")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 6
Targeting myeloid-derived suppressor cells in the treatment of hepatocellular carcinoma: current state and future perspectives
Authors Lu LC , Chang CJ, Hsu CH
Received 15 December 2018
Accepted for publication 16 February 2019
Published 7 May 2019 Volume 2019:6 Pages 71—84
DOI https://doi.org/10.2147/JHC.S159693
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Li-Chun Lu,1–3 Chun-Jung Chang,1,2 Chih-Hung Hsu1–3
1Graduate Institute of Oncology, National Taiwan University College of Medicine, Taipei, Taiwan; 2Departments of Oncology, National Taiwan University Hospital, Taipei, Taiwan; 3Department of Oncology, National Taiwan University Cancer Center, Taipei, Taiwan
Abstract: Systemic therapy for advanced hepatocellular carcinoma (HCC) has been focusing on overcoming tumor angiogenesis and immunosuppression. Myeloid-derived suppressor cells (MDSCs) promote both angiogenesis and immunosuppression in the tumor microenvironment (TME). Multiple clinical studies have demonstrated the prognostic implications of and suggested the translational significance of MDSCs in patients with HCC. In preclinical HCC models, targeting MDSCs has been shown to enhance antitumor efficacy of sorafenib or immune checkpoint inhibitors. Reversing the protumor effects of MDSCs could be achieved by depleting MDSCs, blocking MDSC trafficking and migration into TME, and inhibiting the immunosuppressive functions of MDSCs. To date, these strategies have not yet been validated to be clinically useful in patients with malignancy including HCC. Future studies should focus on identifying specific markers for human MDSCs and developing combination approaches incorporating MDSC-targeting therapy in the treatment of HCC.
Keywords: myeloid-derived suppressor cells, MDSCs; hepatocellular carcinoma, immunosuppression, angiogenesis, immunotherapy, immune checkpoint inhibitor
Introduction
Primary liver cancer, of which the vast majority of cases are hepatocellular carcinoma (HCC), has been the leading cause of cancer mortality worldwide for decades. The GLOBOCAN 2018 database estimated that 841,000 new cases of primary liver cancer and 782,000 deaths from it would occur annually, making it the sixth most commonly diagnosed and fourth most lethal malignancy worldwide.1 The carcinogenesis of HCC is attributed to chronic inflammation of the liver, predominantly as a result of hepatitis B virus (HBV) and hepatitis C virus (HCV) virus infections, as well as liver cirrhosis. Owing to significant advances in the primary prevention of HBV infection through universal vaccinations as well as effective antiviral therapy for HBV and HCV infections, the incidence of HCC is expected to decline between 2035 and 2040.2 Overall, HCC remains a significant malignant disease that will continue to be a major burden on global health for decades to come.
Treatment of HCC is commonly directed by disease stage.3 For patients with early-stage localized HCC, curative-intent treatment modalities include resection, ablation, and liver transplantation, whereas for patients with intermediate-stage localized HCC, image-guided transcatheter tumor therapies such as transarterial chemoembolization have provided survival benefits. Unfortunately, the majority of patients with HCC either progress to or develop de novo locally advanced or metastatic diseases and are indicated for systemic therapy.
Recent advances in systemic therapy for HCC
Sorafenib, a multikinase inhibitor with antiangiogenic properties, is the first systemic therapy approved for HCC owing to two positive randomized placebo-controlled phase III trials.4,5 Since 2016, four other antiangiogenic agents, including three multikinase inhibitors and one anti-vascular endothelial growth factor receptor monoclonal antibody, have been demonstrated to provide survival benefits for patients with advanced HCC in phase III clinical trials (Table 1).6–9 As a result, the Food and Drug Administration of the United States (US-FDA) and the regulatory agencies of multiple countries approved lenvatinib as a first-line systemic therapy for HCC, and approved regorafenib and cabozantinib for patients with HCC who have been previously treated with sorafenib.
 | Table 1 Systemic therapy approved or with positive results in phase III trials for advanced hepatocellular carcinoma |
Moreover, immunotherapy with immune checkpoint inhibitors (ICIs) such as monoclonal antibodies that target programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) has become a new paradigm of treatment for multiple cancers, including HCC. Nivolumab, an anti-PD-1 monoclonal antibody, induced a considerable and durable objective tumor response in patients with advanced HCC in a phase I/II study.10 Pembrolizumab, another anti-PD-1 monoclonal antibody, exhibited a similar response rate (RR) to nivolumab in patients with HCC who had previously been treated with sorafenib in a phase II study.11 Nivolumab and pembrolizumab were granted accelerated approval for the treatment of HCC in 2017 and 2018, respectively, by the US-FDA (Table 1).
Overall, two classes of drugs now exist to treat patients with advanced HCC: one targets tumor angiogenesis and the other targets immunosuppression. Tumor angiogenesis and immune evasion are two major “cancer hallmarks”.12,13 Efforts have been ongoing to develop strategies that combine antiangiogenic therapy with PD-1/PD-L1 inhibitors in patients with advanced HCC.14–16 However, new studies that elucidate the mechanisms underlying angiogenesis promotion and immunosuppression of HCC may help inspire the development of new therapeutic strategies in the future.
