")
Back to Journals » OncoTargets and Therapy » Volume 8
Targeting specificity of dendritic cells on breast cancer stem cells: in vitro and in vivo evaluations
Authors Nguyen ST, Nguyen H, Pham VQ, Nguyen GH, Tran C, Phan NK, Pham PV , Phan N
Received 15 November 2014
Accepted for publication 3 January 2015
Published 30 January 2015 Volume 2015:8 Pages 323—334
DOI https://doi.org/10.2147/OTT.S77554
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Sinh Truong Nguyen,1 Huyen Lam Nguyen,1 Viet Quoc Pham,1 Giang Thuy Nguyen,1 Cuong Do-Thanh Tran,1 Ngoc Kim Phan,1,2 Phuc Van Pham1,2
1Laboratory of Stem Cell Research and Application, 2Faculty of Biology, University of Science, Vietnam National University, Ho Chi Minh City, Vietnam
Abstract: Breast cancer is a leading cause of death in women, and almost all complications are due to chemotherapy resistance. Drug-resistant cells with stem cell phenotypes are thought to cause failure in breast cancer chemotherapy. Dendritic cell (DC) therapy is a potential approach to eradicate these cells. This study evaluates the specificity of DCs for breast cancer stem cells (BCSCs) in vitro and in vivo. BCSCs were enriched by a verapamil-resistant screening method, and reconfirmed by ALDH expression analysis and mammosphere assay. Mesenchymal stem cells (MSCs) were isolated from allogeneic murine bone marrow. DCs were induced from bone marrow-derived monocytes with 20 ng/mL GC-MSF and 20 ng/mL IL-4. Immature DCs were primed with BCSC- or MSC-derived antigens to make two kinds of mature DCs: BCSC-DCs and MSC-DCs, respectively. In vitro ability of BCSC-DCs and MSC-DCs with cytotoxic T lymphocytes (CTLs) to inhibit BCSCs was tested using the xCELLigence technique. In vivo, BCSC-DCs and MSC-DCs were transfused into the peripheral blood of BCSC tumor-bearing mice. The results show that in vitro BCSC-DCs significantly inhibited BCSC proliferation at a DC:CTL ratio of 1:40, while MSC-DCs nonsignificantly decreased BCSC proliferation. In vivo, tumor sizes decreased from 18.8% to 23% in groups treated with BCSC-DCs; in contrast, tumors increased 14% in the control group (RPMI 1640) and 47% in groups treated with MSC-DCs. The results showed that DC therapy could target and be specific to BCSCs. DCs primed with MSCs could trigger tumor growth. These results also indicate that DCs may be a promising therapy for treating drug-resistant cancer cells as well as cancer stem cells.
Keywords: dendritic cells, 4T,1 cell line, breast tumor, breast cancer stem cells, verapamil, drug resistance
Introduction
Breast cancer is the most common cancer in women both in developed and in developing countries. According to Global Health Estimates 2013 (WHO), breast cancer caused over 508,000 female deaths worldwide in 2011. In 2013, the average survival period of breast cancer was 5 years, however, this period is lower in developing countries with similar distributions of the stage at diagnosis.1 For many years, the only standardized treatment options for cancer have been surgery, radiotherapy, and chemotherapy. However, many cases are complicated by tumor relapse and resistance to chemotherapy.2 Therefore, it is necessary to develop new therapies that are less toxic and more effective. Because of the importance of cancer stem cells in tumors, many researchers are trying to isolate these cells to study their functional properties and evaluate whether they can effectively treat cancer. Recently, there have been many reports showing the prospective isolation of cancer stem cells in numerous malignancies, including breast,3 brain,4 colon,5 head and neck,6 pancreatic,7 melanoma,8 hepatic carcinoma,9 lung,10 prostate,11 and ovarian tumors.12 These cancer stem cells have therefore become targets for cancer treatment.
In recent years, dendritic cell (DC)-based therapy has shown promise as a cancer treatment. DCs were first discovered by Steinman and Cohn,13 and are professional antigen-presenting cells that have the ability to activate both innate and adaptive immune responses. DCs have the unique ability of cross-presentation, because they process and present peptide fragments on the surface of MHC class I and MHC class II molecules.14 After maturation, DCs migrate to the draining lymph node and activate naïve T cells. Immature DCs are more efficient than mature DCs at capturing and processing antigens. However, mature DCs are more efficient at activating and stimulating T cells.15–18 These mature DCs are more efficient than immature DCs at homing to lymph nodes.19,20 Immature DCs can be generated in vitro in the presence of cytokines GM-CSF and IL-4, and then mature when primed in vitro with tumor-specific antigens used for cancer treatment.21,22
To date, some studies have used DC-specific antigens to treat breast tumors and reported that DC treatment is effective for reducing tumor mass.23–25 These results have opened the door for DC therapy as a novel approach in breast cancer treatment. However, these studies targeted tumor or cancer cells. In order to improve the efficiency of this therapy, some recent studies developed DC therapy targeting cancer stem cells,26 such as breast27 and glioblastoma cancer stem cells.28 More importantly, targeting glioblastoma cancer stem cells by DC therapy was permitted in a clinical trial (NCT00846456).
However, to the best of our knowledge, no study has addressed the specific effects of DCs on cancer stem cells or stem cells. This study evaluates the specificity of DC therapy primed with breast cancer stem cells (BCSCs) in breast cancer treatment. We investigated the specific inhibition of DCs and induced cytotoxic T lymphocytes (CTLs) in vitro and in vivo.
Materials and methods
4T1 culture
Murine 4T1 mammary gland tumor cells, which are spontaneously metastatic tumor cells derived from BABL/c mice, were purchased from the American Type Culture Collection (ATCC). Murine 4T1 mammary gland tumor cells are comparable to human stage IV breast cancer. The tumor cells were cultured in RPMI 1640 medium (Sigma-Aldrich, St Louis, MO, USA) and supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) as well as 1× antibiotic mycotic (Sigma-Aldrich) in 25 mL cell culture flasks. The culture medium was regularly changed at 3-day intervals.
Verapamil cytotoxicity assay
The 4T1 cells were cultured in culture medium supplemented with various levels of verapamil from 0 to 100 μg/mL. The cytotoxicity effect of verapamil on 4T1 cells was measured by the xCELLigence Real-Time Cell Analyzer (RTCA) (Hoffmann-La Roche Ltd., Basel, Switzerland). This system monitored cellular events such as cell number, adhesion, viability, and morphology in real time by measuring the change in electrical impedance as the living cells interacted with the biocompatible microelectrode surface in the E-plate well. The final cell density was 5 cells per well in 200 μL RPMI-10% FBS. After 24 hours of culture to allow cell adhesion and spreading, all of the RPMI-10% FBS was removed, transferred, and supplemented with 0, 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100 μg/mL verapamil (Sigma-Aldrich). Each concentration of verapamil was repeated in three different wells. The highest concentration of verapamil in which 4T1 cells could survive was used to select verapamil-resistant 4T1 cells for further experiments.
