")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
Targeting Impaired Antimicrobial Immunity in the Brain for the Treatment of Alzheimer’s Disease
Authors Fulop T , Tripathi S , Rodrigues S, Desroches M, Bunt T, Eiser A, Bernier F, Beauregard PB, Barron AE, Khalil A, Plotka A , Hirokawa K, Larbi A, Bocti C, Laurent B, Frost EH , Witkowski JM
Received 3 February 2021
Accepted for publication 16 April 2021
Published 4 May 2021 Volume 2021:17 Pages 1311—1339
DOI https://doi.org/10.2147/NDT.S264910
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Tamas Fulop,1 Shreyansh Tripathi,2,3 Serafim Rodrigues,3,4 Mathieu Desroches,5,6 Ton Bunt,7 Arnold Eiser,8 Francois Bernier,9 Pascale B Beauregard,10 Annelise E Barron,11 Abdelouahed Khalil,1 Adam Plotka,12 Katsuiku Hirokawa,13 Anis Larbi,14 Christian Bocti,15 Benoit Laurent,16 Eric H Frost,17 Jacek M Witkowski12
1Research Center on Aging, Geriatric Division, Department of Medicine, Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, Quebec, Canada; 2Cluster Innovation Centre, North Campus, University of Delhi, Delhi, 110007, India; 3Ikerbasque, The Basque Foundation for Science, Bilbao, Spain; 4Mathematical Computational and Experimental Neuroscience (MCEN), BCAM - The Basque Center for Applied Mathematics, Bilbao, Spain; 5MathNeuro Team, Inria Sophia Antipolis Méditerranée, Sophia Antipolis, France; 6Department of Mathematics, Université Côte d’Azur, Nice, France; 7Izumi Biosciences, Inc., Lexington, MA, USA; 8Leonard Davis Institute, University of Pennsylvania, Drexel University College of Medicine, Philadelphia, PA, USA; 9Morinaga Milk Industry Co., Ltd, Next Generation Science Institute, Kanagawa, Japan; 10Department of Biology, Faculty of Sciences, Université de Sherbrooke, Sherbrooke, Quebec, Canada; 11Department of Bioengineering, Stanford School of Medicine, Stanford, CA, USA; 12Department of Pathophysiology, Medical University of Gdansk, Gdansk, Poland; 13Institute of Health and Life Science, Tokyo Med. Dent. University, Tokyo and Nito-Memory Nakanosogo Hospital, Department of Pathology, Tokyo, Japan; 14Singapore Immunology Network (SIgN), Agency for Science Technology and Research (A*STAR), Immunos Building, Biopolis, Singapore, Singapore; 15Research Center on Aging, Department of Medicine, Division of Neurology, Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, Quebec, Canada; 16Research Center on Aging, Department of Biochemistry and Functional Genomics, Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, Quebec, Canada; 17Department of Microbiology and Infectious Diseases, Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, Quebec, Canada
Correspondence: Tamas Fulop
Research Center on Aging, Faculty of Medicine and Health Sciences, Université de Sherbrooke, 3001, 12th Avenue North, Sherbrooke, Quebec, J1H 5N4, Canada
Tel +1 819 780 2220
Fax +1 819 829 7141
Email [email protected]
Serafim Rodrigues
Ikerbasque Prof. Dr., Ikerbasque, The Basque Foundation for Science Bilbao, Spain and BCAM - The Basque Center for Applied Mathematics, Mathematical, Computational and Experimental (MCEN) Research Group, Alameda de Mazarredo 14, Bilbao, Bizkaia, Basque-Country, 48009, Spain
Tel +34 946 567 842
Email [email protected]
Abstract: Alzheimer’s disease (AD) is the most common form of dementia and aging is the most common risk factor for developing the disease. The etiology of AD is not known but AD may be considered as a clinical syndrome with multiple causal pathways contributing to it. The amyloid cascade hypothesis, claiming that excess production or reduced clearance of amyloid-beta (Aβ) and its aggregation into amyloid plaques, was accepted for a long time as the main cause of AD. However, many studies showed that Aβ is a frequent consequence of many challenges/pathologic processes occurring in the brain for decades. A key factor, sustained by experimental data, is that low-grade infection leading to production and deposition of Aβ, which has antimicrobial activity, precedes the development of clinically apparent AD. This infection is chronic, low grade, largely clinically silent for decades because of a nearly efficient antimicrobial immune response in the brain. A chronic inflammatory state is induced that results in neurodegeneration. Interventions that appear to prevent, retard or mitigate the development of AD also appear to modify the disease. In this review, we conceptualize further that the changes in the brain antimicrobial immune response during aging and especially in AD sufferers serve as a foundation that could lead to improved treatment strategies for preventing or decreasing the progression of AD in a disease-modifying treatment.
Keywords: Alzheimer’s disease, mild cognitive impairment, neuroinflammation, antimicrobial immunity, brain, treatment
Introduction
Alzheimer’s disease (AD) is the most frequent neurodegenerative disease leading to clinical dementia; however, the cause is still nebulous despite the important research effort invested to understand the disease. The most prevalent hypothesis is the amyloid cascade hypothesis, which states that the deposition of amyloid-beta (Aβ) as plaques is the cause of neurodegeneration. All clinical trials targeting this as the causal factor have failed, suggesting that we should understand the real, underlying, and treatable factors of this disease to find new treatment targets. For decades, alternative explanations for AD pathogenesis have been proposed, the most important being the vascular, the metabolic, the oxidative stress, and the infection hypotheses. In this review we describe the various putative causes of AD, with a special focus on the infection hypothesis. We also discuss how targeting the impaired antimicrobial defense of the brain may slow the progression or even prevent AD.
What is Alzheimer’s Disease?
Clinically
The clinical manifestations of AD appear quite late in life as one of the most important risk factors for the late onset of AD is aging but the pathology of AD starts decades before that.1,2 Several stages can be defined before the onset of full-blown clinical manifestation of the disease that involves memory and language changes resulting in altered everyday functions. The preclinical stage is characterized by the subjective memory complaint. There is still a controversy whether it exists but when it is present in a family occurrence context (ie, one parent is suffering from overt AD), it could have a certain prognostic value.3 The most recognized prodromal stage called mild cognitive impairment (MCI) is when the cognitive problems may be revealed by tests, but the person is still functioning perfectly. However, not all patients will progress from the preclinical or prodromal stages to the full-blown disease state.4 There should be a complex constellation of factors such as genetic, immunological, and environmental factors to progress from one stage to the other. These prodromal stages are the best time/targets for prevention or disease-modifying treatments; however, the lack of real biomarkers needs to be addressed before the development of any treatment.5
Pathologically
AD is a neurodegenerative disease with a very long development history that can last decades.6 AD is likely a syndrome as it seems that many different causes can lead to its development. The initiating event is not precisely defined, nevertheless several events have been incriminated such as an acute brain injury (fall or sport trauma), a vascular injury, a metabolic injury, or infection.7 The common action between each of these triggering events is an acute inflammation as well as the production of Aβ following the amyloidogenic processing of the cellular amyloid precursor protein (APP). This acute inflammation is at the very beginning a protective process, meant to contain the damaging effects of this injury.8 The proper characteristic of this inflammation is to produce an immune reaction which will normally eliminate the damaging effects of the insult. This will mobilize the innate immune system first to produce pro-inflammatory cytokines as well as antimicrobial peptides. Usually with the blow-up of this inflammatory process, the insult will be resumed. However, because of genetic, environmental, metabolic reasons as well as the persistence of the insult, the acute inflammation is not completely resolved, but instead it may become chronic with the maintenance of the low-grade inflammatory signals which are the pro-inflammatory cytokines, free radicals, and antimicrobial peptides. This persistent chronic inflammatory process can lead, decades later, to AD with the characteristic pathological hallmarks including amyloid plaques, neurofibrillary tangles with intracellular hyperphosphorylated tau protein, synaptic loss and neuroinflammation.9–12
Immunosenescence and Inflammaging: A Nutshell Description
Immunity evolves with aging and it was suggested that changes in immune functions with the concomitant occurrence of inflammaging could be responsible for the age-related diseases such as cardiovascular diseases, neurodegenerative diseases, malignancies, and frailty syndrome.13–18 Age-related immune functionality has been extensively investigated and the most important paradigm states that the decrease in T cell function characterizes immunosenescence.19–23 The innate immune response is also affected.24–26 Whether age-related immune changes act alone, or through inflammaging, is not clearly elucidated, as precise biomarkers of these phenomena are still missing.27–32 As inflammation in AD mainly concern the innate immune system either in the brain or at the periphery, we will be mainly considering it below.
