")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Targeting Indoleamine Dioxygenase and Tryptophan Dioxygenase in Cancer Immunotherapy: Clinical Progress and Challenges
Authors Peng X, Zhao Z, Liu L, Bai L, Tong R, Yang H, Zhong L
Received 7 May 2022
Accepted for publication 3 August 2022
Published 8 August 2022 Volume 2022:16 Pages 2639—2657
DOI https://doi.org/10.2147/DDDT.S373780
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Xuerun Peng,1,* Zhipeng Zhao,1,* Liwen Liu,2 Lan Bai,1 Rongsheng Tong,1 Hao Yang,3 Lei Zhong1
1Department of Pharmacy, Personalized Drug Therapy Key Laboratory of Sichuan Province, Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, Sichuan, 610072, People’s Republic of China; 2Department of Obstetrics and Gynecology, Fengrun District People’s Hospital, Tangshan, Hebei, 063000, People’s Republic of China; 3POWERCHINA Chengdu Engineering Corporation Limited, Chengdu, Sichuan, 610072, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lei Zhong, Department of Pharmacy, Personalized Drug Therapy Key Laboratory of Sichuan Province, Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, Sichuan, 610072, People’s Republic of China, Email [email protected] Hao Yang, POWERCHINA Chengdu Engineering Corporation Limited, Chengdu, 610072, Sichuan, People’s Republic of China, Email [email protected]
Abstract: Indoleamine 2.3-dioxygenases (IDO1/2) and tryptophan 2.3-dioxygenase (TDO) are the initial and rate-limiting enzymes in tryptophan metabolism, which play an essential role in mediating immunosuppression in tumor microenvironment. Accumulating evidence has indicated that both IDO1 and TDO are highly expressed in many malignant tumors, and their expression is generally associated with reduced tumor-infiltrating immune cells, increased regulatory T-cell infiltration, as well as cancer progression and poor prognosis for malignancies. A large number of IDO1 and TDO inhibitors have been screened or synthesized in the last two decades. Thus far, at least 12 antagonists targeting IDO1 and TDO have advanced to clinical trials. In this account, we conducted a comprehensive review of the development of IDO1 and TDO inhibitors in cancer immunotherapy, particularly their clinical research progress, and presented the current challenges and corresponding solutions.
Keywords: indoleamine 2, 3-dioxygenase, tryptophan-2, 3-dioxygenase, tryptophan metabolism, cancer immunotherapy, immune tolerance
Introduction
Cancer immunotherapy was named as one of the top ten scientific breakthroughs in 2013 by Science based on the therapeutic benefits of chimeric antigen receptor (CAR) T cells and immune checkpoint inhibitors. Thereafter, cancer immunotherapy has entered a stage of rapid development, especially therapeutic targeting with checkpoint blockade, the most successful immunotherapeutic intervention for cancer in recent years. Thus far, total 15 immune checkpoint inhibitors have been approved either as monotherapy or in combination across a range of tumor types,1–3 including 1 cytotoxic T-lymphocyte associated protein 4 (CTLA-4) monoclonal antibody, 10 monoclonal antibodies that block programmed cell death protein 1 (PD-1) and 4 that block programmed cell death ligand 1 (PD-L1) (Figure 1). Although checkpoint blockade is broadly effective and is considered a common denominator for cancer treatment, the response rate to most malignancies is limited to 10%–25%.4 Moreover, patients who initially respond to checkpoint blockade may subsequently develop acquired resistance. These limitations indicate the existence of additional tumor immunosuppressive mechanisms that contribute to immune tolerance to immune checkpoint inhibitors.
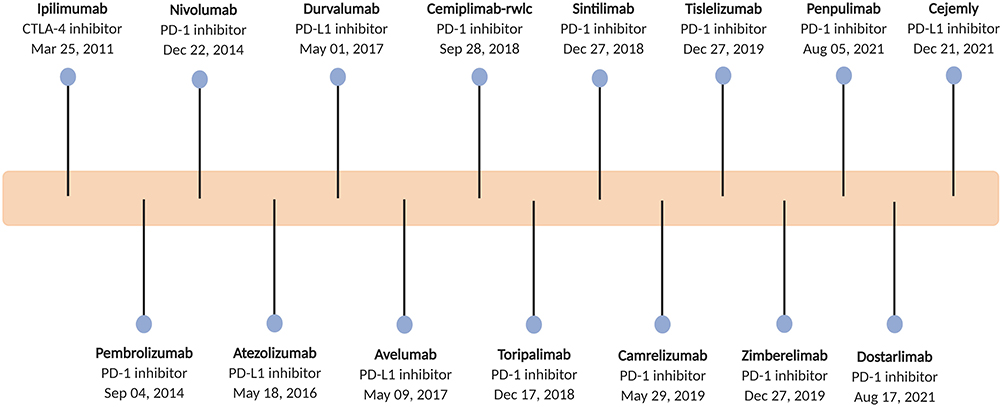 |
Figure 1 Timeline of the approved immune checkpoint inhibitors. |
Accumulating evidence has indicated that L-tryptophan (Trp) metabolism plays an important role in mediating immunosuppression in tumor microenvironment. Trp, available only from dietary intake, is an essential amino acid and vital for cell growth and protein synthesis.5 Trp metabolism is regulated by multiple routes, and more than 95% of free Trp is catabolized through the tryptophan-kynurenine (Trp-Kyn) pathway, which converts Trp into active metabolites, such as L-kynurenine (Kyn), kynurenic acid, 3-hydroxykynurenine, 3-hydroxyanthranilic acid, quinolinic acid and picolinic acid.6 Trp-Kyn metabolic cascade is catalyzed by three enzymes, including indoleamine 2.3-dioxygenase 1 and 2 (IDO1/2), and tryptophan 2.3-dioxygenase (TDO). This enzymatic reaction is the initial and rate-limiting step in Trp metabolism. Both IDO1 and IDO2 genes were identified on chromosome 8 and conserved in mammals.7,8 They have 43% sequence identity at the amino acid level.9 The expression of IDO1 and IDO2 is restricted to eukaryotes, and there are significant differences in expression profile and substrate specificity between them.10 Under physiological state, IDOs are weakly or not constitutively expressed in most tissue cells. For instance, IDO1 is expressed constitutively in a restricted set of normal cells, including the endothelial cells of placenta and lung, the epithelial cells of female genital tract, and the mature dendritic cells.11 When exposed to inflammatory stimuli, such as interferon (IFN-α, IFN-β, or IFN-γ), tumor necrosis factor-α (TNF-α), or interleukins for IDO1 and IFN-γ or interleukin-10 (IL-10) for IDO2,10,12,13 the inducible expression of IDO1 is ubiquitous, whereas IDO2 is expressed primarily in placenta, liver, kidney, and dendritic cells.14 IDO1 plays a pivotal role in immune regulation and the retro-control of immune responses, which are mediated mainly by Trp catabolites. In contrast, IDO2 is less well studied for its low catalytic activity on Trp, and its physiological role remains unclear.14,15 TDO is a tetrameric enzyme broadly distributed in both bacteria and eukaryotes and is highly conserved across distinct species.16,17 TDO expression is selective in the liver.18 It has a different structure from IDOs, and displays higher substrate specificity on L-Trp compared with IDO1, which can catalyze a variety of indoleamines, such as L-Trp, D-Trp, tryptamine, and serotonin.19
IDOs/TDO and Cancer Immune Tolerance
Abnormal constitutive expression of IDO1 has been observed in a variety of cancer types.11,20 IDO1 expression is generally related to reduced tumor-infiltrating immune cells, increased regulatory T-cell infiltration, as well as cancer progression and poor prognosis in many malignancies.21 These characteristics indicated a negative regulation of IDO1 on the recruitment of antitumor immune cells. In an ovarian cancer concerning study, high IDO1 expression was identified in 56.7% of tested cases, and associated with a decreased number of CD8+ tumor-infiltrating lymphocytes as well as impaired progression-free survival and overall survival. Overexpressing IDO1 in ovarian cancer cells did not affect the proliferation, migration, invasion, and chemosensitivity of paclitaxel, but promoted tumor peritoneal dissemination in xenograft models, which could be abrogated by IDO1 inhibitor 1-methyl-tryptophan (1-MT).22 Pflügler et al discovered an immune tolerance mechanism in colorectal cancer (CRC) based on Stat1-dependent expression of IDO1 in Paneth cells. IDO1-positive Paneth cells, discovered in the stem cell niche of intestinal crypts and tumors, deplete Trp and increase local Kyn levels, producing an immune-tolerant microenvironment. Simultaneous loss of IDO1 and Stat1 activity will improve immune cell composition in CRC.23 The expression and function of IDO2 in human tumors are not yet clear. Although IDO2 expression has been observed at high levels in several human malignancies, there are not many tumor types involved, and its role in tumor progression remains far from being understood.24 Moreover, there are two high-prevalent polymorphisms of IDO2 gene, including IDO2 R248W and IDO2 Y359X, which prominently decrease IDO2 activity `in approximately half of the individuals.14,25 These findings, along with its weak catalytic efficiency for Trp, do not yet support IDO2 as a reliable target for cancer immunotherapy. TDO expression was detected in a substantial proportion of human tumors, with the case for 100% of hepatocarcinomas, 50% of melanomas, and 41% of bladder carcinomas, etc.26 In a mouse tumor model, TDO-expressing tumor cells had the ability to prevent their rejection by T lymphocytes, eventually resulting in progressive tumors and mouse death.26 It has also been reported that TDO facilitated the proliferation, migration, and invasion of tumor cells, and high expression of TDO was closely related to malignant progression, unfavorable prognosis, and poor survival of patients with malignant tumors.27,28 In addition, TDO plays a dominant role in elevating Kyn expression in some tumors, which restrains antitumor immune responses and promotes tumor cell survival and motility.29 Taken together, these studies provide the proof-of-concept for IDO1 and TDO as attractive therapeutic targets for cancer immunotherapy.
Trp consumption and Kyn metabolites accumulation regulated by IDOs and TDO create an immunosuppressive microenvironment characterized by decreased effector T (Teff) cells and natural killer (NK) cells, as well as increased functional myeloid-derived suppressor cells (MDSCs) and regulatory T (Treg) cells, which are the primary factors contributing to immune tolerance of tumor cells. Trp metabolism-mediated immunosuppressive mainly involves the following molecular mechanisms (Figure 2). First, Trp consumption results in the accumulation of uncharged Trp-tRNA, which binds to and activates the stress-response kinase general control nonderepressible-2 (GCN2).30 Activated GCN2 induces the cell cycle arrest of Teff cells and promotes differentiation and activity of Treg cells, forming an immunosuppressive microenvironment.30,31 Second, Trp depletion suppresses the activity of immune-regulatory kinases mammalian target of rapamycin complex 1 (mTORC1) and protein kinase C theta (PKC-θ), along with the activation of autophagy, which can limit T-cell function in tumor microenvironment.32,33 Finally, accumulation of Kyn metabolites activates aryl hydrocarbon receptor (AHR), a transcription factor essential for tumor immune regulation. AHR activation could result in the generation of immune-tolerant Treg cells and dendritic cells, as well as the suppression of antitumor immune responses through classical IDO1/TDO-Kyn-AHR pathway.34 Meanwhile, activated AHR also promotes tumor immune tolerance by upregulating PD-1 expression. Blockade of Kyn-AHR can boost the efficacy of antitumor adoptive T cell therapy.35 In addition to Trp metabolism-mediated immunosuppressive mechanisms of IDOs and TDO, enzyme-independent approaches have also been reported in recent years. IDO1 upregulated the expression of complement factor H (CFH) and its isoform factor H-like protein 1 (FHL-1) in glioblastoma cells via its nonenzymic activity. FHL-1 expression markedly reduced tumor-infiltrating CD8+ T cells and increased MDSCs and Treg cells, thereby promoting immune suppression and reducing survival in mice with syngeneic brain tumor.36 Moreover, IDO also functions as a signaling molecule in response to transforming growth factor-β (TGF-β).37 Due to the complexity of tumor heterogeneity and microenvironment, all these mechanisms may act synergistically or differentially in different tumor types, resulting in an IDOs/TDO-mediated immunosuppressive environment.
