")
Back to Journals » OncoTargets and Therapy » Volume 10
Targeting the androgen receptor in triple-negative breast cancer: current perspectives
Authors Mina A, Yoder R, Sharma P
Received 29 June 2017
Accepted for publication 30 August 2017
Published 20 September 2017 Volume 2017:10 Pages 4675—4685
DOI https://doi.org/10.2147/OTT.S126051
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Dr XuYu Yang
Alain Mina,1 Rachel Yoder,2 Priyanka Sharma1
1Division of Medical Oncology, Department of Internal Medicine, University of Kansas Medical Center, Westwood, 2University of Kansas Cancer Center, Kansas City, KS, USA
Abstract: Triple-negative breast cancer (TNBC) is an aggressive subtype associated with frequent recurrence and metastasis. Unlike hormone receptor-positive subtypes, treatment of TNBC is currently limited by the lack of clinically available targeted therapies. Androgen signaling is necessary for normal breast development, and its dysregulation has been implicated in breast tumorigenesis. In recent years, gene expression studies have identified a subset of TNBC that is enriched for androgen receptor (AR) signaling. Interference with androgen signaling in TNBC is promising, and AR-inhibiting drugs have shown antitumorigenic activity in preclinical and proof of concept clinical studies. Recent advances in our understanding of androgenic signaling in TNBC, along with the identification of interacting pathways, are allowing development of the next generation of clinical trials with AR inhibitors. As novel AR-targeting agents are developed and evaluated in clinical trials, it is equally important to establish a robust set of biomarkers for identification of TNBC tumors that are most likely to respond to AR inhibition.
Keywords: triple-negative breast cancer, androgen signaling, targeted therapy, biomarkers, prognosis
Introduction
Of the 250,000 new cases of breast cancer expected in the United States in 2017, 15% will be diagnosed as triple-negative breast cancer (TNBC).1–5 Accelerated growth, high recurrence rates, and frequent metastasis characterize the aggressiveness of TNBC and result in poor long-term patient survival.2,3 TNBC is defined by the lack of estrogen and progesterone receptors as well as absence of human epidermal growth factor 2 (HER2) overexpression/amplification. Despite successful use of targeted therapies in other subtypes of breast cancer, similar approaches in TNBC have not reached clinical practice. Because of the lack of targeted therapy, ~30%–40% of patients with early-stage TNBC develop metastatic disease and succumb to the cancer despite receiving standard multiagent adjuvant chemotherapy.6,7
Variable response to treatment has been a major challenge in developing targeted therapies in TNBC, where it points to an underlying heterogeneity within the subtype. Advances in gene expression profiling have revealed several complementary TNBC classification systems that may be associated with response to therapy.5,8–11 New classifications have isolated a subset of TNBC that are enriched for AR expression.5,12 Given the tremendous clinical success of targeting estrogen receptor in hormone-positive breast cancer, AR positivity in TNBC may constitute a clinically targetable signaling pathway. In support of this approach, numerous preclinical studies have validated the use of AR modulation in limiting cell proliferation, and there are ongoing clinical trials evaluating the safety and efficacy of AR antagonists in breast cancer. The following review explores the role of AR in tumorigenesis and progression, and its role not only as a prognostic and predictive tool but also as a potential therapeutic target in TNBC.
Hormone signaling in tumorigenesis
Estrogen receptor (ER) and progesterone receptor (PR) can stimulate tumor growth and metastasis in breast cancer. Antihormone therapies, such as tamoxifen, aromatase inhibitors, and selective ER degraders, are efficacious in hormone-positive breast cancer. A third hormone receptor, AR, is present in all subtypes of breast cancer and is attracting attention as a potential therapeutic target in breast cancer.13
The AR is an intracellular steroid receptor that dimerizes and translocates to the nucleus upon binding of androgen ligands, where it binds to androgen response elements (AREs) to promote target gene transcription in a tissue-specific manner (Figure 1). Normal breast development is driven by AR interaction with the Wnt pathway, but AR is also known to regulate genes implicated in metastasis, and androgens have shown independent tumorigenic activity in vitro and in animal models.14–20
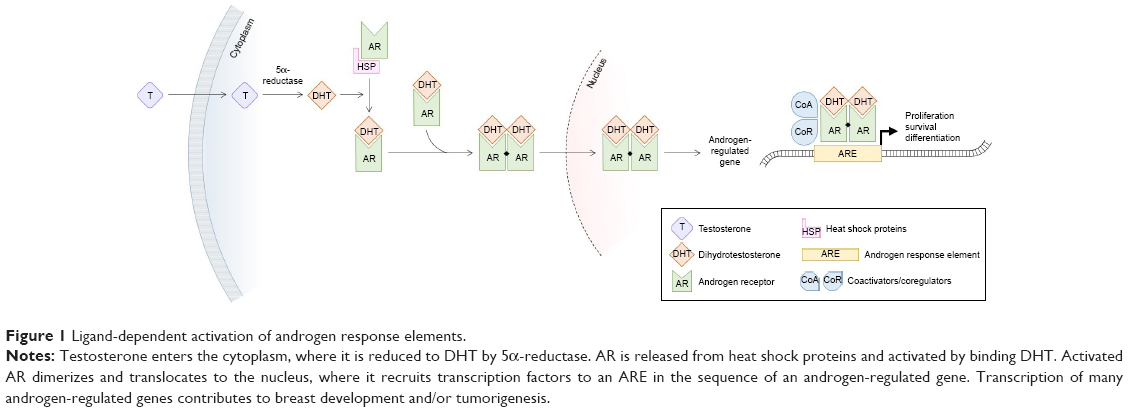 | Figure 1 Ligand-dependent activation of androgen response elements. |
AR also interacts with other intracellular signaling pathways. Despite AR’s demonstrated tumorigenic activity, crosstalk with the ER pathway can have the paradoxical effect of limiting tumor proliferation. The ER regulates gene transcription in a similar manner by binding to estrogen response elements (EREs) in cis-regulatory elements of estrogen-regulated genes.21,22 AR can competitively bind to EREs and coactivators to suppress estrogen-mediated tumor proliferation.18,23 However, in the absence of ER, as is the case in TNBC, AR mainly interacts with AREs and stimulates tumor cell growth in an androgen-dependent manner.24
Androgen signaling in breast cancer
Unlike ER and PR, AR is found in all major breast cancer subtypes and is estimated to be present in 53%–80% of all breast cancers.4,25–31 AR+ breast tumors are diagnosed more commonly in older patients.4,24,29,31,32 A positive AR tumor status appears to be associated with favorable clinical features such as lower tumor-node-metastasis stage, lower nuclear grade, less risk of lymph node involvement, and smaller tumor size at diagnosis.4,24,25,28–33 AR expression significantly overlaps with ER+/PR+ status, lack of HER2 overexpression/amplification, and lower proliferative index.18,24,27,29–32,34,35
In patients unselected for hormone receptor or HER2 expression, AR may be an indicator of favorable prognosis. Some studies have reported association of AR+ breast cancers with better response to endocrine therapy and longer disease-free survival (DFS) and overall survival (OS).4,18,24–27,29,31,32,36,37
A dual role for AR, dependent on the relative strength of ER signaling, has been proposed by some groups, and would explain the varied prognoses among ER+AR+, ER+AR-, ER-AR+, and ER-AR- breast cancer.14 The detrimental effect of discordant AR and ER expression suggests that androgen-mediated proliferation in breast cancer may be regulated by the relative availability of each receptor. When estrogen is low, testosterone is preferentially converted to estradiol, an ER ligand, instead of to 5α-dihydrotestosterone (DHT), an AR ligand, thus translating androgen supply into ER-driven tumorigenesis.20,38–40 Accordingly, a higher tumor AR-to-ER ratio is independently associated with lymph node metastasis and poor survival.20 When the AR-to-ER ratio is low, or when estrogens are available, androgen metabolism will activate AR to compete for EREs. In these circumstances, AR can be antitumorigenic.