Innate Immunity in Aging: The Fate Keeper
The innate immune system is an ancestral immune response assuring the first line of defense against challenges coming from the inside or the outside, mainly pathogenic microorganisms and damaged cells.33 It is a very fast and efficient reaction that determines the subsequent adaptive immune response.34 The important factors of innate immunity are the various phagocytic cells including neutrophils (PMN), monocyte/macrophages, dendritic cells (DC) and natural killer (NK) cells,35–38 as well as the most recent innate-like lymphocytes such as mucosal associated invariant T cells (MAIT).39–41 It is important to mention that the number of pattern recognition receptors (PRR), danger receptors (DR) that sense the pathogen-associated molecular patterns (PAMPs) from pathogens, and damage-associated molecular patterns or alarmins (DAMPs) from damaged cells do not fluctuate significantly during aging.42–46 However, the signaling pathways such as MAPKs, PI3K-akt and JAK-STAT initiated by ligation of PRRs and DRs may be altered and lead to impaired NF-κB nuclear translocation during aging.25,47–49 This alteration may significantly impact some cellular functions in all of the abovementioned cells including phagocytosis, intracellular killing, chemotaxis and free radical production.50–52 In its prime, the innate immune system can return to a quiescent state after neutralizing these aggressions, but with the accumulation of stressors, periods of rest are less and less frequent. Thus, the innate immune cells become more permanently activated even at the “resting” state,53–56 a concept that has been termed “trained innate memory”.57–60 Nevertheless, this permanent antigenic stimulation contributes to a state of low but significant secretion of pro-inflammatory mediators that participate to inflammaging61–64 and lead to a disequilibrium between activation and inhibition. Thus, the innate cells are probably a cornerstone in driving the fate of immune responsiveness in old age resulting in inflammaging27 and as such contributing to the development of AD.65,66
Inflammaging
Inflammaging is characterized by a peculiar presentation, being a sterile (allegedly) inflammatory status that is chronic, systemic, low grade and therefore subclinical for a long time. The level of cytokines often remains within the (high) normal range but is significantly more elevated in older adults than in younger individuals. This is why inflammaging is also referred to as low-grade inflammation. IL-6, TNFα and CRP are often cited in inflammaging-related studies generating a myeloid hypothesis that could explain the association between aging and a low-grade inflammatory state.67,68 In the meantime, the anti-inflammatory mediators such as IL-10, IL-4, IL-13 may also be increased as a tentative measure to control this state.61–69 Latent virus infections such as cytomegalovirus (CMV) infection, as well as commensal bacteria (eg bacteria in gut microbiome dysbiosis), may be reactivated and become harmful and contribute to inflammaging.70–73 Indubitably, the finding that noninfectious agents can strongly contribute to the spreading of inflammatory processes has paved the way to extend the list of mechanisms that fuel inflammaging over time. The senescence-associated secretory phenotype (SASP), that can be acquired by different types of senescent cells, is currently considered as the main noninfectious trigger of inflammaging.74–76 Senescent cells are in a state where they cannot divide, however, the activation of DNA damage-associated responses (DDR) leads these cells to a higher capacity of secretion of pro-inflammatory molecules defining the SASP.77–79 Recent studies suggested also that exosomes secreted by senescent cells (and their cargo) participate to SASP80 and can modulate immune system functions.81 Studies also involved exosomes in AD as the means to propagate Aβ pathology, neuroinflammation and oxidative stress.81–89
Because of the abovementioned relationships, inflammaging is one of the most important links between aging and the age-related neurodegenerative diseases. Therefore, in this context, the production of Aβ represents most probably the consequence of the neuroinflammatory process induced by several chronic situations including chronic infection, as suggested by the AMP nature of Aβ.90–95
Do Chronic Infections Contribute to the AD Pathomechanism?
The most popular hypothesis to explain the origin of AD proposes that deposition of Aβ in senile plaques leads to inflammation and neuron death.96–98 However, attempts to decrease the Aβ load or to prevent its formation have had no effect on AD.99,100 No cure whatsoever exists or seems to be on the horizon.101,102 These facts together question the validity of this mainstream hypothesis.103–105 Therefore, new and bold avenues of research need to be pursued to unravel new pathomechanisms leading to successful prevention and/or treatment of AD,7,106 however integrating the unavoidable Aβ cascade hypothesis. Obviously, the most important risk factor for late onset AD is aging, which is associated with pro-inflammatory conditions that increases the risk of neurodegenerative disorders including AD.107–109 Infection by particular microorganisms as a plausible pathomechanism had been voiced several years ago but did not receive significant attention. The demonstration by Wozniak et al of the presence of HSV-1 viral DNA110–112 and by Miklossy and Miklossy and McGeer of the presence of spirochetes in the AD brain were too instrumental to consider infections as contributors to the pathogenesis of AD.113–116 Furthermore, it is well recognized that periodontitis and gingivitis are linked to a higher risk of AD.117–121 What could be the pathomechanism of this association and are specific pathogens involved? In this context, the role of Porphyromonas gingivalis as the master bacteria orchestrating the whole community of microorganisms inside the mouth has been strongly evoked.120,122 We and other groups have shown that Aβ is a powerful antimicrobial peptide secreted by neurons in response to an attack by microorganisms lends weight to this hypothesis.90–92 However, none of these individual microorganisms has been linked irrefutably with the disease, therefore, we suggest that simultaneous or consecutive infection by several microorganisms fueled by inflammaging together lead to AD pathogenesis.123–125 The most important common characteristic of all these microorganisms is their persistence and the inability of the brain and systemic immune system to clear them. This persistence creates a constant cycle of latency and reactivation that will activate microglia either periodically or constantly in the brain. Concomitantly with other risk factors such as genetics, diet, trauma, this can contribute to neuroinflammation and ultimately after several decades to neurodegeneration.126,127
Neuroinflammation, Inflammaging and Alzheimer’s Disease
The infectious hypothesis provides a plausible stimulus for this neuroinflammation which is considered a hallmark of AD.96,128–135 It also throws light on two other fundamental facts: (1) Neuroinflammation is not only the consequence of Aβ deposition (as stated by the amyloid hypothesis) but it is also the cause for Aβ deposition, and (2) Aβ is not only a “harmful” molecule that aggregates to form plaque and induce neuroinflammation, but it is also a basic element of the innate immune defense and thus a “beneficial” molecule,13 at least at the beginning. Ultimately, as the reactivation of latent pathogens (HSV-1) and new infections become more frequent, the chronic production of Aβ increases, but its antimicrobial effect may be blunted by loss of active Aβ through its recruitment to plaque formation. Consequently, inflammation becomes chronic, endocytosis and clearance of Aß by microglia is overwhelmed, and ultimately the deposition proceeds and results in senile plaque formation.124 The deposition of plaque may be the initiator of a chronic, harmful neuroinflammatory process that finally destroy the neighboring neurons. This process pursues unnoticed, then becomes visible clinically only when a threshold is crossed.
Local neuroinflammation may continue at a low level throughout life with little negative effect. However, when exacerbated by reactivation of infections combined with other insults such as oxidative stress, the acute inflammatory response results in unbalanced production of cytotoxic mediators difficult to control or stop.27,136–141 Microbial metabolites may also fuel neuroinflammation. The enhanced neuroinflammatory process damages neurons and alters the blood-brain barrier (BBB). These mediators also induce peripheral inflammation and then return to further stimulate local neuroinflammation.142–144 This progressive pro-inflammatory situation is exacerbated with age, creating a vicious cycle of local and systemic inflammatory responses leading to activation of cytotoxic microglia, unbalanced cytokine production, Aβ accumulation and irreversible brain damage.
Experimental Data Substantially Support the Infection Hypothesis of AD
The infection hypothesis was proposed decades ago when it became clear that there should be some triggering events at some points of the disease progression.7,145–147 It is noteworthy that Oskar Fisher, in the same epoch as Alois Alzheimer, had already evoked this possibility.148 Early evidence was done on HSV-1 viruses. The group lead by Ruth Itzhaki has identified the HSV-1 DNA in the plaques of fully developed AD brains.149,150 An epidemiological Taiwanese study recently showed that HSV-1 antiviral treatments may interfere with the development of AD in contrast to those who did not get them.151 The Lovheim group could make the association between the ApoE4 genotype, the susceptibility to HSV-1 infection and the occurrence of AD.152 The virus could remain latent for many years, especially in the in neurons and in the trigeminal ganglia,153,154 and then reactivate each time when the immune defense is diminished by stress, diseases, or other infections.155,156 The virus can easily gain access to the brain by the trigeminus nerve and the olfactory system. Other herpes viruses like the HHV6 and HHV7 are also involved.157 In this period of COVID-19, it has become evident that the brain might be affected either directly or indirectly by the respiratory SARS-CoV2 virus.158 However, the long-term effects are unknown but may lead to AD-like neurodegenerative disease decades later.159,160
Concerning the bacteria which may be involved in triggering, strong evidence exist for P. gingivalis,122 Borrelia burgdorferi113,114 and Chlamydia pneumoniae.161 All these bacteria themselves, their remnant or products (LPS or gingipain) have been directly found in the brain.122 P. gingivalis as a cornerstone bacterium can migrate from the mouth to the brain by the trigeminal nerve or the olfactive pathway.162 Its presence in the brain of AD patients was recently demonstrated.122 These bacteria were found directly in the amyloid plaques suggesting that the plaques may be a sort of biofilm.114 Epidemiological studies also strongly suggest a correlation between periodontitis occurrence and AD.163
Another major source for the microbial contribution to AD is the gut–brain axis.8,164–166 It is well known that when the gut microbiota is perturbed several psychological, psychiatric and cognitive problems, mostly acute, may arise.167,168 These are mostly acute processes. The gut–brain axis provides a bidirectional communication via cytokines, hormones, and neurotransmitters.169,170 In case of neurodegeneration, an alteration in the normal composition of the gut microbiota caused by infection, age or diet171,172 may result in an inflammatory process in the brain by either direct migration of the pathological microbes to the brain,173 via the vagus nerve,166,174,175 or via the inflammatory products originating from these microorganisms such as LPS, lipoteichoic acid or Escherichia coli K99pili.176–180 Evidence also suggests that the co-localization of LPS and other bacterial fragments in amyloid plaques175,177 may contribute to the neuroinflammation.181 It was also shown that production of short-chain fatty acids (SCFAs) by microbiota can activate brain microglia which causes neuroinflammation and neuronal damage in an AD model but may be also protective by decreasing BBB permeability.182–184 Therefore, the (eubiotic) microbiome may have also protective effect on the brain neurons.185 It should be strongly emphasized that the majority of the data discussed here were done in animal models, with the exception of the 2019 P. gingivalis results published by Dominy et al, which had human data.122
Brain Antimicrobial Immunity
The antimicrobial immunity of the brain is complex, and data are quite scarce. In the periphery, it is composed by natural defense lines, cells and mediators. In the context of the brain, the innate immunity has been the most studied because of the microglia existence and the Aβ-triggered neuroinflammation.186 The relationship between the antimicrobial immunity and the infection hypothesis is becoming slowly unraveled.145 This immunity seems very adequate and efficient at the beginning to eradicate the invaders, but considering the persistence of the aggressors it becomes more harmful.187,188 In the context of AD, the innate immune system (via microglia) plays an important role in the neuroinflammation. We will mainly consider this part of the immunity in this review; however, we will also succinctly mention the adaptive part.