Stemness of verapamil-resistant 4T1 cells
The stemness of verapamil-resistant 4T1 cells was evaluated by assays including mammosphere culture, ALDH expression, and in vivo tumorigenesis. The verapamil-treated and -untreated 4T1 cells were placed in culture flasks in serum-free DMEM-F12, supplemented with 10 ng/mL basic fibroblast growth factor, 20 ng/mL epidermal growth factor, 5 ng/mL insulin, and 0.4% bovine serum albumin at 37°C, 5% CO2 in a humidified chamber. The culture flasks were placed vertically. Culture medium was changed at 3-day intervals. After 10 days of culture, the number and diameter of spheres were determined using an inverted microscope at 20× magnification. ALDH expression was evaluated using an ALDEFLUOR kit (Stemcell Technologies, Vancouver, British Columbia, Canada) according to the manufacturer’s instructions. Immune-deficient mice were used to test the tumorigenicity of BCSC candidates. Immunodeficient mice were administered busulfan intramuscularly through abdominal muscles, and cyclophosphamide intravenously via the tail vein at doses of 20 and 200 mg/kg, respectively.
Bone marrow-derived mesenchymal stem cell isolation and proliferation
Mesenchymal stem cells (MSCs) were isolated from murine bone marrow. Mononuclear cells were cultured in medium DMEM/F12 supplemented with 10% FBS and 1% antibiotic mycotic (Sigma-Aldrich). MSC candidates were subcultured for five passages, and the stemness of MSCs was checked according to Dominici’s guidelines.29 Cells were evaluated by marker expression of CD44, CD73, CD90, CD105, CD14, CD34, and CD45 by flow cytometry; cells were then differentiated into adipocytes, osteoblasts, and chondrocytes.
Antigen generation
Both verapamil-resistant 4T1 cells and MSCs at a concentration of 5×106 cells/mL were suspended in PBSA. Cells were rapidly frozen at −196°C and thawed at 37°C. This freeze–thaw process was repeated 3 times. Protein was harvested by PRO-PREP instructions (Intron Biotechnology, Seongnam-si, Gyeonggi-do, Korea). In brief, after centrifugation, the cell pellet was suspended in 1 mL cold PRO-PREP solution. The mix was centrifuged at 13,000 rpm at 4°C. The supernatant containing the expected protein was collected and stored at −80°C. Protein concentration was determined by Bradford assay.
Generation of DCs from mouse bone marrow-derived mononuclear cells
Mouse bone marrow cells were harvested by flushing the marrow cavities of the femur and tibia bones of male mice with medium under aseptic conditions. The harvested marrow was depleted of erythrocytes and cultured only in complete RPMI-10% FBS. On day 2, the cultured medium was changed with RPMI-10% FBS, 100 ng/mL granulocyte monocyte colony-stimulating factor (GM-CSF), and 50 ng/mL IL-4 (Santa Cruz Biotechnology Inc., Dallas, TX, USA). Half of the medium was replaced with fresh medium containing the same cocktail of cytokines every 2 days. On day 8, antigens were added into the cultured medium at a concentration of 500 ng/mL. On day 10, nonadherent fractions of DCs were harvested. Adherent fractions of DCs were also harvested by incubating with 0.25% Trypsin/EDTA (Sigma-Aldrich). Then, the numbers of harvested DCs were determined by automatic counting using a Nucleocounter® (ChemoMetec A/S, Allerod, Denmark).
Mature DCs generated from mouse bone marrow were directly stained using fluorescent-conjugated monoclonal antibodies, including anti-CD40, CD80, CD83, and CD86, for 30 minutes at 4°C in the dark. Cells were then analyzed using FACSCalibur flow cytometry (BD Biosciences, San Jose, CA, USA) with CellQuest software.
In vitro evaluation of DC-based vaccination
To evaluate the effects of DCs on BCSCs, we developed a system using xCELLigence RTCA equipment. xCELLigence RTCA was used to evaluate cell proliferation and cytotoxicity on the basis of changes in electrical impedance on the surface of the E-plate, a plate with electric nodes on the surface allowing measurement of changes in impedance (Table 1).
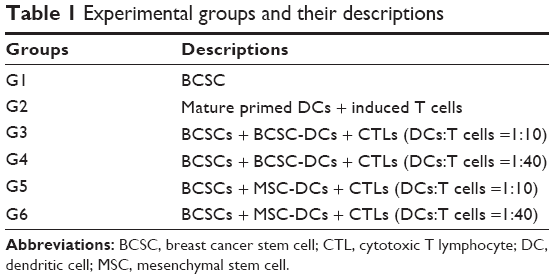 | Table 1 Experimental groups and their descriptions |
We observed differences in adherence of BCSCs, DCs, and CTLs. BCSCs were strongly attached to the surface of the E-plate, while DCs and lymphocytes were weakly attached. Thus, on the basis of BCSC proliferation on the E-plate with or without DCs or CTLs, we could determine the cytotoxic effects of this therapy on target cells. From 0 to 24 hours, verapamil-resistant cells (VRCs) were cultured as adherent cells in the E-plate (96 wells) in groups 1 and 3–6, while group 2 had DCs and CTLs added. After 24 hours, cells in groups 1–6 had fresh medium added. Only fresh culture medium was added to groups 1 and 2, while DC-induced CTLs were added to groups 3–6. Before this, mature DCs (BCSC-DCs and MSC-DCs) were incubated with CTLs at different ratios of DCs and CTLs, ie, 1:10 and 1:40, over the course of 24 hours. These mixtures of DCs and CTLs were added to the E-plate wells containing BCSCs. Finally, the E-plates were then placed on the xCELLigence instrument to monitor BCSC proliferation. CTLs were prepared according to a previously published study.30
Breast tumor-bearing mouse models and in vivo assay
The male mice were housed in an animal maintenance facility. The soda bedding was changed at 4-day intervals. All experiments on animals were performed in accordance with the guidelines approved by the Ethics Committee of Stem Cell Research and Application Laboratory, University of Science, VNU-HCM. Fifty μL of verapamil-resistant 4T1 cells (106 cells) were injected into the mammary pads of 8-week-old mice.