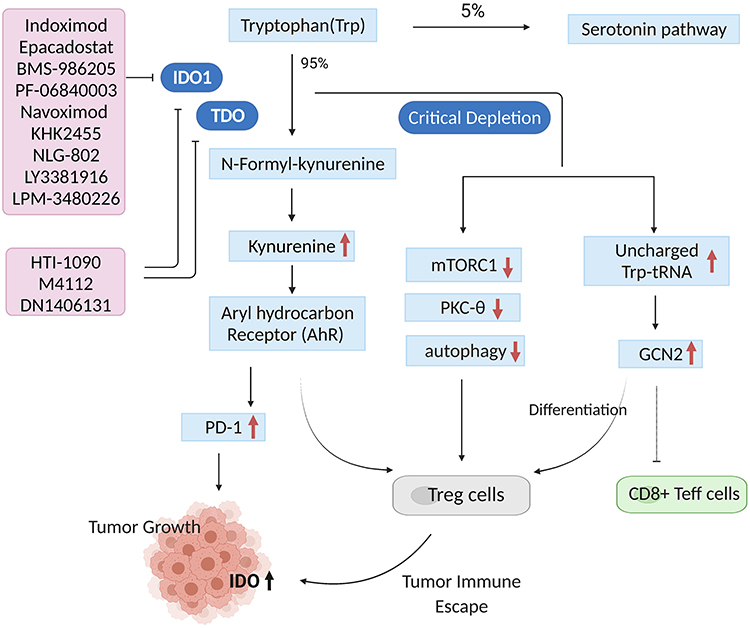 |
Figure 2 Trp metabolism-mediated immunosuppressive mechanisms of IDOs and TDO (created with biorender.com). More than 95% of free Trp is catabolized through the tryptophan-kynurenine (Trp-Kyn) pathway, which leads to decreased Trp levels and increased Kyn levels. Trp depletion results in the accumulation of uncharged Trp-tRNA and activation of the stress-response kinase (GCN2), thus inducing cell cycle arrest of CD8+ Teff cells and promoting differentiation and activity of Treg cells. Trp consumption restrains the activity of immune-regulatory kinases mTORC1 and PKC-θ, along with the activation of autophagy, which will limit T-cell function in tumor microenvironment. Kyn metabolites accumulation activates AHR, and then results in the generation of Treg cells. Meanwhile, activated AHR also promotes tumor immune tolerance by upregulating PD-1 expression. These mechanisms work together to form an immunosuppressive microenvironment. |
Targeting IDOs and TDO in Cancer
In the last two decades, hundreds of IDOs and TDO inhibitors have been screened or synthesized by academic institutions or pharmaceutical industries. These inhibitors could be divided into indole analogues, inhibitors with an imidazole, tetrazole or triazole scaffold, inhibitors with quinone or iminoquinone, hydroxylamine derivatives and others.19,38 To date, at least 12 antagonists targeting IDO1 and TDO have entered clinical trials, including indoximod, epacadostat, BMS-986205, PF-06840003, navoximod, KHK2455, HTI-1090, NLG-802, LY3381916, LPM-3480226, DN1406131, and M4112 (Figure 3).
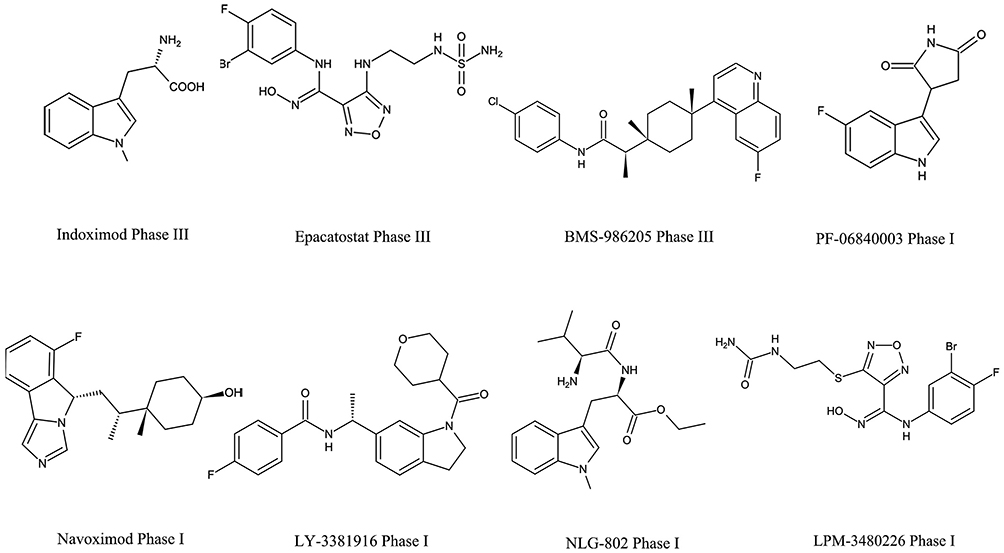 |
Figure 3 The disclosed structure of IDO1 and TDO inhibitors tested in the clinic. |
Indoximod
Indoximod, also called 1-methyl-D-tryptophan (D-1MT) or NLG-8189, is the most representative tryptophan analogue inhibitor developed by Newlink Genetics. It was first studied as a simple racemic compound 1-methyl-D, L-tryptophan with anti-tumor activity in preclinical studies.39 Muller et al then discovered that 1-MT combined with paclitaxel had a significant synergistic effect and elicited regressions of otherwise intractable tumors.40 Nakamura et al investigated the therapeutic potential of indoximod in combination with cyclophosphamide in mouse non‑Hodgkin lymphoma models and also confirmed a conspicuous synergistic interaction.41 In immunodeficient recombination activating gene 1 (RAG1) knockout mice, the anti-tumor activity of 1-MT was lost, indicating that its efficacy is immune-mediated and mainly based on provoking T cell response and attacks.42 Moreover, in IDO-expressing dendritic cells of both human and mouse, the D isomer was prominently more effective in relieving IDO-mediated suppression of T-cell proliferation and in combination with chemotherapy.43
Phase I Studies
Several phase I clinical trials evaluated the safety and toxicity profile of indoximod monotherapy or in combination with chemotherapy in patients with advanced cancer (Table 1). The phase I trial of indoximod monotherapy (NCT00567931) was conducted in 48 patients with solid malignancies. The maximal tolerance dose (MTD) was not reached at the highest dosage (2000 mg twice/day). Pharmacokinetic data showed that indoximod plasma Cmax and AUC plateaued at doses above 1200 mg. C-reactive protein (CRP) level increased at multiple dosage levels, as did autoantibody titers to tumor-associated antigens. The best response was stable disease (SD) >6 months in 5 patients.44 Indoximod combined with docetaxel was assessed in 27 patients with solid tumors (NCT01191216).45 Preliminary data showed the combination was well tolerated. The dose-limited toxicities (DLTs) included grade 3 dehydration, hypotension and mucositis; and grade 5 colitis. The frequent adverse events (AEs) were nausea, infection, hyperglycemia, anemia and fatigue. Moreover, 4 partial responses were observed among 22 evaluable patients.45 Another trial (NCT02835729) assessed indoximod in combination with cytarabine and idarubicin in patients with newly diagnosed acute myeloid leukemia (AML). The most common indoximod-related AEs included abdominal pain, diarrhea, hyperhidrosis, headache, fatigue, nausea, asthenia, and vomiting, which occurred in more than 5% of patients. In the intention-to-treat analysis, 11 of 13 patients (85%) achieved morphologic complete response (CR). Seven of 9 patients (78%) in the per-protocol analysis achieved morphologic CR. These data proved that indoximod had favorable toxicity and high CR rate to the standard of care chemotherapy for newly diagnosed AML.46 In addition, the safety and preliminary efficacy of indoximod plus temozolomide-based therapy in children with newly diagnosed diffuse intrinsic pontine glioma were also evaluated in the NCT02502708 trial.47 It is also the first-in-children clinical trial for indoximod. The most frequent AEs related to indoximod were diarrhea, thrombocytopenia, nausea, fatigue and vomit. The overall survival (OS) was 62% in 12 months and the estimated median OS was 14.5 months.47
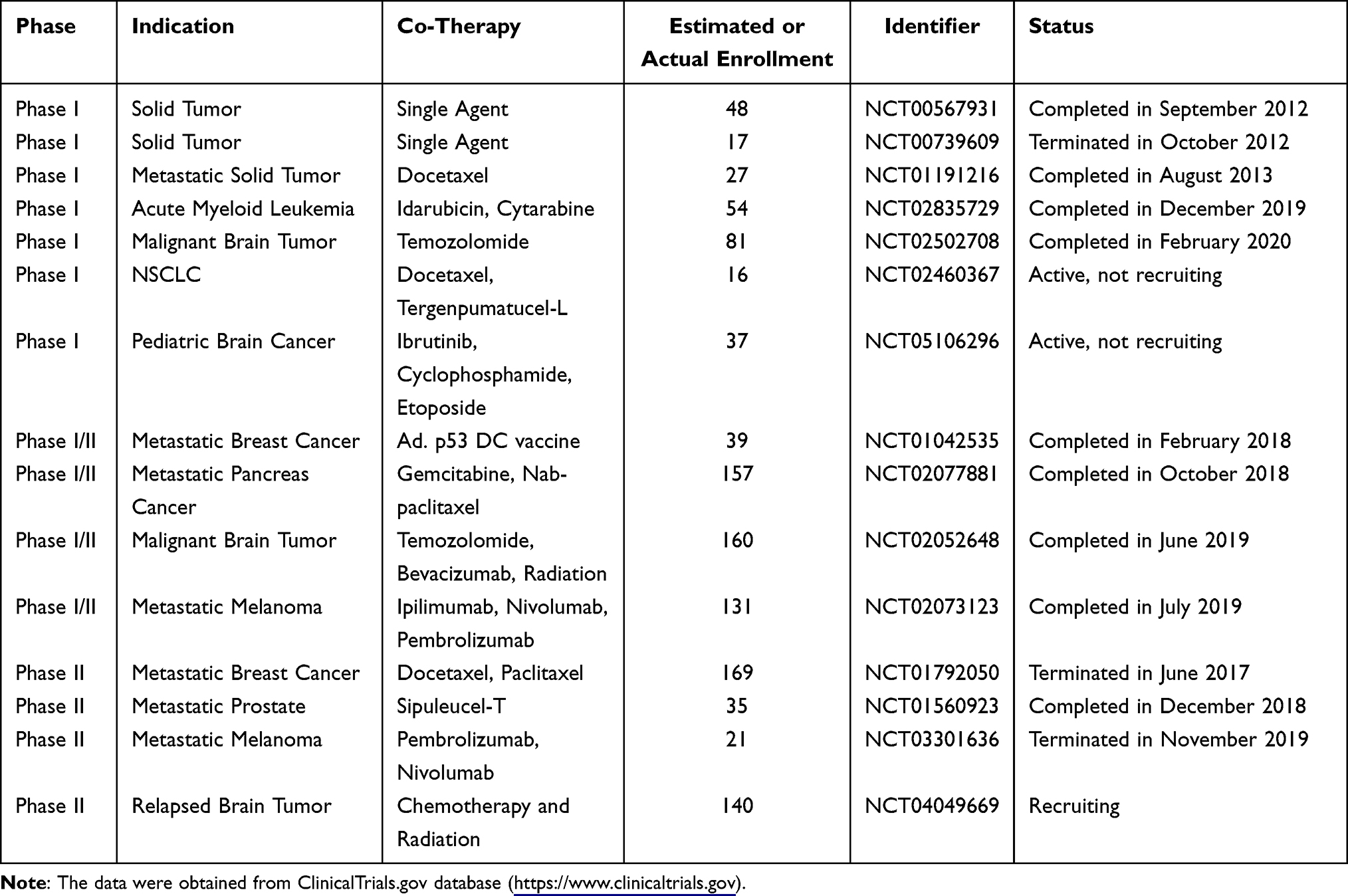 |
Table 1 Clinical Trials Testing Indoximod in Oncological Indications |
Phase I/II Studies
Soliman et al reported the results of an indoximod-related phase I/II trial (NCT01042535) which enrolled 39 patients with metastatic solid tumors.48 The combination of indoximod with Ad.p53-DC vaccination therapy was well tolerated. The attributable AEs were mostly grade 1/2, including fatigue, nausea, transient lymphopenia, anemia, and anorexia, which occurred in about 20% of patients. No DLTs occurred during the study. Of the total study population, immune responses were detected in 7 out of 23 evaluable patients. The median progression-free survival (PFS) and median OS were 13.3 weeks and 20.71 weeks, respectively. 40% of patients had stable disease or better on imaging.48 The preliminary efficacy of indoximod plus immune checkpoint inhibitors for patients with metastatic or stage III/IV melanoma was evaluated in phase I/II study (NCT02073123). A total of 131 patients were enrolled in phase II portion. The combination was well tolerated, and no high-grade autoimmune side effects were observed in this combination. In the 89 efficacy evaluable patients, the objective response rate (ORR) was 51%, including 18 CR patients and 27 partial response (PR) patients. 70% of evaluable population achieved disease control rates (DCR), with the median duration of response of 33 months. The median PFS in these responders was 12.4 months.49 Moreover, indoximod combined with chemotherapeutic agents, such as gemcitabine/nab-paclitaxel and temozolomide, were also assessed in phase I/II trials (NCT02077881, NCT02052648). Both combination regimens were well tolerated. No indoximod-related serious AEs occurred.50,51 The median OS was 10.9 months, and the overall response rate was 46% (48/104) in the NCT02077881 trial.50 The best responses included 1 subject with an ongoing PR at 15 months and SD in 4 subjects lasting 4–11 months in the NCT02052648 trial.51
