogene. 1994;9(7):2019–2027. | |
Ponzetto C, Bardelli A, Zhen Z, et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994;77(2):261–271. | |
Fixman ED, Fournier TM, Kamikura DM, Naujokas MA, Park M. Pathways downstream of Shc and Grb2 are required for cell transformation by the Tpr-MET oncoprotein. J Biol Chem. 1996;271(22):13116–13122. | |
Koch A, Mancini A, El Bounkari O, Tamura T. The SH2-domian-containing inositol 5-phosphatase (SHIP)-2 binds to c-MET directly via tyrosine residue 1356 and involves hepatocyte growth factor (HGF)-induced lamellipodium formation, cell scattering and cell spreading. Oncogene. 2005;24(21):3436–3447. | |
Boccaccio C, Ando M, Tamagnone L, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998;391(6664):285–288. | |
Organ SL, Tong J, Taylor P, et al. Quantitative phospho-proteomic profiling of hepatocyte growth factor (HGF)-MET signaling in colorectal cancer. J Proteome Res. 2011;10(7):3200–3211. | |
Hammond DE, Hyde R, Kratchmarova I, Beynon RJ, Blagoev B, Clague MJ. Quantitative analysis of HGF and EGF-dependent phosphotyrosine signaling networks. J Proteome Res. 2010;9(5):2734–2742. | |
Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6(8):637–645. | |
Montesano R, Matsumoto K, Nakamura T, Orci L. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell. 1991;67(5):901–908. | |
Andermarcher E, Surani MA, Gherardi E. Co-expression of the HGF/SF and c-MET genes during early mouse embryogenesis precedes reciprocal expression in adjacent tissues during organogenesis. Dev Genet. 1996;18(3):254–266. | |
Song W, Majka SM, McGuire PG. Hepatocyte growth factor expression in the developing myocardium: evidence for a role in the regulation of the mesenchymal cell phenotype and urokinase expression. Dev Dyn. 1999;214(1):92–100. | |
Maina F, Klein R. Hepatocyte growth factor, a versatile signal for developing neurons. Nat Neurosci. 1999;2(3):213–217. | |
Canadas I, Rojo F, Taus A, et al. Targeting epithelial-to-mesenchymal transition with MET inhibitors reverts chemoresistance in small cell lung cancer. Clin Cancer Res. 2014;20(4):938–950. | |
Nagai T, Arao T, Furuta K, et al. Sorafenib inhibits the hepatocyte growth factor-mediated epithelial mesenchymal transition in hepatocellular carcinoma. Mol Cancer Ther. 2011;10(1):169–177. | |
Lu KV, Chang JP, Parachoniak CA, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22(1):21–35. | |
Lorenzato A, Olivero M, Patane S, et al. Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res. 2002;62(23):7025–7030. | |
Di Renzo MF, Olivero M, Martone T, et al. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000;19(12):1547–1555. | |
Rong S, Segal S, Anver M, Resau JH, Vande Woude GF. Invasiveness and metastasis of NIH 3T3 cells induced by MET-hepatocyte growth factor/scatter factor autocrine stimulation. Proc Natl Acad Sci U S A. 1994;91(11):4731–4735. | |
Rahimi N, Tremblay E, McAdam L, Park M, Schwall R, Elliott B. Identification of a hepatocyte growth factor autocrine loop in a murine mammary carcinoma. Cell Growth Differ. 1996;7(2):263–270. | |
Navab R, Liu J, Seiden-Long I, et al. Co-overexpression of MET and hepatocyte growth factor promotes systemic metastasis in NCI-H460 non-small cell lung carcinoma cells. Neoplasia. 2009;11(12):1292–1300. | |
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. | |
Koochekpour S, Jeffers M, Rulong S, et al. MET and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997;57(23):5391–5398. | |
Kong DS, Song SY, Kim DH, et al. Prognostic significance of c-MET expression in glioblastomas. Cancer. 2009;115(1):140–148. | |
Liu W, Fu Y, Xu S, et al. c-MET expression is associated with time to recurrence in patients with glioblastoma multiforme. J Clin Neurosci. 2011;18(1):119–121. | |
Abounader R, Ranganathan S, Kim BY, Nichols C, Laterra J. Signaling pathways in the induction of c-MET receptor expression by its ligand scatter factor/hepatocyte growth factor in human glioblastoma. J Neurochem. 2001;76(5):1497–1508. | |