Blood–Brain Barrier
The BBB is the first line of defense against many noxious elements coming from the periphery including infectious agents (pathogens) and activated immune cells. The BBB is a semipermeable interface between the brain parenchyma and cerebral circulation consisting of endothelial cells, astrocytes, pericytes and a basal lamina. The BBB is predisposed to filtrate, retain and destroy the microorganisms.189 Macrophages and endothelial cells as part of the BBB can eliminate the infectious agents. However, when the attacks become more frequent, an inflammatory process alters BBB permeability, leading microorganisms and their products to pass more freely from the blood into the brain.190 Astrocytes are also part of the brain antimicrobial defense as they have a very important neuroprotective function by assuring the BBB integrity and as such decreasing the passage of inflammatory cells from the periphery.191–193 However, when they become activated as A1 astrocytes they are mediating the neuroinflammation either themselves via the production of cytokines and chemokines194 or by making the BBB more permeable to peripheral inflammatory mediators.195
Cellular Defense
As part of the innate immune system of the brain, microglia which are the macrophages of the brain are the most important cellular defense.9,196,197 They are mostly from embryonic origin but some of them may originate from the monocytes getting to the brain.198–200 Microglia very efficiently get rid of invaders and detect synaptic anomalies as they are always patrolling the brain.194,201,202 At the same time, they are maintained in a quiescent state by their interaction with neurons via CXCR1203 or CD200L.204 When activated through different pathways including the TREM2-DAP12,196,200,205 they can phagocytose and kill intracellularly all types of microorganisms. Interestingly, mutations in the TREM2 have been identified as risk factor for AD.206 They are also able to migrate and proliferate and they exhibit two distinct phenotypes but are able to be very plastic.207,208 The type 1 microglia are very pro-inflammatory and secrete pro-inflammatory cytokines to help clearing the aggressors, while the type 2 microglia are anti-inflammatory and are able to mitigate the inflammation and repair the tissue damages. Recently a third microglia type was described and called disease associated microglia (DAM)209–212 and its functions are related to a stepwise activation manner implicating or not TREM2.213,214 The DAM are somehow specific for the neurodegenerative state and participate at the beginning to the clearance of Aβ and later to neurodegeneration. However, most of the data concerning microglia in AD are in relation to Aβ peptide independently of its form (monomeric, polymeric, fibrillar or aggregated as plaques).215,216 Through their activation via CD36, CD14, CD47 and TLRs (especially TLR4), microglia produce pro-inflammatory cytokines and chemokines.217–219 The role of microglia is well established in various cerebral infections220,221 however, there are no data related to the infection hypothesis of AD at the exception of one study that reported the relation between microglia and some changes of microbiota and its derivatives including LPS, as well as related to the genetic background such as ApoE4.222 Therefore, it would be interesting to assess how microglia could behave at different stages of AD.
As the aggression in the brain persists microglia become more activated, producing chronically pro-inflammatory mediators that participate in the neuronal destruction.196,223 Furthermore, they possess receptors which react to the overproduced Aβ.224,225 The most important receptors are the TLRs, in particular TLR4,199,226,227 which initiate intracellular signaling pathways leading to the activation of the NF-κB, the inflammasome and the antiviral molecular machinery.228 At the end of this process, activated microglia become senescent and only produce pro-inflammatory neurotoxic mediators such as TNFα.229–231 Morphologically senescent microglia show cytoplasmic hypertrophy and pseudopodia reduction135,232,233 in contrast to the stationary microglia which can always scan the milieu for invaders with their extended pseudopodia.234 These data point to the need of a timely regulation of the brain innate immune response to exploit the beneficial potential and decrease the inflammatory action.197
It is of note that the role of the adaptive immunity in the antimicrobial defense of the brain is much less understood than that of innate immunity.235 In AD, the changes in the peripheral adaptive system are well established.236,237 T cells can be found normally in meningeal, perivascular space and choroid plexus, but the resident T cells in the parenchyma are rare. When T and B cells can be found in brain parenchyma, it means that the BBB is compromised. However, recent studies found a small population of resident, tissue specific, memory CD4+ and CD8+ T cells in human brains.238–241 Studies performed mainly in animal models suggested that T cells may modulate the microglia phenotype and activation state.242–244 Most of the data on the adaptive immunity role in AD came from mouse models. They indicate that, when T cells are solicited because of the infection, they can enter the brain and contribute either to the neuroinflammation or to the antimicrobial defense of the brain.245 The better understanding of their role is of the utmost importance for further immunotherapies in AD.
Soluble Mediators
The activated microglia and astrocytes secrete cytokines, chemokines and reactive oxygen species. Interleukin-1 (IL-1β), tumor necrosis factor-α (TNF-α), IL-6, IL-10, chemokines and free radicals are the most important. These mediators are very useful at the beginning of an infection as they drive the innate and the adaptive immune responses.246 They prime the microglia for better anti-infectious response, stimulate antigen presenting cell differentiation and prime the adaptive immunity. During chronic stimulation, the production of these pro-inflammatory mediators becomes uncontrolled and leading to the constant activation of the innate immune system and to tissue destruction.247 The activation of inflammasome via NLRP3 and NLRP1 in AD largely contribute to the production of pro-inflammatory cytokines of the IL-1 family.228,248–250
One of the most efficient antimicrobial and antiviral system in the brain is the interferon pathway leading to production and secretion of various interferons.251–254 In the interferon family, the most important members are type I and type III acting on different receptors but with similar cellular effects.255,256 The interferon regulatory factors (IRF3, IRF7) regulate IFN production.257,258 A recent study by Romagnoli et al154 found that decreased mRNA levels of IRF7, MED23, IL28B and IFN-α were present in human AD brain hippocampus and temporal cortex samples, with a genetic background of the patients (eg ApoE ε4 and IRF7 A alleles) that could worsen mRNA levels and affect brain immune efficiency. Thus, in the early phase of the infection, this downregulation would favor the decrease of microglia and astrocyte activation and as such mitigate brain damage. While it becomes detrimental when the system cannot eradicate the aggression; the inflammation continues, and neurodegeneration is occurring. However, the exact role and contribution of this important antimicrobial defense pathway just starts to be elucidated as the recent COVID-19 disease revealed the gaps in our understanding especially with its neurological manifestations.
Antimicrobial peptides (AMP) are also part of the immune defense against pathogens. They can efficiently fight the infections. Aβ is not only a harmful by-product, but also could have important physiological roles.33,35,259 The knowledge about the physicochemical properties by which Aβ may exert its antimicrobial action is emerging but still remains partially understood. It became evident that targeting Aβ at preclinical and prodromal stage may be very harmful as demonstrated by many clinical trials targeting Aβ. All the pharmacological attempts to block its production by inhibiting the BACE or by directly targeting any physical Aβ form will not lead to any clinical and cognitive improvement.100–102 Therefore, this discovery gave a new impetus to the infection hypothesis of AD.103 This also highlighted that in the brain there may be other antimicrobial peptides protecting against invasion. LL-37 and defensin-1 are well-known AMP260 and several neuropeptides may also play this role including GLP1 and PACAP.261,262 Furthermore, it was shown in 2017 by De Lorenzi et al that LL-37 can bind to Aβ peptide and form a nontoxic complex.260
Together the brain antimicrobial immune defense is very efficient but the concomitant chronic insults, inflammaging, genetic, epigenetic, and environmental factors lead to neuroinflammation resulting in neurodegeneration. Thus, it is very important to understand the upstream events resulting in neuroinflammation which lead to the final common step in the pathology of AD—the uncontrolled Aβ production initiating and maintaining a vicious chronic inflammatory circle which may serve target to treatments.
What About Interventions?
In the past decades, there have been numerous valuable randomized clinical trials (RCTs), mainly targeting Aβ. For example, the most recent ones—with solanezumab263 or verubecestat264 have been unsuccessful. Thus, we need new treatments targeting other pathomechanisms of the disease such as the neuroinflammation.265,266
By understanding the syndromic nature of AD, it would be very difficult to design just one treatment, but one pathological process seems to be common to all specific causes: neuroinflammation. An optimal treatment for AD could be an agent specifically targeting neuroinflammation. As we are specifically interested here in the antimicrobial defense in the brain, we will consider the fight against infections and inflammation in a chronic setting.5 We will describe what treatment options exist and what is in the pipeline considering these pathological processes.
Prevention/treatment of Infections
The simplest intervention would be to find a prevention for the most important agents involved in the development of AD. We could develop efficient vaccines against the putative pathogens. The recent development of vaccines against SARS-CoV2 gives hope that we could develop a vaccine against the herpes virus HSV-1 and other Herpesviridae such as HHV6, HHV7 or CMV. If this is not possible, an intermittent secondary prevention treatment in all individuals, and more specifically in APOE4 homozygote carriers,267,268 should be initiated with acyclovir, an antiviral treatment that shows almost no side effects. This assumption at least in a recent Taiwanese epidemiological study received strong support.151 Therefore, strong arguments exist for the use of somehow intermittent antiviral treatment in individuals who are the most susceptible to carry lifelong infections with HSV-1, herpes zoster269 or show signs of reactivation measured by IgM serum level. This is a cheap, affordable treatment with great potential. There is presently an ongoing phase II study with valacyclovir in mild AD patients.270 It is of note that this treatment should be efficacious in the preclinical stage preventing its development or at the prodromal stage (MCI) delaying or preventing the progression to full AD. The results of the mentioned study should be known very soon (during 2021) which could hopefully change our present practice. There are other antiviral drugs which could become therapeutics in AD prophylaxis if appropriate clinical trials are carried out. Penciclovir, foscarnet, valacyclovir, Bay57-1293 and bioflavonoids derived from the leaves of Ginkgo biloba have been proposed for an eventual use in prevention or at least in stopping progression in AD (Tables 1 and 2). All these substances have demonstrated powerful antiviral activity in vitro and also in animal models, but data on their clinical trials are missing.