Phase II Studies
Based on the promising results on safety and responses from the phase I trial in metastatic breast cancer patients, indoximod was evaluated in a randomized placebo-controlled phase II trial (NCT01792050). However, this trial was discontinued in June 2017 because it failed to show any evidence of efficacy. Researchers found that indoximod did not improve median PFS over placebo (6.8 months vs 9.5 months). The OS (19.5 vs 20.6 months) was also not statistically significant.52 Subjects without a heavily pre-treatment could be considered for the next step. Moreover, indoximod in combination with Sipuleucel-T (a therapeutic cancer vaccine) was evaluated in patients with metastatic castration-resistant prostate cancer (NCT01560923). Thirty-five patients completed therapy. No difference in prostate-specific antigen progression nor difference in the primary endpoint of immune response to the immunizing protein PA2024 was observed between the treatment arm and placebo arm.53 In the treatment arm, the median radiographic PFS was 10.3 months, vs 4.1 months in the placebo arm. The combination therapy was well tolerated and had improved radiographic and clinical progression.53 Several other clinical trials evaluating indoximod monotherapy and combination therapy are still active or recruiting subjects (Table 1).
Epacadostat
Epacadostat, initially known as INCB024360, was a highly selective and orally active IDO1 inhibitor developed by Incyte.54 It effectively interfered with Trp metabolism mainly by competing with Trp to bind to the catalytic domain of IDO1 and had high specificity against IDO1 enzyme over IDO2 and TDO, with IC50 value of approximately 10 nM on IDO1.55 In the co-cultures of human allogeneic lymphocytes with dendritic cells (DCs) or tumor cells, IDO inhibition by epacadostat could result in enhanced proliferation of T and NK cells, production of IFN and CD86 positive DCs, and reduced DC apoptosis and conversion to regulatory T (Treg)-like cells.56 In tumor-bearing syngeneic mice, epacadostat inhibited the Kyn level up to 90% in immunocompetent mice instead of immunocompromised mice, indicating that the efficacy of epacadostat relied on functional immunity.57 In murine melanoma models, epacadostat treatment promoted the proliferation of tumor-infiltrating CD8+ T cells and IL-2 production, thereby significantly enhancing T cell activity. Furthermore, it showed synergistic effects in combinatorial immunotherapy with anti-CTLA4 mAb or anti-PD-L1 mAb.58 Based on the impressive preclinical data, a total of 63 clinical trials associated with epacadostat are conducted to evaluate its safety and efficacy in monotherapy or combination therapy in various malignancies (Table 2).
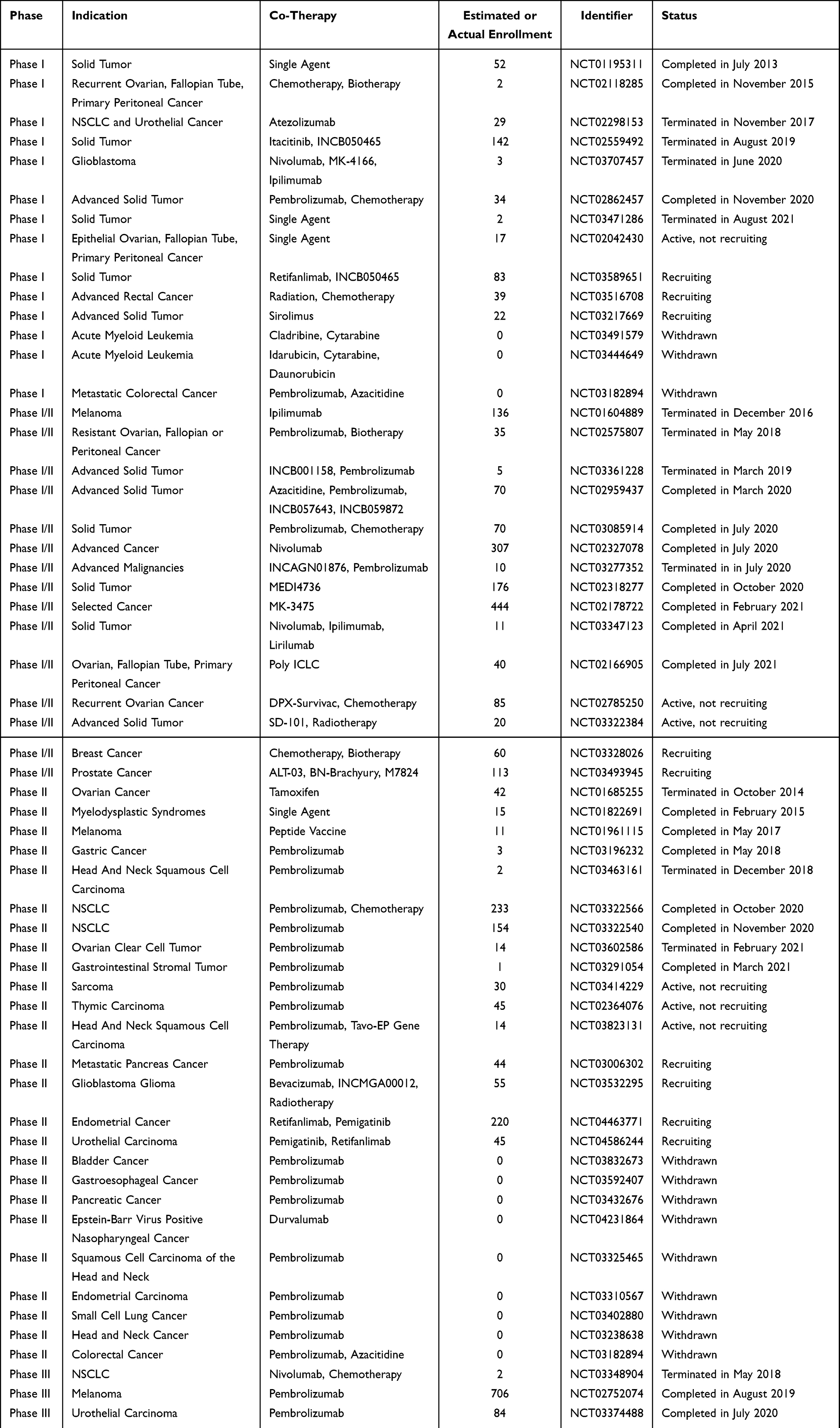 |
Table 2 Clinical Trials Testing Epacadostat in Oncological Indications |
Phase I Studies
The first-in-human phase I trial of epacadostat recruited 52 patients with advanced solid tumors (NCT01195311).59 Two DLTs were observed in this study. The most frequent AEs (>30% of patients) included fatigue, nausea, decreased appetite, vomiting and constipation. Pharmacokinetics (PK) analysis showed that the maximum observed concentration could be reached at approximately 2 hours after administration of epacadostat, and the half-lives ranged from 2.4 hours to 3.9 hours at all doses. The best overall response was SD observed in 18 patients, and 7 of them achieved SD for no less than 16 weeks.59 Although the objective response of epacadostat as monotherapy was not satisfactory, IDO1 inhibition was achieved at tolerable dosages. Epacadostat combined with immune checkpoint inhibitors in patients with advanced solid tumors was reported in two studies (NCT02298153, NCT02862457). In both trials, DLTs were observed in a very handful of subjects, and most of the patients experienced TRAEs.60,61 The PKs of epacadostat and atezolizumab/pembrolizumab were not affected by each other. The efficacy analysis of NCT02862457 trial demonstrated that a reduction in target lesion size from baseline could be detected in 53.3% of patients (8/15). Four patients achieved PR (ORR: 26.7%), and 2 of them had an ongoing response after >200 days.61 Notably, NCT02298153 was terminated in 2017, approximately 2.5 years after initiating the trial, due to the slow recruitment and diverging development strategies of atezolizumab and epacadostat.
Phase I/II Studies
Several phase I/II trials were conducted to assess the safety, tolerability, and efficacy of epacadostat in combination with immune checkpoint inhibitors in select advanced cancers and advanced solid tumors. Among them, epacadostat in combination with anti-CTLA-4 antibody ipilimumab in unresectable or metastatic melanoma (NCT01604889) was terminated based on the evolving immunotherapy landscape, particularly anti-PD-1/anti-PD-L1 antibody-based combination strategies for melanoma therapy.62 Epacadostat plus PD-1 antibody nivolumab (NCT02327078) was well tolerated, and no DLTs were observed.63 In preliminary phase II cohort expansion, 205 patients were enrolled. The DCRs were 70% (16/23), 100% (8/8)/64% (14/22), 28% (5/18)/36% (4/11), and 24% (6/25) in patients with squamous cell carcinoma of the head and neck (SCCHN), melanoma (epacadostat 100/300 mg), ovarian cancer (OVC, epacadostat 100/300 mg), and CRC, respectively. ORRs were 75% (6/8), 11% (2/18)/18% (2/11), and 4% (1/25) in patients with melanoma, OVC (epacadostat 100/300 mg), and CRC, respectively.63 The efficacy outcomes are promising, particularly in SCCHN and melanoma. Updated outcomes specifically aiming at advanced melanoma showed the ORR of 62% and DCR of 78%.64 Naing et al reported the interim results of phase I/II trial (NCT02318277) of epacadostat plus durvalumab in patients with advanced solid tumor. The combination regimen was well tolerated, but no objective responses were observed.65 Another study (NCT02178722) explored the combination of epacadostat with pembrolizumab. Sixty-two participants were enrolled. TRAEs occurred in 52 patients (84%), and the most common TRAEs (>20%) included rash, fatigue, arthralgia, pruritus, and nausea.66 The PK parameters of epacadostat and pembrolizumab were comparable to their monotherapies reported previously.59,67 Moreover, objective responses occurred in 25 patients (8 CRs and 17 PRs), including responders with melanoma, non-small cell lung cancer (NSCLC), urothelial carcinoma (UC), renal cell carcinoma (RCC), endometrial adenocarcinoma and SCCHN.