Myeloid-derived suppressor cells: dual tumor-supporting effects by promoting immunosuppression and angiogenesis
Myeloid-derived suppressor cells (MDSCs) play a critical role in the immune tumor microenvironment (TME). MDSCs represent a heterogeneous population of immature myeloid cells with various states of differentiation and are distributed in the bone marrow, spleen, peripheral blood, and tumor tissues. MDSCs have various functions that support tumor growth, including the suppression of T and NK cells and the promotion of angiogenesis. Therefore, targeting MDSCs is a potential strategy for enhancing the current treatment of cancers.17
In both humans and mice, MDSCs have two major types: monocytic MDSCs (M-MDSCs) and granulocytic or polymorphonuclear MDSCs (PMN-MDSCs). M-MDSCs share morphological characteristics with monocytes, whereas PMN-MDSCs present morphological characteristics of neutrophils (Table 2). In most cancers, PMD-MDSCs are predominant, representing approximately three-fourths of all MDSCs. In mice, MDSCs are defined using surface markers CD11b and Gr1, and Gr1 has shared epitopes with Ly6C and Ly6G, which are expressed in monocytic cells and granulocytes, respectively. Therefore, M-MDSCs are defined as CD11b+Ly6G−Ly6Chigh cells and PMN-MDSCs are defined as CD11b+Ly6GhighLy6Clow cells in mice. In human, MDSCs generally lack HLA-DR expression, and M-MDSCs are defined as CD11b+CD14+CD15−HLA-DRlow/− cells, whereas PMN-MDSCs are defined as CD11b+CD14−CD15+HLA-DR− or CD11b+CD14−CD66b+ cells. Moreover, an alternative immature cell subset in humans that is defined by a lack of lineage markers, including CD3, CD14, CD15, CD19, CD56, and HLA-DR, and the expression of CD33 is named early-stage MDSCs.18 Recently, Condamine et al identified lectin-type oxidized LDL receptor-1 (LOX-1) as a new marker for PMN-MDSCs in humans, further facilitating the discrimination of human PMN-MDSCs from mature neutrophils.19
 | Table 2 Two major types of myeloid-derived suppressor cells and their immunosuppressive functions |
Immunosuppression is the primary feature of MDSCs. Although MDSCs suppress diverse immune cells, their main immunosuppressive mechanisms are the inhibition of T cells and NK cells and induction of regulatory T cells (Treg). The major factors involved in MDSC-mediated immunosuppression include arginase (ARG1), inducible nitric oxide synthase (iNOS), reactive oxygen species (ROS), TGF-β, IL-10, COX2, indoleamine 2,3-dioxygenase (IDO), and others.17,20 ARG1 depletes L-arginine and leads to cell cycle arrest in the G0-G1 phase of tumor-infiltrating T cells. Depleted L-arginine, increased NO production by iNOS, and increased ROS all result in the downregulation or desensitization of the T-cell receptor and induction of T-cell anergy. IDO degrades L-tryptophan and leads to the suppression of T and NK cells and activation of Treg.20 Studies have also suggested that the immunosuppressive mechanisms of MDSCs may vary at different sites. In peripheral lymphoid structures, PMN-MDSCs have a high level of ROS production and suppress T cells function in an antigen-specific manner. By contrast, M-MDSCs suppress not only antigen-specific but also nonspecific T-cell responses by expressing various factors such as ARG1, NO, TGF-β, and IL-10. In TME, because of hypoxia, ROS levels in PMN-MDSCs are substantially reduced; however, the levels of ARG1 and other factors responsible for nonspecific T-cell suppression are increased.17
Additionally, MDSCs influence TME by inducing tumor angiogenesis through the production of several angiogenic factors and vascular-modulating enzymes.21 For example, Bv8 (bombina variegata peptide 8, a homolog of endocrine-gland-derived vascular endothelial growth factor), produced by MDSCs through granulocyte colony-stimulating factor (G-CSF)-dependent STAT3 signaling, was demonstrated to promote angiogenesis and hematopoietic cell mobilization.22 Actually, MDSC accumulation in TME was associated with tumor refractoriness to anti-VEGF treatment; anti-G-CSF therapy or anti-Bv8 therapy could enhance the responsiveness of anti-VEGF treatment.23,24 Moreover, matrix metalloproteinase-9 (MMP-9)-expressing CD11b+ myelomonocytic cells have been shown to be critical for the formation of tumor vasculature. Tumor growth could be inhibited in MMP-9 knockout mice or by the deletion of MMP-9 in CD11b+Gr1+ MDSCs.25,26 In addition, MDSCs could acquire endothelial cell properties in TME and directly incorporate into tumor endothelium.26
Clinical significance of MDSCs in human HCC
Previous studies have demonstrated the role of MDSCs in various chronic liver diseases such as HBV or HCV-related hepatitis, and non-alcoholic fatty liver disease (NAFLD).27–30 MDSCs were shown to inhibit T cells and moderate HBV-related liver damage during viral replication through ARG1-dependent manner.27 MDSCs may protect liver from detrimental necroinflammation, but also contribute to persistence of HBV infection.31 HCV infection could induce MDSCs, which suppressed T cells and antiviral NK cell responses via ROS and ARG1.28,29 Moreover, MDSCs accumulated in the livers of NAFLD mice and had strong suppression effect on T cells, which was dependent on NO production by iNOS.30
Clinical studies of MDSCs in HCC have mainly focused on analyzing M-MDSCs in the peripheral blood of patients with HCC, probably because the cryopreservation process may negatively affect PMN-MDSCs.32 Several groups have studied M-MDSCs, defined by CD14+HLA-DR−/low cells, in the peripheral blood mononuclear cells (PBMCs) of patients with HCC. They found that these MDSCs increased in the PBMCs of patients with HCC compared with patients with only hepatitis or cirrhosis and healthy controls.33–35 Moreover, a high frequency of MDSCs in PBMCs has been associated with aggressive tumor features and poor clinical outcomes after hepatectomy, local ablation, or hepatic arterial infusion chemotherapy.34–36 Another report defined MDSCs as CD33+HLA-DRlow/−CD11b+CD14+ cells, and found PD-L1+ MDSCs to be increased in the PBMCs of patients with HCC. In addition, tumor-infiltrating leukocytes contained markedly higher percentages of PD-L1+ MDSCs than liver-infiltrating leukocytes and PBMCs.37 Mechanistically, M-MDSCs isolated from the PBMCs of patients with HCC have been proven to be immunosuppressive, as well as shown to have high ARG1, suppress autologous T cell proliferation, inhibit autologous NK cell cytotoxicity, and induce Tregs when cultured ex vivo.33,38
Other studies have utilized various markers to define MDSCs in circulation in patients with HCC. Kalathil et al measured multiple immunosuppressive factors in HCC, and found that the frequency of CD14−HLA-DR−CD11b+CD33+ MDSCs, Tregs, and PD-1+-exhausted T cells as well as immunosuppressive cytokines levels were increased in the peripheral blood of patients with HCC compared with healthy donors. Combined depletion of MDSCs, Tregs, and PD-1+-exhausted T cells in PBMCs isolated from patients with advanced HCC restored the production of granzyme B by CD8+ T cells in vitro.39 Recently, Nan et al employed a novel marker, LOX-1, to analyze PMN-MDSCs in patients with HCC and determined that LOX-1+CD15+ cells were significantly increased in the PBMCs of patients with HCC compared with patients with hepatitis or cirrhosis and healthy controls. The levels of LOX-1+CD15+ PMN-MDSCs in circulation were associated with those identified in HCC tissues. LOX-1+CD15+ PMN-MDSCs suppressed the proliferation and interferon (IFN)-γ production of T cells in vitro, whereas the LOX-1−CD15+ PMNs did not.40
Table 3 summarizes the results of studies on MDSCs in patients with HCC, emphasizing the immunosuppressive activities and prognostic implications of MDSCs.