Tumors formed after 2 days. The animals with tumors were divided into 4 groups of 4 mice each. The control group (group 1) was intravenously injected with RPMI 1640. The second group (group 2) was intravenously injected with DCs primed with mouse MSC-derived antigen (MSC-DC). Group 3 was treated with DCs primed with verapamil-resistant 4T1-derived antigen (BCSC-DC) by injection into the tumor. The tumor lengths were measured daily. Survival, symptoms of pain, and inflammation were monitored. Any mice expressing abnormal symptoms were isolated from the groups and closely observed. After the last experiment, all mice were euthanized to collect and measure the tumors.
CD4 and CD8 analysis
Two days after treatment, blood was collected for analysis at 5-day intervals. Blood from healthy mice (receiving no inoculation) was also harvested and used as a control group. Forty microliters of blood was extracted from the tail and immediately mixed with anticoagulant. Whole blood was incubated with the fluorescent-conjugated antibodies anti-CD4-FITC and anti-CD8-PE (BD Biosciences) at 4°C for 30 minutes. Red blood cells were removed from the sample using a lysis buffer, Pharm Lyse (BD Biosciences). Lymphocytes were harvested after centrifugation at 500 rcf and washed 2 times with FACSflow. The pellet was resuspended in 200 μL FACSflow buffer and analyzed by FACSCalibur (BD Biosciences).
Statistical analysis
The differences in mean tumor size and percentage of T cells in blood from control and experimental mice were analyzed using Graphpad Prism 6.1 Software. P-values less than 0.05 were considered significant.
Results
Verapamil-resistant cells enriched stem cell populations
In groups with 10–40 μg/mL verapamil, cancer cells proliferated more slowly than in the control (0 μg/mL verapamil). Alternatively, at 60–100 μg/mL levels of verapamil, cancer cells were completely inhibited after 20 hours of treatment, and cell growth rates dramatically decreased and the cells died (Figure 1A). At 50 μg/mL verapamil, cancer cells were also inhibited after 20 hours of treatment, but there were a few living cells, so these populations continued to develop and formed verapamil-resistant cell populations. This concentration of verapamil was used to develop VRCs for the next experiment.
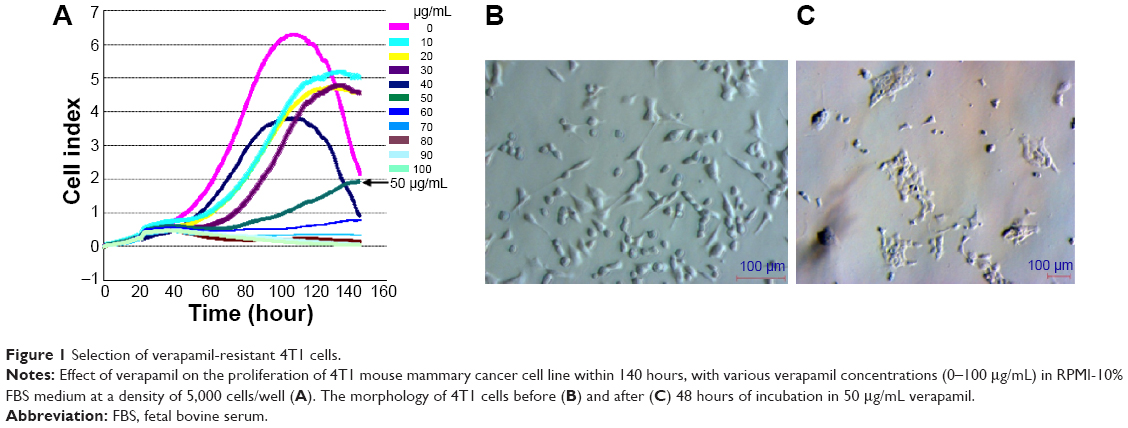 | Figure 1 Selection of verapamil-resistant 4T1 cells. |
In the T-25 flask, 4T1 cancer cells were treated with 50 μg/mL verapamil in RPMI-10% FBS. There was a significant change in cell morphology and cell viability (Figure 1B). Cell membranes were not transparent, and the cells had a tendency to contract. Several 4T1 cells died and floated in the medium culture within 24 hours of exposure to verapamil, and the cell death ratio dramatically increased after 48 hours. Only the cells that could tolerate and resist verapamil survived and proliferated (Figure 1C).
ALDH expression in VRCs was analyzed. The results showed that these cells expressed ALDH (Figure 2A–C), and could form mammospheres in serum-free medium (Figure 2D). In the in vivo tumorigenesis assay, this population formed tumors with 105 cells in mice (Figure 2E–G).
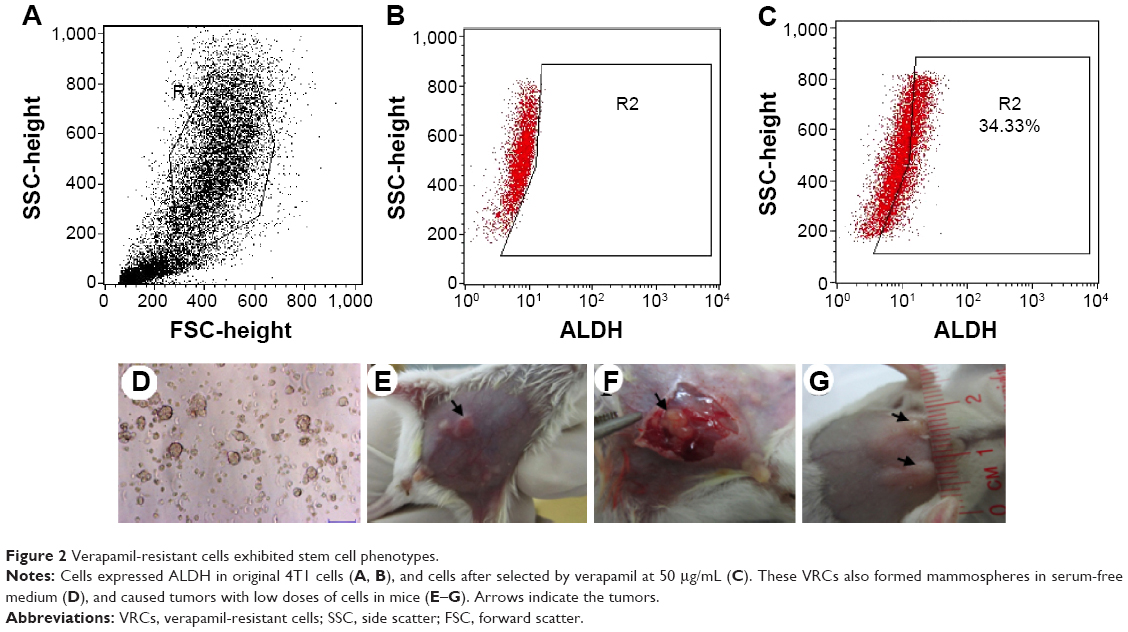 | Figure 2 Verapamil-resistant cells exhibited stem cell phenotypes. |
Mesenchymal stem cells derived from murine bone marrow
After 24 hours of incubation, some mononuclear cells attached to the flask surfaces and exhibited the fibroblast-like shape (Figure 3H). These cells rapidly proliferated after 96 hours. These cells exhibited some properties of MSCs, such as positive expression of CD44, CD73, CD90, and CD105 (Figure 3D–G), but no expression of CD14, CD34, and CD45 (Figure 3A–C). These cells also successfully differentiated into adipocytes that stained with Oil red, and osteoblasts that stained with Alizarin red (Figure 3I, J). These cells continuously proliferated for five passages and were used for further experiments.