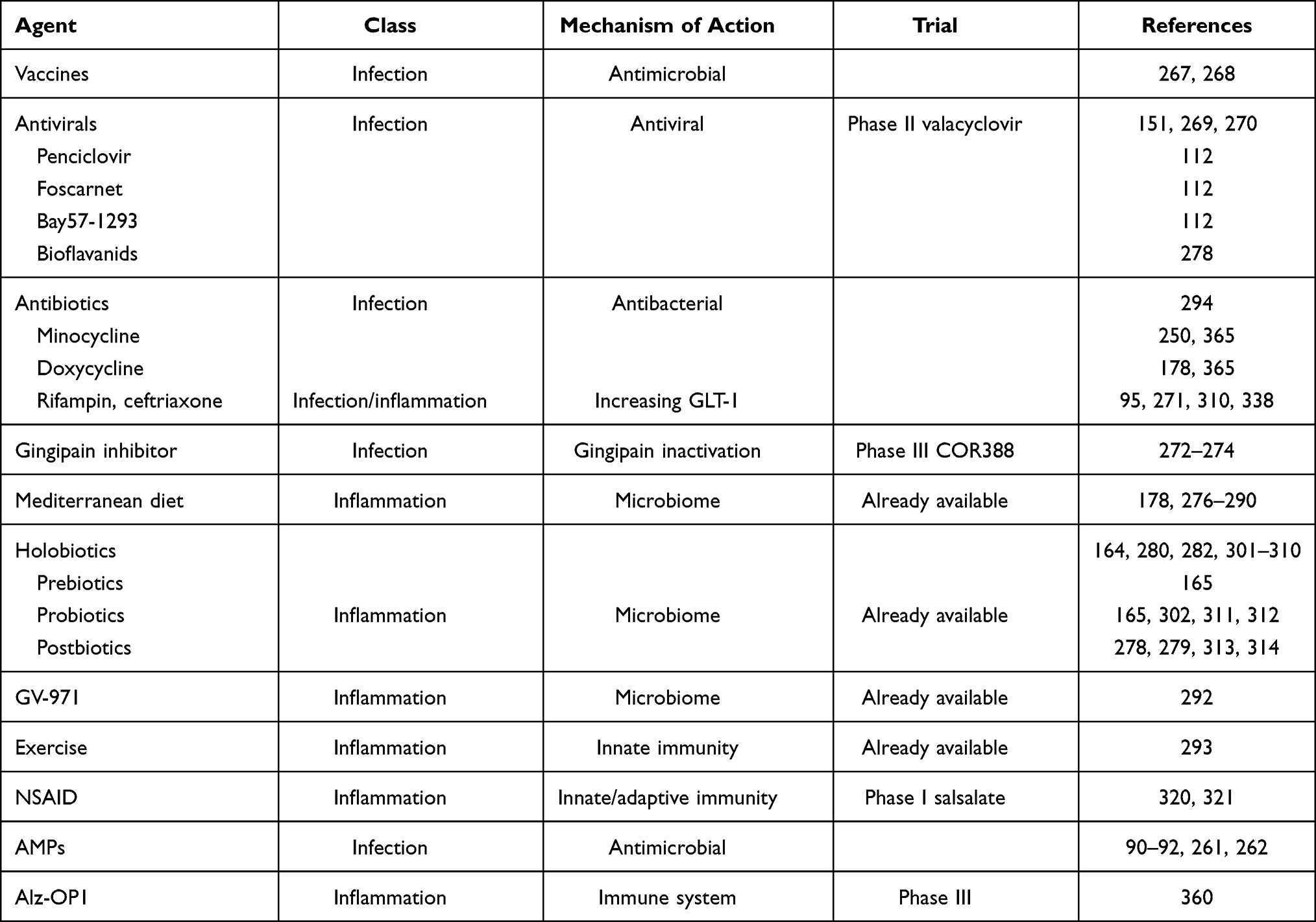 |
Table 1 Prevention Therapies (Potential Therapeutics for Modulating Inflammation/antimicrobial Immune Defense) |
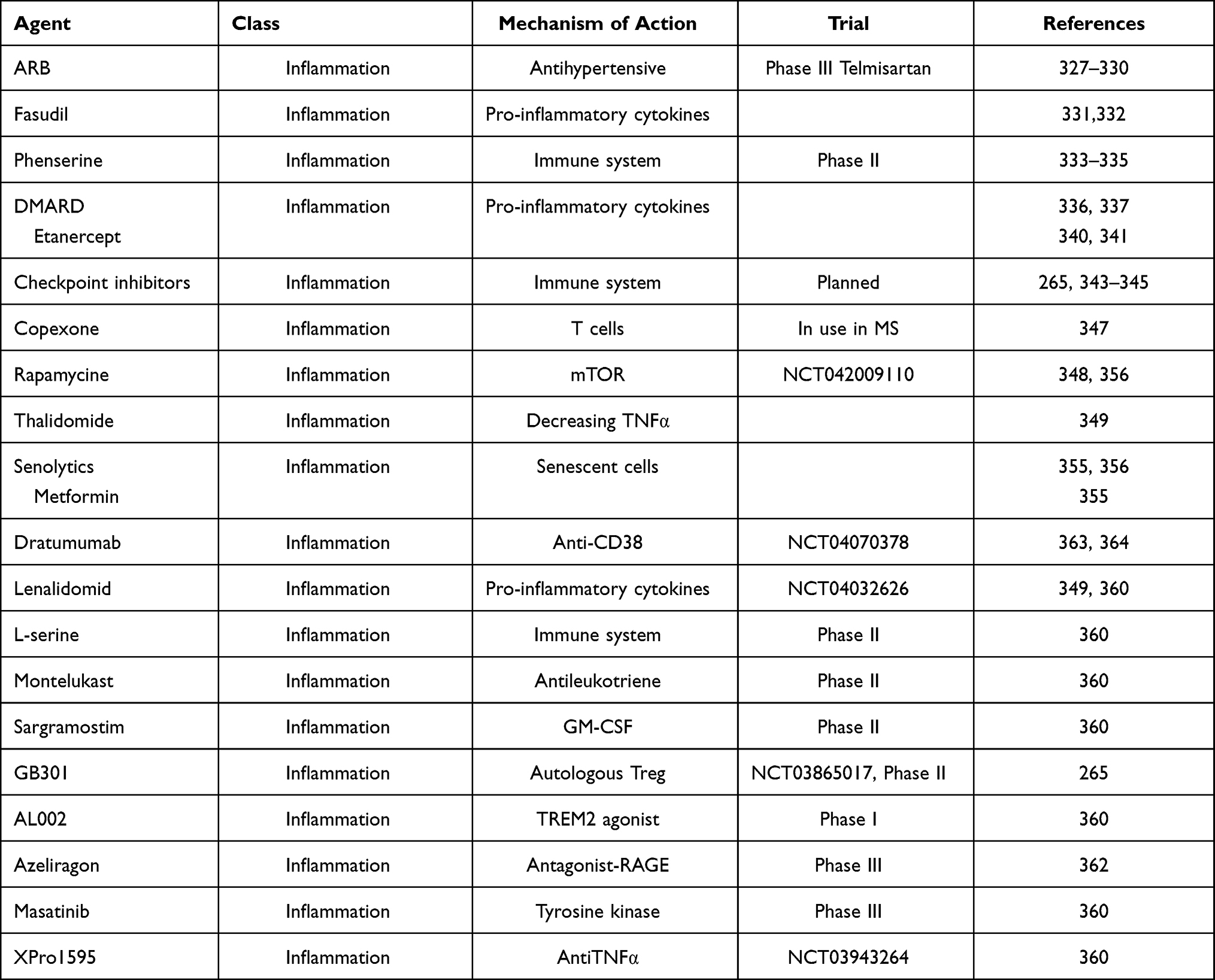 |
Table 2 Disease-modifying Treatments (Potential Therapeutics for Modulating Inflammation/antimicrobial Immune Defense) |
The same tactic may be also used for other pathogens like P. gingivalis. A vaccine neutralizing gingipain, toxic proteases from P. gingivalis could be efficient to prevent AD. Repeated courses of antibiotics may be also envisaged for bacteria, spirochetes, and chlamydia but all the different strategies using antibiotics have been unsuccessful in any RCTs carried out until now. Tetracycline antibiotics (minocycline or doxycycline) or rifampin were ineffective.271 Even the reasons may be multiple (treatment time, dose, pathways used, brain transport), these failures unfortunately dried the antibiotic treatment pipeline for AD. Hope was revived with the development of potential gingipain inhibitors, as gingipain, is the virulence factor of P. gingivalis and plays a crucial role in the colonization of the host and in the inactivation of the antimicrobial defense of the host and as such ensures the pathogenicity and survival of P. gingivalis.272,273 The developed gingipain inhibitors COR286, COR271 and COR388 were found to induce the bacterial death and reduce the bacterial burden in animal models.274 Unfortunately, there is no epidemiological data for antibiotics lifelong use and AD development like it exists for antivirals or for nonsteroidal anti-inflammatory drugs (NSAIDs). This would be worthwhile to perform275 but keeping in mind that a possible antimicrobial resistance may develop.
The occurrence of dysbiosis/pathobiont276 may be prevented since the earliest period of life by a diet maintaining gut microbiota health.8 In this context, a balanced anti-inflammatory diet such as the Mediterranean or the Asian diet may be successful candidates.277–283 In contrast to the western diet, these diets contain many beneficial products maintaining a balanced inflammatory milieu even during aging when the inflammaging is very frequent. Many components such as polyphenols, short chain fatty acids, flavonoids, proteins, vitamins B, curcumin and oligoelements (including selenium, copper, cobalt, magnesium) assure a healthy homeodynamic milieu decreasing the pro-inflammatory, pro-oxidant and epigenetic modulatory effects of the internal and external challenges.178,280–292 These diets have not only the advantage to decrease the propensity for chronic inflammations, but also reinforce the adaptive immune response.172,293–295 Recently GV-971, a new drug meant to regulate gut flora imbalance and reshape immune homeostasis, was approved in 2019 in China. GV-971 can prevent the infiltration of the peripheral immune cells into the brain, inhibit neuroinflammation and prevent the progression of AD.296 Beside stabilizing the gut microbiota, it is reducing the increased circulating phenylalanine/isoleucine shown in AD patients and known to increase neuroinflammation. Together all these interventions alone or in a multimodal way considering their beneficial effects concur to improve cognitive functions in AD patients.
We should also mention the beneficial effect of regular physical activity. This has been shown to increase the cerebral flow, the production of antioxidants, and to reinforce our antimicrobial immunity. It is needless to say that most probably any of these interventions alone will be enough to prevent or treat the very early stages of AD, but a multimodal intervention combining all of them may be efficient. Some of these components were already involved in the original FINGER trial.297 Together, as appealing as this antimicrobial therapeutic approach could be, the lack of real knowledge and insight into pathogenesis related to microbes preclude a judicious utilization of the antimicrobial agents.298 More studies are needed to confirm their efficacy without any doubt on a long-term basis.
