")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 11
Targeting stem cells in the realm of drug-resistant breast cancer
Received 29 September 2018
Accepted for publication 29 January 2019
Published 7 March 2019 Volume 2019:11 Pages 115—135
DOI https://doi.org/10.2147/BCTT.S189224
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Pranay Dey,1,2 Maitreyi Rathod,1,2 Abhijit De1,2
1Molecular Functional Imaging Lab, Advanced Centre for Treatment, Research and Education in Cancer, Tata Memorial Centre, Navi Mumbai, India; 2Molecular Functional Imaging Lab, Homi Bhabha National Institute, Mumbai, India
Abstract: Since its first documentation, breast cancer (BC) has been a conundrum that ails millions of women every year. This cancer has been well studied by researchers all over the world, which has improved the patient outcome significantly. There are many diagnostic markers to identify the disease, but early detection and then subclassification of this cancer remain dubious. Even after the correct diagnosis, more than half the patients come back with a more aggressive and metastatic tumor. The underpinning mechanism that governs the resistance includes over-amplification of receptors, mutations in key gene targets, and activation of different signaling. A plethora of drugs have been devised that have shown promising results in clinical settings. However, in recent times, the role played by cancer stem cells in disease progression and their interaction in mediating the resistance to cellular insults have come into the limelight. As breast cancer stem cells (BCSCs) are dormant in nature, it is highly likely that they fail to directly respond to the cytotoxic drugs which are meant for ablating rapidly proliferating cells. Furthermore, the absence of well-characterized, drug-able surface markers to date, has limited the application of targeted therapies in complete eradication of the disease. In this review, our intent is to discuss versatile therapeutics in practice followed by discussing the upcoming therapy strategies in the pipeline for BC. Furthermore, we focus on the roles played by BCSCs in mediating the resistance, and therefore, the aspects of new therapeutics against BCSCs under development that may ease the burden in future has also been discussed.
Keywords: chemoresistance, cancer stem cell, BCSC, tumor microenvironment, breast cancer, conventional therapies, TRAIL therapy
Introduction
The first reported case of any kind of cancer was that of a breast cancer (BC) in around 1600 BC in Egypt, which is not surprising as the organ allowed easier identification. BC, since its first documentation, has been well-studied but is still a leading cause of deaths in women worldwide.1 Extensive research has shown that this is due to the heterogeneous nature of the disease itself, which predicts the therapeutic response. Based on the presence or absence of the well-established biomarkers on the surface of BC cells, they are classified into luminal A, luminal B, HER2, and triple negative breast cancer (TNBC).2 Each subclass has diverse risk factors for incidence, disease progression, therapeutic response, and favored organ sites of metastases. Out of these subtypes, TNBC is the most aggressive subtype and shows high metastatic potential. This can be attributed to the high number of cancer stem cell (CSC) population present within the tumor. There is a plethora of therapeutic modalities administered to BC patients on the basis of their initial diagnosis. Landmark discoveries like radical mastectomy, lumpectomy, radiation implants, tamoxifen, trastuzumab, and so on have remarkably helped in improving disease outcome, for a wide number of patients (Figure 1). However, >50% of treated patients come back with a more aggressive disease. This aggressive behavior and resistance to the predicted therapies have marked a major challenge for BC clinics. Reports have shed a light onto the various mechanisms that might be responsible for these resistance mechanisms. Knowledge of the signaling cascades has led to the development of numerous molecules with potential to reduce the burden of the resistance encountered in BC. Even though chemotherapy and radiotherapy have undergone substantial improvements and refinements in efficacy and administration in the past decade, orthodox cancer treatments remain futile for many patients, predominantly whose cancer has been diagnosed at a later stage.3 Delay in cancer diagnosis reduces the overall treatment efficacy mainly due to the increased likelihood of the manifestation of metastatic disease, but also partly because more advanced disease requires more intensive treatment, which may, itself, cause treatment intolerance. In this review, first we discuss the conventional chemotherapeutics in practice followed by the novel therapeutics that are being developed against drug-resistant BC. Further, we focus on the role of BCSCs in mediating the resistance. Lastly, aspects of currently developing treatment strategies against BCSCs are discussed.
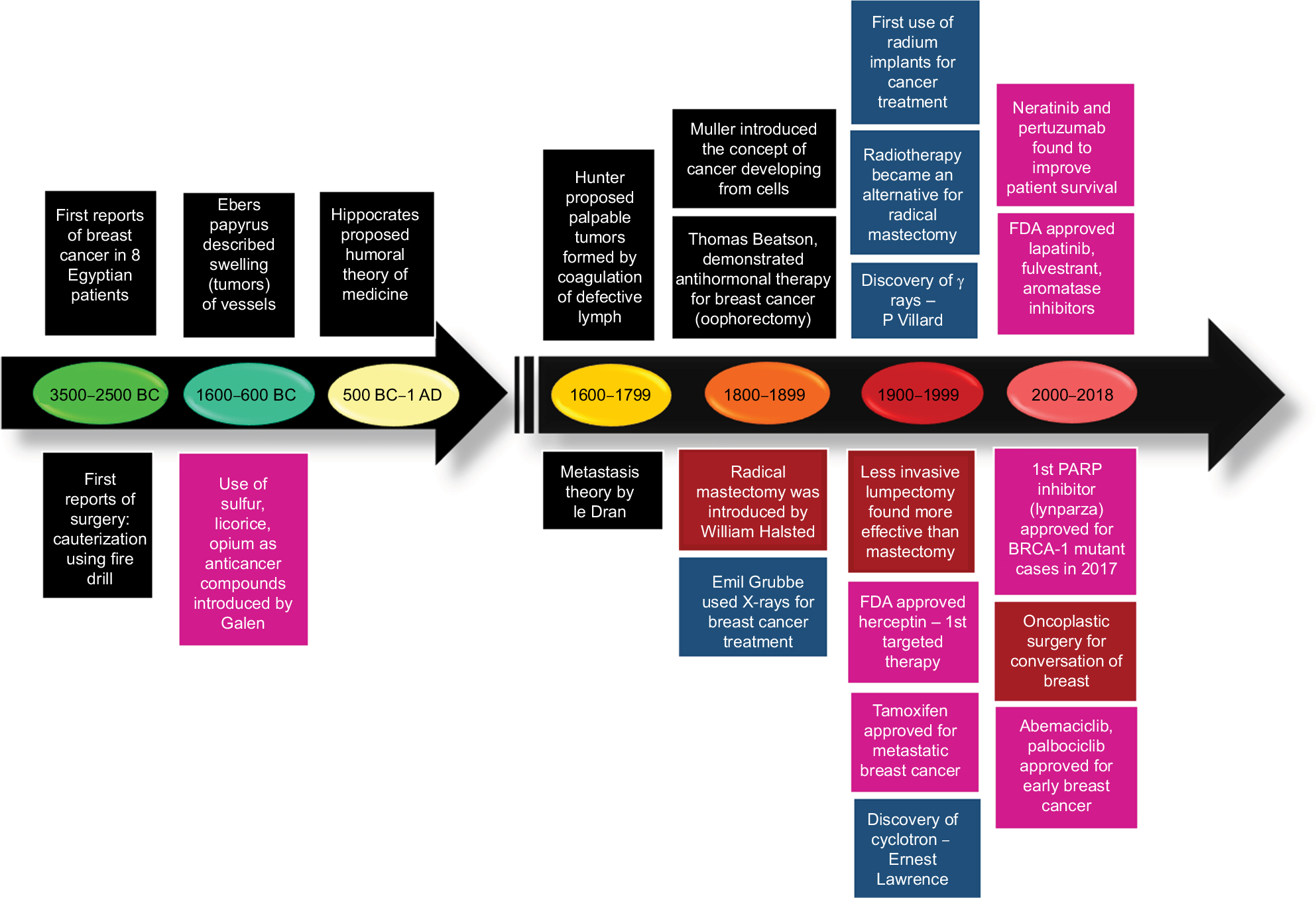 | Figure 1 Image depicting the development of therapeutic regimens against breast cancer. Abbreviation: FDA, US Food and Drug Administration; PARP, poly(ADP-ribose) polymerase. |
BC therapy: conventional approaches
In the genome era, rapid advances in molecular understanding have subdivided BC into ten interclasts.2 However, based on the current clinical practices, BC is known to have four primary subtypes. Luminal BCs are positive for steroid hormone receptors (estrogen receptor [ER] and progesterone receptor [PR]), which is further classified into two groups (A and B). The luminal A (ER+/PR+/HER2−) type tumors are less aggressive than other subtypes and take much longer to grow as well. These cancer cells also respond better to hormonal interventions and have a better prognosis.4 Luminal B subgroup (ER+/PR+/HER2+) typically shows high Ki67 proliferative index marker as well as HER2 expression. BC cells belonging to luminal B subgroup usually show poorer prognosis than luminal A, but respond better to standard chemotherapy. Since patients of this subgroup also show high HER2 expression, targeted therapy for HER2 might also be employed in some cases.4 In HER2+, BCs, which have amplification or overexpression of the HER2/ERBB2 oncogene, are generally treated with anti-HER2 therapies including the antibody drug trastuzumab and small molecule inhibitor lapatinib. Basal-like BC lacks the hormonal receptors as well as HER2 receptor and therefore is often known as triple negative breast cancer (TNBC). Standard chemotherapeutic regimens involving platinum-based drugs are majorly administered for treating TNBCs.
Majority of BC patients (~77%) have hormonal receptor-positive diseases, which comprise 23.7% from ER+/PR+/HER2− (luminal A) and ~53% from ER+/PR+/HER2+ (luminal B). Approximately, 23%–30% of BC patients show HER2 amplification. TNBC represents about 10%–12% of the total BC population.4 Endocrine therapy is currently the gold standard treatment regimen to treat the hormone receptor+ BCs. This therapy works either by making the hormone effect ineffective or by lowering the hormone level itself. Therapeutic drugs prescribed to the patients include 1) tamoxifen, which acts by blocking the estrogen uptake by ER; 2) exemestane, anastrozole, and letrozole that belong to aromatase inhibitor class of drugs, which inhibits the conversion of androgens to estrogens thereby depleting estrogen in the body; 3) leuprolide and goserelin (luteinizing hormone-releasing hormone analogs), these drugs suppress the synthesis of hormone from the ovary; and 4) fulvestrant (a specific ER inhibitor), which makes it suitable for refractory BC patients. Administration of the above drugs for treating hormone receptor+ BC is recommended until there is clinical resistance or metastasis, where chemotherapy is employed.5 As different endocrine drugs work by distinct mechanism, a combinatorial approach can show improved efficacy. However, the effectiveness of this combination treatment has not been proved well in the patient scenario.5 Therefore, the current consensus is that both endocrine therapy-naïve advanced BC and high endocrine-sensitive patients can benefit from the combination endocrine therapy.6
The patient group having HER2 gene amplification or protein overexpression is generally administered molecular targeted therapy; a range of targeted drugs have been approved as single agent or in combination with standard chemo regimen. The receptor-targeted therapeutic agents include 1) trastuzumab (specific anti-HER2 monoclonal antibody [mAb]); 2) ado-trastuzumab emtansine, which is trastuzumab conjugated with emtansine (microtubule inhibitor); 3) pertuzumab (specific anti-HER2 mAb with distinct binding site on HER2 extracellular region compared to trastuzumab); 4) lapatinib, a small molecule inhibitor (TKI) capable of inhibiting both HER2 and epidermal growth factor receptor (EGFR) signaling. The standard regimen for early stage HER2+ cases includes neoadjuvant therapy with a combination of HER2 targeted therapy and chemotherapy.7 Subsequently, this treatment is followed by surgery, radiotherapy, and 1 year of HER2-targeted therapy. Endocrine adjuvant can be added based on the specific receptor status in patient. The successful advent of molecular targeted therapy against HER2+ BC can be seen by the substantial increase in overall survival (OS) of patients from ~1.5–5 years.7
TNBC is aggressive by nature and defiant to treat as well when compared to hormone-positive and HER2+ BC. TNBC can be further subdivided into six subtypes based on transcriptomic heterogeneity and response to chemotherapy. These subtypes are mesenchymal (M), a mesenchymal stem-like (MSL), basal-like (BL1 and BL2), a luminal androgen receptor (LAR), and an immunomodulatory (IM) type.8 Both M and MSL subtypes have enhanced expression of factors regulating epithelial–mesenchymal transition (EMT), but intriguingly only the MSL subtype has diminished expression of genes involved in proliferation. The BL1 subtype is categorized by augmented expression of cell cycle and DNA damage repair genes, while the BL2 subtype shows higher expression of growth factor receptors and myoepithelial markers. The LAR subtype is regulated by the androgen receptor (AR) and characterized by luminal gene expression. The IM subtype comprises of BC cells encoding immune checkpoint regulatory genes such as programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1), antigens, and immunomodulatory cytokines. Detailed analysis shows activation of immune signal transduction pathways in this subtype, which is likely from both the tumor cells and infiltrating lymphocytes.8
Until now, standard chemotherapy remains the mainstay of treatment in TNBCs. The absence of the receptors precludes the application of targeted therapies against advanced stage disease. The only US Food and Drug Administration (FDA)-approved therapy is chemotherapy drugs such as anthracycline, taxane, and platinum drugs with or without bevacizumab.9 The median OS of patients with metastatic disease ranges between 9 months and 1 year.9 Given the suboptimal treatment outcome with standard therapeutic agents, identification of novel targets and therapy is the need of the hour.