Androgen signaling in TNBC
Association of AR status with clinical-pathological characteristics in TNBC
Using immunohistochemical (IHC) assessment, AR is present in 13%–37% of TNBC, serving as the sole hormone receptor in these cases.4,24,25,30,33,34,41–44 As ER and PR are absent in TNBC, the biological and therapeutic role of AR independent of other hormone receptors can be studied in this subtype. Association of AR with clinical and pathological features and ultimately prognosis in TNBC is not completely understood. AR positivity in TNBC is associated with older age at presentation, coinciding with the high circulating levels of androgens seen in postmenopausal breast cancer patients.45 In TNBC, some studies have shown AR positivity to be associated with higher nuclear grade, higher tumor stage, and lymph node metastases, though others have found an association with lower nuclear grade or have failed to note any associations with clinical-pathological features.20,41–44,46 Conflicting reports of a higher vs lower proliferation index in AR+ TNBC have also been made.42,44 Several reports have noted overlap between AR positivity and apocrine histological features or apocrine gene expression signature in TNBC.30,34,47,48
Meta-analyses led by Qu and Wang, encompassing over 4,000 cases of TNBC, demonstrated AR+ status to be associated with better DFS and OS.26,49 Another meta-analysis by Gonzalez-Angulo et al, however, could only note a nonsignificant trend of better DFS and OS, and numerous other studies have observed no difference or a negative impact of AR status on outcomes.20,24,25,31,32,41,42,50
At present, there is no standardized method or cutoff for detection of AR expression, and AR assessment is not part of routine pathological testing for breast cancer. The majority of published literature has utilized AR IHC nuclear staining for determination of AR positivity, yet tissue processing methods and choice of AR antibody are not consistent among published literature on this topic. Thus, studies assessing the clinical correlations and prognostic impact of AR are limited by their retrospective nature, variability in techniques and cutoffs (>1% to ≥10% nuclear staining by IHC) used to determine AR positivity, and variations in the clinical and treatment characteristics of patient cohorts being evaluated. In summary, the prognostic value of AR expression in TNBC is not yet clear, and is likely complicated by lack of standardized testing methodology and heterogeneity within the patient population.
AR+ subtype presents a unique clinical course
Recent efforts in molecular characterization of TNBC have resulted in its classification into additional subtypes. Seminal gene expression profiling studies by Perou et al categorized breast cancer into four intrinsic molecular subtypes.8 The basal-like subtype comprises a group of tumors characterized by low or absent ER expression, very low prevalence of HER2 overexpression/amplification, and expression of genes usually found in the basal or myoepithelial cells of the human breast. Although the majority of TNBCs fall into the basal-like intrinsic subtype, the overlap between immunohistochemically defined TNBC and basal-like intrinsic subtype is not complete.50 Various studies demonstrate that 70%–80% of TNBC are basal-like and 20%–30% of non-TNBC are basal-like by molecular profiling.51–53 Proportion of non-basal-like subtypes within TNBC may be influenced by age at breast cancer diagnosis; Prat et al demonstrated a higher incidence of non-basal TNBC in women over 60 years (26%) of age as compared to those who are 40 years or younger (4.3%).53 Approximately 7% of unselected TNBC tumors classify as luminal A or B subtype on intrinsic molecular profiling.53 There may be a link between TNBC noted to be luminal on intrinsic profiling and AR overexpression; it is postulated that TNBCs classified as luminal A may be enriched with AR overexpression.5
TNBC is a diverse entity for which additional subclassifications beyond basal and non-basal may be needed. Using gene expression from publically available data sets, Lehmann et al classified TNBC initially into seven molecular subtypes, and recently refined the classification into four molecular subtypes: basal-like 1, basal-like 2, mesenchymal, and luminal androgen receptor-like (LAR) based on gene expression profiles.11 Based on identification of cell lines corresponding to each subtype, they also demonstrated that these subtypes may be responsive to different targeted therapies.5 The methodology of Lehmann et al’s molecular classification has recently been adapted to a RNA-seq platform to better fit individual clinical samples.54
Approximately 16% of TNBCs classify as LAR molecular subtype (Figure 2). Retrospective studies have demonstrated LAR subtype to be associated with clinical-pathologic features, treatment response, and outcomes. LAR subtype tumors have lower pathological grade and are diagnosed in women of older ages compared to all other TNBC types. LAR tumors also demonstrate significant enrichment of axillary lymph node metastasis and preferential distant metastasis to bone.11
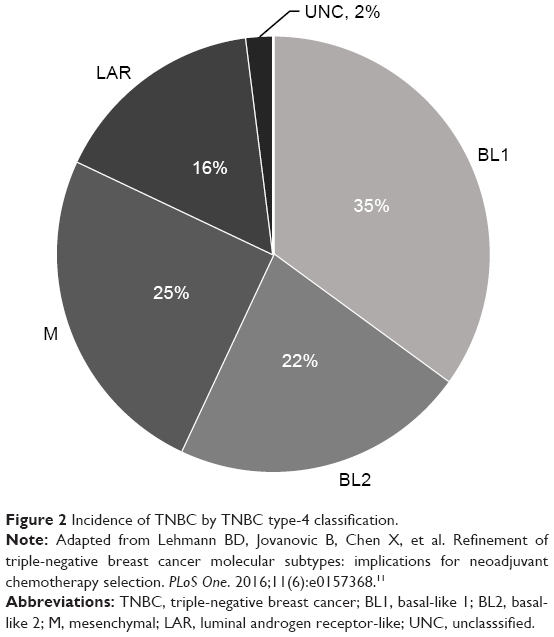 | Figure 2 Incidence of TNBC by TNBC type-4 classification. |
LAR tumors respond to neoadjuvant chemotherapy at much lower rates; recent studies have demonstrated that the LAR molecular subtype is associated with lower pathological complete response (pCR) rates compared to other TNBC subtypes.11,55 Similar associations of positive AR IHC expression with low pCR rates have also been reported in TNBC.4,56 Furthermore, some studies have also demonstrated better DFS in spite of low pCR in AR+ TNBC.4 The discrepancy between neoadjuvant chemotherapy response and survival in the LAR subtype mimics the observations noted for ER-positive and/or luminal A breast cancer57,58 and further supports the notion of biological similarities between AR+/LAR subtype and luminal AR+/ER+ breast cancers.