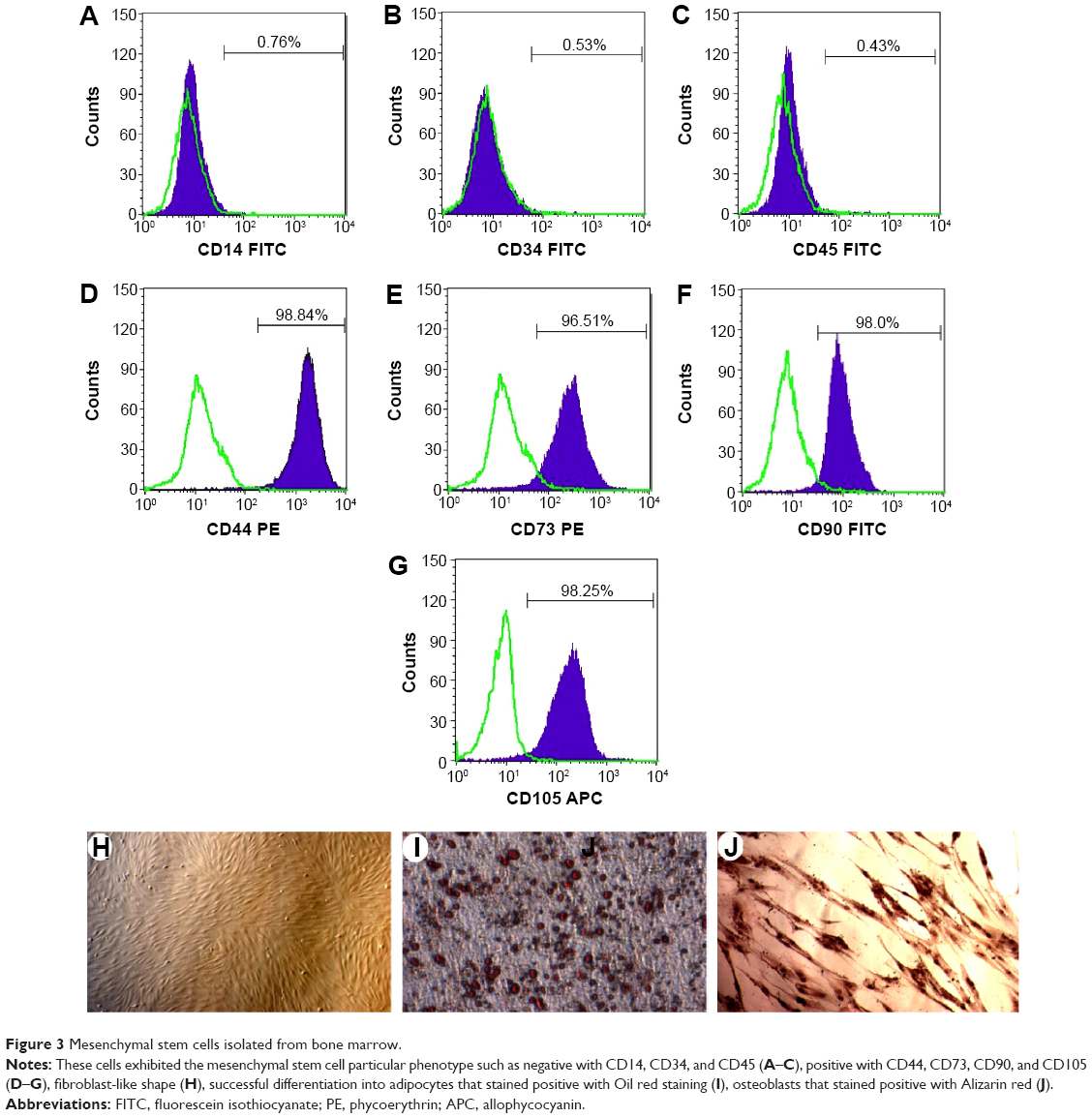 | Figure 3 Mesenchymal stem cells isolated from bone marrow. |
Production of functional dendritic cells from bone marrow-derived mononuclear cells with breast cancer stem cell and mesenchymal stem cell-derived antigens
Bone marrow-derived mononuclear cells were cultured in RPMI medium supplemented with GM-CSF and IL-4 for 7 days to generate immature DCs. Morphological observation on day 7 showed formation of dendrites that are typical of DCs (Figure 4A, B). On day 7, DCs still adhered to the culture flask bottom. These iDCs (immature dendritic cells) expressed the typical phenotypes of DCs, including the presence of CD80, CD86, and CD40 (Figure 4D–F) and the absence of CD14 (Figure 4C). Mature iDCs were induced by adding antigens into the culture medium with cytokines. After 3 days in the medium supplemented with antigens from both BCSCs and MSCs, DCs detached from the flask bottom and floated in the medium. Morphological observation showed that there are veils around the DCs.
 | Figure 4 Dendritic cells were generated from mouse bone marrow. |
Mouse bone marrow-derived DCs collected on day 10 of culturing (3 days after adding antigens) included populations of cells that expressed surface molecules that are typical for DCs. As shown in Figure 4C–F, the DCs expressed CD80 (94.42%), CD86 (87.41%), and CD40 (63.33%). The mouse bone marrow-derived DC populations expressed a few of the lineage markers for monocytes, including CD14 (2.65%). The DC populations generated in this study were mostly CD86, which is phenotypically characteristic of mature DCs, suggesting that mouse bone marrow cells cultured in a medium containing GM-CSF, IL-4 cytokine, and the antigens could successfully generate DCs that displayed typical DC surface antigens.