Mitigatory/modulatory Treatment of Neuroinflammation
There are many ways to intervene in the mitigation or modulation of neuroinflammation.299 These may be pharmacological or nonpharmacological, direct or indirect interventions.300–302 Among the nonpharmacological interventions, the diet and exercise are the most prominent but indirect measures. Multiple pharmacological means already exist to intervene at the neuroinflammation level such as the mentioned NSAIDs. Even controversial, this approach has been shown efficient when patients with rheumatoid arthritis have been treated and developed much less AD.303 It is still questionable whether this effect is direct or indirect. Recently, it was shown that the blockade of peripheral myeloid EP2 (receptor of prostaglandin E2) restored the glucose metabolism, decreased the age-related inflammatory state, and reversed cognitive decline in aging mice.304 These results suggest that regulating the immunometabolism of macrophages/microglia may have neuroinflammation modulatory action leading to better cognition.
Specific Dietary Components
Besides the general diet described above, some nutriments can be directly used to mitigate the neuroinflammation. The microbiota has been targeted by pre-, pro-, and postbiotics as potential complementary therapeutic approach for AD.164,284,286,305–313 Of note, the probiotic may play a prominent role as they can efficiently regulate or even reset the alterations in various microbiota of the organism which could even have direct anti-inflammatory effect. Probiotics may restore the homeostatic equilibrium among pathogenic and beneficial microbes in the holobiota, especially decreasing bacteria that produce glutamate with excitotoxicity effects.314 Few studies have investigated the use of multispecies probiotic treatment in AD with conflicting results. In one study, 12 weeks of probiotic treatment improved the cognitive status,315 while in another the supplementation for one month changed the microbiome but had no effect on cognition.316 A recent study found a significant improvement in cognition with a probiotic cocktail.306 Two ways exist to manipulate the microbiota; the first, more futuristic, uses precision diets based on the specificities of each microbiome, while the other, already available, use bulk diet interventions to restore the healthy microbiome of an individual. Before we can define either the quantitative or qualitative microbial changes as well as the metabolic changes in AD, it will be difficult to implement a generalized microbiota modulating diet to modulate neuroinflammation in AD.164
Ketone (medium chain triglyceride) supplements have also been shown to improve the cognitive functions in MCI subjects.317,318 Ketone bodies are known to act on the microbiome by restoring its equilibrium, but more importantly by reducing the activation of the inflammasome which is one of the contributors to neuroinflammation. Recently, several case studies have been published on the use of ketogenic diet in APOE4 allele carrying MCI and mild AD individuals with a significant increase in their cognitive performance such as the significant increase of their score of MoCA test.319–323 Involvement of vitamins like vitamin D and vitamins B may also modulate the inflammatory state of the brain by acting on the scavenger receptors.324 Some oligoelements such as zinc may also regulate the chronic inflammation in the brain. Amino acids such as glycine or leucine may also be immunoregulatory by an anti-inflammatory effect leading to decreased microglia activation.325 There is a long-lasting debate on the omega-3 docosahexaenoic acid (DHA) efficiency in the treatment of AD at different stages.326 In a study on MCI patients, the combination of omega-3 supplementation with antioxidants, vitamin D3 and resveratrol showed beneficial effects on MMSE (mini mental state Examination) improvement via modulation of the innate immune response.327,328
Anti-inflammatory Drugs/cytokines
There are presently no direct anti-inflammatory drugs recommended or used in AD. Epidemiological and observational studies strongly suggest a decreased relative risk of developing AD with nonselective NSAIDs,329,330 however the RCT studies did not confirm these observations. One of the main problems is the timing of their utilization, since it cannot be to early neither too late. These treatments should not compromise the natural defense of the brain immune response but should act before the immune response could become harmful by becoming chronic. In this context, biomarkers would make the difference, however we currently do not have any of them in the pipeline to be targeted for intervention. The role of IL-10 which may be appealing to become an efficient target, however, its role was questioned as it can be inflammatory at some point of the AD development.331 In contrast, if exposed to Il-1β, TGFβ1 or IL4, microglia may acquire an anti-inflammatory phenotype by expressing arginase-1 what will increase its phagocytic capacity towards Aβ.332 Many omics and other high throughput-based studies were carried out without really much success to find target biomarkers but brought important scientific data for the future development.333 Nevertheless, some anti-inflammatory treatment already exists in the pipeline and some others may become interesting.313,334,335
For a long time, the use of angiotensin receptor blockers (ARBs), including candesartan, telmisartan or losartan, showed a reduction of neuroinflammation, but only in animal models.336 In humans there are mainly epidemiological evidence that ARBs may be efficient in AD treatment.337 In the ONTARGET trial using telmisartan vs ACE inhibitor, the telmisartan group showed less decrease in MMSE than the control group.338 However, it is difficult to establish what is the mechanism as the reduction of the hypertension recognized as a risk factor for AD could be also the cause of the ARB success.339
Another putative repurposed drug could be fasudil which is a selective inhibitor of rho kinase (ROCK) 1 and 2 and a powerful vasodilator.340 This drug has anti-inflammatory properties that decrease the IL-1 and TNFα production in a rat model.341 More studies are needed in humans to confirm these results. Another promising compound may be phenserine which is a cholinesterase inhibitor.342 In preclinical models, this drug suppressed IL-1 production, to protect against free radicals and reduce excitotoxicity, resulting in decreased neuroinflammation.343 A small phase II study showed good tolerability and some cognitive benefits but was very much underpowered.344 Other drugs which may also have promising applications are disease-modifying antirheumatic drugs (DMARDs) which are used in other inflammatory diseases such as rheumatoid arthritis.265,345
Other repurposed molecules could be considered such as the β-lactam antibiotic ceftriaxone which increases the expression of astrocytic glutamate transporter 1 (GLT1) resulting in decreased excitotoxicity and neuroinflammation by detoxifying the brain from glutamate.314,346 Ceftriaxone may also play an anti-inflammatory and antioxidant role.347 This could open new avenues of investigations with similar compounds. It would be also interesting to study whether they have also direct antibacterial properties in AD. Other compounds found beneficial in PD, namely salbutamol and trifusal (platelet aggregation inhibitor), are antioxidant by blocking the cyclooxygenase 1 and anti-inflammatory by modulating among others, NF-κB. RCT human studies in different phases of AD are badly warranted with all these drugs. Considering the role of the pro-inflammatory cytokines, it would be legitimate to think that the drugs developed for other chronic inflammatory diseases could be beneficial for AD too. However earlier studies on anti-TNF treatment failed. A recent study indicated that patients with rheumatoid arthritis are at increased risk for AD but those using the etanercept had a lowered risk of AD.348 These findings initiated a new study using etanercept as disease-modifying therapy (DMT) for AD.349
Reinforcement of the Antimicrobial Defense by Stimulating the Immune System (Immunomodulation)
An efficient approach could be to potentiate the initial immune and inflammatory responses in order to reinforce the antimicrobial defense. One of the first events in the immune fight against pathogens is the production of interferons (IFN) that modulate the inflammatory response, eradicate the pathogen, and prime the immune system to become more efficient. Anti-CSF-1R treatment may enhance the production of type-I IFN as demonstrated in cancer.350 The production of other pro-inflammatory cytokines, the complement system and the free radical production may also be enhanced by this treatment. Sometimes drugs approved for other diseases (eg cancer) may be repurposed for other chronic inflammatory diseases such as AD. This could be the case for the tyrosine kinase inhibitors (eg dasatinib), immune checkpoint inhibitors (eg PD1, PD1L, CTLA-4) in non-T cells such as dendritic cells351 which can increase the innate immune response and prime the adaptive immune response. In animal studies, checkpoint inhibitors enhanced the cognitive performance352–354 but this treatment appears nonconclusive in human studies.
Some immunomodulating agents may also be considered for AD treatment.266 Copaxone used in multiple sclerosis to boost the T cell immune response could modulate the microglia response.355 Rapamycin, an mTOR inhibitor used as anti-aging drug, could be considered as an immunomodulatory drug in case of AD as preclinical data showed that it can maintain BBB integrity and decrease Aβ pathology.356 Thalidomide and its derivatives are immunomodulatory drugs decreasing TNFα levels and regulating microglia and astrocytes activation in preclinical studies.357
Importantly, the use of therapeutics enhancing the antimicrobial efficiency of the brain immune response should be very tightly controlled in power and time as prolonged stimulation will lead to chronic inflammation, cellular senescence, chronic neuroinflammation and neuronal/synaptic damage.
Geroprotectors and Senolytics
Aging, the most important risk factor for AD development, is associated with changes in the immune system which contribute to the decreased antimicrobial defense. It is conceivable that modulation of the immune changes with aging could be a viable strategy for AD prevention and treatment, and toward this approach, geroprotectors may be useful.358 In animal studies, young blood was shown to influence the cognitive status359 and the results from these studies served as a model for a human phase 1 clinical trial demonstrating the feasibility and the innocuity of a such treatment.360 Identical considerations can be given to mTOR inhibitors which demonstrated immune modulating effects in older subjects by increasing the influenza vaccine efficacy.361 IL-7 and thymosin β4 treatments were also proposed in this sense.362,363 However, this type of treatment should be envisaged from very early ages giving the long-term development of AD. Therefore, each of the proposed treatments can be looked at from two possible points of view. One is prophylaxis, where the envisaged drugs, vaccines or other, would have to be applied to young populations, prior to the onset of any symptoms. The other would be aiming at stopping or at least delaying the disease progression when it is already manifested. The latter would likely be easier to be accepted, even if less effective, but new studies try to implement more the prevention type of interventions early at life.362,363
The use of senolytics at a precise timescale may also be rewarding since senescent cells via the SASP phenotype are suggested to be the major mediators of aging and inflammaging, and microglia and astrocytes may adopt a senescent phenotype over time. Among these senolytics, metformin, which is used in the treatment of type 2 diabetes, could be considered. A phase 3 clinical trial is underway to explore whether metformin can improve CNS glucose metabolism or decrease the senescent cell charge (NCT0062019; NCT01965756). In retrospective epidemiological studies, metformin showed a reduced risk of cognitive impairment.364 Rapamycin and other agents modulating/inhibiting the mTOR pathway could act as senolytics.365
Disease Modifying Treatment: Present and Future
Currently, there is no DMT available for AD but the abovementioned treatments may become DMTs and there are more molecules in the pipeline.