Androgen signaling drives the LAR phenotype
Gene ontologies for LAR subtype are heavily enriched in hormonally regulated pathways including steroid synthesis, porphyrin metabolism, and androgen/estrogen metabolism. LAR subtype also shows enhanced activity of AR at the transcript level and correlations with nuclear AR IHC staining and protein levels.5 Tumors within the LAR group are also found to express downstream AR targets and coactivators (DHCR24, ALCAM, FASN, FKBP5, APOD, PIP, SPDEF, and CLDN8).5 AR expression in LAR subtype tumors is responsible for tumor cell viability and survival, as suggested by the significantly reduced ability of LAR cell lines to form colonies after knockdown of AR expression.5
Androgen signaling is a targetable pathway in TNBC
Preclinical in vitro and xenograft studies have demonstrated that LAR cell lines are sensitive to AR inhibition.5,14,19,20 AR expression is also noted in cell lines representing other TNBC subtypes beyond the LAR subtype. AR inhibition can thus be a potential therapeutic strategy for other TNBC subtypes. Recent studies showed that AR inhibition with enzalutamide and bicalutamide significantly reduces baseline proliferation, anchorage-independent growth, migration, and invasion, and increases apoptosis in LAR and three non-LAR TNBC molecular subtypes (mesenchymal-like, mesenchymal stem-like, and basal-like 2).19,59 Enzalutamide inhibits DHT-driven tumor growth in ER-negative (MDA-MB-453) xenografts by increasing apoptosis.20 Thus, the preclinical studies suggest that antagonism of the androgen signaling pathway could be a potential therapeutic approach for TNBC.
Androgen receptor targeting
Preclinical evidence of efficacy
Early androgen signaling inhibitors were first investigated as part of standard-of-care androgen deprivation therapy (ADT) for prostate cancer. Wong and Xie validated the suspected association between androgen exposure and mammary cancer in rats, demonstrating a role of androgens in inducing histological transformation, which was reversed with the androgen-blocking agent flutamide.60 Subsequent studies in breast cancer employed the fourfold more potent agent bicalutamide, which in prostate cancer is capable of inducing accessory sex organ regression with minimal effect on serum hormone levels.61 Bicalutamide has shown a paradoxical effect in breast cancer depending on ER expression, inducing apoptosis in ER- tumors and reversing androgen-driven cell death in ER+ tumors.20,62–64 Lehmann et al have described bicalutamide sensitivity in the LAR molecular subtype of TNBC.5 Bicalutamide’s apoptotic effects in other subtypes of TNBC have also been demonstrated in preclinical studies.40,59 In a noteworthy case report by Arce-Salinas et al, a patient with metastatic AR+ TNBC whose disease had progressed under heavy systemic chemotherapy achieved complete clinical response in chest wall disease after 4 months of oral bicalutamide therapy.65 These preclinical data, along with the decades of safety and tolerability studies in prostate cancer, prompted a surge of clinical trials employing AR antagonists in breast cancer and TNBC in particular.
Bicalutamide is a nonsteroidal antiandrogen, which competitively inhibits the binding of androgens with AR.66 It is commonly used in the treatment of locally advanced and metastatic prostate cancer, either as monotherapy or combined with a gonadotropin-releasing hormone agonist.67,68 Whereas bicalutamide inhibits transcription of AR-regulated genes by assembling corepressors rather than coactivators, the second-generation nonsteroidal antiandrogen enzalutamide has a fivefold greater affinity for AR and prevents nuclear translocation of ligand-bound AR.69–71 As described earlier, bicalutamide promotes proliferation in ER+ breast cancer, perhaps due to AR competitively binding to EREs in the nucleus, an effect which enzalutamide prevents by inhibiting nuclear entry.20 Enzalutamide was shown to inhibit ER-mediated mammary tumor growth in ER+ as well as in ER- cancers, which supports the proposed mechanism of action. Work by Barton et al suggested that AR inhibition may be effective even in low AR-expressing TNBC, as evidenced by enzalutamide-induced apoptosis in one LAR and three non-LAR TNBC subtype cell lines.19 AR inhibition alone may be insufficient in cases of advanced or chemoresistant TNBC. Abiraterone acetate is a selective inhibitor of cytochrome P450 17α-hydroxylase/17,20-lyase (CYP17), reducing adrenal and tumor androgen biosynthesis.72 Castrate-resistant, taxane-resistant prostate cancer exhibits good clinical response to abiraterone acetate in conjunction with prednisone, an effect that is dependent on AR, at least in part.73–76 Use of CYP17 inhibitors to reduce androgen availability may increase the potency of agents targeting the AR in breast cancer.
Kwilas et al evaluated the ability of AR inhibition to reduce the growth and improve the immune-mediated killing of breast cancer cells with differing expression of the ER and AR.77 They reported that while AR expression was required for the growth inhibitory effects of enzalutamide on breast cancer cells, both enzalutamide and abiraterone improved the sensitivity of breast cancer cells to immune-mediated lysis independent of detectable AR expression. Reduction in osteoprotegerin was noted to mediate the increase in sensitivity of AR- TNBC cells to immune-mediated killing. This data further supports investigation of AR inhibition in the AR+ TNBC and also in AR- TNBC, especially in combination with immunotherapy. In a mouse model of prostate cancer, the combination of enzalutamide and immunotherapy resulted in a significantly higher OS as opposed to each individual treatment.78 These findings expand the treatment potential of enzalutamide and other androgen antagonists to both AR+ and AR- TNBC.
Clinical activity of antiandrogen monotherapy
Bicalutamide
AR status in TNBC patients has been utilized as a biomarker for preselection of patients for antiandrogen therapy trials. A Translational Breast Cancer Research Consortium Phase II study was the first to evaluate antiandrogen therapy in TNBC patients selected by AR status (NCT00468715).62 Eligibility for the trial required either the primary or a metastatic tumor to be positive for AR (IHC >10% nuclear staining). Twelve percent (51 of 424) of screened patients demonstrated AR positivity. Patients received bicalutamide 150 mg orally daily. Among 26 evaluable patients, although there were no complete or partial responses, the 6-month clinical benefit rate (CBR) was 19%. Bicalutamide was well tolerated with no grade 4/5 treatment-related adverse events observed. This study demonstrated proof of principle for the efficacy of androgen blockade in a select group of patients with ER-/PR-/AR+ breast cancer.