Phase II Studies
Epacadostat was compared with tamoxifen in ovarian cancer subjects with carbohydrate antigen 125 (CA-125) elevation following complete remission after first-line chemotherapy in a phase II study (NCT01685255). Forty-two women were enrolled, but all of them discontinued the study primarily because of disease progression and AEs.68 In the interim analysis, median PFS was 3.75 months in epacadostat group versus 5.56 months in tamoxifen group. The lack of evidence of superiority for epacadostat together with slow accrual of subjects resulted in termination of this study.68 Another single-center phase II study of epacadostat plus pembrolizumab had been evaluated in patients with advanced sarcoma (NCT03414229). The most common TRAEs were grade 1 or 2, including fatigue, rash and alanine aminotransferase (ALT) elevation. Three patients ceased the therapy due to grade 3 TRAEs. Limited efficacy was observed in evaluable patients, with 1 confirmed PR (3%), 13 SDs (45%), and 15 disease progressions (52%). The median PFS was 8 weeks and the median OS was not available.69 Additionally, multiple related phase II clinical trials have been conducted in recent years, particularly in patients with melanoma, SCCHN, NSCLC and UC. Although some of these trials have been completed, few data have yet been disclosed.
Phase III Studies
A total of 5 phase III trials of epacadostat-related combination therapy have been conducted in patients with melanoma, UC, NSCLC, or SCCHN. Currently, only one phase III study has disclosed the research data. This trial (NCT02752074) aimed at comparing epacadostat/placebo plus pembrolizumab in participants with unresectable stage III or IV melanoma.70 Seven hundred and six subjects were enrolled to receive treatment with epacadostat plus pembrolizumab (n = 354) or placebo plus pembrolizumab (n = 352). Despite favorable safety and tolerability, epacadostat did not improve patient outcomes in the combination therapy. No statistically significant differences were observed in PFS (4.7 vs 4.9), OS (74.4 vs 74.1) at 12 months, and ORR (34% vs 32%) between two groups.70 Several hypotheses have been proposed to explain the discrepancy in efficacy between the early clinical trials for epacadostat and this ECHO-301 trial, including the differences in treatment populations, inappropriate dosage of epacadostat, and incomplete inhibition of intratumoral Kyn.71
BMS-986205
BMS-986205, developed by Bristol Myers Squibb Company, is an orally irreversible and highly selective IDO1 inhibitor. As the only suicidal IDO inhibitor, it was regarded as a best-in-class IDO1 inhibitor and exhibited inhibitory effects via a unique mechanism of binding the heme cofactor site.72 BMS-986205 showed potent cellular activity in multiple cell models. In xenograft models, it effectively reduced kynurenine levels and restored T-cell proliferation with high oral bioavailability and favorable PK characteristics.73 Importantly, BMS-986205 could decrease serum Kyn ≥ 45% even at a low dose of 25 mg, with >60% mean reduction at the 100/200 mg QD in human whole blood.73
Four BMS-986205-related trials have been completed, but only one report is currently available. Luke et al presented the interim results of phase I/II trial evaluating the safety, tolerability, and efficacy of BMS-986205 in combination with nivolumab in advanced bladder cancer (NCT02658890). A total of 516 patients were enrolled in this trial.74 The most common AEs were fatigue and nausea. TRAEs occurred in 57% of patients. 4% of patients ceased treatment due to TRAEs and less than 1% of patients died due to TRAEs. Among the 27 patients with immuno-oncology naive advanced bladder cancer, the ORR was 37%, including 3 CRs and 7 PRs, and the DCR was 56%. The ORR was observed in 50% of patients with PD-L1 expression ≥1% (n = 14) and 30% of patients with PD-L1 expression <1% (n = 10).74 In addition, several trials are conducted to investigate the safety and preliminary effects of BMS-986205 in combination with chemotherapeutic drugs, CTLA-4 inhibitors, tyrosine kinase inhibitors, PARP inhibitors or vaccines in different types of tumors, most of which are recruiting patients (Table 3).
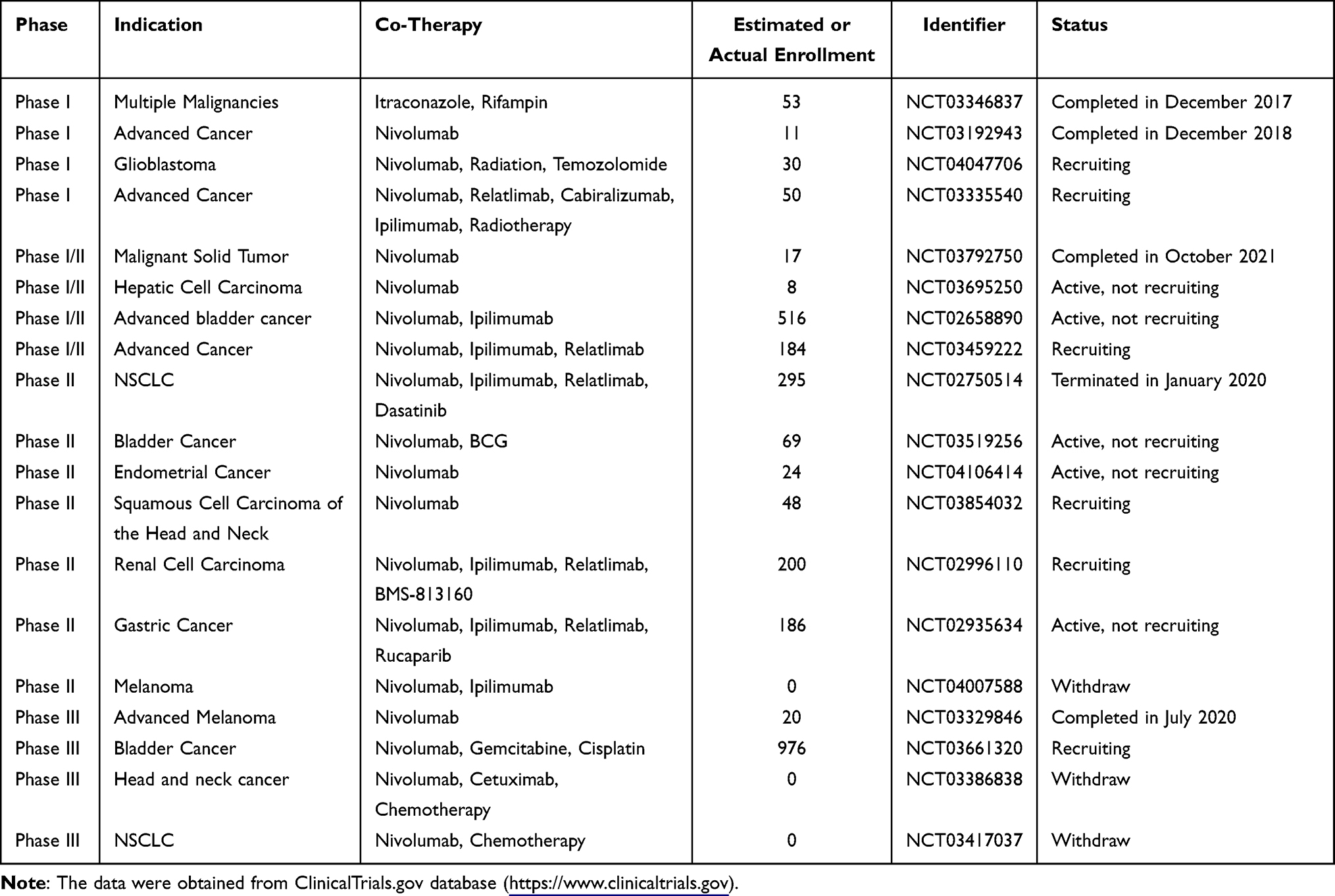 |
Table 3 Clinical Trials Testing BMS-986205 in Oncological Indications |
PF-06840003
PF-06840003/EOS200271 was a highly selective and non-competitive IDO1 inhibitor co-developed by Pfizer and iTeos. The structure of PF-06840003 differed from that of most IDO1 inhibitors. It achieved the inhibitory function without coordinating to heme iron atom but had a novel binding mode.75 Notably, the R-enantiomer of PF-0684003 was more potent than S-enantiomer. Preclinical experiments revealed that R-enantiomer could be converted into S-enantiomer in the blood, resulting in a loss or reduction of inhibitory effect.75 Therefore, PF-06840003 is a racemic mixture of active and inactive enantiomers, which spontaneously epimerize to each other in plasma for maximum efficiency. In preclinical studies, PF-06840003 exerted anti-tumor efficacy by regulating Trp/Kyn balance and T-cell function in vitro and in vivo.76 Gomes et al reported that PF-06840003 reduced the level of kynurenine metabolite by about 80% in syngeneic tumor model and inhibited tumor growth in both monotherapy and in combination with PD-L1 inhibitors. The combination induced a higher proportion of IFN-γ-secreting T cells to improve treatment efficacy.77 Besides, PF-06840003 had a favorable predicted human pharmacokinetic profile, with a bioavailability of 64%. It could pass through the blood–brain barrier (BBB) in rats, suggesting potential therapeutic effects for brain metastatic tumor.77
The safety, tolerability, and preliminary efficacy of PF-06840003 in patients with malignant gliomas have been evaluated in a phase I study (NCT02764151) (Table 4). Increasing doses of PF-06840003 were administered to a total of 17 patients. Serious AEs were observed in 4 patients, one of which was TRAE. The DLT rate was 12.5% at the highest dose of PF-06840003, so the MTD was not reached in this study. The median Tmax ranged from 1.5 to 3.0 hours following PF-06840003 dosing, and the elimination half-life was approximately 2 to 4 hours. Disease control occurred in 47% of patients (8/17), and SD was the best response. The mean duration of SD before progression/discontinuation was 32.1 weeks (12.1–72.3).78 Although PF‑06840003 was well tolerated with pharmacodynamics (PD) effect and clinical benefit in the glioma patients enrolled in this trial, the data were limited by the small sample size, nonrandomized design and low ORR. Further large-scale randomized clinical studies are still needed to support PF-06840003 development.