If we consider the composition of the microbiome to explain AD pathogenesis, we should also ponder why many older subjects do not acquire AD. These individuals might possess in their gut bacteria that are metabolically and immunologically active which may produce either beneficial small molecules specifically targeting the brain. The issue is worth investigation by last-generation techniques such as artificial intelligence, transcriptomic, systems biology and complex system approach which would allow us to probe this question.366
Another treatment avenue to explore is the AMP antimicrobial characteristics.90–93 Protein analysis comparing known AMPs and Aβ confirmed structural homology between Aβ and a specific family of bacteriocins.367 Bacteriocins are traditionally synthesized by bacteria against other bacteria.368 Aβ also has structural similarities with another AMP called LL-37. This implies that both can efficiently destroy microbes but also form cytotoxic soluble oligomers and insoluble fibrils.92 Thus, it is conceivable that in the future, Aβ structure and properties may serve as a template for advanced computational models to develop new more powerful specific AMPs.
Small molecules targeting neuroinflammation (ie Masitinib, ALZT-OP1, COR388, telmisartan, sumifilam, neflamapimod, azeliragon, DNL758 and GC021109) are in phase 3 clinical trials. Except for ALZT-OP1, all these molecules target the mild-to-moderate stages of AD. ALZT-OP1 targets the early stage of the disease with results being available between 2020 and 2024.369 A recent article by Cummings et al370 reviewed the AD drug development pipeline and stated that among the drugs in development many are disease-modifying agents or repurposed drugs. Four drugs targeting the inflammation/infection/immunity (17.6% of all agents) are currently in phase 3 clinical trials. The first is ALZT-OP1 (cromolyn + ibuprofen) aiming to increase the clearance of Aβ by modulating the microglia activation; the next is azeliragon, a RAGE antagonist, aiming to reduce Aβ load and neuroinflammation in the brain;371 the third is masitinib, a tyrosine kinase inhibitor, aiming to reduce the Aβ charge and tau phosphorylation; and the last is COR388, a bacterial protease inhibitor targeting gingipain, aiming to reduce neuroinflammation and hippocampal neurodegeneration. Other potential candidates did not seem to lead to conclusive results and were halted.335
There are also several phase 2 clinical trials targeting inflammation/infection/immunity, including four using biologics and seven small molecules. One is using curcumin and aerobic yoga to exploit their antioxidant and anti-inflammatory properties and target neuroinflammation. Daratumumab (NCT04070378) is a monoclonal antibody targeting CD38 which is expected to have immunomodulatory effects by decreasing the microglial activity. CD38 is a glycoprotein found on the surface of many immune cells including CD4+, CD8+, B lymphocytes and natural killer (NK) cells. CD38 also functions in cell adhesion, signal transduction and calcium signaling. The CD38 role is controversial and depends on the cell types, the aggression and the moment of the immune stimulation. However, it may play a determinant role in the modulation of inflammatory processes such as in neuroinflammation.372,373 Dasatinib and quercetin are respectively a tyrosine kinase inhibitor and a flavonoid antioxidant, with strong senolytic activity. They both can decrease inflammation and increase immune response. To downmodulate the immune system, GB301 is a trial comprising of isolating autologous Tregs from AD patients, expanding them and reinjecting them expecting the promotion of immune homeostasis and decrease of neuroinflammation. Lenalidomid, an antineoplastic and immunomodulatory molecule, is expecting to reduce the pro-inflammatory cytokines TNFα, IL-6 and IL-8 and to modulate both the innate and the adaptive immune responses to decrease neuroinflammation (NCT04032626). L-serine, a naturally occurring dietary amino-acid decreasing neuroinflammation, is expected to play a role in brain neuron preservation, similar to montelukast, a leukotriene receptor antagonist that reduces inflammatory pathways and neuronal injury. Sargramostim (GM-CSF) is expected to modulate neuroinflammation by stimulating the right immune response that will remove Aβ and improve synaptic functions. The infection/inflammation modulating agents rifaximin (antibiotic) and valacyclovir (antiviral) studies have been already discussed. All these studies are expected to be completed in the coming years with the hope that some of them may be pushed to phase 3 trials and ultimately become a disease-modifying drug.
There are also some potential drugs in phase 1 trials, including AL002 (monoclonal antibody targeting TREM2 receptors), AL003 (monoclonal antibody targeting SIGLEC-3: CD33), J1J-40346527 (CSF-1R antagonist), salsalate (NSAID), rapamycin (NCT042009110 the CARPEDIEM) and XPro1595 (TNF inhibitor: NCT03943264). They are all designed to mitigate neuroinflammation either by decreasing the microglia activation or increasing microglia functionality for Aβ phagocytosis and clearance.
The discovery that MMP13 and PI3K participate in Aβ production at a later stage of the neuroinflammation could stimulate the use of multistage treatment involving the mitigation of neuroinflammation and the modulation of the MMP13 pathway. This can also apply to many unique treatments that could be more efficient in combination using multi-hit targets. Among all these molecules mentioned above, none of them have proven a substantial efficacy during the trials but we should wait until the completion of these trials to know whether any of them could become a disease-modifying treatment.
Ways to Find New Treatments for AD: What Could Help to Accelerate the DMT Discovery?
To develop new treatments, we could investigate the molecular pathways underlying the pathogenesis of the disease, but it is not presently the case for AD. Another possibility is the combinatorial chemistry which can lead to the discovery of new molecules. Nowadays, one promising way is to repurpose or reorient drugs toward the treatment of AD as many of them are already being used in practice for other indications and revealed to be harmless.
An alternative pathway to drug development, as it was also revealed in the search for COVID-19 treatment, is via the use of computational methods and artificial intelligence (AI).374 In particular, an interesting and novel direction is computational drug design under the infection hypothesis and antimicrobial protection hypothesis of AD.7,95 From the experimental side, receptor-ligand binding assays will be required to quantify binding affinity and kinetics, conformations of targets, binding thermodynamics between Aβ, AMPs and glycoproteins of AD-related microorganisms. This should also include nuclear magnetic resonance, surface plasmon resonance and isothermal titration calorimetry. These experimental data would provide invaluable information to narrow down the drug search space during the computational screening of novel AMPs. Moreover, these computations and experiments should be coupled to research strategies that shy away from transgenic animal models that do not recapitulate human AD. This is possible due to recent technological leaps in stem cell research, which enable lab-grown human mini-brains that reproduce the hallmarks of AD.375,376 The mini-brains (as an alternative AD model) allow for testing of various invivo-based hypotheses and to gather complementary and complex information that perhaps is missed in transgenic animal models. For example, biofilm experiments, neural tissue based on multiomics data from patients and deceased frozen brains can in principle be recreated in mini-brains and tested. Altogether, this framework provides clear targets for the design of AMPs with high-therapeutic efficacy against AD. Indeed, since Aβ is a powerful antimicrobial peptide that targets and neutralizes AD pathogens, then it is reasonable to consider the development of a cocktail of novel and more powerful AMPs based on Aβ template and possibly other peptides (eg LL-37) but without their negative physical-chemical properties. Taking all this together, we can envisage a multistage closed-loop framework between in silico drug screening and drug testing in mini-brains as follows: stage one should involve data mining in existing databases, antimicrobial activity prediction via rational design377 and quantitative structure–activity relationship (QSAR) should generate analogs with improved activity. This step should also incorporate novel computational methods based on topological data analysis (TDA), which enable us to extract topological and geometrical invariants from candidate molecules (see378 for a brief introduction to TDA). The overall aim of this stage is to extract the microscopic structure/features of a molecule characterized by physical-chemical descriptors (polarizability, dipole moment, number of atoms, hydrophobicity, toxicity, etc) and uniquely map it to macroscopic experimental observables (ie activity of the molecule, for example, binding kinetic and thermodynamic parameters). With TDA one can go beyond and include geometrical and topological features of the molecule associated with primary, secondary, tertiary structures (and more) of the molecule. Stage two, should consider state-of-the-art molecular simulations to determine the mechanism of action of AMPs (in particular Aβ) against AD pathogens. Stage three, should combine information gained from steps 1 and 2, and with further determination of physical-chemical descriptors of the generated analogs and Aβ, these can be used to train and screen potential AMP candidates via advanced machine learning. This step should include optimization by means of an evolutionary algorithm, which runs in closed-loop process by bootstrapping the experimental assay (eg mini-brain) to the peptide synthesis process and further interactive in silico prediction by machine learning. During this stage the screened AMPs should be tested against user-desired property (eg IC50), as well as multiomics analysis. In this way AMP sequences can be ranked in terms of the desired property and those of poorest quality are rejected, allowing a new population to be selected. The added value of multiomics is that it departs from traditional experimental studies, which are usually carried out to isolate the effects of a single mechanism and not to investigate the interactions of many mechanisms. This leads to a set of results that are conflicting, difficult to interpret or understand the interactions of the underlying mechanisms leading to the pathogenesis of a disease. Overall, the proposed closed-loop framework based on advanced data analysis and state-of-the-art in silico drug screening provides a systematic and holistic screening of AMPs with high-therapeutic efficacy against AD pathogens. Moreover, it has the potential of accelerating drug design and reducing the overall cost of drug development, which aligns with the National Alzheimer’s Project Act that articulates the ultimate goal of preventing or effectively treating AD by the year 2025.379
Below, we briefly compare Aβ42 (in an apolar microenvironment380) and LL-37381 to provide a glimpse of only a very small part of the proposed closed-loop computational framework in order to sway the AD community about the validity of this research pathway. A key part of screening novel AMPs will involve comparing AMPs and search for new amino acid sequences with improved physical-chemical properties. One step in this direction is to employ traditional primary amino acid sequence alignments and secondary protein alignment (ie 3D structural superposition), as shown in Figure 1 (left panels A–E), which compares Aβ42 and LL-37. Panel A, depicts the 3D secondary structures of Aβ42 in an apolar environment, which appear to depict formation of α-helices (see380). Panel B shows LL-37 and panel C illustrates the 3D alignment and superposition of the two peptides, showing that they possess similarities in their secondary structures. Panel D shows primary sequence alignment and indicates a minor level of amino acid sequence homology between the two peptides. However, by performing sequence alignment followed by 3D structural superposition (of secondary structures) we observe that there is a significant portion of their amino acid sequence that aligns. In Figure 1 (right panels A–E) we use TDA to characterize the topological invariants of the two peptides. For the sake of brevity, we will not explain the method in great detail but rather refer the reader to our recent article that gives an insight of TDA and how it can be applied to high-dimensional and multiscale data.371 However, in brief, TDA extracts topological and geometrical features that persist across spatial scales (hence beyond classical network analysis). These persistent features correspond to invariances of the data and are summarized in specific diagrams as shown in Figure 1 (right panel, A and B). These invariances can in principle be related to primary, secondary, tertiary (and so on) structures of proteins. In Figure 1 (right panel, C–E) we compare the topological invariances of Aβ42 and LL-37 via an appropriate distance called bottleneck distance. The results show that there is some level of topological similarities between these two peptides and we envisage that such information could be used as input features to machine learning algorithms to screen for new AMPs.