Enzalutamide
A recent Phase II clinical trial evaluated single-agent enzalutamide in women with advanced AR+ TNBC (AR IHC >0%) (NCT001889238). Of the 75 patients who were evaluable, CBR was 35% and 29% at 16 and 24 weeks, respectively.79 This trial also reported on the positive association of gene signature (PREDICT AR) for identification of TNBC patients most likely to benefit from this approach.50 PREDICT AR positive status was noted in 50% of patients with metastatic TNBC in this study, and patients with positive signature experienced higher CBR at 16 and 24 weeks compared to those lacking this gene signature.50,79 AR IHC, on the other hand, did not correlate with response to enzalutamide in this study. This indicates that further development and refinement of biomarkers for identification of patients most likely to benefit from antiandrogen therapy are needed.
CYP17 inhibitors
Abiraterone acetate plus prednisone treatment was evaluated in a Phase II proof-of-concept trial for advanced AR+ TNBC (NCT01842321).80 AR positivity for this trial was set at ≥10% IHC expression, and 37% of tested samples met this criterion. Among 30 evaluable patients, the 6-month CBR was 20% (one complete response and five patients with stable disease for ≥6 months). Although the study did not meet its predefined end point of a 25% CBR, abiraterone acetate may be effective for a selected subset of AR+ TNBC. Indeed, several ongoing Phase I–II trials are investigating the efficacy of CYP17 inhibitors, alone or combined with other pathway inhibitors (Table 1).
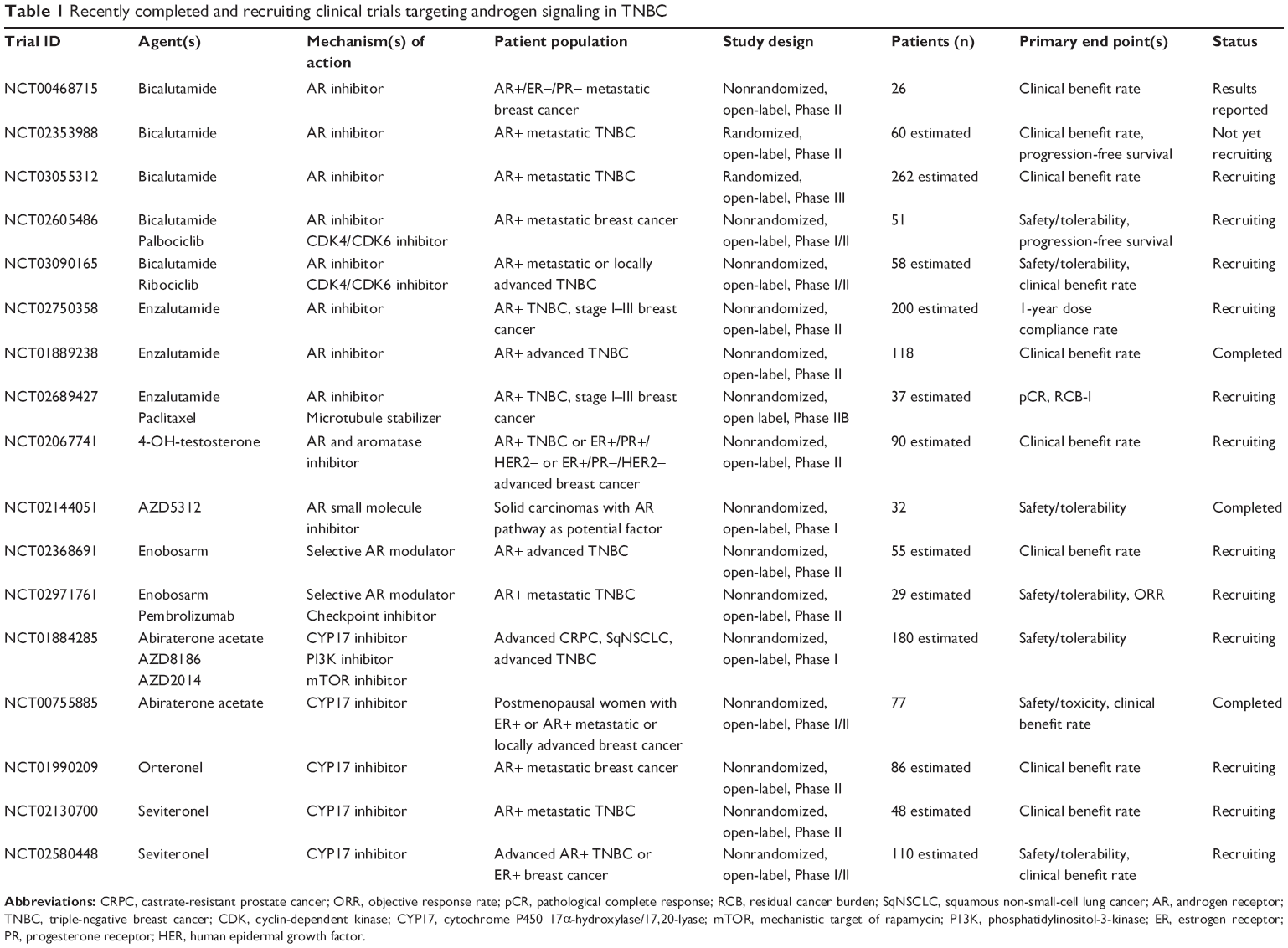 | Table 1 Recently completed and recruiting clinical trials targeting androgen signaling in TNBC |
Combination therapy
Preclinical data suggest that AR dependency may coexist with other oncogenic aberrations, suggesting potential value of combining AR targeting with other targeted agents.
AR inhibitors plus CDK4/CDK6 inhibitors
Cyclin-dependent kinases (CDK) 4 and 6 promote proliferation by removing retinoblastoma (RB) protein-driven suppression of cell cycle progression. CDK4/6 inhibitors such as palbociclib inhibit proliferation by arresting the cell cycle in the G1 phase.81 Although RB protein is commonly lost in TNBC, RB has been associated with AR expression in TNBC, and sensitivity to palbociclib has been reported in three LAR cell lines.82,83 Combination therapy of CDK4/6 inhibitors and androgen deprivation treatment is being evaluated in two ongoing Phase I/II clinical trials (Table 1).
AR inhibitors plus PI3K inhibitors
Phosphatidylinositol-3-kinase (PI3K) and the downstream components Akt and mechanistic target of rapamycin (mTOR) are recognized as promising targets for treatment of breast cancer.84,85 Activating mutations of PIK3CA are seen in ~40% of AR+ TNBC tumors, as compared to 4% of AR- TNBC tumors.86 Though PIK3CA inhibitors display antiproliferative effects on tumors with elevated PI3K activity, AR knockdown alone can allow cells to bypass the tumor suppressor activity of phosphate and tensin homolog (PTEN), which can promote proliferation.17 Combination therapy with a PI3K inhibitor and AR inhibitor has an additive apoptotic effect in AR+ TNBC cell lines.84,86 Combination of the mTOR inhibitor rapamycin and the antiandrogen enzalutamide has also shown additive effect in LAR TNBC cell lines and in a LAR xenograft model.87 Based on this preclinical evidence, clinical investigation of antiandrogen therapy with drugs targeting PI3K/mTOR pathway is underway. An ongoing Phase I study is assessing combinations of abiraterone with PI3K inhibitor or mTOR inhibitor in metastatic TNBC (NCT01884285).