Even with the development of so many different agents, the BC patient scenario is still very disappointing. This can be attributed to the innate biology of the cancer cells to outsmart the current therapies. Over the past decade, it has been identified that cancer cells employ various strategies to overcome the cytotoxic effects such as activation of other signaling pathways, altered metabolism, change in the cell cycle machinery, and epigenetic changes to name a few. This knowledge has led to the development of agents with potential to overcome the resistance of BC cells (Figure 2). In the following sections, we discuss some of the promising therapeutic strategies that are being investigated to treat drug-resistant BC.
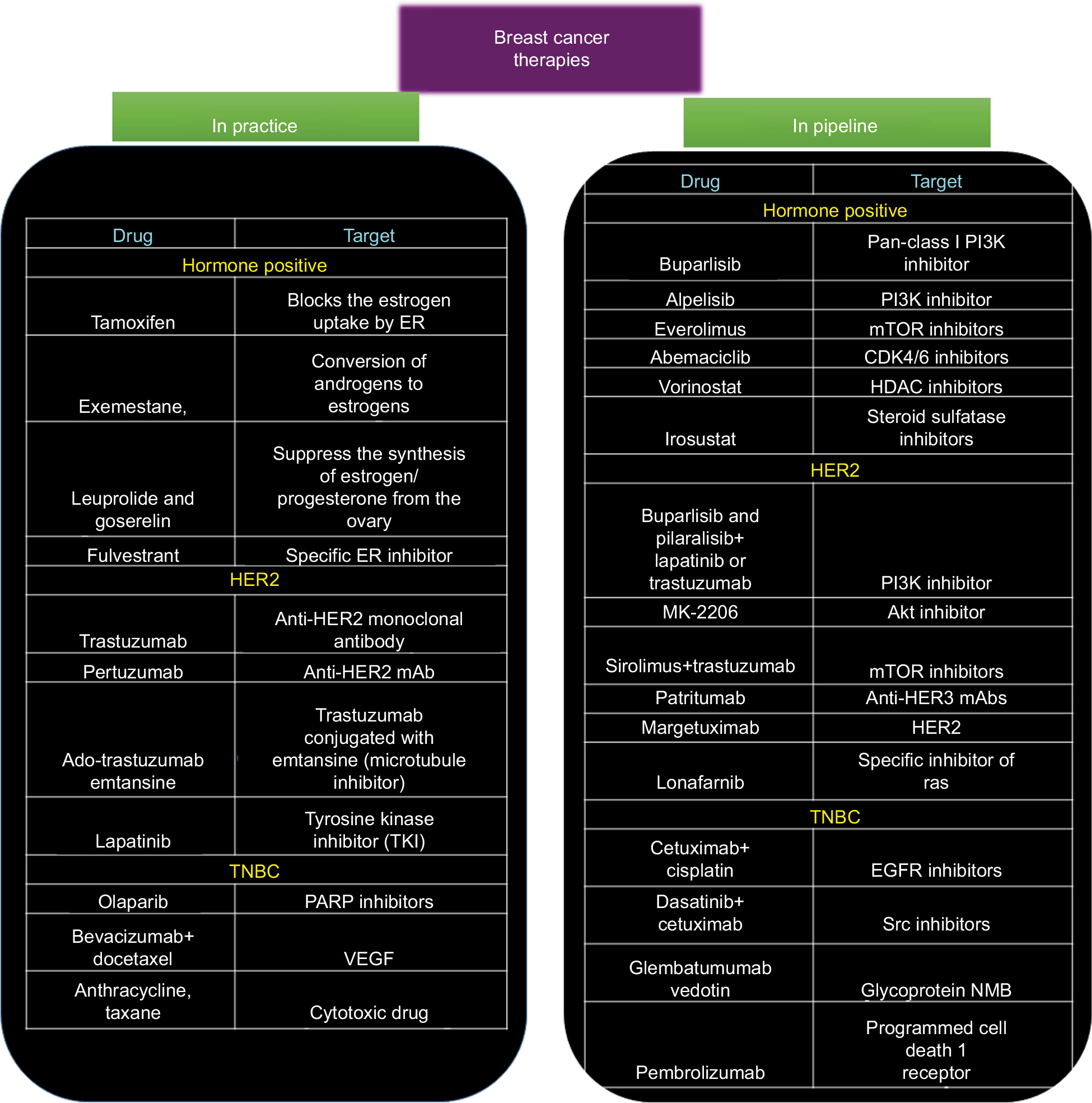 | Figure 2 Snapshot of various chemotherapeutic modalities that are being prescribed in clinics and the novel therapeutic drugs that are being developed. Abbreviations: EGFR, epidermal growth factor receptor; ER, estrogen receptor; HDAC, histone deacetylase; mAb, monoclonal antibody; mTOR, rapamycin; PARP, poly(ADP-ribose) polymerase; PI3K, phosphatidylinositol 3-kinase; TNBC, triple negative breast cancer; VEGF, vascular endothelial growth factor. |
BC therapy: developments in the challenging dogma
Hormonal therapy-resistant BC
Development of resistance in hormone receptor-positive BC against their targeted treatment agents is now a well-established phenomenon. Resistant cancer is often metastatic in nature and the underpinning genomic alterations occur majorly in ER cascade. However, other signaling pathways might also get activated and are involved. Mammalian target of phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/rapamycin (mTOR) (PI3K–Akt–mTOR) signaling circuit is considered as one of the prime contributing factors to the resistance in a variety of cancers including BC with hormonal drug resistance.10 This signaling cascade is reported to be overactivated in almost 70% of BC, with PIK3CA (PI3K catalytic subunit p110α) being the frequently mutated and/or amplified genes.11 In addition, activation of escape pathways like HER2 signaling as well as altered cell cycle kinetics has been observed to mediate resistance against ER therapies Thus, to tackle ER+ metastatic BC, there is a need to develop novel therapeutic approach with a potential to either minimize or reverse drug resistance. To address the conundrum of resistance, spectrum of different chemotherapeutic agents has been developed.
PI3K inhibitors
Combinatorial therapies targeting both hormonal receptors and PI3K/AKT/mTOR pathways have been appraised to reverse the resistance to hormonal therapies. Combinatorial treatment with PI3K inhibitors and aromatase inhibitors has been employed as a second-line of treatment for advanced luminal A cases. Buparlisib (a pan-class I PI3K inhibitor) was reported to considerably improve progression-free survival (PFS) in patients, specifically in those having PIK3CA mutation. However, PI3K inhibitors such as pilaralisib,12 voxtalisib,12 and buparlisib13 cannot be employed in treating patients due to their high toxicity. Recently, taselisib and alpelisib are also under Phase III trials (NCT02340221 and NCT02437318, respectively) and are reported to be efficacious primarily due to their high selectivity and lesser toxicity. These α-specific PI3K inhibitors showed promising results in patients harboring PIK3CA mutations.14 Irrespective of PIK3CA status, both taselisib15 and pictillisib16 in combination with letrozole or anastrozole were found to augment antitumor effects in early luminal A patients when employed as neoadjuvant treatment. Buparlisib and alpelisib are currently under Phase II efficacy investigation (NCT01923168).
mTOR inhibitors
Everolimus (derived from sirolimus) has been approved by the US FDA for treating ER/PR + advanced BC in combination with exemestane. Everolimus has also been employed in combination with letrozole, but its clinical efficacy was a failure as it could not reverse the resistant BC.17 Another derivative of sirolimus, that is, temsirolimus, was a complete defeat as it could hardly show any clinical benefits either as first-line therapy in combination with letrozole or as a single agent in second-line therapy in advanced ER/PR + BCs.18
Cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitors
It is known that cancer cells have aberrant cell cycle machinery that aids them to divide and proliferate infinitely. Thus somehow, inhibiting the cell cycle in these cancer cells might provide a way to overcome the resistance in cancer cells. The proteins involved in regulating cell cycle belong to cyclin-dependent kinase (CDK) family. In addition, CDK4/6 has been shown to regulate the cell cycle progression by its reversible interaction with cyclin D1. Thus, among the emerging therapies against CDKs, CDK4/6 inhibitors such as abemaciclib, ribociclib, and palbociclib are the most promising candidates. These CDK4/6 inhibitors block the phosphorylation of retinoblastoma protein, resulting in the downregulation of E2F-response genes to mediate cell cycle arrest at the G1-S stage. These small molecule inhibitors have also been reported to dephosphorylate the forkhead box protein M1 (transcription factor), causing inhibition in cellular proliferation.19 Interestingly, it was observed that hormone-resistant tumors are still dependent on CDK4/6-cyclin D1 for their growth and proliferation.20
Promising results have led to the FDA approval of combination treatment using ribociclib and palbociclib along with aromatase inhibitor as the first-line treatment for ER+/PR+/HER2− advanced BC. They have been reported to significantly improve the PFS in advanced BC patients by 10 months and the PFS rate by 20% after 18 months, respectively, compared to letrozole alone.21 Abemaciclib has been shown to prolong the median PFS by 7 months,22 when employed as second-line treatment in combination with fulvestrant in ER+/PR+/HER2− advanced BC. To assess the efficacy of ribociclib and abemaciclib alone, they are currently in Phase III trials (NCT02422615 and NCT02246621). Although the mechanism of action of these CDK4/6 inhibitors is alike, abemaciclib showed greater monotherapy response and induced lesser neutropenia as compared to other inhibitors, primarily due to its more specific CDK4 inhibition.21 In addition to the conventional/direct approach of targeting the signaling cascade involved in mediating resistance, there has been development of inhibitors which have the potential to reverse the resistance.
Histone deacetylase (HDAC) inhibitors
Resistance to conventional hormonal therapy has also been attributed to the histone deacetylation-mediated loss of ER expression in ER+ patients.1 In line with this observation, application of HDAC inhibitors has been shown to upregulate the expression of ERα and aromatase, thereby aiding in suppression of the signaling governed by ER.23 Entinostat when combined with exemestane and vorinostat in combination with tamoxifen has shown promising antitumor activity when employed as second-line treatment for ER+/PR+ advanced BC in combination with, compared to, their monotherapy counterparts.23
Steroid sulfatase inhibitors
Steroid sulfatase also known as arylsulfatase C is a sulfatase enzyme involved in the metabolism of steroids. Interestingly, the enzymatic activity of steroid sulfatase was reported to be substantially increased in ERα-positive BC cells.24 Thus, inhibiting the enzymatic activity reduces the estrogenic steroids and suppresses tumor growth. Encouraging results were reported from the Phase II trial of the combinatorial treatment using irosustat along with conventional aromatase inhibitor.25 SR16157 (dual-acting steroid sulfatase inhibitor) which is a direct inhibitor of steroid sulfatase and releases ERα modulator has also been assessed for its effects in hormone-dependent BC.26
Resistant BC against HER2 targeted therapy
The growing reports of primary and acquired resistance to lapatinib or trastuzumab are alarming and severely hampering its clinical significance as HER2+ BC therapeutics. Thus, identifying the resistance mechanisms and discovery of potential therapeutic agents to tackle the resistance is the need of the hour. Various groups have identified and verified numerous therapeutic agents that might play an important role in the fight against metastatic HER2+ BC.
PI3K/Akt/mTOR inhibitors
The aberrant activation of PI3K/Akt/mTOR pathway is also considered to be mediator of resistance in HER2+ BC, thus combining the inhibitors of this pathway with HER2 targeted candidates and studying the efficacy of the drugs is an area of active research. Buparlisib and pilaralisib (pan-class I PI3K inhibitors), when administered with lapatinib,27 trastuzumab,28 or trastuzumab and paclitaxel,29 was proven to be efficacious and safe in patients having HER2+ advanced disease. In addition, MK-2206 (an Akt inhibitor) also showed promising antitumor activity when combined with trastuzumab and paclitaxel28 or trastuzumab30 alone in patients with HER2+ advanced BC. To directly target the mTOR, everolimus was combined with trastuzumab and vinorelbine; however, the clinical outcome of advanced HER2+ BC patients did not improve.31 Surprisingly, this combination demonstrated better anticancer activity than trastuzumab alone in HER2+ patients who are hormone receptor negative.31 Recent drugs such as sirolimus32 and ridaforolimus33 when administered in combination with trastuzumab have demonstrated promising results in refractory HER2+ BC.