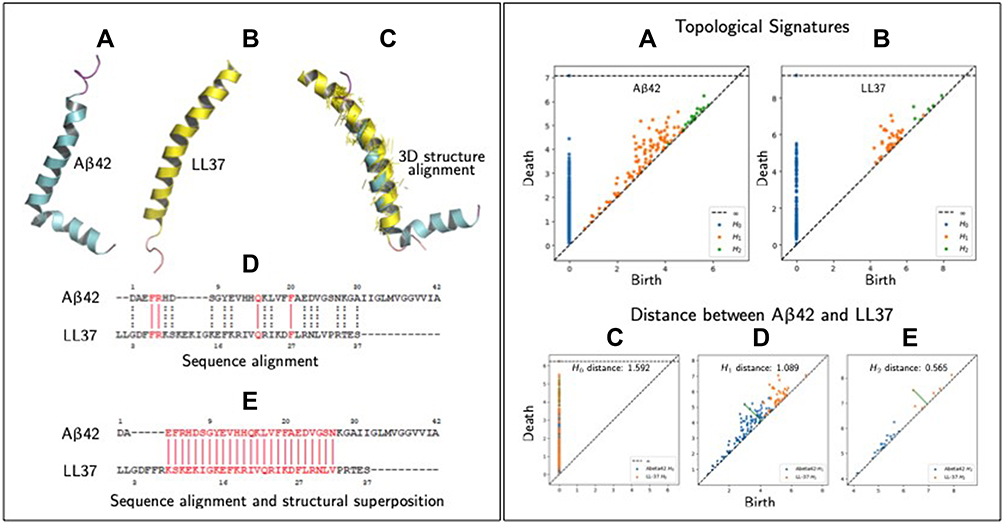 |
Figure 1 Similarities measures between peptides (specifically Aβ42 and LL-37). Left panel, (A) 3D structure of Aβ42 in an apolar environment; data from PDB (RCSB Protein Data Bank, http://www.rcsb.org, PDB ID 1IYT) shown using PyMol software. (B) 3D structure of human host defense cathelicidin LL-37 (RCSB Protein Data Bank, PDB ID 2K6). (C) Structural superposition/alignment of 3D structures of Aβ42 and LL-37 represented in blue and yellow colors, respectively. The yellow colored lines represent actual alignments the algorithm has predicted shown using PyMol. (D) Sequence alignment of Aβ42 and LL-37 using the Clustal Omega shareware (http://expasy.org/proteomics). Identical amino acid residues are indicated by vertical solid red lines and amino acids possessing similar properties, by dashed vertical dotted black lines. (E) Sequence alignment of Aβ42 and LL-37 using PyMol alignment plugin using method “super” whose algorithms can be looked at (http://pymolwiki.org/index.php/Align). Vertical red lines represent the sequence that gets aligned/superimposed in the 3D structure as shown in (C). Right panel, (A) Topological signatures of Aβ42, which persist (birth/death) across scales. The invariants (H0,1,2) are computed with RIpser software (https://ripser.scikit-tda.org/en/latest/), where the input is the peptide as a point cloud. In this case we generated the point cloud in which each point represents one the centroid of the amino acid residue. (B) Topological signatures of LL-37. (C–E) Compares three topological signatures of Aβ42 and LL-37 using bottleneck distances, which shows some level of topological similarities. |
Given the context of infection hypothesis and antimicrobial protection hypothesis of AD we also computed the antimicrobial and antiviral activity of Aβ42 and LL-37, as shown in Tables 3 and 4 respectively. Specifically, in Table 3, we find that various amino-acid subsequences of Aβ42 show antimicrobial activity. However, so far, we found that only a subsequence that overlaps between the Turn and C-terminus region of Aβ42 has antiviral activity. Note that previous studies have suggested that the C-terminus region of Aβ42 has also some similarity with a virus fusion domain.380 Although these results are under an apolar environment, we cannot dismiss the possibility that microenvironments may be formed in the brain due to different conditions and thus these results may inform future AMPs design. Table 4 outlines the antimicrobial activity of various amino acid subsequences of LL-37. However, we were unable to compute the antiviral activity but this could possibly be related to the fact existing online databases need to be updated with novel AMPs that have been found to have antiviral properties.
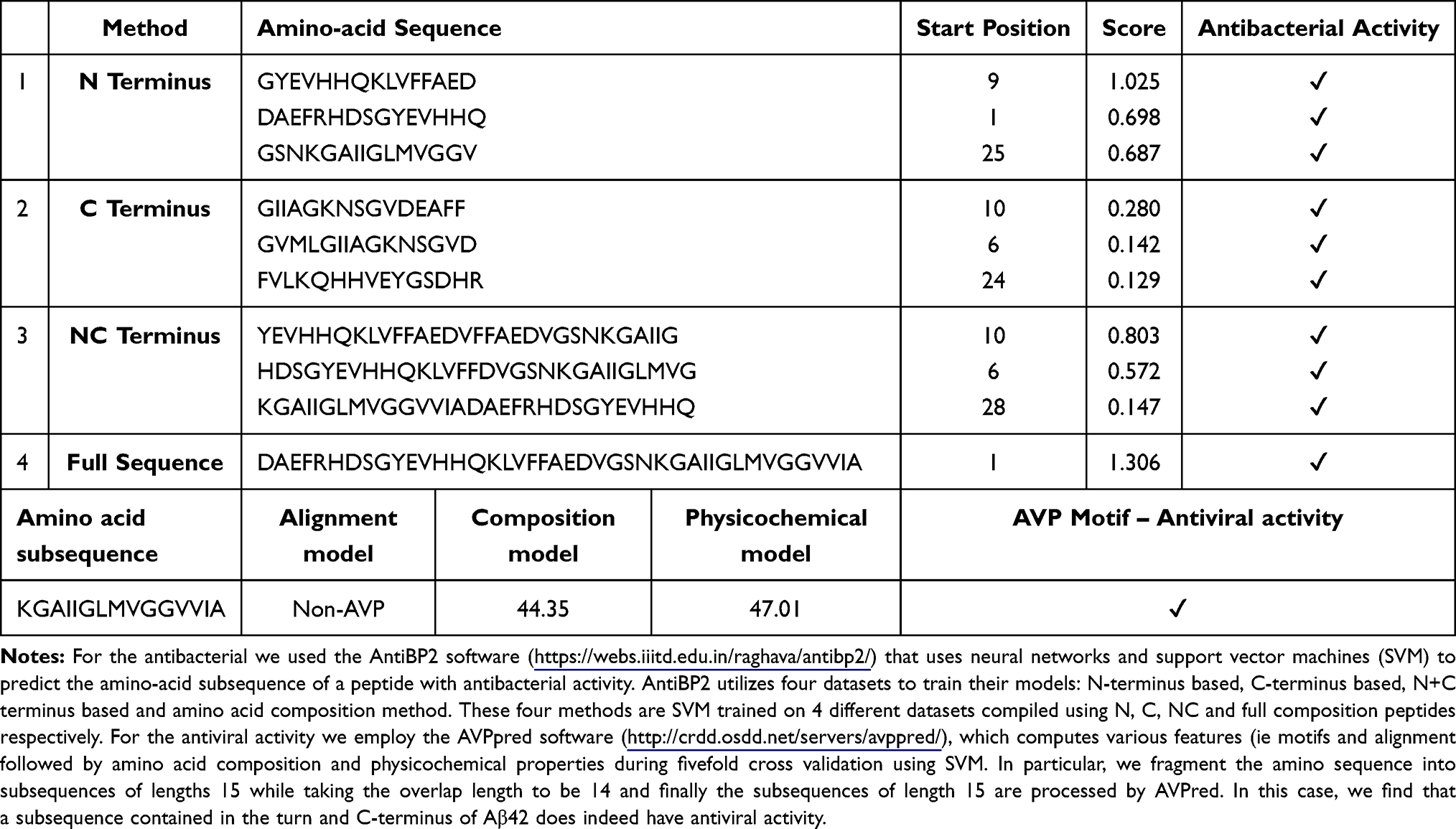 |
Table 3 Antibacterial and Antiviral Activity of Aβ42 |
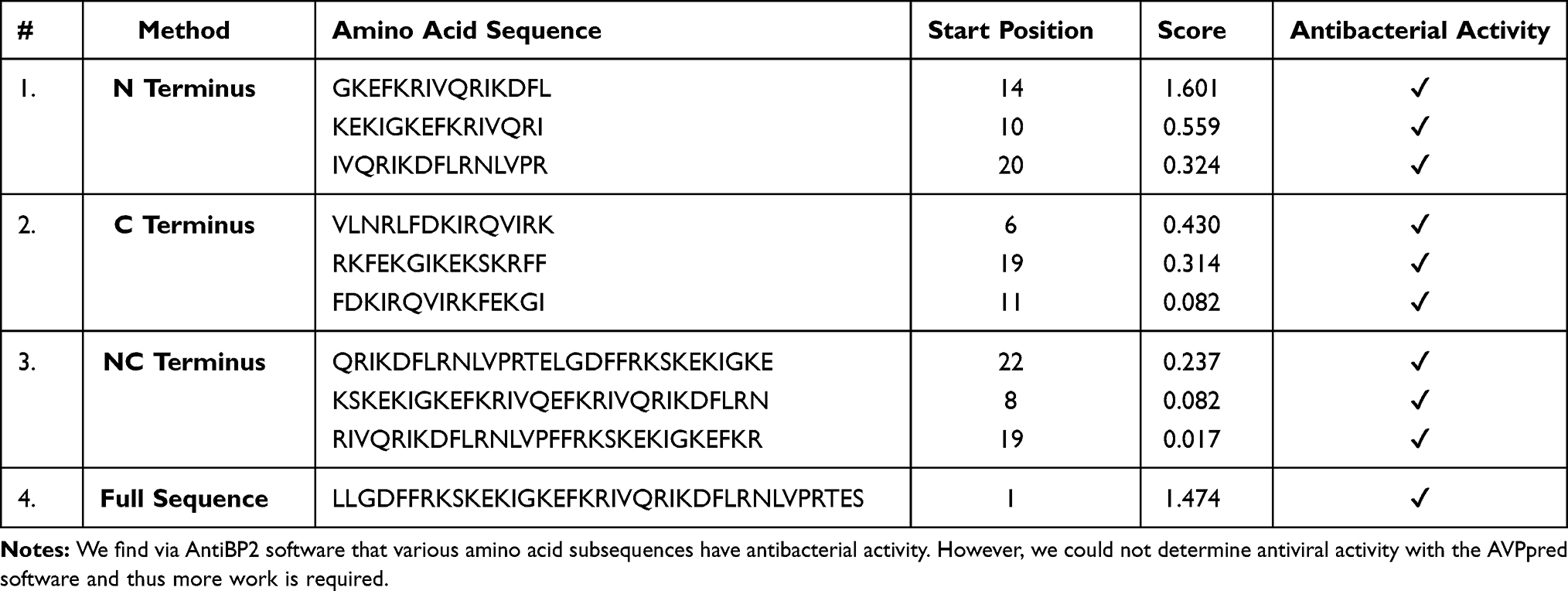 |
Table 4 Antibacterial LL-37 |
Conclusions
It is clear that AD cannot be linked to any specific microbe. According to the emerging infection hypothesis, this is a polymicrobial induced inflammatory disease in which these microbes may play a role in the initiation and the progression of AD; see Figure 2 and Table 5 for a summary. If we successfully link at least a subgroup of AD subjects to underlying chronic or recurrent infections, this could open the way to treat at a preclinical stage of AD to delay or stop the progression. There are several candidates for these treatments but at the end of the road a very few are chosen. However, those chosen could make the difference by decreasing microbial load and reinforcing the immune defense at an early stage.