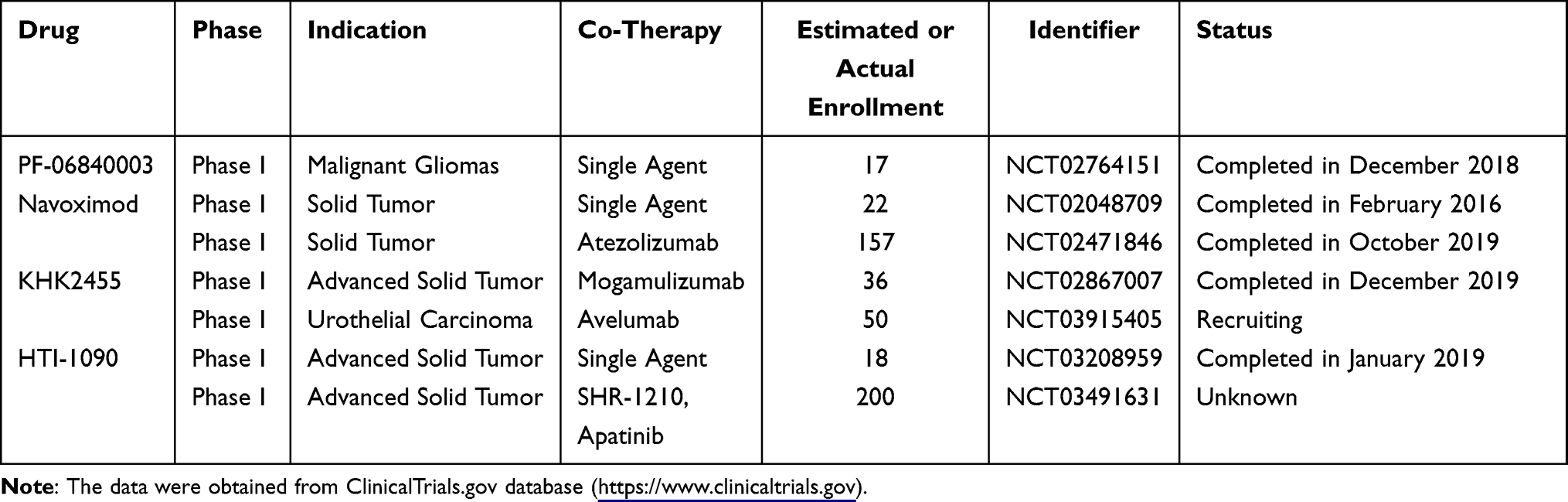 |
Table 4 Clinical Trials of Other IDO1/TDO Inhibitors in Oncological Indications |
Navoximod
Navoximod, also known as GDC-0919, NLG-919 or RG-6078, was developed by Newlink Genetics.79 It is a potent, selective and non-competitive IDO1 inhibitor with an EC50 value of 75 nM on IDO1. Its oral bioavailability was >70%, and a single administration could reduce plasma and tissue Kyn by 50% in mice.80 Spahn found that navoximod attenuated immune suppression in tumors by inhibiting Trp metabolism and had the ability to cross BBB.81 Navoximod combined with PD-L1 inhibitors enhanced the depth and duration of responses compared with single-use of PD-L1 inhibitor.82 Navoximod effectively blocked the IDO-induced T cell inhibition and restored robust T cell responses with an ED50 value of 80 nM in human monocyte-derived DCs and 120 nM in mouse DCs.80,83 In B16F10 mice, navoximod plus pmel 1/vaccine reduced tumor volume by up to 95% compared with the control group receiving pmel-1/vaccine alone.80,83 The benefits observed in preclinical treatment facilitate its progress in clinical studies.
Several phase I trials of navoximod have been completed (Table 4). Nayak-Kapoor et al reported a phase Ia study of navoximod in patients with recurrent solid tumors (NCT02048709).84 MTD was not reached, and DLT occurred in 1 subject with metastatic RCC. The frequent AEs occurring in ≥30% of patients included fatigue, decreased appetite, cough, pruritus, and nausea. PK and PD assays showed a rapid absorbability of navoximod (Tmax: ~1 hour). The average half-life across all doses was approximately 12 hours, and the modulation of plasma Kyn was consistent with the half-life. Of enrolled patients, SD observed in 36% of patients (8/22) was the best response.84 Navoximod combined with atezolizumab was also assessed in a phase I study (NCT02471846). Six dose levels of navoximod (50–1000 mg) plus atezolizumab were administered to patients (n = 157).85 MTD was not identified, and a DLT (grade 3 sepsis syndrome) was observed to be associated with study drugs. Thirty-five patients (22%) experienced grade ≥3 TRAEs. The PKs of two drugs in combination therapy were consistent with those of individual drugs. In the dose-escalation stage, 6 patients (9%) achieved the best response of PR and 11 patients (17%) achieved the SD.85 Of note, it did not benefit patients significantly from adding navoximod to atezolizumab.
KHK2455
KHK2455 was a novel, highly selective and durable effective IDO1 inhibitor. Its structure has not been reported. Currently, only 2 KHK2455-related clinical trials are in progress or have been completed (Table 4). The first clinical trial of KHK2455 (NCT02867007) aimed at evaluating KHK2455 in combination with mogamulizumab (an anti-CCR4 monoclonal antibody) in subjects with advanced solid tumors.86 Thirty-six patients received KHK2455 at escalating dose levels, followed by a combination of mogamulizumab. One patient experienced DLT (Grade 3 gastrointestinal necrosis). The most common TEAEs (>30%) were drug eruption, nausea, infusion-related reaction, fatigue, headache and vomiting. The PK and ex vivo stimulation assays showed that the plasmatic Kyn concentration reduced by 70.5% at 100 mg dose and >95% inhibition in Kyn production was observed at ≥10 mg KHK2455. Six patients achieved durable RECIST disease stabilization for over 6 months, one of whom even lasted for ≥2 years. The median OS was 13.4 months and 30% of individuals survived for ≥2 years.86 Another active phase I trial of KHK2455 plus avelumab is conducted in adult patients with advanced bladder cancer (NCT03915405). It is currently recruiting subjects.
HTI-1090 (SHR9146)
HTI-1090, also referred as SHR9146, is a novel highly potent dual inhibitor against both IDO1 and TDO. It had favorable safety profiles and oral bioavailability in preclinical studies.87 HTI-1090 have been evaluated in monotherapy (NCT03208959) or combination therapy (NCT03491631) for patients with solid tumors (Table 4). The results of the combination therapy have been disclosed recently. Twenty-three eligible patients were treated with SHR9146 (100, 200, 400, 600 mg BID) plus SHR-1210 (200 mg Q2W) with (cohort A)/without (cohort B) apatinib (250 mg QD).88 Two patients experienced DLTs: grade 4 hypercalcemia (with apatinib) and grade 3 rash (without apatinib). The most frequent grade ≥3 TRAEs were hypercalcemia, fatigue and nausea. No fatal AEs occurred. In evaluable patients, the ORR and DCR in cohort A were 21.4% (3/14) and 42.9% (6/14), respectively, compared with 33.3% (3/9) and 77.8% (7/9), respectively, in cohort B.88 These data demonstrated the acceptable safety profile and promising anti-tumor activity of this combination treatment in patients with advanced solid tumors. Further evaluation is needed to validate the safety and efficacy.
Other IDOs/TDO Inhibitors
Several structurally optimized inhibitors targeting IDO1 and TDO have also entered clinical trials, including NLG-802, LY3381916, LPM-3480226, DN1406131, and M4112 (Table 5). NLG802 is a prodrug of indoximod and has improved safety profile and oral bioavailability.89,90 Preclinical data showed that increasing the dose of indoximod above the currently achievable levels resulted in better anti-tumor efficacy; this could be achieved by NLG802 due to the enhanced bioavailability. In a melanoma tumor model, NLG802 significantly promoted the anti-cancer response of tumor-specific pmel-1 T cells.90 A phase I clinical trial (NCT03164603) of NLG802 monotherapy in patients with advanced solid tumors has been completed, but relevant results were not available. LY3381916 is an indoline derivative. As a potent and selective IDO1 inhibitor, LY3381916 inhibited IDO1 activity in cell models, with IC50 values of 7 nM and >20 µM against IDO1 and TDO, respectively.91 The preclinical PK/PD modeling indicated that a once daily dosing of LY3381916 could maintain >90% inhibitory effects for >24 hours. In preclinical tumor models, LY3381916 enhanced the activity of anti-PD-L1 antibody LY3300054, which was related to the improved T cell response. It also had significant central nervous system (CNS) penetration in rodents.91 Based on this study, a phase I clinical trial (NCT03343613) was conducted to explore LY3381916 monotherapy and in combination with LY3300054 in patients with advanced solid tumors. However, this trial was terminated in May 2020 probably due to the limited response of this combination.92 M4112 is a potent and highly selective IDO1 and TDO2 dual inhibitor. It markedly decreased the Kyn/Trp ratio in a dose-dependent manner in vitro, particularly in liver tissue.93 In a phase study, 15 patients with advanced solid malignancies were treated with M4112 (NCT03306420). M4112 monotherapy was well tolerated, but no objective responses were observed. The best response was SD in 9 patients lasting ≥16 weeks.93 This trial was terminated in February 2020 due to the insufficient pharmacodynamic effect. Similar structural modifiers included LPM3480226 and DN1406131, both of whom are IDO1/TDO dual inhibitors. They are now active in phase I clinical trials.
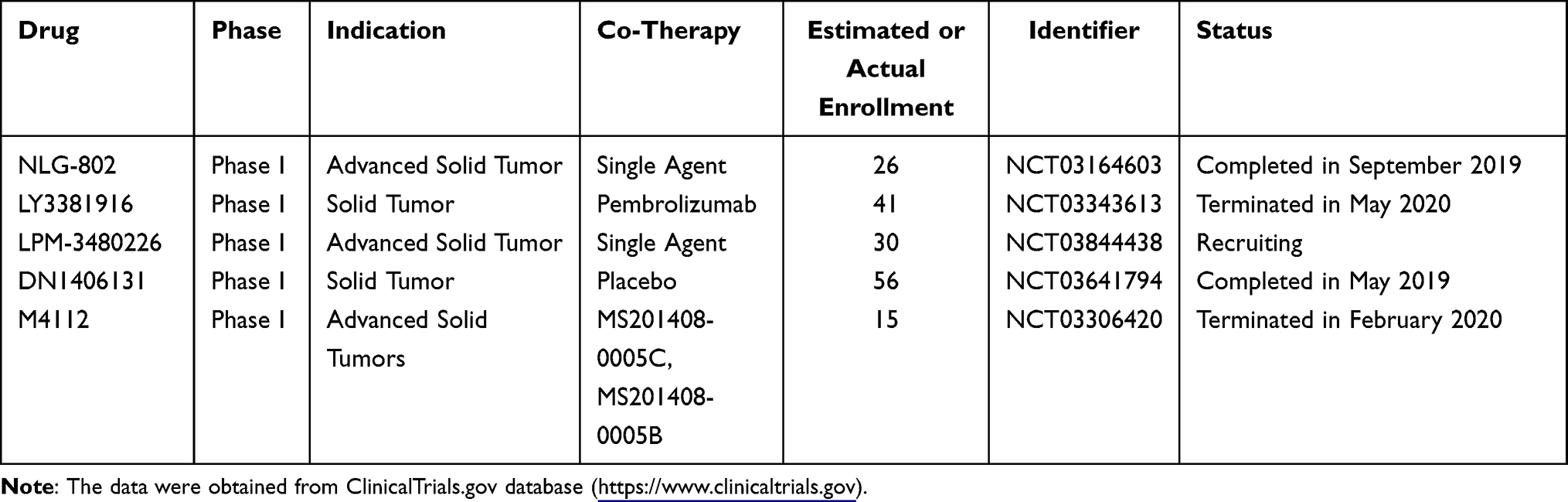 |
Table 5 Clinical Trials of Structurally Optimized Inhibitors Targeting IDO1 and TDO |
In addition to the above IDO1/TDO inhibitors that have advanced to clinical evaluation, there are still a large number of potential IDOs and TDO inhibitors in preclinical and biological testing stages (Supplementary Table 1). For example, Hamilton et al synthesized a series of inhibitors targeting IDO1 and identified IACS-9779 as the hit compound. IACS-9779 inhibited IDO1 activity in HeLa cells with an IC50 value of 1.7 nM. It demonstrated an acceptable safety margin in rodent toxicology and dog cardiovascular studies and was suitable for human evaluation.94 F04, a novel phosphoramidite-containing compound, is an IDO1 and TDO dual target inhibitor, with IC50 values of 94 nM and 2.6 nM on IDO1 and TDO, respectively. In the cellular and mice models, F04 significantly decreased Kyn/Trp ratio and suppressed tumor progression.95 He et al synthesized a series of IDO1/2 dual inhibitors. Among all the tested compounds, 4t was the highly potent IDO1/2 inhibitor, with IC50 values of 28 nM and 144 nM against IDO1 and IDO2, respectively. In CT26 xenograft mouse models, 4t showed a stronger anti-tumor effect than epacadostat.96 Moreover, the antibiotic salinomycin was also reported to be a potent inhibitor of kynurenine synthesis, with IC50 values of 3.36 μM, 4.13 μM and 4.66 μM in MCF-7, HeLa and MDA-MB-231, respectively, in a cell-based biochemical test. Mechanism studies revealed that salinomycin inhibits kynurenine synthesis mainly through suppressing IDO1 expression and its catalytic activity by blockade of JAK/STAT and NF-κB pathways.97,98
Conclusion
Immune escape is one of the pivotal hallmarks of cancer. It has been strongly supported by the impressive efficacy and disease remission achieved in some patients treated with various immune checkpoint inhibitors. In this paper, we present the functions and mechanisms of IDOs and TDO in immune regulation and the research progress of corresponding inhibitors for cancer immunotherapy. Despite the general tolerance and moderate PK properties of IDOs and TDO inhibitors tested in clinical trials, they are seriously challenged by the efficacies, particularly the failure of epacadostat in combination with pembrolizumab for patients with advanced melanoma in the ECHO-301 phase III trial, which increases the uncertainty of the development of such drugs. Currently, in order to cope with this challenge, some strategies should be applied.