Inhibitors targeting HER-family receptors
Receptor ligands switching between HER-family members (HER1 [EGFR], HER3, or HER4) can activate the signaling cascade, this phenomenon is reported to make trastuzumab redundant.34 In addition, HER2/HER3 heterodimers have also been associated with trastuzumab resistance.35 Thus suppression of the HER-family members may be promising to deal with the conundrum of resistance in HER2+ advanced BC. In view of this, an irreversible TKI, neratinib was developed that has the ability to inhibit HER1/HER2/HER4. It has been reported that administration of neratinib after trastuzumab adjuvant therapy has significantly improved the 2-year invasive disease-free survival in HER2+ patients.36 Preclinical study has also shown encouraging antitumor activity of an anti-HER3 mAb (patritumab) by inhibiting HER2/HER3 heterodimers. Further, it was shown that it is efficacious and has lower toxicity in patients with advanced HER2+ disease.37
A novel mAb margetuximab (HER2 targeting) was also assessed in a first Phase I trial for its antitumor activity and was found to be well tolerated and had promising activity even as a single therapeutic agent.38 Further, this antibody inhibitor is currently undergoing trials to test its efficacy as a single agent (NCT02492711) and/or in combination with pembrolizumab (targets PD-1 receptor of lymphocytes) (NCT02689284).38 Among other ongoing efforts, trastuzumab is conjugated with emtansine (microtubule inhibitor) which utilizes the specificity of trastuzumab for targeting HER2+ BC cells and microtubule cytotoxicity for killing the cells.39 It has been approved as a second-line treatment for lapatinib/trastuzumab-relapsed/refractory HER2+ BC patients.40
Immunotherapy
The use of immune system against tumor cells serves as an area of extensive research with the aim to develop a vaccine against cancer. One of the first devised immunotherapeutic agents was nelipepimut-S, derived from the extracellular region of HER2. It has been extensively analyzed as a potential vaccine to prevent relapse in high-risk BC patients.41 The combinatorial application of nelipepimut-S and trastuzumab in HER2+ early BC is studied in Phase IIb clinical trial (NCT02297698). Recombinant HER2 protein (dHER2) was also studied for the potential vaccine and exhibited immunogenicity to augment T-cell-mediated response against HER2+ BC.42 Follow-up studies are being carried out to elucidate its role as monotherapy in HER2+ advanced BC as well as advanced BC refractory to trastuzumab or lapatinib.43
TNBC
Among the BC subtypes, TNBC has fewer choice of therapeutic drugs, primarily due to the lack of well-characterized molecular targets. Therefore, the need of the hour is to identify novel targets and develop effective agents against these targets to achieve improved clinical benefits. Till now the agents developed against TNBCs are primarily based on drug repurposing.
Anti-angiogenic agents
Vascular endothelial growth factor (VEGF), which is a key angiogenic factor implicated in various cancers, has been reported to be higher in TNBC as compared to non-TNBC BC.44 A well-known anti-VEGF mAb, bevacizumab, demonstrates suppression of tumor neovasculature growth and inhibits metastasis. It was also reported in a Phase III trial that supplementation of bevacizumab to docetaxel (first-line chemotherapy) resulted in improved response rate.45 It was also observed that the combination of bevacizumab and docetaxel had minimal or no side effects when compared to docetaxel alone.
Poly(ADP-ribose) polymerase (PARP) inhibitors
A major breakthrough toward the understanding of the heterogeneity of the TNBCs came in the form of detection of a subclass of sporadic TNBC that has deficiency in the homologous-repair pathway, which is a characteristic of BRCA1/2-mutated BC. In line with this observation, the therapeutic drugs administered to these patients incorporate PARP inhibitors or platinum drugs (DNA targeting) like carboplatin46 along with conventional chemotherapy.47 BRCA1/2 genes are responsible for encoding tumor-suppressor genes that are involved in repairing DNA double-stranded breaks via homologous recombination. Whereas, PARP enzymes repair the single-stranded breaks. Patients harboring germline BRCA1/BRCA2 mutation (gBRCA+) benefit the most after administration of PARP inhibitors, probably due to synthetic lethality.48
When considering PARP inhibitors, olaparib seems to be a success story. In addition to olaparib, other inhibitors targeting PARP such as talazoparib is currently in Phase III trial (NCT01945775). Talazoparib has shown promising preclinical results, which can be attributed to its strong affinity to DNA by trapping PARP–DNA complexes.49 It has also demonstrated strong anticancer activity as a monotherapeutic drug in advanced gBRCA+ BC.50 Rucaparib (Phase II, NCT02505048) and niraparib (Phase III, NCT01905592) are being explored in case of gBRCA+ advanced BC patients as a single agent as well as in combination with standard chemotherapy (niraparib: Phase I/II, NCT02657889; rucaparib: Phase II, NCT01074970).
The decision of using either PARP inhibitors or carboplatin in TNBC is generally determined by three DNA-based homologous recombination deficiency scores, which highly correlates with the germline genetic defects in BRCA1/2.51 However, none of the abovementioned therapeutic agents is beneficial against all TNBC because of the heterogeneous nature of TNBC. Thereby, an urgent need for the identification and characterization of novel clinically important molecular biomarkers for further refinement of the in-practice treatment approaches.
EGFR inhibitors
The EGFR has been reported to be overexpressed in TNBC. Therefore, numerous clinical trials are underway to evaluate the antitumor activity of cetuximab in combination with platinum-based drugs like cisplatin in metastatic TNBC patients.52,53 Identification of a subpopulation of TNBC patients that might respond well to EGFR inhibitors is an area of active research efforts.54 Low expression of α-crystalline B chain, lack of KRAS expression, and higher expression of PTEN in tumors might be correlated with favorable response.54
SRC inhibitors
SRC is a non-receptor signaling kinase which is a downstream molecule of several growth factor receptors such as PDGFR, EGFR, HGFR, and IGF-1R which have been reported to be deregulated in TNBC. Dasatinib, when tested as monotherapy for TNBC in Phase II trial (CA180059), showed substandard result.55 However, when tested in cell lines, dasatinib in combination with anti-EGFR mAb: cetuximab and cisplatin showed synergistic antitumor activity in different TNBC cell lines.56 The combination of three drugs resulted in more prominent induction of apoptosis and inhibition of MAPK and EGFR phosphorylation than all the other combinations.56 In addition, cancer cell migration and invasiveness were also substantially suppressed by dasatinib as well as combination treatment with dasatinib, cetuximab, and cisplatin in TNBC cell lines.56 Thus, clinical investigations are required to further access the use of dasatinib-containing amalgamations in TNBC patients that have tumors expressing both EGFR and c-Src.
Monoclonal antibodies
Glembatumumab vedotin is a mAb conjugated with a cytotoxic drug aimed at targeting glycoprotein NMB-overexpressing (gpNMB+) TNBC.57 gpNMB is a transmembrane protein that has been linked with tumor invasion and promote metastasis and is overexpressed in about 40% of TNBC.58 Phase II trial done on gpNMB+ advanced TNBC patients showed significant improvement in PFS and OS in glembatumumab vedotin-treated patients as compared to conventional therapy.59
Breast cancer stem cells (BCSCs): the troublemakers
BC is widely understood as a heterogeneous disease which in turn contributes to therapy failure and disease progression.60 There is not only intratumoral heterogeneity, that is, diversity within a tumor in context to phenotypic, functional, and genetic variations, but also intertumoral diversity, that is, the diversity between primary and metastasized tumor. To explain the intratumoral heterogeneity, two theories have been put forward. The first one was clonal evolution theory/stochastic theory, introduced by Peter Nowell, according to which cancer is an evolutionary process in which most neoplasms arise from single cell and progression of tumor results from stepwise accumulation of mutations within original clones following selection of more aggressive subclones. Accordingly, each dominant subclone possess similar tumorigenic potential.61
The second theory proposed is CSC theory. According to this hypothesis, only a small population of cells, called CSCs, are capable of self-renewal and have the potential to initiate tumor. In CSC model, cancers originates from the malignant transformation of a stem or progenitor cells through the deregulation of self-renewal program or from transformation of committed cells through dedifferentiation of mature cells that gain a self-renewal potential.62
The first CSCs from solid tumors were identified in breast tumors,63 subsequently CSCs were isolated from other organs. Al-Hajj et al were the first to identify a subpopulation of BC which had the potential to form tumors in immune-deficient Nonobese Diabetic (NOD)/Severe Combined Immunodeficiency (SCID) mice.63 They used a set of cell surface markers to isolate cells with increased tumorigenic capacity. In particular, cells that were CD44+CD24lowEpCAM+ and lineage negative (cells lacking markers CD2, CD3, CD10, CD16, CD18, CD31, CD64, and CD140b), isolated from one primary breast tumor and eight metastases, were able to form heterogeneous tumors eight out of nine times and were termed as BCSCs. Surprisingly, as few as 200 CD44+CD24lowEpCAM+lin- cells transplanted into NOD/SCID mice could form tumors with 100% efficiency, while CD44−CD24+EpCAM− cells could not form tumors. Different subtypes of BC constitute different proportion of BCSCs contributing to different disease outcome. Among the BC subtypes, the highest amount of CSCs was observed in patients with TNBC (basal) subtype and has been correlated with its aggressiveness.64
With momentous discovery of CSCs, their pivotal role in driving key processes during cancer development such as tumor growth, metastasis, recurrence, as well as treatment resistance was established. However, the signaling pathway that regulates CSCs and that might be involved in promoting the resistance toward the conventional therapies remains largely elusive. Hedgehog, Notch, and Wnt pathways have been shown to play crucial role in promoting resistance to therapy. These pathways are generally involved in the development of embryo and adult tissue homeostasis. Deregulation of the Notch and Hedgehog pathways, which normally regulates stem cell self-renewal and differentiation, results in BCSC phenotype.65 The Wnt pathway plays an important role in maintaining and preserving undifferentiated state of stem cells.66 Hedgehog pathway, which is an embryonic development organizer pathway, is also deregulated in BC, thereby activating Gli1 and Ptch1 genes (positive modulators of the hedgehog pathway) and thus leading to BCSC proliferation.67 The Notch pathway is involved in cell differentiation during both embryogenesis and adulthood. Notch pathway deregulation activates genes important for regulating proliferation and apoptosis inhibition in cancer cells.68 The transcription factors targeted by Notch signaling include CDKN1A, cyclinD1, c-myc, and HES-related repressor protein. These pathways have been reported to be activated in BCSCs.69 In addition, other transcriptional factors involved in maintaining the potency of BCSCs have also been identified. The transcriptional factors such as Sox2, Oct4, and Nanog act as master regulators of pluripotency and maintain the undifferentiated state of BC cells.70 Of the basal-like breast carcinomas, 43% exhibit higher Sox2 expression, indicating a less differentiated phenotype.71 Another member of the Sox family, Sox4, induces changes associated with the EMT process that is responsible for increased invasiveness and mobility of cancer cells in vivo.72 Recently, the ability of BCSCs to undergo EMT has been scrutinized, leading to the identification of partial EMT. Reports suggest that the circulating tumor cells (CTCs) survive in blood by exhibiting both epithelial and mesenchymal (E/M) phenotypes. The CTCs employ the collective cell migration properties of the epithelial cells and enhance their attachment to the extracellular matrix by achieving mesenchymal properties.73 This significantly enhances the chance of survival and promote distant metastasis. Several evidences suggest that the expressions of Oct3/4, Nanog, and Sox2 are strongly associated with different CSCs, including BCSCs.74
Apart from the genes that maintain the potency of stem cells, the BCSCs can be distinguished based on the following unique features:
- Presence of classical cell surface marker such as CD44+CD24−, in addition CD133, CD44+ CD49 fhi CD133/2hi. CD49f and CD61 have also been introduced as BCSC marker.75 These markers can be detected by flow cytometer, via employing specific mAbs.
- High expression of BC resistance protein 1, also known as ATP-binding cassette (ABC) transporter G family ABCG2 or CD338.75 This can be tested by using orthodox side population assay.
- Ability to form mammospheres in suspension culture and the overexpression of aldehyde dehydrogenase-1 (ALDH1).75
Chemoresistance to the conventional therapies can be divided into two main groups, namely intrinsic resistance due to genetic alterations and extrinsic resistance including microenvironment influences (Figure 3).76 Intrinsic resistance includes overexpression of ABC transporter, overexpression of ALDH1, enhanced DNA repair mechanism, an altered cell cycle, and resistance to apoptosis. The extrinsic cause of resistance includes all microenvironment influences such as hypoxia or EMT.