AR inhibitors plus neoadjuvant chemotherapy
AR+ TNBC is associated with relative resistance to conventional neoadjuvant chemotherapy as demonstrated by lower rates of pCR.5,11,55 This observation raises the question of whether combining AR inhibition with chemotherapy would improve response to chemotherapy in AR+ TNBC. A Phase II clinical trial is currently underway to assess rates of pCR or near-pCR in early-stage AR+ (≥10%) TNBC patients treated with enzalutamide and weekly paclitaxel (NCT02689427).
Future direction
The 6-month CBR of 19%–29% observed with antiandrogen monotherapy in clinical trials to date (NCT00468715, NCT001889238, and NCT01842321) is relatively modest. However, this degree of activity is not very different from early experience of targeting ER in metastatic breast cancer where diethylstilbestrol monotherapy yielded response rates of 4%–21%.114,115 Subsequently, decades of research that involved development of more efficacious agents to target ER and standardization of techniques to accurately identify ER-positive disease led to improved success in clinical trials. The low response rates seen with current AR targeting in clinical trials could also be related to resistance (primary or secondary) to antiandrogen therapy. In prostate cancer, failure of ADT has been linked to amplification of AR and/or increased expression of AR variants such as AR-V7 that lack the C-terminal ligand-binding domain and are thus constitutively active.73,88,89 Constitutively active AR variants have recently been detected in breast cancers, and have been shown to induce in vitro proliferation in the presence of enzalutamide.90 The antibody used in most clinical trials (AR441, Dako North America Inc, Carpinteria, CA, USA) is antigenic to the receptor’s N-terminus, implying that the total AR detected may be comprised of both full-length AR and ligand-independent truncated AR variants. The development of IHC antibodies that detect AR’s C-terminal ligand-binding domain may improve patient selection in future clinical trials by allowing quantitation of both full-length AR and truncated variants. Recent preclinical studies suggest that AR variant antagonists (HSP90 inhibitors, ROR-γ inhibitors) may reverse ADT resistance in tumors with constitutively active AR.91,92 Similarly, other agents that block dimerization, nuclear translocation, or DNA binding could prove effective against full-length and truncated AR isoforms alike. Future clinical trials targeting AR in TNBC will benefit from a better understanding of ADT resistance and the ability to further select patients who will benefit from antiandrogen therapy.
A second potential source of ADT resistance in AR+ TNBC is membrane-initiated androgen signaling. While the classical model of AR signaling requires intracellular AR ligand binding, nuclear translocation, and ARE recognition (Figure 1), nongenomic AR activity may also play a role in androgen resistance. Actin skeleton reorganization, decreased cell motility, and increased apoptosis have been ascribed to membrane AR signaling in breast cancer cell lines.93–95 Further investigation is required, though, as activated membrane AR has also been shown to promote cell viability in other cell types.96,97 The discovery of nongenomic androgen activity suggests the possibility of modulated androgen signaling independently of intracellular AR activity. Various approaches are being explored in preclinical studies, including albumin-conjugated androgens that only activate membrane AR and cannot enter the cytosol and conversely, agents that inhibit membrane AR phosphorylation and downstream signaling.98,99 In summary, membrane-initiated androgen signaling may partially complement and partially compete with genomic AR activity, and though its role is incompletely understood, membrane AR represents a potentially targetable marker in TNBC and other cancers.
Studies using knockout rodent models have revealed a complex relationship between AR and the immune response. Intracellular AR is differentially expressed in immune cell subpopulations, and androgen signaling can suppress B and T lymphocyte development and conversely stimulate neutrophil production.100,101 In prostate cancer, AR-dependent cell lines are highly susceptible to TNF-α-induced apoptosis, and exposure to TNF-α induces hypersensitivity to androgen signaling.102,103 The cytokine interleukin-6 upregulates AR transcription and induces ligand-independent AR activation, which may promote inflammation and tumor growth.104–106 Mediators of these responses include NF-κB, MAPK, STAT3, and PI3K/Akt/mTOR.102,104–111 Additionally, androgen signaling downregulates expression of certain toll-like receptors, which may inhibit the immune response and promote proliferation.112
The AR is thus involved in an intricate web of both pro- and anti-tumor signaling with other pathways. These complex interactions are incompletely understood, yet they illustrate the need for additional biomarkers to evaluate androgen dependency in patients. While standardized methods for detecting AR in breast tumor samples are important for establishing its predictive and prognostic value, patients with similar tumor AR expression may not respond homogeneously to antiandrogen therapy due to coregulated signaling pathways. As illustrated in the above section ‘Combination therapy’, several clinical trials are underway to evaluate combinations of targeted therapy.
Summary
Androgen targeting has demonstrated early promise and is worthy of further evaluation in appropriately selected TNBC. Evaluation of AR targeting in clinical trials has thus far utilized AR IHC expression (with various cutoffs) as selection criteria. Preclinical studies investigating this therapeutic approach have, on the other hand, utilized AR IHC and gene expression-defined subtypes (luminal or LAR subtypes). It unlikely that IHC positivity for AR alone will accurately identify patients likely to respond to AR modulating therapies, due to the complexity of interacting signaling pathways.79,113 Several gene expression-defined subtypes to identify tumor AR dependency have come forth in recent years (PREDICT AR, LAR subtype, intrinsic luminal subtype).5,11,50,53,54 However, these need to be evaluated in context of prospective trials before being translated to routine clinical use. Additional translational efforts should also focus on changes in tumor AR dependency under pressures of standard chemotherapy and whether primary or metastatic tumor androgen dependency is most likely to correspond to antiandrogen treatment response.
Use of single-agent AR inhibitors have exhibited modest efficacy in clinical trials. Recent advances in our understanding of androgenic signaling in TNBC, along with the identification of interacting pathways, are allowing development of next generation of clinical trials with AR inhibitors. As novel AR-targeting agents are developed and evaluated in clinical trials, it is equally important to establish a robust set of biomarkers for identification of TNBC tumors that are androgen dependent. Once achieved, this approach should guide successful study design and accrual efforts (Figure 3).