In vitro selective inhibition of breast cancer stem cell-dendritic cells and mesenchymal stem cell-dendritic cells on breast cancer stem cells
The results show that a mixture of DCs and CTLs effected BCSC proliferation that changed the CI (cell index) value recorded by the xCELLigence system (Figure 5). BCSCs normally proliferated over time in group 1. Group 2 demonstrated that a mixture of DCs and CTLs increased impedance slightly in the first 24 hours and was stable thereafter. From 0 to 24 hours (before adding CTLs), the proliferation rates in groups 1 and 3–6 were similar. After adding induced CTLs, from 24 to 63 hours, the effects of induced CTLs in all groups on BCSC proliferation were nonclear. However, from 63 to 98 hours, proliferation rates of groups 3–6 were somewhat different. BCSC proliferation in groups 3–6 was inhibited when a mixture of DCs and CTLs was added (Figure 5A).
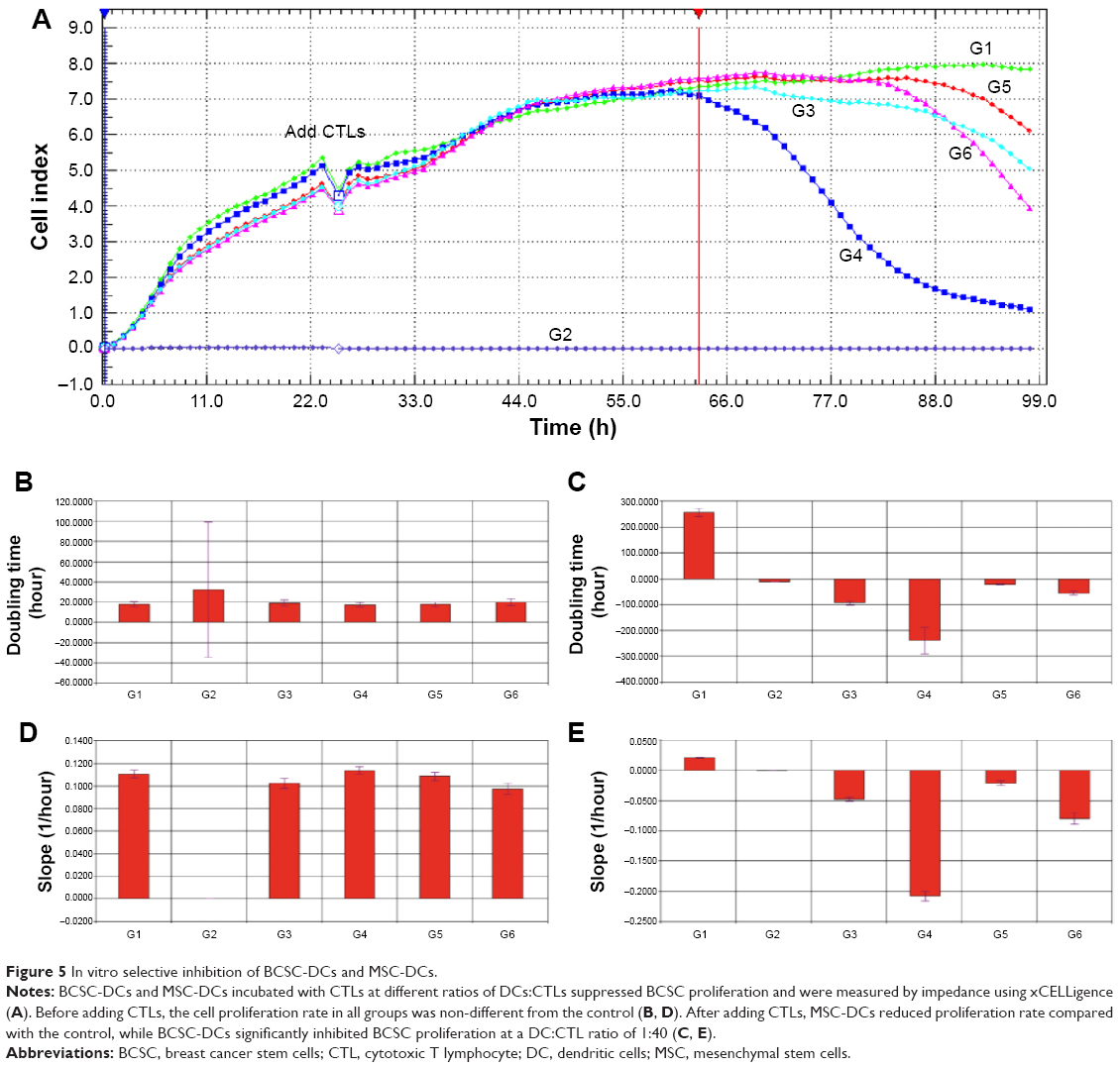 | Figure 5 In vitro selective inhibition of BCSC-DCs and MSC-DCs. |
Differences in ratios of DCs:CTLs also altered inhibition effects. Overall, there was increased BCSC inhibition in both the BCSC-DC and the MSC-DC groups with increased amounts of CTLs. When we compared the inhibition effects between the BCSC-DC and MSC-DC groups, we observed that although the DC-CTL mixture inhibited BCSC proliferation in both BCSC-DCs and MSC-DCs, BCSC-DC groups significantly decreased BCSC proliferation compared with MSC-DC (groups 5 and 6) groups and the control (group 1). There were also differences in inhibition of BCSC-DCs on BCSC proliferation at different ratios of DCs:CTLs, with the strongest inhibition occurring at a ratio of 1:40. These results are also supported by results from slope and doubling time analyses (Figure 5B–E).
In vivo selective inhibition of breast cancer stem cell-dendritic cells and mesenchymal stem cell-dendritic cells on tumors
Although there is no significant difference in daily tumor size between the 4 groups, we observed differences in increasing and decreasing tumor size between treated and control (RPMI 1640) groups (Figure 6A). On the basis of linear regression, the slopes of the control and the MSC-DC groups were positive, and the slopes of the BCSC-DC groups were negative (Figure 5B). This suggests that the tumor size of the groups treated with DCs primed by BCSC antigens decreased faster than that of those treated with DCs primed by MSC antigens and the control group. Slope values (Δy/Δx) were 0.003159±0.005604, 0.02555±0.004868, and −0.003324±0.005297 in the control, MSC-DC, and BCSC-DC groups, respectively. The slopes were significantly different (P=0.0001337).