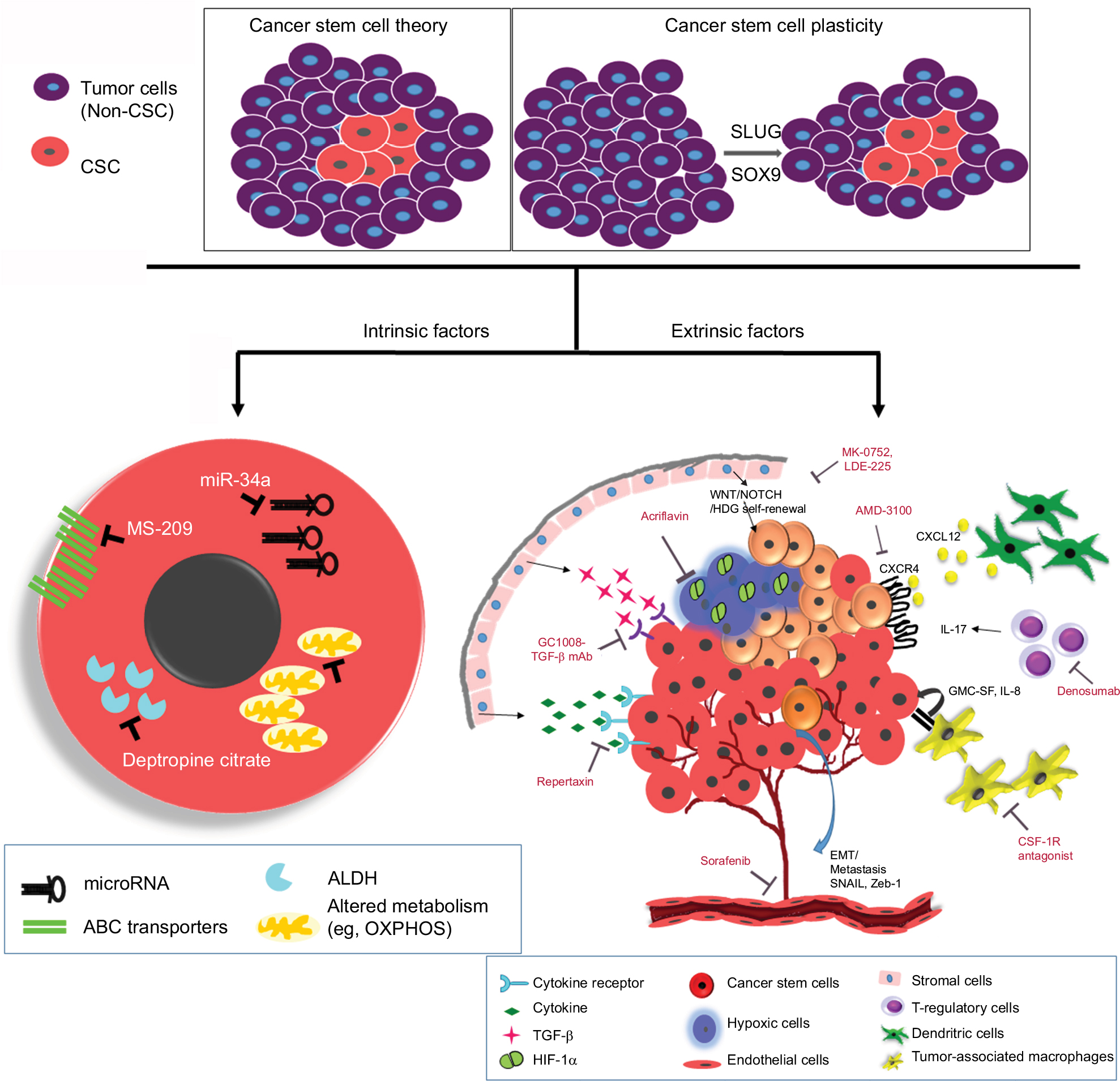 | Figure 3 Schematic diagram showing intrinsic and extrinsic factors that aid the breast cancer stem cells to evade and survive against therapeutic insults. Notes: Two widely accepted theories regarding the origins of CSCs are highlighted. According to “Cancer stem cell theory,” an inherent subpopulation of dormant cancer cells that are pluripotent in nature with the ability to repopulate the depleted pool of cancer cells following therapy. The “cancer stem cell plasticity theory” describes that therapeutic insults and EMT triggers a few breast cancer cells to undergo a switch, converting epithelial cells to pluripotent CSCs. The interplay of microenvironment and intrinsic cascades of CSCs aids them to elude the conventional therapy. Intrinsic factors depict microRNAs, drug transporters, ALDH, and altered cellular metabolism as pivotal processes that can be targeted. Further, extrinsic factors include a plethora of various components from the tumor microenvironment. Some of the important players being hypoxia, angiogenesis, EMT, immune cells, and stromal cells promoting proliferative signaling like cytokines, TGF-β, and self-renewal signals like Wnt/Notch/Hedgehog. Some of the inhibitors/mAbs against these resistance promoting factors have also been depicted. Abbreviations: ABC, ATP-binding cassette; ALDH, aldehyde dehydrogenase; CSC, cancer stem cell; EMT, epithelial–mesenchymal transition; GMC-SF, granulocyte-macrophage colony-stimulating factor; HIF-1α, hypoxia inducible factor-1alpha; OXPHOS, oxidative phosphorylation; TGF-β, transforming growth factor beta. |
Intrinsic factors of resistance
The small pool of cells, that is, BCSCs that evade chemotherapy is possibly because of the presence of ABC transporters. Increased level of ABCG2 in BCSCs was shown to enable rapid expulsion of cytotoxic drugs, conferring cellular resistance to antitumor drugs.77 Increased levels of P-glycoprotein which belongs to ABC transport family also confer resistance to antineoplastic drugs by manipulating several cellular processes like the p53 network which plays a role in mediating chemoresistance. New tumors arising from BCSCs show a chemoresistant phenotype and are often accompanied by activating mutations.75 Hu et al78 observed that Akt signaling altered the subcellular localization of BCRP, thereby regulating drug efflux activity in CSCs. Inhibitors of PI3K, blocked Akt signaling, resulted in the suppression of cancer cell proliferation, but also enhanced the sensitivity of chemoresistant cells.78
Aldefluor assays indicated that highly tumorigenic BC cells were ALDH positive. These BCSCs had the similar differentiation and self-renewal properties when compared to CSC.79 ALDH1A1 and ALDH3A1 are important in the protection and the differentiation of CSCs via the conversion of retinol to retinoic acid.80 ALDH1 has the ability of metabolizing toxic chemotherapeutic agents into nontoxic molecules, particularly cyclophosphamide class, by converting aldophosphamide to carboxyphosphamide and thus eliminating the lethal effects of the acrolein and phosphoramide mustard (metabolite of cyclophosphamide).80,81 It has been observed that metastatic breast tumors overexpress ALDH.82
Alteration of cell cycle kinetics is another alternative intrinsic mechanism of resistance reported in BCSC.83,84 This feature aids the BCSC to evade death due to chemotherapeutic agents targeting rapidly dividing cells.76,81 This quiescent state of BCSC is also responsible for relapsed disease after a long-periods of time. A dexterous DNA repair mechanism in the BCSCs is another example of intrinsic resistance mechanism.84 BCSC uses the augmented activity of ChK1 and ChK2 allowing them to escape from mitotic catastrophe and to repair their damaged DNA proficiently.84,85 This state of dormancy and robust DNA repair mechanism contributes to the resistance of BCSC against standard chemotherapeutic regimes.
In recent times, miRNAs are also shown to govern and regulate the BC resistance against the standard therapies. miRNAs are short, non-coding RNAs that regulate crucial biological processes and are frequently deregulated in cancer. Suppression of miR200c has been shown to promote tumorigenicity of BCSCs and normal mammary stem cells. In addition, it was shown that suppression of miR-200c triggers migration and invasion of cancer cells in the neighboring tissues.86 Loss of miR-205 in BCSC populations has been shown to result in drug resistance properties.87 Another report demonstrated that miR-141 is inhibited in BCSCs, contributing to the dedifferentiation of BC cells into stem-like cells which in turn enhances the stem population.88 Alternatively, reduced expression of miR-34a in human BC resulted in inhibition of stem cell properties. A report has shown that miR-34a regulates Notch-1 pathway in sustaining stem cell properties of BCSC populations, thereby suggesting that the miR-34a/Notch-1 pathway might be a potential therapeutic target for treating BC.89 Further, enhanced expression of let-7 miRNA has been shown to be involved in tumorigenesis of BCSCs. Other report has revealed that isoform let-7c along with Wnt signaling cascade regulates BCSC renewal in vivo.90 In addition, miR-1 has been shown to be associated with Wnt signaling pathway which is critical for the aggressiveness of BC.91 Most of the knowledge gained in recent times has provided a foundation for the future development of miRNA-based therapy against BCSCs.
Extrinsic cause of resistance
The interaction between the microenvironment and CSC is a dynamic process resulting in continuous remodeling of both.84 EMT plays a crucial role in chemoresistance and aids in cancer metastasis.92 Augmented drug efflux, suppressed apoptotic signaling pathways, and slow cellular proliferation are associated with EMT, and this contributes to the resistance of BC cells against anticancer drugs.93 Gefitinib or erlotinib, prescribed to BC patients with high EGFR, often relapses. EMT-associated transcription factor Snail is reported to enhance the expression of AXL receptor tyrosine kinase. Signals transduced by AXL allow the BC cells to override the cytostatic effects of EGFR inhibitors. EMT also triggers other processes that enable the BC cells to elude the lethal effect of cytotoxic T cells. Elevated expression of PD-L1 is one such major evasion mechanism employed by BC cells.75 PD-1, an inhibitory immune-checkpoint receptor, expressed by cytotoxic T cells recognizes the PD-L1 on the cancer cells and diminishes their function. Further, enhanced secretion of thrombospondin-1 by mesenchymal cells induces the development of regulatory T cells within the tumor microenvironment that suppresses the cytotoxic T cells.