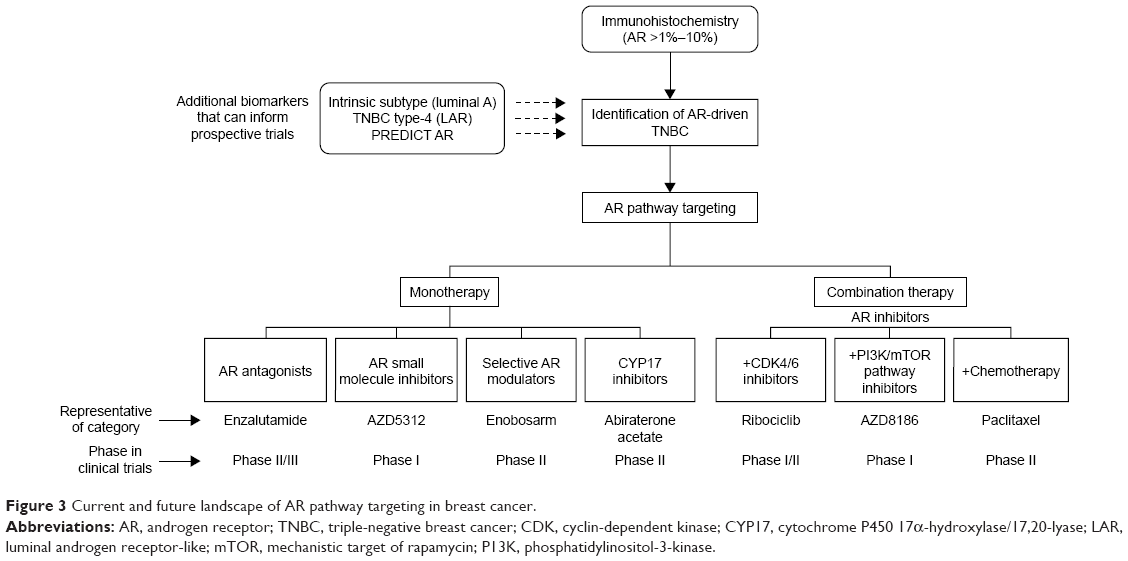 | Figure 3 Current and future landscape of AR pathway targeting in breast cancer. |
Though the role of androgen signaling in TNBC is complex, AR emerges as a potential therapeutic target for a subset of patients with this aggressive disease that otherwise lacks molecular targets. Antiandrogen agents are well tolerated, with acceptable and well-established safety profiles. Several antiandrogen agents are being evaluated in clinical trials either as single agents or in combination with other pathway inhibitors and cytotoxic compounds. The prospective development of standardized and reproducible methods for identifying AR-dependent tumors will allow investigators to tailor clinical trials to the correct subject population and allow clinicians to better predict patients’ response to these therapies in future.
Disclosure
The authors report no conflicts of interest in this work.
References
American Cancer Society. Cancer Facts and Figures 2017. Atlanta, GA: American Cancer Society; 2017. | ||
Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–4434. | ||
Bauer KR, Brown M, Cress RD, et al. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer registry. Cancer.2007;109(9):1721–1728. | ||
Loibl S, Muller BM, von Minckwitz G, et al. Androgen receptor expression in primary breast cancer and its predictive and prognostic value in patients treated with neoadjuvant chemotherapy. Breast Cancer Res Treat. 2011;130(2):477–487. | ||
Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. | ||
Haffty BG, Yang Q, Reiss M, et al. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol. 2006;24(36):5652–5657. | ||
Tan DS, Marchio C, Jones RL, et al. Triple negative breast cancer: molecular profiling and prognostic impact in adjuvant anthracycline-treated patients. Breast Cancer Res Treat. 2008;111(1):27–44. | ||
Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. | ||
Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100(14):8418–8423. | ||
Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. | ||
Lehmann BD, Jovanovic B, Chen X, et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One. 2016;11(6):e0157368. | ||
Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232(2):142–150. | ||
Rampurwala M, Wisinski KB, O’Regan R. Role of the androgen receptor in triple-negative breast cancer. Clin Adv Hematol Oncol. 2016;14(3):186–193. | ||
Ni M, Chen Y, Lim E, et al. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011;20(1):119–131. | ||
Liu YN, Liu Y, Lee HJ, et al. Activated androgen receptor downregulates E-cadherin gene expression and promotes tumor metastasis. Mol Cell Biol. 2008;28(23):7096–7108. | ||
Naderi A, Hughes-Davies L. A functionally significant cross-talk between androgen receptor and ErbB2 pathways in estrogen receptor negative breast cancer. Neoplasia. 2008;10(6):542–548. | ||
Wang Y, Romigh T, He X, et al. Differential regulation of PTEN expression by androgen receptor in prostate and breast cancers. Oncogene. 2011;30(42):4327–4338. | ||
Peters AA, Buchanan G, Ricciardelli C, et al. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. 2009;69(15):6131–6140. | ||
Barton VN, D’Amato NC, Gordon MA, et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. 2015;14(3):769–778. | ||
Cochrane DR, Bernales S, Jacobsen BM, et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014;16(1):R7. | ||
Bourdeau V, Deschenes J, Metivier R, et al. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol Endocrinol. 2004;18(6):1411–1427. | ||
Carroll JS, Meyer CA, Song J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38(11):1289–1297. | ||
Lanzino M, De Amicis F, McPhaul MJ, et al. Endogenous coactivator ARA70 interacts with estrogen receptor alpha (ERalpha) and modulates the functional ERalpha/androgen receptor interplay in MCF-7 cells. J Biol Chem. 2005;280(21):20421–20430. | ||
Aleskandarany MA, Abduljabbar R, Ashankyty I, et al. Prognostic significance of androgen receptor expression in invasive breast cancer: transcriptomic and protein expression analysis. Breast Cancer Res Treat. 2016;159(2):215–227. | ||
Hu R, Dawood S, Holmes MD, et al. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin Cancer Res. 2011;17(7):1867–1874. | ||
Qu Q, Mao Y, Fei XC, et al. The impact of androgen receptor expression on breast cancer survival: a retrospective study and meta-analysis. PLoS One. 2013;8(12):e82650. | ||
Vera-Badillo FE, Templeton AJ, de Gouveia P, et al. Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J Natl Cancer Inst. 2014;106(1):djt319. | ||
Collins LC, Cole KS, Marotti JD, et al. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses’ Health Study. Mod Pathol. 2011;24(7):924–931. | ||
Elebro K, Borgquist S, Simonsson M, et al. Combined androgen and estrogen receptor status in breast cancer: treatment prediction and prognosis in a population-based prospective cohort. Clin Cancer Res. 2015;21(16):3640–3650. | ||
Park S, Koo J, Park HS, et al. Expression of androgen receptors in primary breast cancer. Ann Oncol. 2010;21(3):488–492. | ||
Park S, Koo JS, Kim MS, et al. Androgen receptor expression is significantly associated with better outcomes in estrogen receptor-positive breast cancers. Ann Oncol. 2011;22(8):1755–1762. | ||
Gonzalez-Angulo AM, Stemke-Hale K, Palla SL, et al. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin Cancer Res. 2009;15(7):2472–2478. | ||
He J, Peng R, Yuan Z, et al. Prognostic value of androgen receptor expression in operable triple-negative breast cancer: a retrospective analysis based on a tissue microarray. Med Oncol. 2012;29(2):406–410. | ||
Safarpour D, Pakneshan S, Tavassoli FA. Androgen receptor (AR) expression in 400 breast carcinomas: is routine AR assessment justified? Am J Cancer Res. 2014;4(4):353–368. | ||
Agoff SN, Swanson PE, Linden H, et al. Androgen receptor expression in estrogen receptor-negative breast cancer. Am J Clin Pathol. 2003;120(5):725–731. | ||
Bryan RM, Mercer RJ, Bennett RC, et al. Androgen receptors in breast cancer. Cancer. 1984;54(11):2436–2440. | ||
Castellano I, Allia E, Accortanzo V, et al. Androgen receptor expression is a significant prognostic factor in estrogen receptor positive breast cancers. Breast Cancer Res Treat. 2010;124(3):607–617. | ||
Hickey TE, Robinson JL, Carroll JS, et al. Minireview: the androgen receptor in breast tissues: growth inhibitor, tumor suppressor, oncogene? Mol Endocrinol. 2012;26(8):1252–1267. | ||