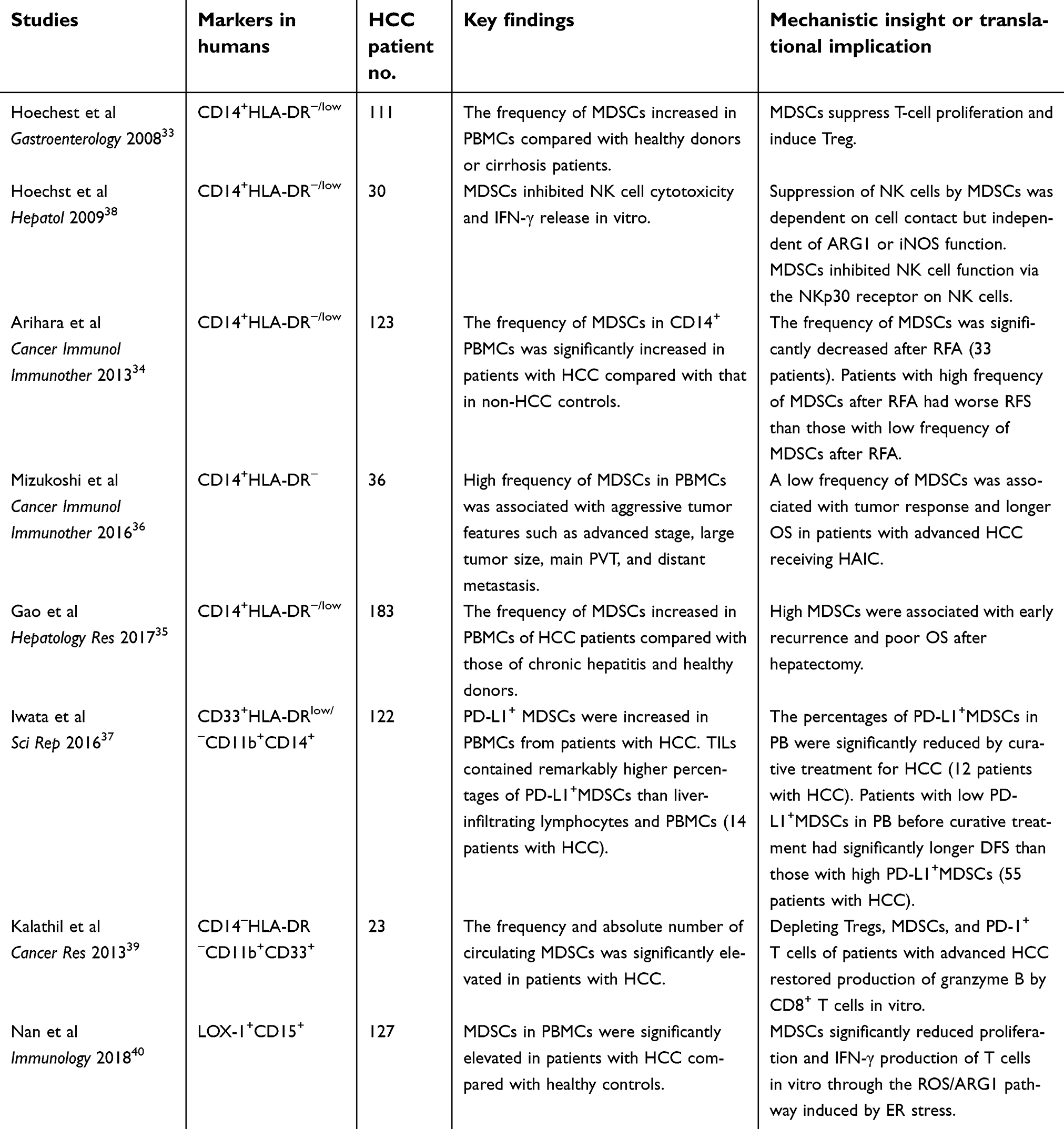 | Table 3 Previous clinical studies on myeloid-derived suppressor cells in hepatocellular carcinoma |
Biological significance of MDSCs in experimental models of HCC
Multiple mouse HCC models have demonstrated that MDSCs are present at an increased level in tumor-bearing mice and accumulate in HCCs. In orthotopic and subcutaneous tumors derived from the mouse HCC cell line RIL-175, MDSCs rapidly expanded in the liver, spleen, and blood, whereas in slow-growing diethylnitrosamine-induced HCC and MYC-expressing spontaneous HCC models, MDSCs were increased in more advanced stages.41 Some studies using transplantable HCC models have demonstrated that MDSCs not only inhibit T and NK cells but also suppress the T cell stimulating activity of dendritic cells and alter Kupffer cell function.42,43 Collectively, these results illustrate the roles of MDSCs playing in the development and progression of mouse HCCs.
Other preclinical studies have explored the significance of MDSCs in TME of mouse HCCs while investigating the therapeutic effect of sorafenib. Chen et al demonstrated that sorafenib-induced tumor hypoxia and stroma-derived factor 1 alpha (SDF1-α) expression, which subsequently induced CD11b+Gr1+ MDSC-infiltration in an HCA-1 orthotopic mouse liver cancer model. Furthermore, CD11b+Gr1+ MDSCs mediated the resistance of sorafenib in liver tumors by promoting hepatic stellate cell differentiation and survival and inducing tumor fibrosis. Inhibiting C-X-C receptor type 4 (CXCR4), the receptor of SDF1-α, or targeting Gr-1 improved the therapeutic effect of sorafenib by reducing the growth of mouse HCCs.44 Our group studied another orthotopic HCC model using BNL mouse liver cancer cells and found that tumor-infiltrating Ly6G+ PMN-MDSCs increased in mouse HCCs treated with sorafenib. Ly6G+ MDSCs suppressed T cell proliferation, induced IL-10 or TGF-β-expressing CD4+ T cells, and downregulated the cytotoxic activity of CD8+ T cells. Multiple proinflammatory and proangiogenic factors, including G-CSF, stroma-derived factor (SDF), TGF-β, tumor necrosis factor (TNF)-α, VEGF, IL-1β, and IL-6 were found to be increased in sorafenib-treated mouse orthotopic liver tumors. Targeting MDSCs with anti-Ly6G or anti-IL-6 antibody significantly reduced the frequency of Ly6G+ MDSCs in orthotopic liver tumors, enhanced T cell proliferation, and improved the therapeutic effect of sorafenib.45
Recent studies have investigated the roles of MDSCs in the efficacy of ICIs in mouse HCC models. Chiu et al studied multiple orthotopic mouse HCC models and found tumor hypoxia induced the ectoenzyme, ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2), by stabilizing HIF-1 in cancer cells. ENTPD2 supported the maintenance of MDSCs, and targeting ENTPD2 inhibited tumor growth and enhanced the efficacy of PD-1/CTLA-4 blockade.46 Zhou et al demonstrated that the overexpression of cell cycle-related kinase (CCRK), a cyclin-dependent kinase family member, increased MDSC accumulation and T cell suppression in liver-specific CCRK-inducible transgenic mice. Targeting CCRK or downstream IL-6 signaling reduced tumor-infiltrating MDSCs and increased intratumoral IFN-γ+TNF-α+CD8+ T cells. Furthermore, the inhibition of CCRK enhanced the antitumor effect of anti-PD-L1 therapy.47
Overall, preclinical studies using mouse liver cancer models not only confirmed the roles of MDSCs in tumor formation and progression but also indicated the effects of MDSCs in the treatment efficacies of sorafenib and ICIs against HCCs (Table 4). Most studies have indicated that targeting MDSCs would improve the efficacy of sorafenib or ICIs—the currently approved therapeutic agents—in HCC.