In addition to chemoresistance, EMT process also equips the BC cells to evade cytotoxic effects of radiation. Radio-resistant BC cells that acquire mesenchymal properties have been reported to be more invasive, attributed to enhanced traction forces and membrane ruffling.94 The intricate program of EMT is not only responsible for the development of BCSC, but deeper understanding in the process would help us to develop novel approaches to target cells that evade conventional therapeutic regimens. Paracrine signals from the Notch/Wnt/Hedgehog pathway influence EMT by cytoskeleton rearrangements which results in mesenchymal-like phenotype.95
BCs have the tendency to recruit mesenchymal cells from the normal breast stroma96 or from the bone marrow.97 For instance, mesenchymal stem cells (MSCs) expressing ALDH1 are selectively recruited to areas of actively dividing tumor, where they interact with BCSCs via cytokine loops of CXCL7 and IL-6.97 These cytokine signaling augments the self-renewal of BCSCs.97 In addition, MSCs have also been shown to protect the BCSCs via recruitment of regulatory T cells.98 Immunohistochemical analysis has established the presence of such interacting MSC/BCSC in tumor biopsies of BC patients.97 High ALDH1 expression in BC cells has been shown to be an independent predictor of poor outcome in patients with BC.79 Further, MSCs have the capability to differentiate into adipocytes and tumor-associated fibroblasts, which might also interact with tumor cells and can influence disease progression.99
Gabbiani and Majno were the first who reported the morphological alterations in the stimulated myofibroblasts of dormant tumor- and wound-associated fibroblasts.100 Confirming the observation, in an experimental mouse model, it was demonstrated that acute wounding of the mammary gland by dermal incision augmented BC growth and metastasis.101 While the exact underpinning mechanisms remain elusive, it is believed that paracrine signals from the developing tumors induce epigenetic changes in the neighboring stromal fibroblasts.102 Certainly, the expression profile of cancer-associated fibroblasts (CAFs) is similar to that of wound-associated fibroblasts; this profile has been linked with poor outcome of patients.103,104 A report suggests that transforming growth factor beta (TGF-β; growth factor) may be involved in regulating the epigenetic changes, leading to fibroblast activation.105 In addition, cytokines like CXCL12 (also known as SDF-1) released by BC-associated fibroblasts might help in the proliferation of cancer cells, which expresses CXCR4 (SDF-1 receptor).106 The high levels of free SDF-1 in serum has been correlated with poor outcome in BC patients.107,108
Interleukin-6 (IL-6) and IL-8 have been associated with both chronic inflammation and tumor growth.109,110 Many cell types present in the tumor microenvironment including macrophages, immune cells, and mesenchymal cells have been reported to secrete both IL-6 and IL-8.110 In addition, the high levels of both of these cytokines in serum have been related to poor BC patient outcome.111,112 IL-6 has been shown to promote angiogenesis, tumorigenicity, and metastasis.113 In clinics, correlation of high IL-6 serum levels and poor outcome in BC patients justifies the studies aimed to elucidate the role that these cytokines play in tumorigenesis. A report demonstrated that IL-6 is directly involved in BCSC self-renewal, that was mediated by the IL-6 receptor/GP130 complex via STAT3 activation.114 IL-6 has been shown to be a vital element of positive feedback loop that regulates these MSCs and BCSCs.97 Utilizing relative gene expression profiling, it was identified that CXCR1 (IL-8 receptor) was overexpressed on BCSCs and also IL-8 was able to induce the self-renewal of the BCSCs.115 Further, blocking the receptor activity in mouse xenografts significantly reduced the population of BCSCs, resulting in decreased tumorigenicity and metastasis. The production of inflammatory cytokines such as IL-6 and IL-8 is controlled by the NF-κB signaling pathway.116
Hepatocyte growth factor (HGF), released by mammary stromal cells, might also play an important role in developing mammary tumors.117 HGF serves as a co-stimulatory signal to activate the Wnt pathway during colon carcinogenesis;118 however, involvement of similar pathways in breast carcinogenesis is still unknown. Another vital growth factor released by activated fibroblasts includes fibroblast growth factors (FGFs). It was recently reported that estrogen regulates the BCSC population via paracrine signaling cascade involving FGF9.119 Additional factors such as PDGF, IGF, Wnt, Hedgehog ligands, Notch ligands, and matrix metalloproteinases (MMPs) are released in the tumor microenvironment that controls tumor proliferation, invasion, and metastasis.120–125
Endothelial cells are involved in blood vessel formation and might play an important role in developing the tumor microenvironment via direct interaction with tumor cells. Endothelial cells have been reported to be a significant constituent of normal neuronal and hematopoietic stem cell niches.126,127 It has been observed that cytokines produced by endothelial cells regulate CSCs.128,129 Remarkably, the tumor vasculature is substantially different from the normal vasculature, as exemplified by the differential expression of almost 1,000 genes between them, including JAK3, MMPs, and FGF receptors.128 Even though several pro-angiogenic factors have been recognized, VEGF is the principal facilitator of this process,130 and because of this, it has become the primary target of many anti-angiogenic therapeutics. Bevacizumab and two small molecule multi-kinase VEGF inhibitors, sunitinib and sorafenib, are currently approved for clinical application. Bevacizumab was approved against metastatic BC as it can prolong the time taken to tumor progression.131 However, more recent studies have suggested discouraging the result that the effect is severely limited and that the combination of bevacizumab and cytotoxic chemotherapy failed to increase the OS of patient.132 These results are corroborated with reports in mouse models that application of anti-angiogenic agents might accelerate BC invasion and metastasis.133,134 By studying mouse model of human BC, a report also suggests that these anti-angiogenic agents increase the CSC pool through tissue hypoxia.135 Anti-angiogenesis drugs might also augment tumor growth by stimulating HGF production from tumor-associated stromal cells.136
New approaches to develop therapeutics against BCSCs
As stated above resistance to conventional therapeutic regime such as radiation and cytotoxic chemotherapy has been the motivation behind the development of specific agents capable of targeting the CSC population. BCSCs show enhanced expression of CD44 and ABC transporters promoting survival of these stem cells. These surviving cells again give rise to tumors that have enhanced chemo-tolerance and metastatic ability resulting in relapse. Here we would discuss some of the current approaches used for targeting BCSC. The promising therapies employed against BCSCs have been summarized in Table 1.
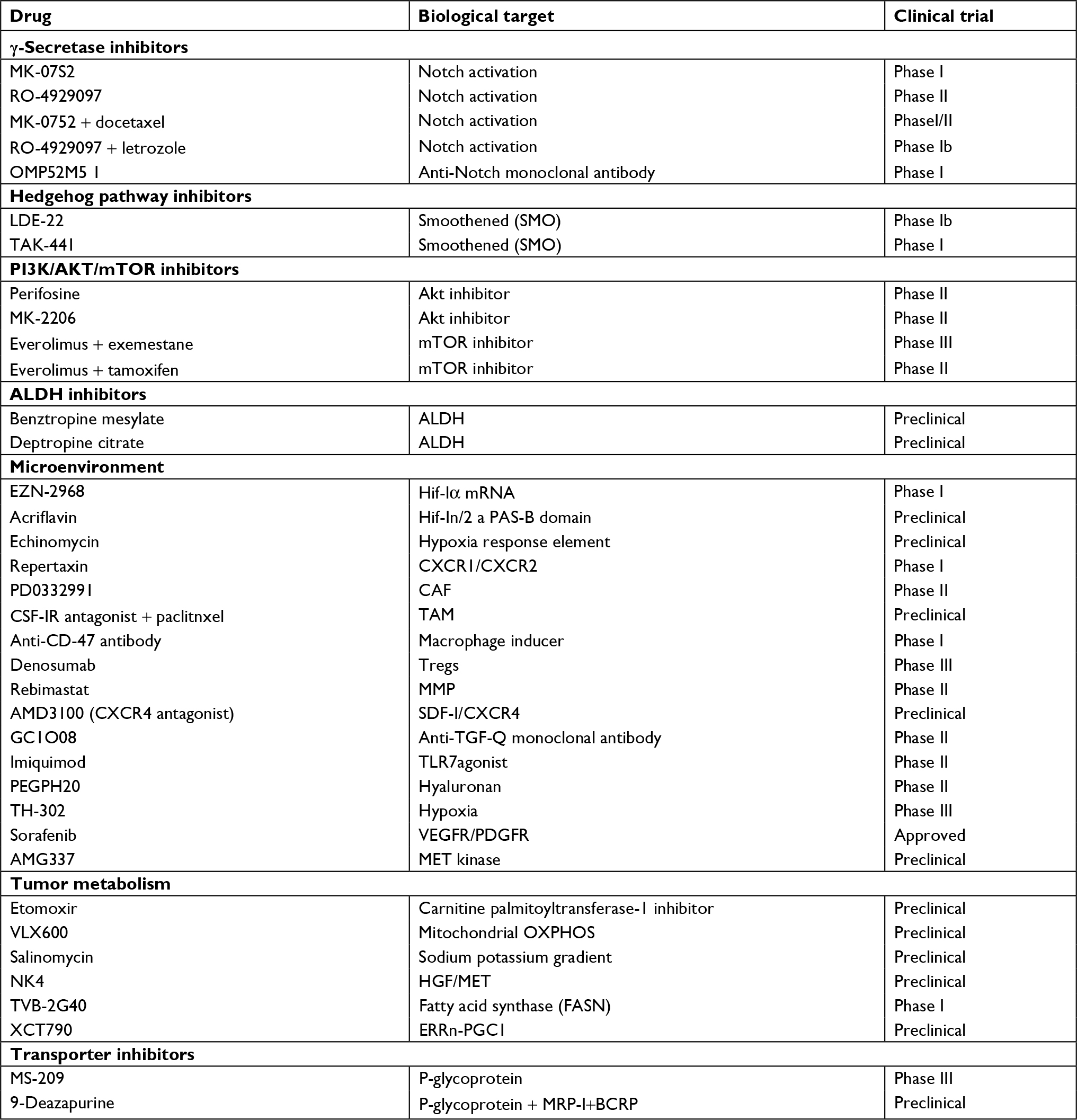 | Table 1 Upcoming therapeutic modalities against breast cancer stem cells Abbreviations: ALDH, aldehyde dehydrogenase; CAF, cancer-associated fibroblast; HGF, hepatocyte growth factor; MMP, matrix metalloproteinase; OXPHOS, oxidative phosphorylation; TAM, tumor-associated macrophage. |
Targeting signaling cascades
There are significant reports of dysregulation of Notch pathway in a substantial fraction of human BCs.137,138 Of the various approaches, one of the most clinically promising candidates is γ-secretase inhibitor. Activation of Notch signaling is regulated by this proteolytic enzyme (γ-secretase), which cleaves Notch receptors and releases the intracellular domain, which in turn acts as a transcription factor and regulates important oncogenic gene functions.139 Therefore, γ-secretase inhibitors were designed for treating BC patients in an early-phase clinical trial. The most severe effect observed has been the gastrointestinal toxicity due to goblet cell hyperplasia, which is an on-target effect of Notch inhibition.140 Although, a moderate dosage along with the administration of high dose of corticosteroids was able to lower the toxicity.141 Combination of γ-secretase inhibitor with taxane chemotherapy has also been clinically investigated in a Phase I trial.141 Other pathway regulating BCSC is the Hedgehog pathway. This pathway has been reported to be active in tumor cells as well as in the tumor stroma.142 Oral Hedgehog inhibitors were clinically tested, and they appear to be fairly nontoxic.143 Phase II clinical studies employing this drug compounds in combination with conventional cytotoxic agents are underway.
Despite the outstanding clinical efficiency of HER2-targeted therapy, almost one third of HER2-positive BC patients do not respond to these agents, and chemoresistance may develop in these patients with chronic exposure. Increasing evidence indicates that resistance may be associated with the activation of other receptor kinases, gain of function mutations of PI3K, loss of PTEN tumor suppressor gene, or truncation of the extracellular domain of HER2.144 These mutations cause aberrant activation of the downstream PI3K/Akt/mTOR pathway and are generally correlated with poor prognosis after conventional trastuzumab therapy.144 Confirmatory evidence has recently shown that the PI3K/Akt/mTOR pathway plays a significant role in regulating BCSC pool. This ensues via Akt activation of the Wnt pathway through phosphorylation of GSK/3β and direct phosphorylation of β-catenin on serine552 amino acid which results in its nuclear transport.145 This observation suggests that suppressing Akt that is downstream of HER2 signaling might efficiently target BCSCs in HER2-resistant tumors. Indeed, perifosine (Akt inhibitor) has demonstrated promising prospect by effectively targeting the BCSC pool in breast tumor xenografts.145 Encouraged by the abovementioned observations, a spectrum of PI3K and Akt selective inhibitors are being clinically investigated, providing us the direct assessment of the effects of these agents in controlling the stem cell population.
Targeting tumor microenvironment
The role of cytokine signaling in maintaining and promoting CSCs is well-documented. Among the cytokines, IL-6 and IL-8 play an important role in the maintenance of BCSC population. These two cytokines promote the inflammatory cascade via NF-κB pathway leading to chronic inflammation and augmentation of tumorigenesis. Interestingly, anti-inflammatory agents such as statins show a decrease in BC risk.146 Statins lower the levels of pro-inflammatory cytokines, as revealed by lowered CRP levels.147 A recent report has demonstrated that antibodies against the CXCR1 (IL-8 receptor) or repertaxin (small molecule CXCR1/CXCR2 inhibitor) has the potential to target BCSC in mouse xenograft models impeding tumor growth and metastasis.145 Repertaxin was initially developed to avert graft rejection and has shown to have promising effect in Phase I trials. Repertaxin was reported to mediate stem cell death in bulk cellular population through bystander effect involving release of Fas cell surface death receptor (FAS) ligand. CXCR1 inhibits FOXO3A localization and FAS ligand expression through AKT signaling. Treatment with repertaxin suppressed AKT, resulting in nuclear FOXO3A and FAS ligand expression. Conventional chemotherapeutics are also known to cause cell death via a bystander effect through FAS ligand, but that in turn induces IL-8 which protects BCSC from FAS ligand. This suggested that repertaxin might cause blockade of this effect and efficiently kill the BCSC population. Repertaxin also suppressed the BCSC population in vitro as well as in tumor xenografts. As a monotherapy, repertaxin had a nonsignificant effect on tumor growth, but drastically reduced tumor volume when employed in combination with docetaxel. Further, repertaxin was able to reduce metastatic lesions and secondary tumor formation. These promising results show that repertaxin can sensitize BCSC to bystander effect via FAS ligand and that CXCR1 blocking might represent a novel approach to targeting and eliminating breast CSCs. Further, mAbs targeting IL-6 or its receptor are currently being assessed in clinical trials for multiple myeloma.148
The downregulation of caveolin-1 (CAV1) in CAFs is a well-studied biomarker which is associated with oncogenic transformation. It has been observed that inhibition of CAV1 in CAFs resulted in hyper-proliferative phenotype of BC cells. The replacement of CAV1 with CAV1 mimetic eliminated the proliferative behavior of the cells.149 It was also reported that CAFs and MSCs sensitized MCF7 cells to the RAD001 (an mTOR inhibitor) and augmented the cytotoxic effect of RAF265 (an RAF inhibitor) on MDA-MB-231 cells through the inhibition of ERK1/2 phosphorylation.150 However, both CAFs and MSCs had no significant effect on the response to TKI258 (a PDGFR/FGFR/VEGFR inhibitor) in BC cell lines.150 This demonstrates that CAFs may not be involved in all the mechanisms of drug resistance, but heterogeneity of CAFs should be taken into account during drug response.