Li Y, Li A, Glas M, et al. c-MET signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci U S A. 2011;108(24):9951–9956. | |
Kim KH, Seol HJ, Kim EH, et al. Wnt/beta-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro Oncol. 2013;15(2):161–171. | |
Date K, Matsumoto K, Shimura H, Tanaka M, Nakamura T. HGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factor. FEBS Lett. 1997;420(1):1–6. | |
Matsumoto K, Nakamura T. Mechanisms and significance of bifunctional NK4 in cancer treatment. Biochem Biophys Res Commun. 2005;333(2):316–327. | |
Michieli P, Mazzone M, Basilico C, et al. Targeting the tumor and its microenvironment by a dual-function decoy MET receptor. Cancer Cell. 2004;6(1):61–73. | |
Burgess TL, Sun J, Meyer S, et al. Biochemical characterization of AMG 102: a neutralizing, fully human monoclonal antibody to human and nonhuman primate hepatocyte growth factor. Mol Cancer Ther. 2010;9(2):400–409. | |
Morotti A, Mila S, Accornero P, Tagliabue E, Ponzetto C. K252a inhibits the oncogenic properties of MET, the HGF receptor. Oncogene. 2002;21(32):4885–4893. | |
Christensen JG, Schreck R, Burrows J, et al. A selective small molecule inhibitor of c-MET kinase inhibits c-MET-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003;63(21):7345–7355. | |
Wang X, Le P, Liang C, et al. Potent and selective inhibitors of the MET [hepatocyte growth factor/scatter factor (HGF/SF) receptor] tyrosine kinase block HGF/SF-induced tumor cell growth and invasion. Mol Cancer Ther. 2003;2(11):1085–1092. | |
Sattler M, Pride YB, Ma P, et al. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res. 2003;63(17):5462–5469. | |
Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-MET and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65(4):1479–1488. | |
Rodig SJ, Shapiro GI. Crizotinib, a small-molecule dual inhibitor of the c-MET and ALK receptor tyrosine kinases. Curr Opin Investig Drugs. 2010;11(12):1477–1490. | |
Surriga O, Rajasekhar VK, Ambrosini G, Dogan Y, Huang R, Schwartz GK. Crizotinib, a c-MET inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol Cancer Ther. 2013;12(12):2817–2826. | |
Dai J, Zhang H, Karatsinides A, et al. Cabozantinib inhibits prostate cancer growth and prevents tumor-induced bone lesions. Clin Cancer Res. 2014;20(3):617–630. | |
Katayama R, Aoyama A, Yamori T, et al. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013;73(10):3087–3096. | |
Cao B, Su Y, Oskarsson M, et al. Neutralizing monoclonal antibodies to hepatocyte growth factor/scatter factor (HGF/SF) display antitumor activity in animal models. Proc Natl Acad Sci U S A. 2001;98(13):7443–7448. | |
Kim KJ, Wang L, Su YC, et al. Systemic anti-hepatocyte growth factor monoclonal antibody therapy induces the regression of intracranial glioma xenografts. Clin Cancer Res. 2006;12(4):1292–1298. | |
Burgess T, Coxon A, Meyer S, et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-MET-dependent human tumors. Cancer Res. 2006;66(3):1721–1729. | |
Martens T, Schmidt NO, Eckerich C, et al. A novel one-armed anti-c-MET antibody inhibits glioblastoma growth in vivo. Clin Cancer Res. 2006;12(20 Pt 1):6144–6152. | |
Xie Q, Bradley R, Kang L, et al. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci U S A. 2012;109(2):570–575. | |
Zhang YW, Staal B, Essenburg C, et al. MET kinase inhibitor SGX523 synergizes with epidermal growth factor receptor inhibitor erlotinib in a hepatocyte growth factor-dependent fashion to suppress carcinoma growth. Cancer Res. 2010;70(17):6880–6890. | |
Rath P, Lal B, Ajala O, et al. In vivo c-MET pathway inhibition depletes human glioma xenografts of tumor-propagating stem-like cells. Transl Oncol. 2013;6(2):104–111. | |
Lal B, Goodwin CR, Sang Y, et al. EGFRvIII and c-MET pathway inhibitors synergize against PTEN-null/EGFRvIII+ glioblastoma xenografts. Mol Cancer Ther. 2009;8(7):1751–1760. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.