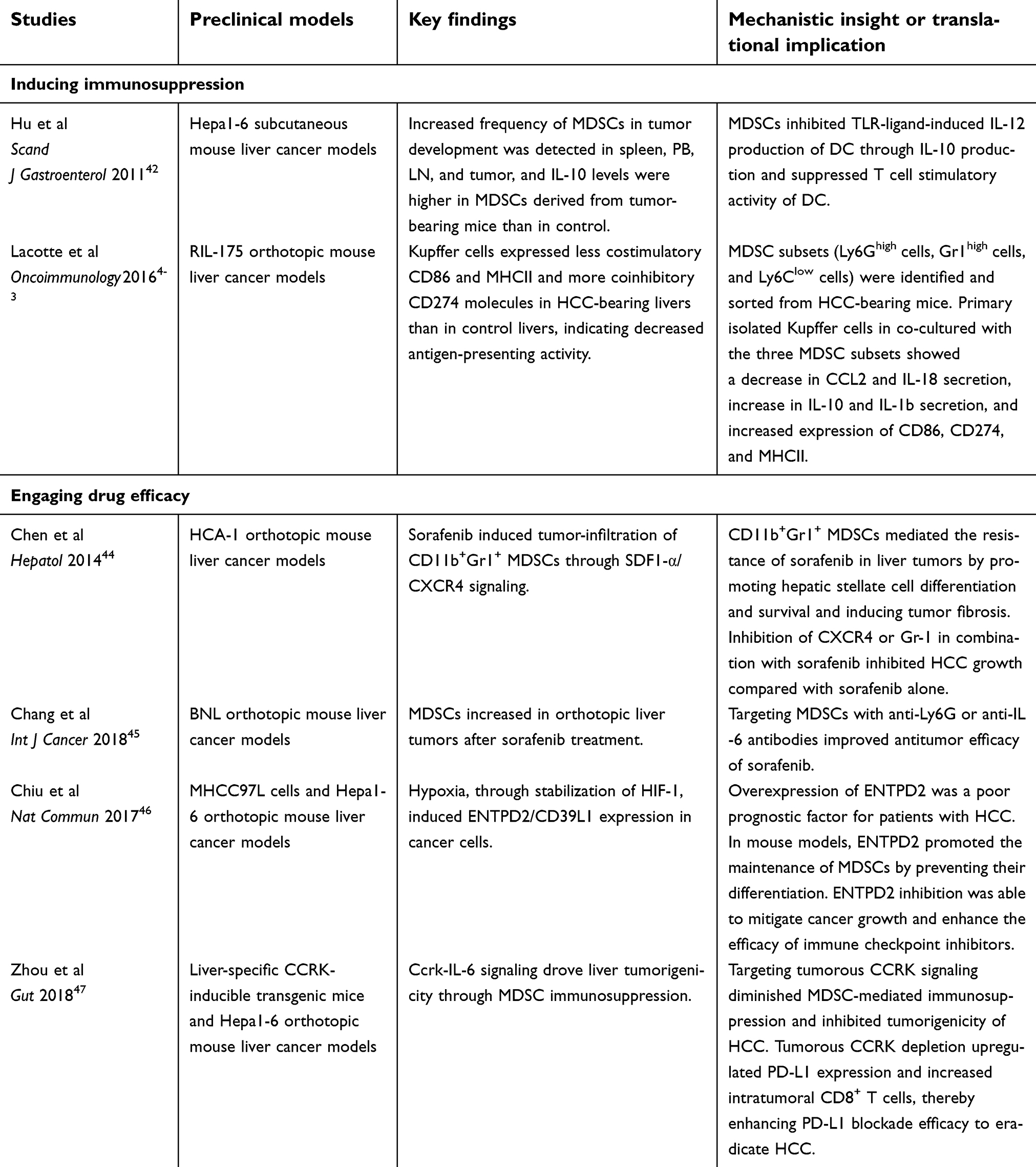 | Table 4 Recent preclinical studies of myeloid-derived suppressor cells in experimental hepatocellular carcinoma models |
Targeting MDSCs in the treatment of human HCC: clinical evidence to date
Numerous preclinical studies have investigated targeting MDSCs as a therapeutic strategy to improve tumor control in experimental animal models. Reversing the protumor effects of MDSCs could be achieved by depleting MDSCs, blocking MDSC trafficking and migration into TME, and inhibiting the immunosuppressive function of MDSCs (Figure 1). The scientific rationales and potential approaches of targeting MDSCs as cancer treatment have been previously reviewed by several groups.17,48–50 Herein, we discuss the clinical data concerning HCC by focusing on agents or approaches that have been directly or indirectly implicated in targeting MDSCs in preclinical studies.
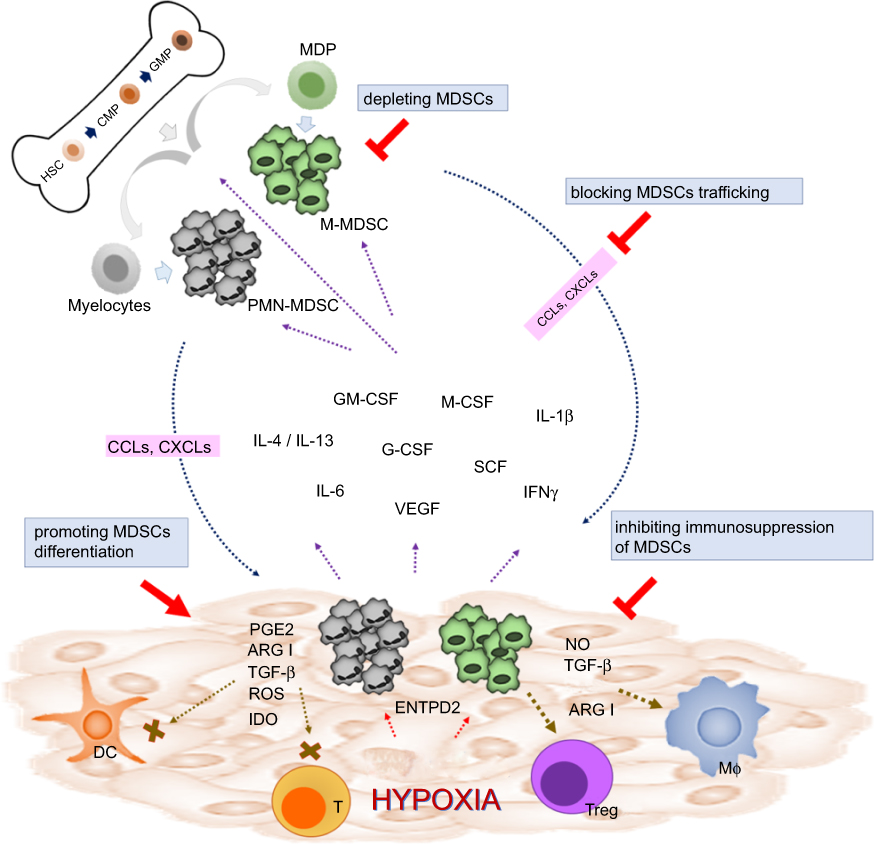 | Figure 1 Strategies of targeting MDSCs in cancers. In physiological condition, HSCs differentiate into CMPs and GMPs, which subsequently become mature granulocytes or monocytes. In pathological condition such as malignancy, multiple tumor-derived factors affect the differentiation of myeloid cells, leading to the generation M-MDSCs and PMN-MDSCs. Both types of MDSCs migrate to the tumor site through the interaction of chemokine receptors and ligands (CCLs or CXCLs). In TME, MDSCs are activated and can support tumor growth by suppressing antitumor response of T cells through various mechanisms such as ARG1, iNOS, ROS, TGF-β, IL-10, and IDO. MDSCs can also promote macrophage polarization and induce Tregs and tolerogenic DCs. Reversing the protumor effects of MDSCs could be achieved by depleting MDSCs, promoting MDSC differentiation, blocking MDSC trafficking and migration into TME, and inhibiting the immunosuppressive function of MDSCs. Abbreviations: HSC, hematopoietic stem cells; CMP, common myeloid progenitor; GMP,granulocyte-macrophage progenitor; MDP, macrophage/dendritic cell progenitors; MDSCs, myeloid-derived suppressor cells; M-MDSCs, monocytic myeloid-derived suppressor cells; PMN-MDSCs, polymorphonuclear myeloid-derived suppressor cells; GM-CSF, granulocyte-macrophage colony-stimulating factor; M-CSF, macrophage colony-stimulating factor; VEGF, vascular endothelial growth factor; G-CSF, granulocyte colony-stimulating factor; SCF, stem cell factor; IFN-γ, interferon-γ; PGE2, prostaglandin E2; ARG1, arginase; IDO, indoleamine 2,3-dioxygenase; NO, nitric oxide; DC, dendritic cell, T, T cell; Treg, regulatory T cell; Mϕ, macrophage; ENTPD2, ectonucleoside triphosphate diphosphohydrolase 2; CCLs, CC chemokine ligands; CXCLs, C-X-C chemokine ligands; TME, tumor microenvironment; iNOS, inducible nitric oxide synthase. |
Depletion of MDSCs
The number of MDSCs of cancer-bearing hosts could be reduced by inhibiting the myelopoiesis of bone marrow and inducing apoptosis of MDSCs; both these effects are commonly induced by chemotherapeutic agents. Indeed, several chemotherapeutic agents, including gemcitabine, doxorubicin, paclitaxel, and 5-fluorouracil (5-FU), have been investigated in preclinical studies and demonstrated to reduce the number of MDSCs in circulation and in TME.51–54 Clinical trials conducted a decade or two ago, most of which were small-scale single-arm phase II trials, demonstrated objective tumor RRs ranging from 0% to 33% for the aforementioned agents in patients with advanced HCC.55–62 However, the successful use of systemic chemotherapy in the treatment of HCC has been hampered by the inevitable toxicities associated with maximum tolerated dose-type chemotherapy and the poor toleration by patients with HCC because of impaired organ function and decreased bone marrow reserves.