Tumor and adjoining stromal cells are known to secrete CCL2, which is an essential chemoattractant for macrophages. CCL2 and its receptor, CCR2, are involved in monocyte recruitment onto the tumor periphery. Lu et al observed that overexpression of CCL2 promotes both bone and lung metastases in BC. Targeting the tumor-derived CCL2 via a neutralizing mAb reduced metastasis to bone and lung.151 Adverse side effects of anti-CCL2 therapy have raised serious concern as it has shown to aggravate metastasis via increasing macrophage recruitment within weeks of treatment termination.
Adipocytes are one of the main components of the breast and have been shown to play a role in tumor development. In line with this observation, a few chemopreventive agents have been tested for their efficacy against BC cells. Sulforaphane, which is a compound present in broccoli, has been extensively studied and shown promising results.152 However, the limited understanding about the role of adipocytes in promoting tumorigenesis is limiting the scope of available options to target these adipocytes. Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) is a well-known death receptor that can mediate ligand (TRAIL-R1, TRAIL-R2)-induced apoptosis, in tumor cells. TRAIL-mediated therapies are in Phase I clinical trials for TNBC.153 It has been reported that TRAIL can be a valuable tool for targeting patients who have limited treatment options.153 However, there are some tumors that show resistance to TRAIL therapy, with CSC being major contributors in therapy resistance. The overexpression of anti-apoptosis proteins like c-FLIP can lead to the resistance of anti-TRAIL therapy. Thus combining the c-FLIP inhibition with anti-TRAIL antibodies can lead to an effective eradiation of CSC and thus overcoming therapy resistance.154
Also, the macrophages and monocytes express a large amount of TRAIL receptors, that is, TRAIL-1R and TRAIL-2R. Thus, recombinant TRAIL therapy can help in selectively inducing apoptosis of the tumor-associated macrophages (TAM), which form a primary signaling arm for tumor microenvironment. Therefore, TRAIL therapy can provide dual benefits all together, where it can selectively eliminate tumor cells and also control the pro-tumor signals coming from TAM present in the microenvironment (Figure 4).155
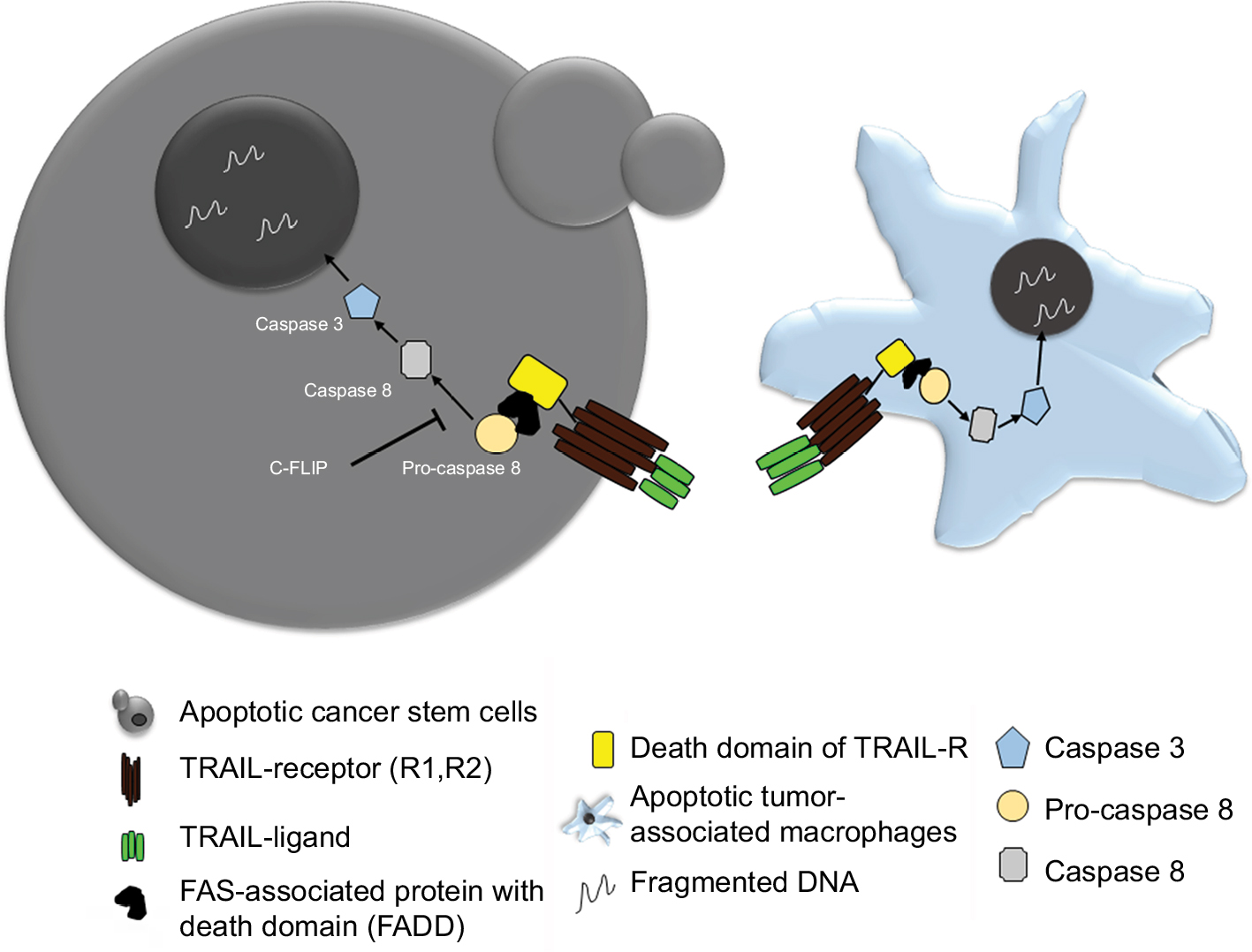 | Figure 4 Schematic showing TRAIL therapy against CSC and TAM. Notes: Recombinant TRAIL binding to its receptor on BCSCs as well as TAMs induces cell death via apoptosis. Abbreviations: BCSC, breast cancer stem cell; CSC, cancer stem cell; FAS, Fas cell surface death receptor; TAM, tumor-associated macrophage; TRAIL, tumor necrosis factor-related apoptosis inducing ligand. |
Targeting CSC metabolism
CSCs show a distinct dependency on glucose and mitochondrial metabolism. It is shown that the multipotent cells rely majorly on glycolysis. The stem cell pool of basal-like BC, which is CD44+/EPCAM+, is dependent on aerobic glycolysis. Overexpression of FBP1, which promotes gluconeogenesis and inhibits glycolysis, reduces the number of spheroids in basal-like BC.156 A well-known regulator for mitochondrial metabolism is BCL-2 protein, which forms a complex with Bcl-2-associated death promoter and glucokinase. Inhibition of BCL-2 activation can lead to the inhibition of oxidative phosphorylation (OXPHOS) leading to the reduction of CSC depending on OXPHOS.157 Increase in mitochondrial activity can promote metastasis and confer resistance to DNA damage in BC.158 A transcription factor peroxisome proliferator-activated receptor gamma, co-activator 1 alpha (PPARGC1A, also known as PGC-1α), is an important target for cancer cell metabolism as it couples with OXPHOS and supports migration and invasion of cells. This factor has been reported to be highly expressed in BCSCs and its inhibition lead to decreased stemness.159 Fatty acid oxidation is another major arm of supporting tumor cell growth and proliferation. It has been reported that various stem cell pools rely on fatty acid oxidation.160 NANOG is known to repress OXPHOS and activate fatty acid oxidation. Thus using etomoxir, a carnitine palmitoyltransferase-1 inhibitor, could reduce the spheroid formation ability of BC in vitro and also reduced in vivo tumorigenic potential.161 A mitochondria inhibitor VLX600 has been reported to target the quiescent cell pool within the tumor, in vivo.162 Salinomycin, which is an antibiotic extracted from Streptomyces albus has been shown to reduce the stemness by targeting the Wnt pathway, which is crucial for maintaining stem cell proliferation.163 Wnt signaling is a known regulator of cell metabolism, where a study has shown that using a therapeutic approach of administering Wnt antagonist frizzled-related protein 4 caused metabolic reprogramming, which led to apoptosis of CSC under variable glucose conditions.164 Recent reports suggest that targeting iron metabolism can be fruitful in targeting CSC since altered iron metabolism can cause increase in ROS and oxidative stress.165
Nano-therapeutics against CSC
Nanoparticle (NP)-mediated therapy is an effective strategy of drug delivery for cancer therapeutics. NPs are also being employed for targeting stem cell subpopulations within tumor bulk, where CSC marker-targeted NPs offer an advantage of specificity and precision. Thus using biocompatible polymers like liposome, PLGA, and so on, which are coated with antibodies/aptamers against BCSC-specific markers, can help in specific delivery of chemotherapeutic drug, RNAi, or antibodies to the stem cell population. BCSCs generally show enhanced expression of CD44, and studies have shown that paclitaxel- and salinomycin-loaded liposomal NP coated with CD44 antibody can target the CD44+ CSC population of MDA-MB-231 cells.166 Iron oxide magnetic NPs coated with CD44 antibody and loaded with gemcitabine have been used for targeting the stem cell population in BC.167 These particles have been shown to have an added advantage of hyperthermia. NPs containing a combination of chemotherapeutic agents along with autophagy inhibitor chloroquine (CQ) are an upcoming line of therapy which can target the tumor bulk as well as CSC pool within a tumor.168 Doxorubicin and CQ NP have shown to reduce the ALDH high population of MDA-MB-231 cells.
Administration of decitabine, a DNA hypermethylation inhibitor encapsulated in NP made with polyethylene glycol, could sensitize the tumor bulk and CSC population to chemotherapy. Also, when these NPs were combined with doxorubicin, they could reduce the ALDH+ population in mammospheres of MDA-MB-231 cells.169 Anticancer drugs are frequently being incorporated into liposomes, for efficient drug delivery. An anticancer compound ESC8 was used along with dexamethasone (Dex)-associated liposome (DX), to form ESC8-entrapped liposome named DXE, showed promising results in reducing the drug-resistant cell population.170 Drug targets against Notch, TGF-β, and Wnt/β-catenin pathways are also being used in combination with NPs.
NPs are also used to deliver siRNA to tumor. A cationic lipid-based polymer was developed along with an siRNA and TGF-βR-I receptor inhibitor LY364947.171 Similarly, a cationic liposomal delivery of miR-34a could reduce the expression of CSC markers ALDH and CD44, thereby delaying tumor growth.172 An interesting observation made by a group showed that graphene oxide (carbon nanomaterial) itself has the potential to induce differentiation of stem cells and reducing their in vitro sphere forming ability, thereby it can be a good source to target tumor bulk as well as the CSC population.173 Carbon nanotubes are capable of mediating a thermal effect, and they have been studied in BC cells where the BCSCs were found to be sensitive to these carbon nanotube thermal therapy.174 Further, conjugation of these carbon-based nanomaterials with stem cell receptor targeting can help achieve specificity. Also, novel methods of gene delivery using NPs are being effectively used to reduce tumor burden. An interesting study showed that specific gene delivery targeting the glucocorticoid receptor using a cationic liposome has the potential to reduce tumor growth in vivo.175
Conclusion
BC is a complex and heterogeneous disease, a culmination of a variety of cells that exert influence on one another, thereby making the disease management complex. Cellular/clonal heterogeneity within tumors and disease relapse are the major threats from the clinical point of view. We have now started to understand the quiescent, self-renewable pool of cells within a tumor population, the so-called BCSCs, which can govern the therapy resistance, metastasis, and disease relapse. Also the stem cells have unique mechanisms to withstand drug/radiation insults, for example, presence of large number of drug efflux pumps, and enhanced DNA repair machinery and thus posing big in terms of future developments of cancer therapeutics.