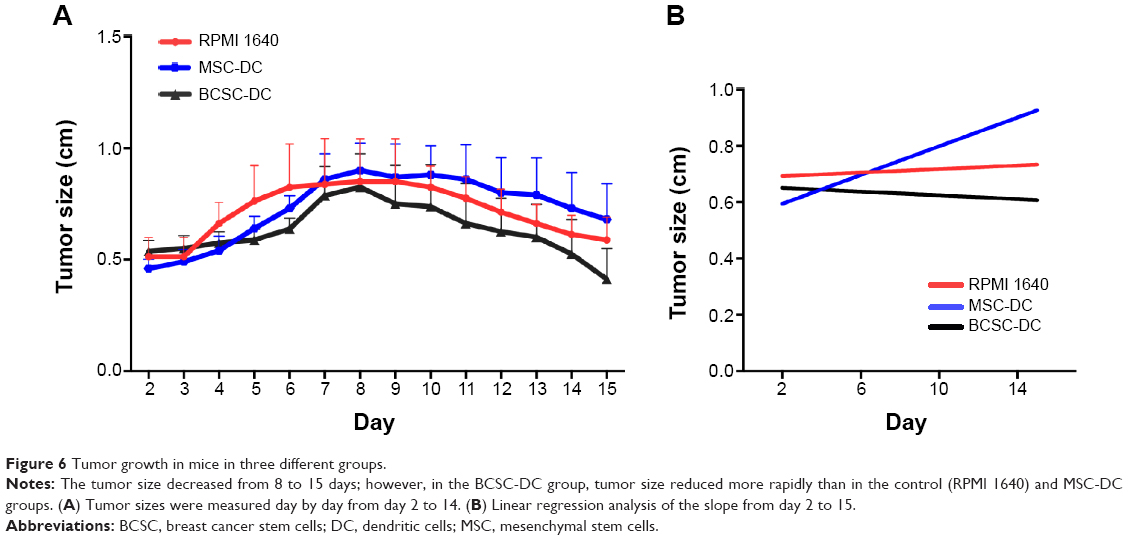 | Figure 6 Tumor growth in mice in three different groups. |
The data can provide more evidence about changing tumor size when comparing tumor size between days 2 and 15 (Figure 7). Tumor size decreased 23% in the BCSC-DC groups. In contrast, tumor size increased 14% in the control group; in particular, in the MSC-DC group, tumor size increased 47%. This indicates that therapeutic treatment with DCs primed by BCSC-derived antigens is effective in decreasing tumor size. Moreover, DCs primed by MSC antigen actually caused tumor mass to increase.
 | Figure 7 Tumor mass harvested from mice on day 15. After sacrificing mice, tumors were collected by surgery. |
T cell responses in mice after DC immunization
In order to elucidate the therapeutic effect of DCs on the immune system, mouse blood was harvested, and the percentage of CD4 and CD8 T cells was analyzed. Two days after inoculation with BCSCs, mice were injected with therapeutic DCs. Four days later, blood was harvested for evaluation. Assessment of lymphocytes was repeated 2 times, on days 7 and 13, after treatment. The results show that the amount of CD4 in normal mouse blood barely changed. However, there was a significant change in the treated groups. In mice with tumors that received no treatment (the control group), CD4 decreased by day 7 compared with day 4 (23% it decreased); in contrast, the amount of CD4 slightly increased, but nonsignificantly, in all of the DC-treated groups by day 7. By day 13, CD4 increased in both the non- and the DC-treated groups. In summary, the amount of CD4 in the nontreated group did not significantly increase over time (the start to the end of the experiment), but significantly increased in all of the DC-treated groups (P<0.05).
Similar to CD4, the amount of CD8 also changed in all mice. However, in contrast to CD4, the amount of CD8 increased by day 7. By day 13, the amount of CD8 significantly increased in treated groups compared with day 4 (P<0.05).
Discussion
Targeting cancer stem cells is considered an important approach in cancer treatment. However, the similarity between stem cells and cancer stem cells can cause mistargeting between stem cells and cancer stem cells, even though targeting cancer stem cells in glioblastoma was approved for clinical trial. Prior to this study, there was no comprehensive study that evaluated the cross-presentation of DCs between stem cells and cancer stem cells. In this study, we used BCSC and MSC models to investigate this concern, and MSCs from bone marrow were used because they are an important stem cell source in the body.
In the first step, BCSCs were enriched by the selection of verapamil-resistant 4T1 cells. In fact, one of the most common characteristics of cancer stem cells is anti-tumor drug resistance.31–33 Thus, the anticancer drug assay is a functional assay that has been applied to enrich cancer stem cells.34–36 This method is based on the overexpression of adenosine triphosphate-binding cassette (ABC) transporters on the membrane of cancer stem cells, such as P-glycoprotein (Pgp), multidrug resistance associated-protein 1 (MRP1), breast cancer resistance protein (BCRP), and multidrug resistance (MDR).
Recent studies have shown that increased expression of these transporters accounted for resistance of those cells to many anticancer drugs.37 These ABC transporters play an important role in normal physiology by protecting cells from toxic xenobiotics and endogenous metabolites. Therefore, in high concentrations of anticancer drugs, overexpression of these ABC transporters could help drug-resistant cells pump drugs out of the cells, whereas normal cancer cells would die at such drug concentrations.
Verapamil, an anticancer drug, has been used as a drug target in many different types of cancer. In 1988, Huber et al38 showed an antiproliferative effect of verapamil on growth rates of certain human brain tumor lines. Growth rates were inhibited by 10%–100% with 10–100 μM verapamil. Growth inhibition was accompanied by dose-dependent decreases in DNA, RNA, and protein synthesis that occurred within minutes after the addition of verapamil. A study by Trompier et al39 demonstrated that verapamil behaves as an apoptogen or triggers apoptosis in MRP1-expressing cells after 8–16 hours of treatment. In this study, 50 μg/mL verapamil efficiently selected verapamil-resistant 4T1 cells; this was the maximum concentration of verapamil in which 4T1 could survive and grow. Our study reports, for the first time, the specific verapamil concentration necessary to develop drug-resistant cell populations in the 4T1 mouse mammary cancer cell line.
Overexpression of ABC superfamily multidrug efflux pumps is known to be responsible for chemoresistance.40 These transporters play an important role in normal physiology by protecting cells from toxic xenobiotics and endogenous metabolites. Many clinically used drugs interact with the substrate-binding pocket of these proteins via flexible hydrophobic and hydrogen-bonding interactions. These efflux pumps are expressed in many human tumors, and expression combined with an enhanced capacity for DNA repair and decreased apoptosis contributes to resistance of tumor cells to chemotherapy treatment.41 Therefore, after allowing 48 hours of 4T1 cell exposure to a high concentration of verapamil (50 μg/mL), we expected that the viable remaining cells would overexpress ABC transporters and produce drug resistance.