The first one is to develop dual-target inhibitors. Tumor microenvironment is characterized by complex immune evasion mechanisms. The immune responses induced by immune checkpoint inhibition, such as PD-1 or PD-L1 blockade, may be weakened or neutralized by IDO1 overexpression.99 Likewise, inhibiting IDO1 alone may be insufficient to restrain Trp metabolism and exert immunotherapeutic effects due to the existence of TDO, which also plays a crucial role in Trp-Kyn metabolic cascade in some malignant tumors. Thus, co-suppression of both IDO1 and TDO is required to get rid of Trp metabolism-mediated immunosuppression in tumor microenvironment and improve treatment efficacy. It is a research trend for the development of agents targeting both IDO1 and TDO.100,101 IDO1 and histone deacetylase (HDAC) dual-target inhibitors were also reported for cancer treatment.102,103 Such inhibitors have advantages of both immunotherapeutic and epigenetic drugs. Meanwhile, HDACs inhibition can benefit immunotherapy via multiple mechanisms.104,105 Thus, co-targeting IDO1 and HDAC may have synergistic effects. In addition to directly target IDO1 or TDO, inhibiting AHR pharmacologically might enhance the anti-tumor efficacy of immune cells, so developing AHR antagonists is also an alternative strategy for its essential modulatory effects in IDO1/TDO-mediated Trp metabolism and subsequent immunosuppression.106 Further assessment of the potential synergistic effects of AHR inhibitors plus immune checkpoint blockade is also warranted.
Second, more efforts should be made to identify biomarkers to help select patients who will benefit from IDO1 or TDO inhibition. IDO1 or TDO expression in tumor samples, as well as the serum Trp and Kyn levels are the most recently reported biomarkers for pharmacodynamic evaluation. However, the immunosuppression effects of IDO1 or TDO in tumor microenvironment do not only rely on its expression, and Trp depletion and Kyn accumulation do not always happen simultaneously. Therefore, their practicality and reliability for patient selection need to be further explored.59,85 Xie et al developed a11 C-l-1MTrp positron emission tomography imaging technology to accurately delineated IDO1 expression in vivo, and found that IDO1 expression in the mesenteric lymph nodes could serve as a promising biomarker for treatment outcomes in cancer immunotherapy.107 The reliability of this predictor is better than that of IDO1 expression in tumor biopsies, which may be affected by post-translational modifications, sampling variables, and heterogeneous expression in tumor samples.107,108 Luke et al identified IFN-γ signature and tumor TDO2 expression as composite biomarkers for predicting the response to BMS-986205 plus nivolumab.109 Neopterin expression, like IDO1, was induced by IFN-ɣ and also found to be a potential biomarker of immune response in cancer therapy.110 In addition, an in-depth understanding of IDOs/TDO function, the mechanisms of immune escape, and the crosstalk between IDOs/TDO and related signal transduction is of utmost importance to discover precise biomarkers for IDO1 and TDO inhibitors.
Furthermore, combination therapy is still a potent regimen to boost the efficacy of IDOs and TDO inhibitors. Although there is a rational mechanism for the combination of IDO1 inhibitors with immune checkpoint therapy, the clinical data are not satisfactory in some malignancies, such as the failure of ECHO-301 trial.111 Nevertheless, many effective combination therapies related to IDO1 or TDO inhibitors have been observed in preclinical studies, including the combination therapy with DNA-damaging agents, molecular targeted drugs, radiotherapy and chemoradiotherapy, etc.112–114 Evidence continues to grow to support these combinations, and some of them are undergoing clinical evaluation (Tables 1–3). Moreover, IDO1/TDO dual-target inhibitor combined with anti-PD-1 or anti-PD-L1 antibody is also an attractive combination that is worthy to be further explored. Finally, some novel technologies, such as proteolysis targeting chimeras (PROTAC) and engineered kynureninase (PEGylated kynureninase), have been adopted for the immunotherapy targeting IDOs/TDO-Trp-Kyn pathway, but much work remains to be done to advance them to the clinic.115,116
In conclusion, all of the clinically available IDOs and TDO antagonists have shown good tolerability either alone or in combination therapies. Although they are currently challenged by the efficacy of some tumors, apparent regulation mechanism and aggregated evidence still strongly support IDO1 and TDO as promising targets for cancer immunotherapy, and there are also many related clinical studies in progress. With an in-depth understanding of tumor immune evasion and the technological innovation of drug research and development, more potent and specific IDOs/TDO inhibitors, more accurate and reliable predictors, and better rationalized therapeutic combinations and trial designs will be discovered or applied, which can substantially boost the development of IDOs and TDO inhibitors.
Acknowledgments
This work was supported by the Basic Research Fund of Science & Technology Department of Sichuan Province (2021YJ0134), the Key Research and Development Program of Science and Technology Department of Sichuan Province (2019YFS0514), the Open Project of Radiation Oncology Key Laboratory of Sichuan Province (2020FSZLX04), the Fundamental Research Funds for the Central Universities (ZYGX2019J106), and the National Key Research and Development Program of China (2020YFC2005500).
Disclosure
Hao Yang is affiliated with POWERCHINA Chengdu Engineering Corporation Limited, Chengdu, 610072, Sichuan, China. The authors report no other conflicts of interest in this work.
References
1. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers. 2020;12(3):E738. doi:10.3390/cancers12030738
2. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21(1):28. doi:10.1186/s12943-021-01489-2
3. Kaplon H, Chenoweth A, Crescioli S, Reichert JM. Antibodies to watch in 2022. MAbs. 2022;14(1):2014296. doi:10.1080/19420862.2021.2014296
4. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37(4):443–455. doi:10.1016/j.ccell.2020.03.017
5. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435–5440. doi:10.1158/0008-5472.CAN-12-0569
6. Mellor AL, Munn DH. Tryptophan catabolism and regulation of adaptive immunity. J Immunol. 2003;170(12):5809–5813. doi:10.4049/jimmunol.170.12.5809
7. Burkin DJ, Kimbro KS, Barr BL, Jones C, Taylor MW, Gupta SL. Localization of the human indoleamine 2,3-dioxygenase (IDO) gene to the pericentromeric region of human chromosome 8. Genomics. 1993;17(1):262–263. doi:10.1006/geno.1993.1319
8. Ball HJ, Sanchez-Perez A, Weiser S, et al. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene. 2007;396(1):203–213. doi:10.1016/j.gene.2007.04.010
9. Prendergast GC, Metz R, Muller AJ, Merlo LMF, Mandik-Nayak L. IDO2 in immunomodulation and autoimmune disease. Front Immunol. 2014;5:585. doi:10.3389/fimmu.2014.00585
10. Pantouris G, Serys M, Yuasa HJ, Ball HJ, Mowat CG. Human indoleamine 2,3-dioxygenase-2 has substrate specificity and inhibition characteristics distinct from those of indoleamine 2,3-dioxygenase-1. Amino Acids. 2014;46(9):2155–2163. doi:10.1007/s00726-014-1766-3
11. Théate I, van Baren N, Pilotte L, et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res. 2015;3(2):161–172. doi:10.1158/2326-6066.CIR-14-0137
12. Banzola I, Mengus C, Wyler S, et al. Expression of indoleamine 2,3-dioxygenase induced by IFN-γ and TNF-α as potential biomarker of prostate cancer progression. Front Immunol. 2018;9:1051. doi:10.3389/fimmu.2018.01051
13. Sun T, Chen XH, Tang ZD, et al. Novel 1-alkyl-tryptophan derivatives downregulate IDO1 and IDO2 mRNA expression induced by interferon-gamma in dendritic cells. Mol Cell Biochem. 2010;342(1–2):29–34. doi:10.1007/s11010-010-0465-y
14. Metz R, Duhadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007;67(15):7082–7087. doi:10.1158/0008-5472.CAN-07-1872
15. Fatokun AA, Hunt NH, Ball HJ. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: characteristics and potential roles in health and disease. Amino Acids. 2013;45(6):1319–1329. doi:10.1007/s00726-013-1602-1
16. Yuasa HJ, Ball HJ, Ho YF, et al. Characterization and evolution of vertebrate indoleamine 2, 3-dioxygenases IDOs from monotremes and marsupials. Comp Biochem Physiol B Biochem Mol Biol. 2009;153(2):137–144. doi:10.1016/j.cbpb.2009.02.002
17. Zhang Y, Kang SA, Mukherjee T, et al. Crystal structure and mechanism of tryptophan 2,3-dioxygenase, a heme enzyme involved in tryptophan catabolism and in quinolinate biosynthesis. Biochemistry. 2007;46(1):145–155. doi:10.1021/bi0620095
18. Knox WE, Mehler AH. The conversion of tryptophan to kynurenine in liver. I. The coupled tryptophan peroxidase-oxidase system forming formylkynurenine. J Biol Chem. 1950;187(1):419–430. doi:10.1016/S0021-9258(19)50967-X
19. Wang XX, Sun SY, Dong QQ, Wu XX, Tang W, Xing YQ. Recent advances in the discovery of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. Medchemcomm. 2019;10(10):1740–1754. doi:10.1039/c9md00208a
20. Uyttenhove C, Pilotte L, Théate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–1274. doi:10.1038/nm934
21. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res. 2011;17(22):6985–6991. doi:10.1158/1078-0432.CCR-11-1331
22. Inaba T, Ino K, Kajiyama H, et al. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol Oncol. 2009;115(2):185–192. doi:10.1016/j.ygyno.2009.07.015
23. Pflügler S, Svinka J, Scharf I, et al. IDO1+ Paneth cells promote immune escape of colorectal cancer. Commun Biol. 2020;3(1):252. doi:10.1038/s42003-020-0989-y
24. Panda A, Ganesan S. Genomic and immunologic correlates of indoleamine 2,3-dioxygenase pathway expression in cancer. Front Genet. 2021;12:706435. doi:10.3389/fgene.2021.706435
25. Witkiewicz AK, Costantino CL, Metz R, et al. Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. J Am Coll Surg. 2009;208(5):781–787. doi:10.1016/j.jamcollsurg.2008.12.018
26. Pilotte L, Larrieu P, Stroobant V, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109(7):2497–2502. doi:10.1073/pnas.1113873109