Administration of chemotherapeutic agents in a low-dose and uninterrupted manner is referred to as metronomic chemotherapy. Metronomic chemotherapy was originally described as an antiangiogenic chemotherapy63 and has recently been demonstrated to modulate TME, including through an effect on the immune system.64,65 Notably, Servo et al demonstrated in a mouse melanoma model that an ultralow and nontoxic dose of paclitaxel could reduce MDSC numbers, improve immunosuppressive functions, and prolong the survival of tumor-bearing mice.53 Clinical studies of metronomic chemotherapy have been conducted in patients with advanced HCC, mainly using oral 5-FU preparations either alone or in combination of antiangiogenic agents.66–69 Although the treatments were well tolerated by HCC patients, their RRs were only modest. These trials did not investigate whether metronomic chemotherapy affects MDSCs in circulation or TME.
Previous studies have shown that treatment with sunitinib, a multikinase inhibitor with antiangiogenic activity, decreased the number of circulating MDSCs in patients with cancer.70,71 Multiple preclinical studies have demonstrated that sunitinib was able to deplete the number of MDSCs in circulation as well as in tumors.72,73 Another preclinical study demonstrated that cabozantinib reduced intratumoral PMN-MDSCs and enhanced the therapeutic effect of ICIs in a prostate cancer model.74 With regard to the clinical efficacy in advanced HCC, sunitinib failed to provide similar clinical efficacy as sorafenib as a first-line therapy for advanced HCC in a phase III trial,75 whereas cabozantinib demonstrated significant survival benefits compared with a placebo in patients with HCC who had been previously treated with sorafenib and became an approved agent for advanced HCC.8
A recent preclinical study demonstrated that MDSCs could be selectively targeted by TRAIL receptor 2 (TRAIL-R2/DR5) agonist.76 A phase I trial testing the agonistic TRAIL-R2 antibody DS-8273a in patients with advanced cancer, including HCC, found that DS-8273a eliminated MDSCs without affecting mature myeloid or lymphoid cells, and the decrease in MDSCs was associated with progression-free survival (PFS).77 Another randomized phase II study evaluated tigatuzumab, a humanized monoclonal antibody directed against TRAIL-R2, in patients with advanced HCC.78 Although patients treated with tigatuzumab plus sorafenib had numerically longer median PFS and overall survival than those treated with sorafenib alone, the differences did not reach statistical significance. The combination of tigatuzumab with sorafenib was well tolerated in patients with HCC; however, the effect on MDSCs was not investigated.
Another strategy to reduce the number of MDSCs in TME is to facilitate MDSCs differentiating into dendritic cells and macrophages. This MDSC differentiation strategy can be achieved through the inhibition of retinoic acid signaling using all-trans retinoid acid (ATRA).79 ATRA has been determined in clinical trials to downregulate MDSCs, and a significant reduction of MDSCs was observed in patients with renal cell carcinoma and small-cell lung cancer.80,81 A case report by Hungarian investigators detailed how a patient received ATRA treatment for hematological malignancy and experienced significant tumor remission in liver tumors, which were clinically diagnosed as HCC because of moderately elevated alfa-fetoprotein and the presence of portal vein thrombosis.82 In addition, polyprenoid acid, a synthetic retinoid derivative, has been demonstrated to prevent second primary HCCs in patients who underwent surgical resection for HCC.83,84 Polyprenoid acid may work through multiple mechanisms to achieve its chemopreventive effect on HCC.85,86 However, whether it would affect anticancer immunity or MDSCs is unclear.
Blockade of MDSC trafficking
Entry of MDSCs into TME is critical for their main immunosuppressive function to be manifested. Therefore, inhibiting chemokine receptors may reduce the number of MDSCs in TME. Chemokine receptor CCR2 and the interaction of its ligand CCL2 are required not only for the recruitment of M-MDSCs and tumor-associated macrophages but also for their suppressive function.87,88 A CCR2 inhibitor, PF-04136309, has been tested in combination with FOLFIRINOX chemotherapy in a phase Ib clinical trial of patients with advanced pancreatic cancer. Compared with patients who received FOLFIRINOX alone, those who received chemotherapy plus PF-04136309 had a significantly lower ratio of blood to bone marrow CCR2-positive monocytes, demonstrating the effect of CCR2 blockade on the inhibition of the mobilization of bone marrow-derived monocytes into circulation.89
CCR5 is another chemokine receptor that is expressed in many immune cells, and the CCR5–CCR5 ligand axis was found to be critical for the mobilization of PMN-MDSCs.90 Targeting CCR5+ MDSCs has been demonstrated to prevent MDSC migration and suppress tumor growth in preclinical studies.91,92 Recently, a phase Ib/2 clinical trial testing a small-molecule CCR2/5 dual antagonist, BMS-813160, as monotherapy or in combination with chemotherapy or nivolumab in patients with advanced pancreatic or colorectal cancer began to recruit patients.93 CXCR2 is another chemokine receptor expressed on PMN-MDSCs and tumor-associated neutrophils. Blocking CXCR2 has limited the recruitment of PMN-MDSCs and enhanced the efficacy of anti-PD-1 therapy or chemotherapy in preclinical studies.94,95 However, there has been no clinical development of inhibitors of these chemokine receptors in HCC.