Increased membrane transporter activity could help pump this anticancer drug out of the cells and lead to cell resistance at this drug concentration, whereas dead cells resulted from a lack of transporter activity. Consistent with this hypothesis, Calcagno et al35 concluded that prolonged drug selection of breast cancer cell line MCF-7/ADR in doxorubicin could increase the cell population with stem cell characteristics. Thus, there was a high probability that a number of drug-resistant cells would be present in verapamil-treated cells. More importantly, verapamil-resistant 4T1 cells exhibited BCSC properties. These cells easily formed mammospheres in the serum-free medium, highly expressed ALDH, and caused tumors in both NOD/SCID and immune-deficient mice at low concentrations of cells. In fact, these properties were considered BCSC properties for a long time.3,35,42–44 These verapamil-resistant 4T1 cells were considered BCSCs for this study.
Many studies have shown that DC therapy effectively inhibits breast cancer tumor growth,23–25 indicating that DCs could specifically target BCSCs. In vitro assay demonstrated that BCSC-DCs induced CTLs that suppressed BCSC proliferation, while MSC-DCs induced CTLs that slightly suppressed BCSC proliferation. In fact, decreased proliferation rate of BCSCs treated with MSC-DCs induced CTLs related to nutrient competition in the medium owing to a high concentration of cells in these groups.
We also hypothesized that some MSC-DCs and induced CTLs could die and release granzyme and perforin that suppressed BCSC proliferation. This result was supported by in vivo assay. The data showed that treatment with DCs primed by BCSCs effectively suppress BCSC proliferation in breast tumor-bearing mice. We provided evidence that treating with BCSC-DCs by intravenous injection reduced initial tumor mass by 18%–23%. Conversely, tumors increased by 14% with no DC treatment. The effect of DC treatment was consistent with studies of DC treatment in human systems45–47 and mouse models.48 Interestingly, treatment with DCs primed by allogeneic MSCs (MSC-DCs) increased tumor size up to 47%. This means that immune response to reduce breast tumor did not occur when mice were treated with MSC-DCs.
In this study, besides the reduction of tumor size in the treated groups, the control group also showed decreasing tumor size. This finding is a little different from those of studies that used a more common breast tumor model.48–50 This study used Swiss and mammary tumor 4T1 cell lines to develop a breast tumor model. The 4T1 cell line comes from BALB/c mice, whereas the Swiss mice retained the complete immune system. Therefore, mice would develop an immune response to the graft 4T1 cells, causing reduction of tumor size without any treatments. This may explain the reduction of tumor size in the control group on days 8–9. In treated mice, therapeutic DCs enhanced the immune system, causing a stronger reduction of tumor size. The lymphocyte data from blood after treatment provides more evidence for the effectiveness of DC therapy.
In the control group, there was no significant change in the amount of CD4 by day 7, but there was a significant increase in the amount of CD4 by day 13. This is consistent with the result of reduction of tumor size, which started to shrink by days 8–9 but not before day 7. This demonstrated that after day 7, the amount of CD4 T cells increased and there was efficient tumor reduction. However, the effect is not equal among all the groups. In the nontreated group, the amount of CD4 did not significantly increase by day 13, but there was a significant increase in the groups treated with MSC-DCs and BCSC-DCs by day 13 (P<0.05). Interestingly, although CD4 was high in all of the treated groups, the efficacy in tumor reduction occurred only in groups injected with BCSC-DCs and not MSC-DCs. We found little difference in the amount of CD8 between days 4 and 7 in all groups with tumor-carrying mice. These results show that BCSC-DCs, but not MSC-DCs, could induce immune response that targets tumors. Additionally, consistent with the results of studies that reported that therapeutic DCs enhance and support the response of CD4 T cells, CD8 T cells also kill cancer cells.51,52
It is unclear why treatment with intravenous MSC-DCs caused tumor sizes to increase up to 47%, particularly compared with the BCSC-DC-treated groups. This increase could be caused by MSC-DCs inducing a large amount of immune cells specific to MSCs; therefore, the immune system weakly attacks breast tumor cells, thus facilitating tumor growth. More experiments with DCs primed by other cell lines are needed to understand this finding.
Conclusion
Cancer stem cells in tumors represent cell populations that can cause failure of chemotherapy as a cancer treatment. Targeting cancer stem cells is considered a promising therapy in cancer treatment. This study evaluated the specificity of DC therapy for targeting BCSCs in murine models. Both in vitro and in vivo DCs primed with BCSC-derived antigens inhibited BCSC proliferation in a target cell-specific manner. Although this study is limited by using a Swiss mouse model, therapeutic DCs clearly inhibit tumor size in a specific manner compared with the control group. These results support the application of DC therapy in targeting BCSCs as well as cancer stem cells.
Acknowledgment
This work was funded by a grant from the Ministry of Science and Technology, Vietnam (grant number DTDL.2011-T/30).
Disclosure
The authors report no conflicts of interest in this work.