Labrie F, El-Alfy M, Berger L, et al. The combination of a novel selective estrogen receptor modulator with an estrogen protects the mammary gland and uterus in a rodent model: the future of postmenopausal women’s health? Endocrinology. 2003;144(11):4700–4706. | ||
Mehta J, Asthana S, Mandal CC, et al. A molecular analysis provides novel insights into androgen receptor signalling in breast cancer. PLoS One. 2015;10(3):e0120622. | ||
McGhan LJ, McCullough AE, Protheroe CA, et al. Androgen receptor-positive triple negative breast cancer: a unique breast cancer subtype. Ann Surg Oncol. 2014;21(2):361–367. | ||
Mrklic I, Pogorelic Z, Capkun V, et al. Expression of androgen receptors in triple negative breast carcinomas. Acta Histochem. 2013;115(4):344–348. | ||
Sutton LM, Cao D, Sarode V, et al. Decreased androgen receptor expression is associated with distant metastases in patients with androgen receptor-expressing triple-negative breast carcinoma. Am J Clin Pathol. 2012;138(4):511–516. | ||
Tang D, Xu S, Zhang Q, et al. The expression and clinical significance of the androgen receptor and E-cadherin in triple-negative breast cancer. Med Oncol. 2012;29(2):526–533. | ||
Cauley JA, Lucas FL, Kuller LH, et al. Elevated serum estradiol and testosterone concentrations are associated with a high risk for breast cancer. Ann Intern Med. 1999;130(4 Pt 1):270–277. | ||
Rakha EA, El-Sayed ME, Green AR, et al. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109(1):25–32. | ||
Farmer P, Bonnefoi H, Becette V, et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene. 2005;24(29):4660–4671. | ||
Mills AM, Gottlieb C, Wendroth S, et al. Pure apocrine carcinomas represent a clinicopathologically distinct androgen receptor-positive subset of triple-negative breast cancers. Am J Surg Pathol. 2016;40(8):1109–1116. | ||
Wang C, Pan B, Zhu H, et al. Prognostic value of androgen receptor in triple negative breast cancer: a meta-analysis. Oncotarget. 2016;7(29):46482–46891. | ||
Parker JS, Peterson AC, Tudor IC, et al. A novel biomarker may predict clinical activity from enzalutamide in triple-negative breast cancer. Poster presented at: 2015 ASCO Annual Meeting; May 29–June 2; 2015;Chicago, IL. | ||
Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5(1):5–23. | ||
Bastien RR, Rodriguez-Lescure A, Ebbert MT, et al. PAM50 breast cancer subtyping by RT-qPCR and concordance with standard clinical molecular markers. BMC Med Genomics. 2012;5:44. | ||
Prat A, Adamo B, Cheang MC, et al. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist. 2013;18(2):123–133. | ||
Ring BZ, Hout DR, Morris SW, et al. Generation of an algorithm based on minimal gene sets to clinically subtype triple negative breast cancer patients. BMC Cancer. 2016;16:143. | ||
Masuda H, Baggerly KA, Wang Y, et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. 2013;19(19):5533–5540. | ||
Asano Y, Kashiwagi S, Onoda N, et al. Clinical verification of sensitivity to preoperative chemotherapy in cases of androgen receptor-expressing positive breast cancer. Br J Cancer. 2016;114(1):14–20. | ||
Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384(9938):164–172. | ||
Rouzier R, Perou CM, Symmans WF, et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res. 2005;11(16):5678–5685. | ||
Zhu A, Li Y, Song W, et al. Antiproliferative effect of androgen receptor inhibition in mesenchymal stem-like triple-negative breast cancer. Cell Physiol Biochem. 2016;38(3):1003–1014. | ||
Wong YC, Xie B. The role of androgens in mammary carcinogenesis. Ital J Anat Embryol. 2001;106(2 Suppl 1):111–125. | ||
Furr BJ, Valcaccia B, Curry B, et al. ICI 176,334: a novel non-steroidal, peripherally selective antiandrogen. J Endocrinol. 1987;113(3):R7–R9. | ||
Gucalp A, Tolaney S, Isakoff SJ, et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic breast cancer. Clin Cancer Res. 2013;19(19):5505–5512. | ||
Szelei J, Jimenez J, Soto AM, et al. Androgen-induced inhibition of proliferation in human breast cancer MCF7 cells transfected with androgen receptor. Endocrinology. 1997;138(4):1406–1412. | ||
Toth-Fejel S, Cheek J, Calhoun K, et al. Estrogen and androgen receptors as comediators of breast cancer cell proliferation: providing a new therapeutic tool. Arch Surg. 2004;139(1):50–54. | ||
Arce-Salinas C, Riesco-Martinez MC, Hanna W, et al. Complete response of metastatic androgen receptor-positive breast cancer to bicalutamide: case report and review of the literature. J Clin Oncol. 2016;34(4):e21–e24. | ||
Wirth MP, Hakenberg OW, Froehner M. Antiandrogens in the treatment of prostate cancer. Eur Urol. 2007;51(2):306–313; discussion 14. | ||
Anderson J. The role of antiandrogen monotherapy in the treatment of prostate cancer. BJU Int. 2003;91(5):455–461. | ||
Pilepich MV, Winter K, John MJ, et al. Phase III radiation therapy oncology group (RTOG) trial 86-10 of androgen deprivation adjuvant to definitive radiotherapy in locally advanced carcinoma of the prostate. Int J Radiat Oncol, Biol, Phys. 2001;50(5):1243–1252. | ||
Baek SH, Ohgi KA, Nelson CA, et al. Ligand-specific allosteric regulation of coactivator functions of androgen receptor in prostate cancer cells. Proc Natl Acad Sci U S A. 2006;103(9):3100–3105. | ||
Cheng S, Brzostek S, Lee SR, et al. Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol Endocrinol. 2002;16(7):1492–1501. | ||
Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790. | ||
Bedoya DJ, Mitsiades N. Clinical appraisal of abiraterone in the treatment of metastatic prostatic cancer: patient considerations, novel opportunities, and future directions. Onco Targets Ther. 2013;6:9–18. | ||
Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028–1038. | ||
Azad AA, Eigl BJ, Murray RN, et al. Efficacy of enzalutamide following abiraterone acetate in chemotherapy-naive metastatic castration-resistant prostate cancer patients. Eur Urol. 2015;67(1):23–29. | ||
Romanel A, Gasi Tandefelt D, Conteduca V, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015;7(312):312re10. | ||
Salvi S, Casadio V, Conteduca V, et al. Circulating cell-free AR and CYP17A1 copy number variations may associate with outcome of metastatic castration-resistant prostate cancer patients treated with abiraterone. Br J Cancer. 2015;112(10):1717–1724. | ||