Inhibition of the immunosuppressive function of MDSCs
STAT3 is a critical transcription factor for immunosuppressive activity and proliferation of MDSCs. A STAT3 oligonucleotide inhibitor, danvatirsen (AZD9150), was tested in a phase I clinical trial of patients with advanced HCC (NCT01839604); 39 patients with HCC actually received the study agent in the escalation or expansion cohort, and only one patient in the escalation cohort had a partial response. The most common adverse events were transaminase elevation and thrombocytopenia.96 In a recent phase Ib/2 study testing danvatirsen with or without durvalumab, an anti-PD-L1 antibody, in patients with head and neck squamous cell carcinoma (HNSCC), no responses were reported to monotherapy with danvatirsen; however, a relatively high RR of 23% was reported in the danvatirsen plus durvalumab combination arm.97
Histone deacetylase (HDAC) inhibitors may suppress MDSC function by reducing ARG1, iNOS, and COX-2 levels.98 Entinostat, a class I HDAC inhibitor, was demonstrated to inhibit the immunosuppressive function of both PMN-MDSCs and M-MDSCs in lung cancer and renal cancer mouse models. The antitumor effect of PD-1 blockade was also enhanced by adding entinostat in vivo.98 Several clinical trials have tested various HDAC inhibitors in HCC.99,100 Although these agents were generally tolerated, their activities as single-agent appeared to be low because of low RRs and short PFS. Further, the effect of such therapy on MDSCs was not evaluated.
Phosphodiesterase-5 (PDE-5) inhibition downregulated ARG1 and iNOS activities in several preclinical models.101–103 Tadalafil, the FDA-approved PDE-5 inhibitor, has been tested in HCC mouse models; MDSC suppressor function was reversed and the antitumor effect of cytokine-induced killer-cell therapy was enhanced by the addition of tadalafil.103 Tadalafil has been tested in clinical trials of patients with HNSCC and melanoma, but not in patient with HCC. The treatment was well tolerated, MDSCs were suppressed in circulation and tumor tissues, and T cell immunity was determined to be elevated.104–106
Targeting MDSCs in the treatment of HCC: future perspectives
Ensuring that the targeting MDSCs is a clinically useful therapy is challenging because of the following reasons. First, MDSCs are a heterogeneous group of immature myeloid cells that require multiple markers to define and differentiate their subtypes. In humans, no single-specific marker exists for defining MDSCs or their subtypes. This limitation makes direct demonstration of MDSCs in human HCC tumors and tracking their dynamic changes in humans cumbersome. Lack of specific makers also renders the development of “targeted therapy” for specifically targeting MDSCs difficult in humans. Second, although multiple therapeutic strategies focusing on depleting, inhibiting the trafficking, and downregulating the immunosuppressive function of MDSCs have been proposed in preclinical models, most agents have exhibited multiple biological functions, thereby making the true contribution of targeting MDSCs to therapeutic effects less convincing. Third, the clinical data of potential MDSCs-targeting agents, revealed by preclinical studies, suggest that these strategies when administered alone are of limited efficacy against HCC.
Therefore, future studies should focus on identifying specific markers of and developing reliable assays for detecting human MDSCs and their subtypes. Specific markers will be invaluable for helping to develop more specific approaches for targeting MDSCs. Assays that could reliably detect MDSCs in both circulation and tissues, and in freshly prepared and archival samples, are of great importance for confirming the significance of MDSCs in patients with HCC who undergo therapeutic approaches. Furthermore, future studies should focus on developing combined approaches for treating HCC, especially those that incorporate MDSC-targeting therapy with ICIs or antiangiogenic agents, the two approved therapeutic strategies for treating HCC.
In conclusion, MDSCs play critical roles in promoting immunosuppression and angiogenesis, two major “cancer hallmarks” and two crucial therapeutic targets for HCC. The prognostic significance of MDSCs has been demonstrated in multiple clinical studies of patients with HCC. Thus, targeting MDSCs may be a potential therapeutic strategy for treating HCC. Although multiple preclinical studies have demonstrated the promising therapeutic efficacy of targeting MDSCs, additional well-designed clinical studies incorporating strong immunological, molecular, and biochemical research are warranted for the successful development of targeting MDSCs in the treatment of HCC.
Acknowledgments
This study was supported by grants from the Ministry of Science and Technology, Taiwan (MOST 105-2314-B-002-186-MY3, 106-2314-B-002-210, 107-3017-F-002-002, and 108-3017-F-002-004) and the Ministry of Education, Taiwan (NTU-108L901403).
Disclosure
Dr Chih-Hung Hsu reports grants from the Ministry of Science and Technology, Taiwan, during the conduct of the study; personal fees were received from Ono Pharmaceutical, BMS, MSD, and Eli Lilly, outside the submitted work. All authors report no other conflicts on interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Hsiue EH-CH, Lo W-C, Lin -H-H, Hsu C-H, Cheng A-L. Abstract 286: forecast of the incidence and etiology of liver cancer in Taiwan, Japan, United States, and United Kingdom: toward harmonization of East and West. Cancer Res. 2017;77(13 Supplement):286. doi:10.1158/1538-7445.AM2017-286
3. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi:10.1016/S0140-6736(18)30010-2
4. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857
5. Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. doi:10.1016/S1470-2045(08)70285-7
6. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66. doi:10.1016/S0140-6736(16)32453-9
7. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–1173. doi:10.1016/S0140-6736(18)30207-1
8. Abou-Alfa GK, Meyer T, Cheng AL, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med. 2018;379(1):54–63. doi:10.1056/NEJMoa1717002
9.
10. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. doi:10.1016/S0140-6736(17)31046-2.
11. Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19(7):940–952.
12. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol. 2018;15(5):310–324. doi:10.1038/nrclinonc.2018.9
13. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15(5):325–340. doi:10.1038/nrclinonc.2018.29
14. Xu J-M, Zhang Y, Jia R, et al. Anti-programmed death-1 antibody SHR-1210 (S) combined with apatinib (A) for advanced hepatocellular carcinoma (HCC), gastric cancer (GC) or esophagogastric junction (EGJ) cancer refractory to standard therapy: a phase 1 trial. J Clin Oncol. 2018;36(15_suppl):4075. doi:10.1200/JCO.2018.36.15_suppl.4075
15. Ikeda M, Sung MW, Kudo M, et al. A phase 1b trial of lenvatinib (LEN) plus pembrolizumab (PEM) in patients (pts) with unresectable hepatocellular carcinoma (uHCC). J Clin Oncol. 2018;36(15_suppl):4076. doi:10.1200/JCO.2018.36.15_suppl.4076
16. Stein S, Pishvaian MJ, Lee MS, et al. Safety and clinical activity of 1L atezolizumab + bevacizumab in a phase Ib study in hepatocellular carcinoma (HCC). J Clin Oncol. 2018;36(15_suppl):4074. doi:10.1200/JCO.2018.36.15_suppl.4074
17. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3–8. doi:10.1158/2326-6066.CIR-16-0297
18. Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. doi:10.1038/ncomms12150
19. Condamine T, Dominguez GA, Youn JI, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1:2. doi:10.1126/sciimmunol.aah6817
20. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi:10.1038/nri2506
21. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–631. doi:10.1038/nrc2444
22. Qu X, Zhuang G, Yu L, Meng G, Ferrara N. Induction of Bv8 expression by granulocyte colony-stimulating factor in CD11b+Gr1+ cells: key role of Stat3 signaling. J Biol Chem. 2012;287(23):19574–19584. doi:10.1074/jbc.M111.326801
23. Shojaei F, Wu X, Malik AK, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–920. doi:10.1038/nbt1323
24. Shojaei F, Wu X, Qu X, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106(16):6742–6747. doi:10.1073/pnas.0902280106
25. Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13(3):193–205. doi:10.1016/j.ccr.2007.11.032
26. Yang L, DeBusk LM, Fukuda K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–421. doi:10.1016/j.ccr.2004.08.031
27. Lj P, Us G, Quaglia A, et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat Med. 2015;21(6):591–600. doi:10.1038/nm.3856
28. Goh CC, Roggerson KM, Lee HC, Golden-Mason L, Rosen HR, Hahn YS. Hepatitis C virus-induced myeloid-derived suppressor cells suppress NK cell IFN-gamma production by altering cellular metabolism via arginase-1. J Immunol. 2016;196(5):2283–2292. doi:10.4049/jimmunol.1501881
29. Tacke RS, Lee HC, Goh C, et al. Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology. 2012;55(2):343–353. doi:10.1002/hep.24700
30. Yao L, Abe M, Kawasaki K, et al. Characterization of liver monocytic myeloid-derived suppressor cells and their role in a murine model of non-alcoholic fatty liver disease. PLoS One. 2016;11(2):e0149948. doi:10.1371/journal.pone.0149948
31. Dandri M, Bockmann JH. The challenge of protecting without overprotecting: the two sides of myeloid-derived suppressor cells in hepatitis B viral infection. Hepatology. 2016;63(3):1043–1046. doi:10.1002/hep.28385
32. Duffy A, Zhao F, Haile L, et al. Comparative analysis of monocytic and granulocytic myeloid-derived suppressor cell subsets in patients with gastrointestinal malignancies. Cancer Immunol Immunother. 2013;62(2):299–307. doi:10.1007/s00262-012-1332-3
33. Hoechst B, Ormandy LA, Ballmaier M, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234–243. doi:10.1053/j.gastro.2008.03.020
34. Arihara F, Mizukoshi E, Kitahara M, et al. Increase in CD14+HLA-DR-/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother. 2013;62(8):1421–1430. doi:10.1007/s00262-013-1447-1
35. Gao XH, Tian L, Wu J, et al. Circulating CD14(+) HLA-DR(-/low) myeloid-derived suppressor cells predicted early recurrence of hepatocellular carcinoma after surgery. Hepatol Res. 2017;47(10):1061–1071. doi:10.1111/hepr.12831
36. Mizukoshi E, Yamashita T, Arai K, et al. Myeloid-derived suppressor cells correlate with patient outcomes in hepatic arterial infusion chemotherapy for hepatocellular carcinoma. Cancer Immunol Immunother. 2016;65(6):715–725. doi:10.1007/s00262-016-1837-2
37. Iwata T, Kondo Y, Kimura O, et al. PD-L1(+)MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci Rep. 2016;6:39296. doi:10.1038/srep39296
38. Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799–807. doi:10.1002/hep.23054
39. Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 2013;73(8):2435–2444. doi:10.1158/0008-5472.CAN-12-3381
40. Nan J, Xing YF, Hu B, et al. Endoplasmic reticulum stress induced LOX-1(+) CD15(+) polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology. 2018;154(1):144–155. doi:10.1111/imm.12876
41. Kapanadze T, Gamrekelashvili J, Ma C, et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol. 2013;59(5):1007–1013. doi:10.1016/j.jhep.2013.06.010
42. Hu CE, Gan J, Zhang RD, Cheng YR, Huang GJ. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol. 2011;46(2):156–164. doi:10.3109/00365521.2010.516450
43. Lacotte S, Slits F, Orci LA, et al. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology. 2016;5(11):e1234565. doi:10.1080/2162402X.2016.1234565
44. Chen Y, Huang Y, Reiberger T, et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59(4):1435–1447. doi:10.1002/hep.26790
45. Chang CJ, Yang YH, Chiu CJ, et al. Targeting tumor-infiltrating Ly6G(+) myeloid cells improves sorafenib efficacy in mouse orthotopic hepatocellular carcinoma. Int J Cancer. 2018;142(9):1878–1889. doi:10.1002/ijc.31216
46. Chiu DK, Tse AP, Xu IM, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8(1):517. doi:10.1038/s41467-017-00530-7
47. Zhou J, Liu M, Sun H, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018;67(5):931–944. doi:10.1136/gutjnl-2017-314032
48. Fleming V, Hu X, Weber R, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. 2018;9:398. doi:10.3389/fimmu.2018.00398
49. Medina-Echeverz J, Eggert T, Han M, Greten TF. Hepatic myeloid-derived suppressor cells in cancer. Cancer Immunol Immunother. 2015;64(8):931–940. doi:10.1007/s00262-015-1736-y
50. De Sanctis F, Solito S, Ugel S, Molon B, Bronte V, Marigo I. MDSCs in cancer: conceiving new prognostic and therapeutic targets. Biochim Biophys Acta. 2016;1865(1):35–48. doi:10.1016/j.bbcan.2015.08.001
51. Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFbeta-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med. 2016;14(1):282. doi:10.1186/s12967-016-0867-z
52. Alizadeh D, Trad M, Hanke NT, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014;74(1):104–118. doi:10.1158/0008-5472.CAN-13-1545
53. Sevko A, Michels T, Vrohlings M, et al. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J Immunol. 2013;190(5):2464–2471. doi:10.4049/jimmunol.1202781
54. Vincent J, Mignot G, Chalmin F, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–3061. doi:10.1158/0008-5472.CAN-09-3690
55. Al C, Kh Y, Rl F, et al. Biochemical modulation of doxorubicin by high-dose tamoxifen in the treatment of advanced hepatocellular carcinoma. Hepatogastroenterology. 1998;45(24):1955–1960.
56. Yang TS, Lin YC, Chen JS, Wang HM, Wang CH. Phase II study of gemcitabine in patients with advanced hepatocellular carcinoma. Cancer. 2000;89(4):750–756.
57. Guan Z, Wang Y, Maoleekoonpairoj S, et al. Prospective randomised phase II study of gemcitabine at standard or fixed dose rate schedule in unresectable hepatocellular carcinoma. Br J Cancer. 2003;89(10):1865–1869. doi:10.1038/sj.bjc.6601369
58. Chao Y, Chan WK, Birkhofer MJ, et al. Phase II and pharmacokinetic study of paclitaxel therapy for unresectable hepatocellular carcinoma patients. Br J Cancer. 1998;78(1):34–39.
59. Lai CL, Wu PC, Chan GC, Lok AS, Lin HJ. Doxorubicin versus no antitumor therapy in inoperable hepatocellular carcinoma. A prospective randomized trial. Cancer. 1988;62(3):479–483.
60. Yeo W, Mok TS, Zee B, et al. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97(20):1532–1538. doi:10.1093/jnci/dji315
61. Gebbia V, Maiello E, Serravezza G, et al. 5-Fluorouracil plus high dose levofolinic acid and oral hydroxyurea for the treatment of primary hepatocellular carcinomas: results of a phase II multicenter study of the Southern Italy Oncology Group (G.O.I.M.). Anticancer Res. 1999;19(2B):1407–1410.