27. Zhao Y, Sun J, Li Y, et al. Tryptophan 2,3-dioxygenase 2 controls M2 macrophages polarization to promote esophageal squamous cell carcinoma progression via AKT/GSK3β/IL-8 signaling pathway. Acta Pharm Sin B. 2021;11(9):2835–2849. doi:10.1016/j.apsb.2021.03.009
28. Li S, Li L, Wu J, et al. TDO promotes hepatocellular carcinoma progression. Onco Targets Ther. 2020;13:5845–5855. doi:10.2147/OTT.S252929
29. Sumitomo M, Takahara K, Zennami K, et al. Tryptophan 2,3-dioxygenase in tumor cells is associated with resistance to immunotherapy in renal cell carcinoma. Cancer Sci. 2021;112(3):1038–1047. doi:10.1111/cas.14797
30. Munn DH, Sharma MD, Baban B, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–642. doi:10.1016/j.immuni.2005.03.013
31. Sharma MD, Baban B, Chandler P, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117(9):2570–2582. doi:10.1172/JCI31911
32. Metz R, Rust S, DuHadaway JB, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. OncoImmunology. 2012;1(9):1460–1468. doi:10.4161/onci.21716
33. Chuang HC, Lan JL, Chen DY, et al. The kinase GLK controls autoimmunity and NF-κB signaling by activating the kinase PKC-θ in T cells. Nat Immunol. 2011;12(11):1113–1118. doi:10.1038/ni.2121
34. Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy - challenges and opportunities. Trends Pharmacol Sci. 2018;39(3):307–325. doi:10.1016/j.tips.2017.11.007
35. Liu Y, Liang X, Dong W, et al. Tumor-repopulating cells induce PD-1 expression in CD8+ T cells by transferring kynurenine and AhR activation. Cancer Cell. 2018;33(3):480–494.e7. doi:10.1016/j.ccell.2018.02.005
36. Zhai L, Bell A, Ladomersky E, et al. Tumor cell IDO enhances immune suppression and decreases survival independent of tryptophan metabolism in glioblastoma. Clin Cancer Res. 2021;27(23):6514–6528. doi:10.1158/1078-0432.CCR-21-1392
37. Fallarino F, Grohmann U, Puccetti P. Indoleamine 2,3-dioxygenase: from catalyst to signaling function. Eur J Immunol. 2012;42(8):1932–1937. doi:10.1002/eji.201242572
38. Feng X, Liao D, Liu D, Ping A, Li Z, Bian J. Development of indoleamine 2,3-dioxygenase 1 inhibitors for cancer therapy and beyond: a recent perspective. J Med Chem. 2020;63(24):15115–15139. doi:10.1021/acs.jmedchem.0c00925
39. Friberg M, Jennings R, Alsarraj M, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. 2002;101(2):151–155. doi:10.1002/ijc.10645
40. Brincks E, Adams J, Essmann M, et al. Abstract 3753: indoximod modulates AhR-driven transcription of genes that control immune function. Cancer Res. 2018;78:3753. doi:10.1158/1538-7445.AM2018-3753
41. Nakamura N, Hara T, Shimizu M, et al. Effects of indoleamine 2,3-dioxygenase inhibitor in non-Hodgkin lymphoma model mice. Int J Hematol. 2015;102(3):327–334. doi:10.1007/s12185-015-1835-8
42. Davar D, Bahary N. Modulating tumor immunology by inhibiting indoleamine 2,3-dioxygenase (IDO): recent developments and first clinical experiences. Targ Oncol. 2018;13(2):125–140. doi:10.1007/s11523-017-0547-9
43. Hou DY, Muller AJ, Sharma MD, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67(2):792–801. doi:10.1158/0008-5472.CAN-06-2925
44. Soliman HH, Minton SE, Han HS, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget. 2016;7(16):22928.
45. Soliman HH, Jackson E, Neuger T, Dees E, Antonia S. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. 2014;5(18):8136–8146.
46. Emadi A, Holtzman NG, Imran M, et al. Indoximod in combination with idarubicin and cytarabine for upfront treatment of patients with newly diagnosed acute myeloid leukemia (AML). Haematologica. 2017;102:375.
47. Johnson TS, Aguilera D, Al-Basheer A, et al. 101P - results of the NLG2105 phase I trial using the IDO pathway inhibitor indoximod, in combination with radiation and chemotherapy, for children with newly diagnosed DIPG. Annals Oncol. 2019;30:xi38. doi:10.1093/annonc/mdz451.010
48. Soliman H, Khambati F, Han HS, Ismail-Khan R, Antonia S. A phase-1/2 study of adenovirus-p53 transduced dendritic cell vaccine in combination with indoximod in metastatic solid tumors and invasive breast cancer. Oncotarget. 2018;9(11):10110.
49. Zakharia Y, McWilliams RR, Rixe O, et al. Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J Immunother Cancer. 2021;9(6):e002057. doi:10.1136/jitc-2020-002057
50. Bahary N, Wang-Gillam A, Haraldsdottir S, et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus gemcitabine/nab-paclitaxel for the treatment of patients with metastatic pancreas cancer. J Clin Oncol. 2018;36(15_suppl):4015. doi:10.1200/JCO.2018.36.15_suppl.4015
51. Yousef Z, Howard C, Frank M, et al. Imct-21 updates on Phase 1b/2 combination study of the Ido pathway inhibitor indoximod with temozolomide for adult patients with Temozolomide-refractory primary malignant brain tumors. Neuro-Oncology. 2015;17(suppl_5):v112.
52. Mariotti V, Han H, Ismail-Khan R, et al. Effect of taxane chemotherapy with or without indoximod in metastatic breast cancer: a randomized clinical trial. JAMA Oncol. 2021;7(1):61–69. doi:10.1001/jamaoncol.2020.5572
53. Jha GG, Gupta S, Tagawa ST, et al. A phase II randomized, double-blind study of sipuleucel-T followed by IDO pathway inhibitor, indoximod, or placebo in the treatment of patients with metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol. 2017;35(15_suppl):3066. doi:10.1200/JCO.2017.35.15_suppl.3066
54. Komiya T, Huang CH. Updates in the clinical development of epacadostat and other indoleamine 2,3-dioxygenase 1 inhibitors (IDO1) for human cancers. Front Oncol. 2018;8:423. doi:10.3389/fonc.2018.00423
55. Yue EW, Sparks R, Polam P, et al. INCB24360 (Epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med Chem Lett. 2017;8(5):486–491. doi:10.1021/acsmedchemlett.6b00391
56. Liu X, Shin N, Koblish HK, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115(17):3520–3530. doi:10.1182/blood-2009-09-246124
57. Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017;77(24):6795–6811. doi:10.1158/0008-5472.CAN-17-2285
58. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3. doi:10.1186/2051-1426-2-3
59. Beatty GL, O’Dwyer PJ, Clark J, et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-dioxygenase-1 epacadostat (INCB024360) in patients with advanced solid malignancies. Clin Cancer Res. 2017;23(13):3269–3276. doi:10.1158/1078-0432.CCR-16-2272
60. Hellmann MD, Gettinger S, Chow LQM, et al. Phase 1 study of epacadostat in combination with atezolizumab for patients with previously treated advanced nonsmall cell lung cancer. Int J Cancer. 2020;147(7):1963–1969. doi:10.1002/ijc.32951
61. Doi T, Fujiwara Y, Shitara K, et al. The safety and tolerability of epacadostat alone and in combination with pembrolizumab in patients with advanced solid tumors: results from a first-in-Japanese phase I study (KEYNOTE-434). Invest New Drugs. 2021;39(1):152–162. doi:10.1007/s10637-020-00942-1
62. Gibney GT, Hamid O, Lutzky J, et al. Phase 1/2 study of epacadostat in combination with ipilimumab in patients with unresectable or metastatic melanoma. J Immunother Cancer. 2019;7(1):80. doi:10.1186/s40425-019-0562-8
63. Perez R, Riese M, Lewis K, et al. Epacadostat plus nivolumab in patients with advanced solid tumors: preliminary phase I/II results of ECHO-204. J Clin Oncol. 2017;35:3003. doi:10.1200/JCO.2017.35.15_suppl.3003
64. Daud A, Saleh M, Hu J, et al. Epacadostat plus nivolumab for advanced melanoma: updated phase 2 results of the ECHO-204 study. J Clin Oncol. 2018;36:9511. doi:10.1200/JCO.2018.36.15_suppl.9511
65. Naing A, Powderly IIJ, Falchook G, et al. Abstract CT177: epacadostat plus durvalumab in patients with advanced solid tumors: preliminary results of the ongoing, open-label, phase I/II ECHO-203 study. Cancer Res. 2018;78:CT177–CT177. doi:10.1158/1538-7445.AM2018-CT177
66. Mitchell TC, Hamid O, Smith DC, et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: phase i results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037). J Clin Oncol. 2018;36(32):3223–3230. doi:10.1200/JCO.2018.78.9602
67. Longoria TC, Tewari KS. Evaluation of the pharmacokinetics and metabolism of pembrolizumab in the treatment of melanoma. Expert Opin Drug Metab Toxicol. 2016;12(10):1247–1253. doi:10.1080/17425255.2016.1216976
68. Kristeleit R, Davidenko I, Shirinkin V, et al. A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)–only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol Oncol. 2017:146. doi:10.1016/j.ygyno.2017.07.005
69. Kelly C, Chi P, Dickson M, et al. A phase II study of epacadostat and pembrolizumab in patients with advanced sarcoma. J Clin Oncol. 2019;37:11049. doi:10.1200/JCO.2019.37.15_suppl.11049
70. Long GV, Dummer R, Hamid O, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a Phase 3, randomised, double-blind study. Lancet Oncol. 2019;20(8):1083–1097. doi:10.1016/S1470-2045(19)30274-8
71. Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. Clin Cancer Res. 2019;25(5):1462–1471. doi:10.1158/1078-0432.CCR-18-2882
72. Balog A, an LT, Maley D, et al. Preclinical characterization of linrodostat mesylate, a novel, potent, and selective oral indoleamine 2,3-dioxygenase 1 inhibitor. Mol Cancer Ther. 2021;20(3):467–476. doi:10.1158/1535-7163.MCT-20-0251
73. Siu LL, Gelmon K, Chu Q, et al. Abstract CT116: BMS-986205, an optimized indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor, is well tolerated with potent pharmacodynamic (PD) activity, alone and in combination with nivolumab (nivo) in advanced cancers in a phase 1/2a trial. Cancer Res. 2017;77(13 Supplement):CT116–CT116.
74. Luke JJ, Tabernero J, Joshua A, et al. BMS-986205, an indoleamine 2, 3-dioxygenase 1 inhibitor (IDO1i), in combination with nivolumab (nivo): updated safety across all tumor cohorts and efficacy in advanced bladder cancer (advBC). J Clin Oncol. 2019;37(7_suppl):358. doi:10.1200/JCO.2019.37.7_suppl.358
75. Crosignani S, Bingham P, Bottemanne P, et al. Discovery of a novel and selective indoleamine 2,3-dioxygenase (IDO-1) inhibitor 3-(5-fluoro-1H-indol-3-yl) pyrrolidine-2,5-dione (EOS200271/PF-06840003) and its characterization as a potential clinical candidate. J Med Chem. 2017;60:9617–9629.