References
Lan NH, Laohasiriwong W, Stewart JF. Survival probability and prognostic factors for breast cancer patients in Vietnam. Glob Health Action. 2013;6:1–9. | ||
Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22. | ||
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | ||
Modrek AS, Bayin NS, Placantonakis DG. Brain stem cells as the cell of origin in glioma. World J Stem Cells. 2014;6(1):43–52. | ||
Prasetyanti PR, Zimberlin C, De Sousa EMF, Medema JP. Isolation and propagation of colon cancer stem cells. Methods Mol Biol. 2013;1035:247–259. | ||
Han J, Fujisawa T, Husain SR, Puri RK. Identification and characterization of cancer stem cells in human head and neck squamous cell carcinoma. BMC Cancer. 2014;14:173. | ||
Huang P, Wang CY, Gou SM, Wu HS, Liu T, Xiong JX. Isolation and biological analysis of tumor stem cells from pancreatic adenocarcinoma. World J Gastroenterol. 2008;14(24):3903–3907. | ||
Luo Y, Nguyen N, Fujita M. Isolation of human melanoma stem cells using ALDH as a marker. Curr Protoc Stem Cell Biol. 2013;26: Unit 3.8. | ||
Tomuleasa C, Soritau O, Rus-Ciuca D, et al. Isolation and characterization of hepatic cancer cells with stem-like properties from hepatocellular carcinoma. J Gastrointestin Liver Dis. 2010;19(1):61–67. | ||
Chen YC, Hsu HS, Chen YW, et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS One. 2008;3(7):e2637. | ||
Kasper S. Exploring the origins of the normal prostate and prostate cancer stem cell. Stem Cell Rev. 2008;4(3):193–201. | ||
O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16(12):3113–3120. | ||
Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137(5):1142–1162. | ||
Rodriguez A, Regnault A, Kleijmeer M, Ricciardi-Castagnoli P, Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat Cell Biol. 1999;1(6):362–368. | ||
Jiang W, Swiggard WJ, Heufler C, et al. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 1995;375(6527):151–155. | ||
Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179(4):1109–1118. | ||
Adema GJ, Hartgers F, Verstraten R, et al. A dendritic-cell-derived C-C chemokine that preferentially attracts naive T cells. Nature. 1997;387(6634):713–717. | ||
Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. | ||
De Vries IJ, Krooshoop DJ, Scharenborg NM, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63(1):12–17. | ||
Mackensen A, Krause T, Blum U, Uhrmeister P, Mertelsmann R, Lindemann A. Homing of intravenously and intralymphatically injected human dendritic cells generated in vitro from CD34+ hematopoietic progenitor cells. Cancer Immunol Immunother. 1999;48(2–3):118–122. | ||
Cody V, Shen H, Shlyankevich M, Tigelaar RE, Brandsma JL, Hanlon DJ. Generation of dendritic cells from rabbit bone marrow mononuclear cell cultures supplemented with hGM-CSF and hIL-4. Vet Immunol Immunopathol. 2005;103(3–4):163–172. | ||
Menges M, Baumeister T, Rossner S, et al. IL-4 supports the generation of a dendritic cell subset from murine bone marrow with altered endocytosis capacity. J Leukoc Biol. 2005;77(4):535–543. | ||
[No authors listed]. Vaccine studies demonstrate promise of immunotherapy to treat breast cancer and prevent recurrence. Oncology. 2000;14(10):1415–1416. | ||
Huang X, Ye D, Thorpe PE. Enhancing the potency of a whole-cell breast cancer vaccine in mice with an antibody-IL-2 immunocytokine that targets exposed phosphatidylserine. Vaccine. 2011;29(29–30):4785–4793. | ||
Pecher G, Haring A, Kaiser L, Thiel E. Mucin gene (MUC1) transfected dendritic cells as vaccine: results of a phase I/II clinical trial. Cancer Immunol Immunother. 2002;51(11–12):669–673. | ||
Teitz-Tennenbaum S, Wicha MS, Chang AE, Li Q. Targeting cancer stem cells via dendritic-cell vaccination. Oncoimmunology. 2012;1(8):1401–1403. | ||
Pham PV, Chi HJ, Nguyen NT, et al. Effects of breast cancer stem cell extract primed dendritic cell transplantation on breast cancer tumor murine models. Annu Rev Res Biol. 2011;1(1):1–13. | ||
Vik-Mo EO, Nyakas M, Mikkelsen BV, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013;62(9):1499–1509. | ||
Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. | ||
Schuler T, Kammertoens T, Preiss S, Debs P, Noben-Trauth N, Blankenstein T. Generation of tumor-associated cytotoxic T lymphocytes requires interleukin 4 from CD8(+) T cells. J Exp Med. 2001;194(12):1767–1775. | ||
Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond). 2012;7(4):597–615. | ||
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. | ||
Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. 2013;2(1):3. | ||
Rizzo S, Hersey JM, Mellor P, et al. Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther. 2011;10(2):325–335. | ||
Calcagno AM, Salcido CD, Gillet JP, et al. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst. 2010;102(21):1637–1652. | ||
Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE. Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS One. 2008;3(8):e3077. | ||
Choi CH. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005;5:30. | ||
Huber KR, Schmidt WF, Thompson EA, Forsthoefel AM, Neuberg RW, Ettinger RS. Effect of verapamil on cell cycle transit and c-myc gene expression in normal and malignant murine cells. Br J Cancer. 1989;59(5):714–718. | ||
Trompier D, Chang XB, Barattin R, du Moulinet D’Hardemare A, Di Pietro A, Baubichon-Cortay H. Verapamil and its derivative trigger apoptosis through glutathione extrusion by multidrug resistance protein MRP1. Cancer Res. 2004;64(14):4950–4956. | ||
Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9(1):105–127. | ||
Borst P, de Wolf C, van de Wetering K. Multidrug resistance-associated proteins 3, 4, and 5. Pflugers Archiv. 2007;453(5):661–673. | ||
Van Phuc P, Nhan PL, Nhung TH, et al. Downregulation of CD44 reduces doxorubicin resistance of CD44CD24 breast cancer cells. Onco Targets Ther. 2011;4:71–78. | ||
Pham PV, Phan NL, Nguyen NT, et al. Differentiation of breast cancer stem cells by knockdown of CD44: promising differentiation therapy. J Transl Med. 2011;9:209. | ||
Tanei T, Morimoto K, Shimazu K, et al. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin Cancer Res. 2009;15(12):4234–4241. | ||
Correale P, Cusi MG, Del Vecchio MT, et al. Dendritic cell-mediated cross-presentation of antigens derived from colon carcinoma cells exposed to a highly cytotoxic multidrug regimen with gemcitabine, oxaliplatin, 5-fluorouracil, and leucovorin, elicits a powerful human antigen-specific CTL response with antitumor activity in vitro. J Immunol. 2005;175(2):820–828. | ||
Bai L, Beckhove P, Feuerer M, et al. Cognate interactions between memory T cells and tumor antigen-presenting dendritic cells from bone marrow of breast cancer patients: bidirectional cell stimulation, survival and antitumor activity in vivo. Int J Cancer. 2003;103(1):73–83. | ||
Sato M, Chamoto K, Nishimura T. A novel tumor-vaccine cell therapy using bone marrow-derived dendritic cell type 1 and antigen-specific Th1 cells. Int Immunol. 2003;15(7):837–843. | ||
Abdul Hafid SR, Chakravarthi S, Nesaretnam K, Radhakrishnan AK. Tocotrienol-adjuvanted dendritic cells inhibit tumor growth and metastasis: a murine model of breast cancer. PLoS One. 2013;8(9):e74753. | ||
Coveney E, Wheatley GH 3rd, Lyerly HK. Active immunization using dendritic cells mixed with tumor cells inhibits the growth of primary breast cancer. Surgery. 1997;122(2):228–234. | ||
Radhakrishnan AK, Sim GC, Cheong SK. Comparing the ability of freshly generated and cryopreserved dendritic cell vaccines to inhibit growth of breast cancer in a mouse model. Biores Open Access. 2012;1(5):239–246. | ||
Engleman EG. Dendritic cells: potential role in cancer therapy. Cytotechnology. 1997;25(1–3):1–8. | ||
Galea-Lauri J, Darling D, Mufti G, Harrison P, Farzaneh F. Eliciting cytotoxic T lymphocytes against acute myeloid leukemia-derived antigens: evaluation of dendritic cell-leukemia cell hybrids and other antigen-loading strategies for dendritic cell-based vaccination. Cancer Immunol Immunother. 2002;51(6):299–310. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.