62. Patt YZ, Hassan MM, Lozano RD, et al. Phase II trial of systemic continuous fluorouracil and subcutaneous recombinant interferon Alfa-2b for treatment of hepatocellular carcinoma. J Clin Oncol. 2003;21(3):421–427. doi:10.1200/JCO.2003.10.103
63. Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4(6):423–436. doi:10.1038/nrc1369
64. Chan TS, Hsu CC, Pai VC, et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J Exp Med. 2016;213(13):2967–2988. doi:10.1084/jem.20151665
65. Andre N, Tsai K, Carre M, Pasquier E. Metronomic chemotherapy: direct targeting of cancer cells after all? Trends Cancer. 2017;3(5):319–325. doi:10.1016/j.trecan.2017.03.011
66. Abdel-Rahman O, Abdel-Wahab M, Shaker M, Abdel-Wahab S, Elbassiony M, Ellithy M. Sorafenib versus capecitabine in the management of advanced hepatocellular carcinoma. Med Oncol. 2013;30(3):655. doi:10.1007/s12032-013-0655-z
67. Hsu CH, Shen YC, Lin ZZ, et al. Phase II study of combining sorafenib with metronomic tegafur/uracil for advanced hepatocellular carcinoma. J Hepatol. 2010;53(1):126–131. doi:10.1016/j.jhep.2010.01.035
68. Hsu CH, Yang TS, Hsu C, et al. Efficacy and tolerability of bevacizumab plus capecitabine as first-line therapy in patients with advanced hepatocellular carcinoma. Br J Cancer. 2010;102(6):981–986. doi:10.1038/sj.bjc.6605580
69. Shao YY, Lin ZZ, Hsu C, et al. Efficacy, safety, and potential biomarkers of thalidomide plus metronomic chemotherapy for advanced hepatocellular carcinoma. Oncology. 2012;82(1):59–66. doi:10.1159/000336126
70. Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. doi:10.1158/1078-0432.CCR-08-1332
71. Chen HM, Ma G, Gildener-Leapman N, et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res. 2015;21(18):4073–4085. doi:10.1158/1078-0432.CCR-14-2742
72. Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology. 2015;4(3):e989764. doi:10.1080/2162402X.2015.1008371
73. Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69(6):2514–2522. doi:10.1158/0008-5472.CAN-08-4709
74. Lu X, Horner JW, Paul E, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543(7647):728–732. doi:10.1038/nature21676
75. Cheng AL, Kang YK, Lin DY, et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol. 2013;31(32):4067–4075. doi:10.1200/JCO.2012.45.8372
76. Condamine T, Kumar V, Ramachandran IR, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. 2014;124(6):2626–2639. doi:10.1172/JCI74056
77. Dominguez GA, Condamine T, Mony S, et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin Cancer Res. 2017;23(12):2942–2950. doi:10.1158/1078-0432.CCR-16-1784
78. Cheng AL, Kang YK, He AR, et al. Safety and efficacy of tigatuzumab plus sorafenib as first-line therapy in subjects with advanced hepatocellular carcinoma: a phase 2 randomized study. J Hepatol. 2015;63(4):896–904. doi:10.1016/j.jhep.2015.06.001
79. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67(22):11021–11028. doi:10.1158/0008-5472.CAN-07-2593
80. Mirza N, Fishman M, Fricke I, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66(18):9299–9307. doi:10.1158/0008-5472.CAN-06-1690
81. Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother. 2013;62(5):909–918. doi:10.1007/s00262-013-1396-8
82. Egyed M, Nyaradi A, Boros B, et al. [Successful treatment of hepatocellular carcinoma with All-trans-retinoic acid]. Orv Hetil. 1998;139(14):811–812.
83. Muto Y, Moriwaki H, Ninomiya M, et al. Prevention of second primary tumors by an acyclic retinoid, polyprenoic acid, in patients with hepatocellular carcinoma. Hepatoma Prevention Study Group. N Engl J Med. 1996;334(24):1561–1567. doi:10.1056/NEJM199606133342402
84. Muto Y, Moriwaki H, Saito A. Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. N Engl J Med. 1999;340(13):1046–1047. doi:10.1056/NEJM199904013401315
85. Takai K, Okuno M, Yasuda I, et al. Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. Updated analysis of the long-term follow-up data. Intervirology. 2005;48(1):39–45.
86. Shimizu M, Imai K, Takai K, Moriwaki H. Role of acyclic retinoid in the chemoprevention of hepatocellular carcinoma: basic aspects, clinical applications, and future prospects. Curr Cancer Drug Targets. 2012;12(9):1119–1128.
87. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125(9):3365–3376.
88. Lesokhin AM, Hohl TM, Kitano S, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012;72(4):876–886.
89. Nywening TM, Wang-Gillam A, Sanford DE, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–662.
90. Karin N, Razon H. The role of CCR5 in directing the mobilization and biological function of CD11b(+)Gr1(+)Ly6C(low) polymorphonuclear myeloid cells in cancer. Cancer Immunol Immunother. 2018;67(12):1949–1953.
91. Blattner C, Fleming V, Weber R, et al. CCR5(+) myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. 2018;78(1):157–167.
92. Hawila E, Razon H, Wildbaum G, et al. CCR5 directs the mobilization of CD11b(+)Gr1(+)Ly6C(low) polymorphonuclear myeloid cells from the bone marrow to the blood to support tumor development. Cell Rep. 2017;21(8):2212–2222.
93. Le D, Gutierrez ME, Saleh M, et al. Abstract CT124: a phase Ib/II study of BMS-813160, a CC chemokine receptor (CCR) 2/5 dual antagonist, in combination with chemotherapy or nivolumab in patients (pts) with advanced pancreatic or colorectal cancer. Cancer Res. 2018;78(13 Supplement):CT124–CT124.
94. Highfill SL, Cui Y, Giles AJ, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6(237):237ra267.
95. Nywening TM, Belt BA, Cullinan DR, et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67(6):1112–1123.
96.
97. Cohen EEW, Harrington KJ, Hong DS, et al. 1044OA phase Ib/II study (SCORES) of durvalumab (D) plus danvatirsen (DAN; AZD9150) or AZD5069 (CX2i) in advanced solid malignancies and recurrent/metastatic head and neck squamous cell carcinoma (RM-HNSCC): updated results. Ann Oncol. 2018;29(suppl_8):mdy287.
98. Orillion A, Hashimoto A, Damayanti N, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. 2017;23(17):5187–5201.
99. Yeo W, Chung HC, Chan SL, et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: a multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J Clin Oncol. 2012;30(27):3361–3367.
100. Bitzer M, Horger M, Giannini EG, et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma – the SHELTER study. J Hepatol. 2016;65(2):280–288. doi:10.1016/j.jhep.2016.02.043
101. Serafini P, Meckel K, Kelso M, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203(12):2691–2702. doi:10.1084/jem.20061104
102. Lin S, Wang J, Wang L, et al. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am J Cancer Res. 2017;7(1):41–52.
103. Yu SJ, Ma C, Heinrich B, et al. Targeting the crosstalk between cytokine-induced killer cells and myeloid-derived suppressor cells in hepatocellular carcinoma. J Hepatol. 2018. Epub ahead of print. doi:10.1016/j.jhep.2018.10.040
104. Weed DT, Vella JL, Reis IM, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21(1):39–48. doi:10.1158/1078-0432.CCR-14-1711
105. Califano JA, Khan Z, Noonan KA, et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21(1):30–38. doi:10.1158/1078-0432.CCR-14-1716
106. Hassel JC, Jiang H, Bender C, et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology. 2017;6(9):e1326440. doi:10.1080/2162402X.2017.1326440
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.