76. Tumang J, Gomes B, Wythes M, et al. Abstract 4863: PF-06840003: a highly selective IDO-1 inhibitor that shows good in vivo efficacy in combination with immune checkpoint inhibitors. Cancer Res. 2016;76:4863. doi:10.1158/1538-7445.AM2016-4863
77. Gomes B, Driessens G, Bartlett D, et al. Characterization of the selective indoleamine 2,3-dioxygenase-1 (IDO1) catalytic inhibitor EOS200271/PF-06840003 supports IDO1 as a critical resistance mechanism to PD-(L)1 blockade therapy. Mol Cancer Ther. 2018;14:2530–2542.
78. Reardon DA. A phase 1 study of PF-06840003, an oral indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor in patients with recurrent malignant glioma. Invest New Drugs. 2020;12:1784–1795.
79. Kumar S, Waldo JP, Jaipuri FA, et al. Discovery of clinical candidate (1 R,4 r)-4-((R)-2-((S)-6-Fluoro-5 H -imidazo[5,1- a]isoindol-5-yl)-1-hydroxyethyl) cyclohexan-1-ol (Navoximod), a potent and selective inhibitor of indoleamine 2,3-dioxygenase 1. J Med Chem. 2019;62(14):6705–6733. doi:10.1021/acs.jmedchem.9b00662
80. Mautino MR, Jaipuri FA, Waldo J, et al. Abstract 491: NLG919, a novel indoleamine-2,3-dioxygenase (IDO)-pathway inhibitor drug candidate for cancer therapy. Cancer Res. 2013;73(8 Supplement):491. doi:10.1158/1538-7445.AM2013-491
81. Kesarwani P, Kumar P, Kant S, et al. GDC-0919 modulates tryptophan metabolism in glioblastoma and enhances radiation response. Int J Radiat Oncol Biol Phys. 2017;99(2):E601. doi:10.1016/j.ijrobp.2017.06.2048
82. Spahn J, Peng J, Lorenzana E, et al. Improved anti-tumor immunity and efficacy upon combination of the IDO1 inhibitor GDC-0919 with anti-PD-l1 blockade versus anti-PD-l1 alone in preclinical tumor models. J Immunother Cancer. 2015;3(Suppl 2):303. doi:10.1186/2051-1426-3-S2-P303
83. Mautino M, Link C, Vahanian N, et al. Abstract 5023: synergistic antitumor effects of combinatorial immune checkpoint inhibition with anti-PD-1/PD-L antibodies and the IDO pathway inhibitors NLG-919 and indoximod in the context of active immunotherapy. Cancer Res. 2014;74:5023. doi:10.1158/1538-7445.AM2014-5023
84. Nayak-Kapoor A, Hao Z, Sadek R, et al. Phase Ia study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors. J Immunother Cancer. 2018;6(1):61. doi:10.1186/s40425-018-0351-9
85. Jung KH, LoRusso P, Burris H, et al. Phase I study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 inhibitor (atezolizumab) in advanced solid tumors. Clin Cancer Res. 2019;25(11):3220–3228. doi:10.1158/1078-0432.CCR-18-2740
86. Sahebjam S, Muzaffar J, Yap T, et al. 287 Safety and antitumor activity of indoleamine 2,3-dioxygenase 1 (IDO-1) inhibitor KHK2455 in combination with anti-CCR4 monoclonal antibody mogamulizumab in patients with advanced solid tumors. J Immunother Cancer. 2020;8(Suppl 3):A313–A314. doi:10.1136/jitc-2020-SITC2020.0287
87. Tu W, Yang F, Xu G, et al. Discovery of imidazoisoindole derivatives as highly potent and orally active indoleamine-2,3-dioxygenase inhibitors. ACS Med Chem Lett. 2019:10. doi:10.1021/acsmedchemlett.9b00114
88. Cheng Y, Liu Y, Xu J, et al. A phase I study of an IDO inhibitor (SHR9146) plus camrelizumab and in combination with/without apatinib in patients with advanced solid tumors: safety and efficacy analysis. J Clin Oncol. 2021;39:3101. doi:10.1200/JCO.2021.39.15_suppl.3101
89. Mautino M, Kumar S, Zhuang H, et al. Abstract 4076: a novel prodrug of indoximod with enhanced pharmacokinetic properties. Cancer Res. 2017;77:4076. doi:10.1158/1538-7445.AM2017-4076
90. Kumar S, Jaipuri FA, Waldo JP, et al. Discovery of indoximod prodrugs and characterization of clinical candidate NLG802. Eur J Med Chem. 2020;198:112373. doi:10.1016/j.ejmech.2020.112373
91. Dorsey F, Benhadji K, Sams L, et al. Abstract 5245: identification and characterization of the IDO1 inhibitor LY3381916. Cancer Res. 2018;78:5245. doi:10.1158/1538-7445.AM2018-5245
92. Kotecki N, Vuagnat P, O’Neil BH, et al. A phase I study of an IDO-1 inhibitor (LY3381916) as monotherapy and in combination with an Anti-PD-L1 antibody (LY3300054) in patients with advanced cancer. J Immunother. 2021;44(7):264–275.
93. Naing A, Eder JP, Piha-Paul SA, et al. Preclinical investigations and a first-in-human phase I trial of M4112, the first dual inhibitor of indoleamine 2,3-dioxygenase 1 and tryptophan 2,3-dioxygenase 2, in patients with advanced solid tumors. J Immunother Cancer. 2020;8(2):e000870. doi:10.1136/jitc-2020-000870
94. Hamilton MM, Mseeh F, McAfoos TJ, et al. Discovery of IACS-9779 and IACS-70465 as Potent Inhibitors Targeting Indoleamine 2,3-Dioxygenase 1 (IDO1) Apoenzyme. J Med Chem. 2021;64(15):11302–11329. doi:10.1021/acs.jmedchem.1c00679
95. Feng X, Shen P, Wang Y, Li Z, Bian J. Synthesis and in vivo antitumor evaluation of an orally active potent phosphonamidate derivative targeting IDO1/IDO2/TDO. Biochem Pharmacol. 2019;168:214–223. doi:10.1016/j.bcp.2019.07.011
96. He X, He G, Chu Z, et al. Discovery of the first potent IDO1/IDO2 dual inhibitors: a promising strategy for cancer immunotherapy. J Med Chem. 2021;64(24):17950–17968. doi:10.1021/acs.jmedchem.1c01305
97. Ebokaiwe AP, Njoya EM, Sheng Y, et al. Salinomycin promotes T-cell proliferation by inhibiting the expression and enzymatic activity of immunosuppressive indoleamine-2,3-dioxygenase in human breast cancer cells. Toxicol Appl Pharmacol. 2020;404:115203. doi:10.1016/j.taap.2020.115203
98. Wang H, Zhang H, Zhu Y, Wu Z, Cui C, Cai F. Anticancer mechanisms of salinomycin in breast cancer and its clinical applications. Front Oncol. 2021;11:654428. doi:10.3389/fonc.2021.654428
99. O’Donnell JS, Long GV, Scolyer RA, Teng MWL, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81. doi:10.1016/j.ctrv.2016.11.007
100. Liang H, Li T, Fang X, et al. IDO1/TDO dual inhibitor RY103 targets Kyn-AhR pathway and exhibits preclinical efficacy on pancreatic cancer. Cancer Lett. 2021;522:32–43. doi:10.1016/j.canlet.2021.09.012
101. Ye Z, Yue L, Shi J, Shao M, Wu T. Role of IDO and TDO in cancers and related diseases and the therapeutic implications. J Cancer. 2019;10(12):2771–2782. doi:10.7150/jca.31727
102. Fang K, Dong G, Li Y, et al. Discovery of novel indoleamine 2,3-dioxygenase 1 (IDO1) and Histone Deacetylase (HDAC) Dual Inhibitors. ACS Med Chem Lett. 2018;9(4):312–317. doi:10.1021/acsmedchemlett.7b00487
103. Lin Y, Zhang H, Niu T, Tang ML, Chang J. Discovery of novel indoleamine 2,3-dioxygenase 1 (IDO1) and histone deacetylase 1 (HDAC1) dual inhibitors derived from the natural product saprorthoquinone. Molecules. 2020;25(19):E4494. doi:10.3390/molecules25194494
104. Booth L, Roberts JL, Poklepovic A, Kirkwood J, Dent P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget. 2017;8(47):83155–83170. doi:10.18632/oncotarget.17950
105. Maharaj K, Powers JJ, Mediavilla-Varela M, et al. HDAC6 inhibition alleviates CLL-induced T-cell dysfunction and enhances immune checkpoint blockade efficacy in the Eμ-TCL1 model. Front Immunol. 2020;11:590072. doi:10.3389/fimmu.2020.590072
106. Campesato LF, Budhu S, Tchaicha J, et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat Commun. 2020;11(1):4011. doi:10.1038/s41467-020-17750-z
107. Xie L, Hu K, Duo Y, et al. Off-tumor IDO1 target engagements determine the cancer-immune set point and predict the immunotherapeutic efficacy. J Immunother Cancer. 2021;9(6):e002616. doi:10.1136/jitc-2021-002616
108. Godin-Ethier J, Hanafi LA, Duvignaud JB, Leclerc D, Lapointe R. IDO expression by human B lymphocytes in response to T lymphocyte stimuli and TLR engagement is biologically inactive. Mol Immunol. 2011;49(1–2):253–259. doi:10.1016/j.molimm.2011.08.017
109. Luke J, Siu L, Santucci-Pereira J, et al. 1874OInterferon ɣ (IFN-ɣ) gene signature and tryptophan 2,3-dioxygenase 2 (TDO2) gene expression: a potential predictive composite biomarker for linrodostat mesylate (BMS-986205; indoleamine 2,3-dioxygenase 1 inhibitor [IDO1i]) + nivolumab (NIVO). Annals Oncol. 2019:30. doi:10.1093/annonc/mdz268.001
110. Melichar B, Spisarová M, Bartoušková M, Krčmová LK, Javorská L, Študentová H. Neopterin as a biomarker of immune response in cancer patients. Ann Transl Med. 2017;5(13):280. doi:10.21037/atm.2017.06.29
111. Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol. 2019;41(1):41–48. doi:10.1007/s00281-018-0702-0
112. Monjazeb AM, Kent MS, Grossenbacher SK, et al. Blocking indolamine-2,3-dioxygenase rebound immune suppression boosts antitumor effects of radio-immunotherapy in murine models and spontaneous canine malignancies. Clin Cancer Res. 2016;22(17):4328–4340. doi:10.1158/1078-0432.CCR-15-3026
113. Liu Q, Hua S, Wang X, Chen F, Gou S. The introduction of immunosuppressor (TDO inhibitor) significantly improved the efficacy of irinotecan in treating hepatocellular carcinoma. Cancer Immunol Immunother. 2021;70(2):497–508. doi:10.1007/s00262-020-02697-3
114. Sharma MD, Pacholczyk R, Shi H, et al. Inhibition of the BTK-IDO-mTOR axis promotes differentiation of monocyte-lineage dendritic cells and enhances anti-tumor T cell immunity. Immunity. 2021;54(10):2354–2371.e8. doi:10.1016/j.immuni.2021.09.005
115. Hu M, Zhou W, Wang Y, et al. Discovery of the first potent proteolysis targeting chimera (PROTAC) degrader of indoleamine 2,3-dioxygenase 1. Acta Pharm Sin B. 2020;10(10):1943–1953. doi:10.1016/j.apsb.2020.02.010
116. Triplett TA, Garrison KC, Marshall N, et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat Biotechnol. 2018;36(8):758–764. doi:10.1038/nbt.4180
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.