Tumor microenvironment is another key domain helping in maintenance of CSCs. Various elements like cytokine flux, tumor-associated immune cells, stromal cells, and CAFs impact chemokine receptor signaling, cytoskeletal rearrangements, hypoxia, angiogenesis, as well as cell metabolism. Altered cancer cell metabolism is a consequence of cancer condition where the BCSCs depend particularly on glycolysis. Mitochondrial OXPHOS is an alternative backup for stem cell survival. A large number of preclinical and clinical studies are being conducted on today’s date, targeting eradication of stem cell pool at the tumor site. Studies have also begun to explain the concept of CSC plasticity, where the non-CSCs can revert back to CSCs, and therefore, greater attention is needed as it will be indispensable for CSC management. The advancement in nanotherapeutics and nanomedicine is also greatly changing the face of treatment options by providing novel approaches of combining multimode treatment options. Together, all these dimensions added to BC research is surely going to give us an edge in reducing the impact of therapy resistance and improve disease outcomes in future days.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Curtis C, Shah SP, Chin S-F, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. | ||
Centers for Disease Control and Prevention. Cancers diagnosed at late-stages despite available screening tests [Press release]. Available from: https://www.cdc.gov/media/pressrel/2010/r101124.html. Accessed February 18, 2019. | ||
Haque R, Ahmed SA, Inzhakova G, et al. Impact of breast cancer subtypes and treatment on survival: an analysis spanning two decades. Cancer Epidemiol Biomarkers Prev. 2012;21(10):1848–1855. | ||
Reinert T, Barrios CH. Optimal management of hormone receptor positive metastatic breast cancer in 2016. Ther Adv Med Oncol. 2015;7(6):304–320. | ||
Mehta RS, Barlow WE, Albain KS, et al. Combination anastrozole and fulvestrant in metastatic breast cancer. N Engl J Med. 2012;367(5):435–444. | ||
Wuerstlein R, Harbeck N. Neoadjuvant therapy for HER2-positive breast cancer. Rev Recent Clin Trials. 2017;12(2):81–92. | ||
Lehmann BD, Jovanović B, Chen X, et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One. 2016;11(6):e0157368. | ||
Berrada N, Delaloge S, André F. Treatment of triple-negative metastatic breast cancer: toward individualized targeted treatments or chemosensitization? Ann Oncol. 2010;21(Suppl 7):vii30–vii35. | ||
Miller TW, Hennessy BT, González-Angulo AM, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120(7):2406–2413. | ||
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224. | ||
Blackwell K, Burris H, Gomez P, et al. Phase I/II dose-escalation study of PI3K inhibitors pilaralisib or voxtalisib in combination with letrozole in patients with hormone-receptor-positive and HER2-negative metastatic breast cancer refractory to a non-steroidal aromatase inhibitor. Breast Cancer Res Treat. 2015;154(2):287–297. | ||
di Leo A, Seok Lee K, Ciruelos E, et al. Abstract S4-07: BELLE-3: a phase III study of buparlisib + fulvestrant in postmenopausal women with HR+, HER2–, aromatase inhibitor-treated, locally advanced or metastatic breast cancer, who progressed on or after mTOR inhibitor-based treatment. Cancer Res. 2017;77(4 Supplement): S4-07. | ||
Baselga J, Cortés J, Delaurentiis M, et al. SANDPIPER: Phase III study of the PI3-kinase (PI3K) inhibitor taselisib (GDC-0032) plus fulvestrant in patients (pts) with estrogen receptor (ER)-positive, HER2-negative locally advanced or metastatic breast cancer (BC) enriched for pts with PIK3CA- mutant tumors. J Clin Oncol. 2017;35(15_suppl):TPS1119. | ||
Saura C, de Azambuja E, Hlauschek D, et al. LBA10_PRPrimary results of LORELEI: a phase II randomized, double-blind study of neoadjuvant letrozole (let) plus taselisib versus let plus placebo (Pla) in postmenopausal patients (PTS) with ER+/HER2-negative early breast cancer (EBC). Ann Oncol. 2017;28(suppl_5):mdx440.001. | ||
Schmid P, Pinder SE, Wheatley D, et al. Phase II randomized preoperative Window-of-Opportunity study of the PI3K inhibitor Pictilisib plus anastrozole compared with anastrozole alone in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2016;34(17):1987–1994. | ||
Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–529. | ||
Fleming GF, Ma CX, Huo D, et al. Phase II trial of temsirolimus in patients with metastatic breast cancer. Breast Cancer Res Treat. 2012;136(2):355–363. | ||
Xu H, Yu S, Liu Q, et al. Recent advances of highly selective CDK4/6 inhibitors in breast cancer. J Hematol Oncol. 2017;10(1):97. | ||
Shah AN, Cristofanilli M. The growing role of Cdk4/6 inhibitors in treating hormone receptor-positive advanced breast cancer. Curr Treat Options Oncol. 2017;18(1):6. | ||
Barroso-Sousa R, Shapiro GI, Tolaney SM. Clinical development of the CDK4/6 inhibitors Ribociclib and Abemaciclib in breast cancer. Breast Care. 2016;11(3):167–173. | ||
Sledge GW, Toi M, Neven P, et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875–2884. | ||
Acharya MR, Sparreboom A, Venitz J, Figg WD. Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol Pharmacol. 2005;68(4):917–932. | ||
Stanway SJ, Delavault P, Purohit A, et al. Steroid sulfatase: a new target for the endocrine therapy of breast cancer. Oncologist. 2007;12(4):370–374. | ||
Palmieri C, Stein RC, Liu X, et al. Iris study: a phase II study of the steroid sulfatase inhibitor irosustat when added to an aromatase inhibitor in ER-positive breast cancer patients. Breast Cancer Res Treat. 2017;165(2):343–353. | ||
Rasmussen LM, Zaveri NT, Stenvang J, Peters RH, Lykkesfeldt AE. A novel dual-target steroid sulfatase inhibitor and antiestrogen: SR 16157, a promising agent for the therapy of breast cancer. Breast Cancer Res Treat. 2007;106(2):191–203. | ||
Guerin M, Rezai K, Isambert N, et al. PIKHER2: a phase Ib study evaluating buparlisib in combination with lapatinib in trastuzumab-resistant HER2-positive advanced breast cancer. Eur J Cancer. 2017;86:28–36. | ||
Saura C, Bendell J, Jerusalem G, et al. Phase Ib study of Buparlisib plus trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on trastuzumab-based therapy. Clin Cancer Res. 2014;20(7):1935–1945. | ||
Tolaney S, Burris H, Gartner E, et al. Phase I/II study of pilaralisib (SAR245408) in combination with trastuzumab or trastuzumab plus paclitaxel in trastuzumab-refractory HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2015;149(1):151–161. | ||
Hudis C, Swanton C, Janjigian YY, et al. A phase 1 study evaluating the combination of an allosteric Akt inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013;15(6):R110. | ||
Hurvitz SA, Andre F, Jiang Z, et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015;16(7):816–829. | ||
Acevedo-Gadea C, Hatzis C, Chung G, et al. Sirolimus and trastuzumab combination therapy for HER2-positive metastatic breast cancer after progression on prior trastuzumab therapy. Breast Cancer Res Treat. 2015;150(1):157–167. | ||
Seiler M, Ray-Coquard I, Melichar B, et al. Oral ridaforolimus plus trastuzumab for patients with HER2+ trastuzumab-refractory metastatic breast cancer. Clin Breast Cancer. 2015;15(1):60–65. | ||
Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 2006;8(6):215. | ||
Hettman T, Schneider M, Blum S, et al. Abstract B161: U3-1287 (AMG 888), a fully human anti-HER3 mAb, demonstrates preclinical efficacy in HER2+ and HER2- breast cancer models. Mol Cancer Ther. 2009;8(Supplement 1):B161. | ||
Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17(3):367–377. | ||
Mukai H, Saeki T, Aogi K, et al. Patritumab plus trastuzumab and paclitaxel in human epidermal growth factor receptor 2-overexpressing metastatic breast cancer. Cancer Sci. 2016;107(10):1465–1470. | ||
Bang YJ, Giaccone G, Im SA, et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann Oncol. 2017;28:855–861. | ||
Diéras V, Bachelot T. The success story of trastuzumab emtansine, a targeted therapy in HER2-positive breast cancer. Target Oncol. 2014;9(2):111–122. | ||
Lianos GD, Vlachos K, Zoras O, Roukos DH, et al. Potential of antibody-drug conjugates and novel therapeutics in breast cancer management. Onco Targets Ther. 2014;7:491. | ||
Mittendorf EA, Clifton GT, Holmes JP, et al. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann Oncol. 2014;25(9):1735–1742. | ||
Limentani SA, Campone M, Dorval T, et al. A non-randomized dose-escalation phase I trial of a protein-based immunotherapeutic for the treatment of breast cancer patients with HER2-overexpressing tumors. Breast Cancer Res Treat. 2016;156(2):319–330. | ||
Hamilton E, Blackwell K, Hobeika AC, et al. Phase 1 clinical trial of HER2-specific immunotherapy with concomitant HER2 kinase inhibition [corrected]. J Transl Med. 2012;10:28. | ||
Linderholm BK, Hellborg H, Johansson U, et al. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann Oncol. 2009;20(10):1639–1646. | ||
Robert NJ, Diéras V, Glaspy J, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29(10):1252–1260. | ||
Castrellon AB, Pidhorecky I, Valero V, Raez LE. The role of carboplatin in the neoadjuvant chemotherapy treatment of triple negative breast cancer. Oncol Rev. 2017;11(1). | ||
Robson M, Im SA, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017;377(6):523–533. | ||
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. | ||
Brown JS, Kaye SB, Yap TA. PARP inhibitors: the race is on. Br J Cancer. 2016;114(7):713–715. | ||
Turner NC, Telli ML, Rugo HS, et al. Final results of a phase 2 study of talazoparib (TALA) following platinum or multiple cytotoxic regimens in advanced breast cancer patients (pts) with germline BRCA1/2 mutations (ABRAZO). J Clin Oncol. 2017;35(15_suppl):1007. | ||
Timms KM, Abkevich V, Hughes E, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16(6):475. | ||
Carey LA, Rugo HS, Marcom PK, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012;30(21):2615–2623. | ||
Baselga J, Gómez P, Greil R, et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2013;31(20):2586–2592. | ||
Tomao F, Papa A, Zaccarelli E, et al. Triple-negative breast cancer: new perspectives for targeted therapies. Onco Targets Ther. 2015;8:177. | ||
Finn RS, Bengala C, Ibrahim N, et al. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clin Cancer Res. 2011;17(21):6905–6913. | ||
Kim EM, Mueller K, Gartner E, Boerner J. Dasatinib is synergistic with cetuximab and cisplatin in triple-negative breast cancer cells. J Surg Res. 2013;185(1):231–239. | ||
Rose AAN, Biondini M, Curiel R, Siegel PM. Targeting GPNMB with glembatumumab vedotin: current developments and future opportunities for the treatment of cancer. Pharmacol Ther. 2017;179:127–141. | ||
Rose AA, Grosset AA, Dong Z, et al. Glycoprotein nonmetastatic B is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin Cancer Res. 2010;16(7):2147–2156. | ||
Yardley DA, Weaver R, Melisko ME, et al. Emerge: a randomized phase II study of the antibody-drug conjugate Glembatumumab Vedotin in advanced glycoprotein NMB-Expressing breast cancer. J Clin Oncol. 2015;33(14):1609–1619. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. | ||
Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291. | ||
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100(7):3983–3988. | ||
Chekhun SV, Zadvorny TV, Tymovska YO, et al. CD44+/CD24- markers of cancer stem cells in patients with breast cancer of different molecular subtypes. Exp Oncol. 2015;37(1):58–63. | ||
Muñoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012;6(6):620–636. | ||
Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene. 2009;433(1–2):1–7. | ||
Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–6071. | ||
Rizzo P, Osipo C, Foreman K, et al. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27(38):5124–5131. | ||
Farnie G, Clarke RB. Mammary stem cells and breast cancer--role of Notch signalling. Stem Cell Rev. 2007;3(2):169–175. | ||
Yamanaka S. Induction of pluripotent stem cells from mouse fibroblasts by four transcription factors. Cell Prolif. 2008;41(Suppl 1):51–56. | ||
Rodriguez-Pinilla SM, Sarrio D, Moreno-Bueno G, et al. Sox2: a possible driver of the basal-like phenotype in sporadic breast cancer. Mod Pathol. 2007;20(4):474–481. | ||
Zhang J, Liang Q, Lei Y, et al. SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res. 2012;72(17):4597–4608. | ||
Saitoh M. Involvement of partial EMT in cancer progression. J Biochem. 2018;164(4):257–264. | ||
O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–110. | ||
Chuthapisith S, Eremin J, El-Sheemey M, Eremin O. Breast cancer chemoresistance: emerging importance of cancer stem cells. Surg Oncol. 2010;19(1):27–32. | ||
Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85(9):1219–1226. | ||
Hirschmann-Jax C, Foster AE, Wulf GG, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101(39):14228–14233. | ||
Hu C, Li H, Li J, et al. Analysis of ABCG2 expression and side population identifies intrinsic drug efflux in the HCC cell line MHCC-97L and its modulation by Akt signaling. Carcinogenesis. 2008;29(12):2289–2297. | ||
Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. | ||
Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res Treat. 2012;133(1):75–87. | ||
Economopoulou P, Kaklamani VG, Siziopikou K. The role of cancer stem cells in breast cancer initiation and progression: potential cancer stem cell-directed therapies. Oncologist. 2012;17(11):1394–1401. | ||
Charafe-Jauffret E, Ginestier C, Iovino F, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16(1):45–55. | ||
Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17(3):313–319. | ||
Maugeri-Saccà M, Vigneri P, de Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17(15):4942–4947. | ||
Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26(17):2839–2845. | ||
Li H, Liu L, Guo L, et al. HERG K+ channel expression in CD34+/CD38-/CD123(high) cells and primary leukemia cells and analysis of its regulation in leukemia cells. Int J Hematol. 2008;87(4):387–392. | ||
Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. | ||
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. | ||
Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–10951. | ||
Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65(8):3025–3029. | ||
Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. | ||
Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. 2009;106(33):13820–13825. | ||
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–629. | ||
Desai S, Barai A, Bukhari AB, De A, Sen S. α-Actinin-4 confers radioresistance coupled invasiveness in breast cancer cells through AKT pathway. Biochim Biophys Acta Mol Cell Res. 2018;1865(1):196–208. | ||
Gupta S, Iljin K, Sara H, et al. FZD4 as a mediator of ERG oncogene-induced Wnt signaling and epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2010;70(17):6735–6745. | ||
Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–563. | ||
Liu S, Ginestier C, Ou SJ, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011;71(2):614–624. | ||
Lim PK, Bliss SA, Patel SA, et al. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011;71(5):1550–1560. | ||
Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. | ||
Gabbiani G, Majno G. Dupuytren’s contracture: fibroblast contraction? An ultrastructural study. Am J Pathol. 1972;66(1):131–146. | ||
Stuelten CH, Barbul A, Busch JI, et al. Acute wounds accelerate tumorigenesis by a T cell-dependent mechanism. Cancer Res. 2008;68(18):7278–7282. | ||
Hu M, Yao J, Cai L, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37(8):899–905. | ||
Farmer P, Bonnefoi H, Anderle P, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. 2009;15(1):68–74. | ||
Finak G, Bertos N, Pepin F, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14(5):518–527. | ||
Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6(7):506–520. | ||
Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. | ||
Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107(5):1761–1767. | ||
Chu QD, Panu L, Holm NT, et al. High chemokine receptor CXCR4 level in triple negative breast cancer specimens predicts poor clinical outcome. J Surg Res. 2010;159(2):689–695. | ||
Scheller J, Rose-John S. Interleukin-6 and its receptor: from bench to bedside. Med Microbiol Immunol. 2006;195(4):173–183. | ||
Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14(21):6735–6741. | ||
Chao MP, Alizadeh AA, Tang C, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011;71(4):1374–1384. | ||
Benoy IH, Salgado R, van Dam P, et al. Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin Cancer Res. 2004;10(21):7157–7162. | ||
Ara T, Declerck YA. Interleukin-6 in bone metastasis and cancer progression. Eur J Cancer. 2010;46(7):1223–1231. | ||
Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, let-7 microRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139(4):693–706. | ||
Ginestier C, Liu S, Diebel ME, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120(2):485–497. | ||
Barnes PJ, Karin M. Nuclear factor-κB – a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336(15):1066–1071. | ||
Michieli P, Mazzone M, Basilico C, et al. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell. 2004;6(1):61–73. | ||
Vermeulen L, de Sousa E Melo F, van der Heijden M, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12(5):468–476. | ||
Fillmore CM, Gupta PB, Rudnick JA, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci USA. 2010;107(50):21737–21742. | ||
Dufraine J, Funahashi Y, Kitajewski J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene. 2008;27(38):5132–5137. | ||
Hiscox S, Parr C, Nakamura T, et al. Inhibition of HGF/SF-induced breast cancer cell motility and invasion by the HGF/SF variant, NK4. Breast Cancer Res Treat. 2000;59(3):245–254. | ||
Lebedis C, Chen K, Fallavollita L, Boutros T, Brodt P. Peripheral lymph node stromal cells can promote growth and tumorigenicity of breast carcinoma cells through the release of IGF-I and EGF. Int J Cancer. 2002;100(1):2–8. | ||
Pietras K, Rubin K, Sjöblom T, et al. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002;62(19):5476–5484. | ||
Singer CF, Kronsteiner N, Marton E, et al. MMP-2 and MMP-9 expression in breast cancer-derived human fibroblasts is differentially regulated by stromal-epithelial interactions. Breast Cancer Res Treat. 2002;72(1):69–77. | ||
Dale TC, Weber-Hall SJ, Smith K, et al. Compartment switching of Wnt-2 expression in human breast tumors. Cancer Res. 1996;56(19):4320–4323. | ||
Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. | ||
Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11(1):69–82. | ||
Bhati R, Patterson C, Livasy CA, et al. Molecular characterization of human breast tumor vascular cells. Am J Pathol. 2008;172(5):1381–1390. | ||
Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129(3):465–472. | ||
Holash J, Wiegand SJ, Yancopoulos GD. New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene. 1999;18(38):5356–5362. | ||
Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med Overseas Ed. 2007;357(26):2666–2676. | ||
Petrelli F, Barni S. Bevacizumab in advanced breast cancer: an opportunity as second-line therapy? Med Oncol. 2012;29(1):1–4. | ||
Pàez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. | ||
Ebos JM, Lee CR, Cruz-Munoz W, et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–239. | ||
Santos SJ, Kakarala P, Heath A, Clouthier S, Wicha MS. Abstract 3336: anti-angiogenic agents increase breast cancer stem cells via generation of tumor hypoxia. Cancer Res. 2011;71(8 Supplement):3336. | ||
Shojaei F, Lee JH, Simmons BH, et al. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010;70(24):10090–10100. | ||
Soriano JV, Uyttendaele H, Kitajewski J, Montesano R. Expression of an activated Notch4(int-3) oncoprotein disrupts morphogenesis and induces an invasive phenotype in mammary epithelial cells in vitro. Int J Cancer. 2000;86(5):652–659. | ||
Uyttendaele H, Soriano JV, Montesano R, Kitajewski J. Notch4 and Wnt-1 proteins function to regulate branching morphogenesis of mammary epithelial cells in an opposing fashion. Dev Biol. 1998;196(2):204–217. | ||
Coleman-Vaughan C, Mal A, De A, McCarthy JV. The $γ$-Secretase Protease Complexes in Neurodegeneration, Cancer and Immunity. In Chakraborti S, Dhalla NS, editors. Pathophysiological Aspects of Proteases. Singapore: Springer; 2017:47–87. | ||
Milano J, Mckay J, Dagenais C, et al. Modulation of Notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82(1):341–358. | ||
Real PJ, Ferrando AA. Notch inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia. 2009;23(8):1374–1377. | ||
Tian H, Callahan CA, Dupree KJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci USA. 2009;106(11):4254–4259. | ||
von Hoff DD, Lorusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361(12):1164–1172. | ||
Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–127. | ||
Korkaya H, Paulson A, Charafe-Jauffret E, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7(6):e1000121. | ||
Kochhar R, Khurana V, Bejjanki H, Caldito G, Fort C. Statins to reduce breast cancer risk: a case control study in U.S. female veterans. J Clin Oncol. 2005;23(16_suppl):514–514. | ||
Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352(1):20–28. | ||
Kastritis E, Charidimou A, Varkaris A, Dimopoulos MA. Targeted therapies in multiple myeloma. Targ Oncol. 2009;4(1):23–36. | ||
Mercier I, Casimiro MC, Wang C, et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 down-regulation and Rb tumor suppressor functional inactivation: implications for the response to hormonal therapy. Cancer Biol Ther. 2008;7(8):1212–1225. | ||
Dittmer A, Fuchs A, Oerlecke I, et al. Mesenchymal stem cells and carcinoma-associated fibroblasts sensitize breast cancer cells in 3D cultures to kinase inhibitors. Int J Oncol. 2011;39(3):689–696. | ||
Lu X, Kang Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem. 2009;284(42):29087–29096. | ||
Li Q, Xia J, Yao Y, et al. Sulforaphane inhibits mammary adipogenesis by targeting adipose mesenchymal stem cells. Breast Cancer Res Treat. 2013;141(2):317–324. | ||
Rahman M, Pumphrey JG, Lipkowitz S. The TRAIL to targeted therapy of breast cancer. Adv Cancer Res. 2009;103:43–73. | ||
Piggott L, Omidvar N, Pérez SM, Eberl M, Clarkson RWE. Suppression of apoptosis inhibitor c-FLIP selectively eliminates breast cancer stem cell activity in response to the anti-cancer agent, TRAIL. Breast Cancer Res. 2011;13(5):R88. | ||
Liguori M, Buracchi C, Pasqualini F, et al. Functional TRAIL receptors in monocytes and tumor-associated macrophages: a possible targeting pathway in the tumor microenvironment. Oncotarget. 2016;7(27):41662–41676. | ||
de Francesco EM, Sotgia F, Lisanti MP. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem J. 2018;475(9):1611–1634. | ||
Deshmukh A, Deshpande K, Arfuso F, Newsholme P, Dharmarajan A. Cancer stem cell metabolism: a potential target for cancer therapy. Mol Cancer. 2016;15(1):69. | ||
Snyder V, Reed-Newman TC, Arnold L, Thomas SM, Anant S. Cancer stem cell metabolism and potential therapeutic targets. Front Oncol. 2018;8:203. | ||
Sancho P, Burgos-Ramos E, Tavera A, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22(4):590–605. | ||
Mancini R, Noto A, Pisanu ME, et al. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene. 2018;37(18):2367–2378. | ||
de Francesco EM, Maggiolini M, Tanowitz HB, Sotgia F, Lisanti MP. Targeting hypoxic cancer stem cells (CSCs) with doxycycline: implications for optimizing anti-angiogenic therapy. Oncotarget. 2017;8(34):56126–56142. | ||
Zhang X, Fryknäs M, Hernlund E, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun. 2014;5(1):3295. | ||
Jangamreddy JR, Ghavami S, Grabarek J, et al. Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: differences between primary and cancer cells. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2013;1833(9):2057–2069. | ||
Deshmukh A, Arfuso F, Newsholme P, Dharmarajan A. Regulation of cancer stem cell metabolism by secreted frizzled-related protein 4 (sFRP4). Cancers. 2018;10(2):40. | ||
El Hout M, dos Santos L, Hamaï A, Mehrpour M. A promising new approach to cancer therapy: targeting iron metabolism in cancer stem cells. Semin Cancer Biol. 2018;53:125–138. | ||
Wu D, Si M, Xue HY, Wong HL. Nanomedicine applications in the treatment of breast cancer: current state of the art. Int J Nanomedicine. 2017;12:5879–5892. | ||
Hong IS, Jang GB, Lee HY, Nam JS. Targeting cancer stem cells by using the nanoparticles. Int J Nanomedicine. 2015;10(Spec Iss):251–260. | ||
Sun R, Shen S, Zhang YJ, et al. Nanoparticle-facilitated autophagy inhibition promotes the efficacy of chemotherapeutics against breast cancer stem cells. Biomaterials. 2016;103:44–55. | ||
Li SY, Sun R, Wang HX, et al. Combination therapy with epigenetic-targeted and chemotherapeutic drugs delivered by nanoparticles to enhance the chemotherapy response and overcome resistance by breast cancer stem cells. J Control Release. 2015;205:7–14. | ||
Ahmad A, Mondal SK, Mukhopadhyay D, Banerjee R, Alkharfy KM. Development of liposomal formulation for delivering anticancer drug to breast cancer Stem-Cell-Like cells and its pharmacokinetics in an animal model. Mol Pharm. 2016;13(3):1081–1088. | ||
Zuo ZQ, Chen KG, Yu XY, et al. Promoting tumor penetration of nanoparticles for cancer stem cell therapy by TGF-β signaling pathway inhibition. Biomaterials. 2016;82:48–59. | ||
Pramanik D, Campbell NR, Karikari C, et al. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol Cancer Ther. 2011;10(8):1470–1480. | ||
Fiorillo M, Verre AF, Iliut M, et al. Graphene oxide selectively targets cancer stem cells, across multiple tumor types: implications for non-toxic cancer treatment, via “differentiation-based nano-therapy”. Oncotarget. 2015;6(6):3553–3562. | ||
Burke AR, Singh RN, Carroll DL, et al. The resistance of breast cancer stem cells to conventional hyperthermia and their sensitivity to nanoparticle-mediated photothermal therapy. Biomaterials. 2012;33(10):2961–2970. | ||
Mukherjee A, Narayan KP, Pal K, et al. Selective cancer targeting via aberrant behavior of cancer cell-associated glucocorticoid receptor. Mol Ther. 2009;17(4):623–631. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.