Kwilas AR, Ardiani A, Gameiro SR, et al. Androgen deprivation therapy sensitizes triple negative breast cancer cells to immune-mediated lysis through androgen receptor independent modulation of osteoprotegerin. Oncotarget. 2016;7(17):23498–234511. | ||
Ardiani A, Farsaci B, Rogers CJ, et al. Combination therapy with a second-generation androgen receptor antagonist and a metastasis vaccine improves survival in a spontaneous prostate cancer model. Clin Cancer Res. 2013;19(22):6205–6218. | ||
Traina T, Miller K, Yardley D, et al. Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). J Clin Oncol. 2015;33(suppl):abstr 1003. | ||
Bonnefoi H, Grellety T, Tredan O, et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann Oncol. 2016;27(5):812–818. | ||
Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–1438. | ||
Asghar U, Herrera-Abreu MT, Cutts R, et al. Identification of subtypes of triple negative breast cancer (TNBC) that are sensitive to CDK4/6 inhibition. J Clin Oncol. 2015;33(suppl):abstr 11098. | ||
Trere D, Brighenti E, Donati G, et al. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann Oncol. 2009;20(11):1818–1823. | ||
Cuenca-Lopez MD, Montero JC, Morales JC, et al. Phospho-kinase profile of triple negative breast cancer and androgen receptor signaling. BMC Cancer. 2014;14:302. | ||
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | ||
Lehmann BD, Bauer JA, Schafer JM, et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014;16(4):406. | ||
Robles AJ, Cai S, Cichewicz RH, et al. Selective activity of deguelin identifies therapeutic targets for androgen receptor-positive breast cancer. Breast Cancer Res Treat. 2016;157(3):475–488. | ||
Dehm SM, Schmidt LJ, Heemers HV, et al. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68(13):5469–5477. | ||
Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9(4):401–406. | ||
Hickey TE, Irvine CM, Dvinge H, et al. Expression of androgen receptor splice variants in clinical breast cancers. Oncotarget. 2015;6(42):44728–44744. | ||
Ferraldeschi R, Welti J, Powers MV, et al. Second-generation HSP90 inhibitor onalespib blocks mRNA splicing of androgen receptor variant 7 in prostate cancer cells. Cancer Res. 2016;76(9):2731–2742. | ||
Wang J, Zou JX, Xue X, et al. ROR-gamma drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat Med. 2016;22(5):488–496. | ||
Kallergi G, Agelaki S, Markomanolaki H, et al. Activation of FAK/PI3K/Rac1 signaling controls actin reorganization and inhibits cell motility in human cancer cells. Cell Physiol Biochem. 2007;20(6):977–986. | ||
Kampa M, Nifli AP, Charalampopoulos I, et al. Opposing effects of estradiol- and testosterone-membrane binding sites on T47D breast cancer cell apoptosis. Exp Cell Res. 2005;307(1):41–51. | ||
Pelekanou V, Notas G, Sanidas E, et al. Testosterone membrane-initiated action in breast cancer cells: interaction with the androgen signaling pathway and EPOR. Mol Oncol. 2010;4(2):135–149. | ||
Kousteni S, Bellido T, Plotkin LI, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104(5):719–730. | ||
Migliaccio A, Castoria G, Di Domenico M, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19(20):5406–5417. | ||
Alevizopoulos K, Bacopoulos N, Papadopoulou N, et al. Preclinical studies of MDX-12C, a selective membrane androgen receptor ligand with activity in prostate cancer. J Clin Oncol. 2008;26(15 suppl):abstr 14549. | ||
Malaguarnera R, Sacco A, Morcavallo A, et al. Metformin inhibits androgen-induced IGF-IR up-regulation in prostate cancer cells by disrupting membrane-initiated androgen signaling. Endocrinology. 2014;155(4):1207–1221. | ||
Chuang KH, Altuwaijri S, Li G, et al. Neutropenia with impaired host defense against microbial infection in mice lacking androgen receptor. J Exp Med. 2009;206(5):1181–1199. | ||
Viselli SM, Stanziale S, Shults K, et al. Castration alters peripheral immune function in normal male mice. Immunology. 1995;84(2):337–342. | ||
Chopra DP, Menard RE, Januszewski J, et al. TNF-alpha-mediated apoptosis in normal human prostate epithelial cells and tumor cell lines. Cancer Lett. 2004;203(2):145–154. | ||
Harada S, Keller ET, Fujimoto N, et al. Long-term exposure of tumor necrosis factor alpha causes hypersensitivity to androgen and anti-androgen withdrawal phenomenon in LNCaP prostate cancer cells. Prostate. 2001;46(4):319–326. | ||
Chen T, Wang LH, Farrar WL. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000;60(8):2132–2135. | ||
Hobisch A, Eder IE, Putz T, et al. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998;58(20):4640–4645. | ||
Lin DL, Whitney MC, Yao Z, et al. Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression. Clin Cancer Res. 2001;7(6):1773–1781. | ||
Aarnisalo P, Palvimo JJ, Janne OA. CREB-binding protein in androgen receptor-mediated signaling. Proc Natl Acad Sci U S A. 1998;95(5):2122–2127. | ||
Cinar B, De Benedetti A, Freeman MR. Post-transcriptional regulation of the androgen receptor by mammalian target of rapamycin. Cancer Res. 2005;65(7):2547–2553. | ||
Matsuda T, Junicho A, Yamamoto T, et al. Cross-talk between signal transducer and activator of transcription 3 and androgen receptor signaling in prostate carcinoma cells. Biochem Biophys Res Commun. 2001;283(1):179–187. | ||
Nakajima Y, DelliPizzi AM, Mallouh C, et al. TNF-mediated cytotoxicity and resistance in human prostate cancer cell lines. Prostate. 1996;29(5):296–302. | ||
Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem. 2002;277(9):7076–7085. | ||
Sanchez-Hernandez M, Chaves-Pozo E, Cabas I, et al. Testosterone implants modify the steroid hormone balance and the gonadal physiology of gilthead seabream (Sparus aurata L.) males. J Steroid Biochem Mol Biol. 2013;138:183–194. | ||
Parker J, Peterson A, Tudor I, et al. A novel biomarker to predict sensitivity to enzalutamide (ENZA) in TNBC. J Clin Oncol. 2015;33(15 suppl):abstr 1083. | ||
Carter AC, Sedranks N, Kelley RM, et al. Diethylstilbestrol: recommended dosages for different categories of breast cancer patients. JAMA. 1977;237(19):2079–2085. | ||
Gockerman JP, Spremulli EN, Raney M, Logan T. Randomized comparison of tamoxifen versus diethylstilbestrol in estrogen receptor-positive or -unknown metastatic breast cancer: a Southeastern Cancer Study Group trial. Cancer Treat Rep. 1986;70(10):1199–1203. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.