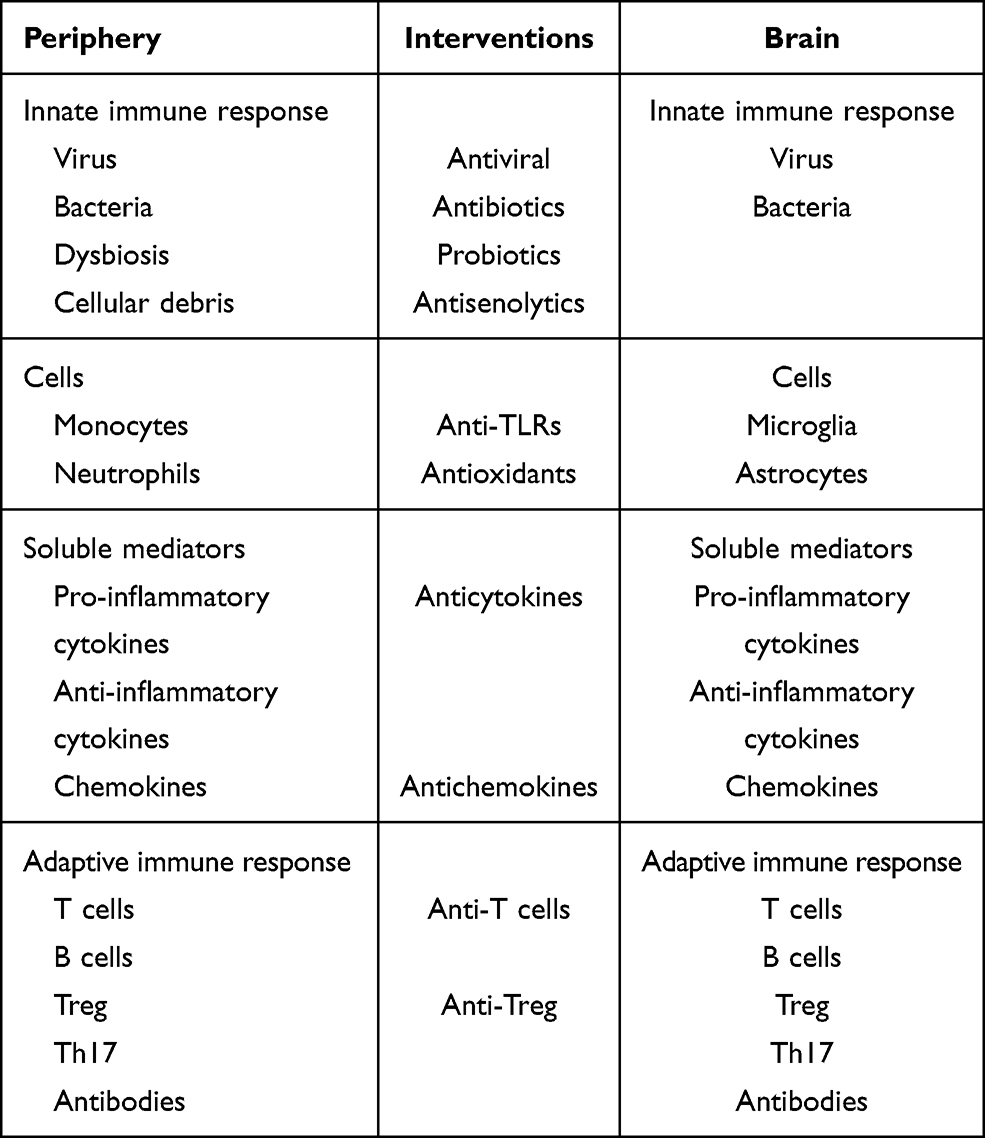 |
Table 5 Summary of the Interventions at the Level of the Periphery and in the Brain |
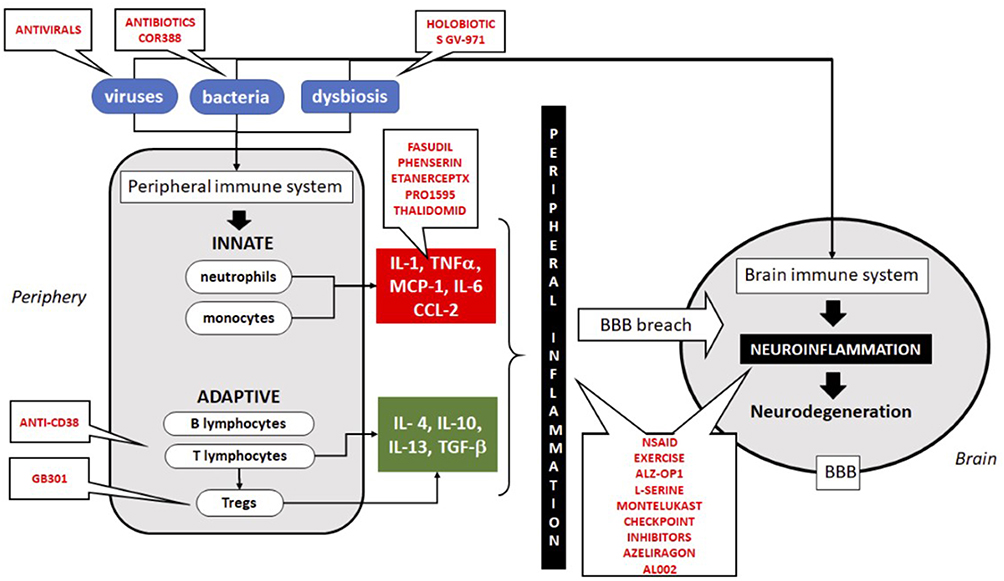 |
Figure 2 Schematic illustration of the immune system implication in neuroinflammation and neurodegeneration and the targets for treatment. All treatments in trial are in red. Abbreviations: IL, interleukins; MCP-1, monocyte chemotactic protein-1; BBB, blood–brain barrier; NSAID, non-steroid anti-inflammatory drugs. |
Furthermore, considering the pathogenesis of AD and its more syndromic nature, it is currently impossible to predict neither the real target nor the moment of an individualized treatment. It is conceivable that more than one treatment could be efficacious which would result in a multimodal intervention, possibly sequentially in time. New ways of thinking are necessary to reinvent the therapeutical approach of AD. Several obstacles should be overcome in designing new drugs such as crossing the BBB, maintaining their activities, and delivering them to the right place. Naturally occurring substances such as flavonoids should be evaluated as well.
We should also be very cautious to use the mice models as templates for humans. There are some similarities, but other models should be used such as 3D brain organoid cultures from human induced pluripotent stem cells (iPSCs). They are also powerful cellular, molecular, genetic, epigenomic techniques to unravel the pathogenetic basis of the disease from human samples. The use of AI techniques is also in constant evolution and could help modeling and find new compounds with potential DMT activities. All these new ways of thinking may lead to promising treatments to alleviate this terrible human disease.
Acknowledgment
The works presented in the article were supported by grants from the Canadian Institutes of Health Research (CIHR) (No. 106634) and No. PJT-162366) to AK and TF, the Société des Médecins de l’Université de Sherbrooke and the Research Center on Aging of the CIUSSS-CHUS, Sherbrooke to TF and EF, the Centre de Recherches Cliniques de l’Université de Sherbrooke to EF, and the FRQS Audace grant to EF, TF, PBB, and J-PBEF; by the Polish Ministry of Science and Higher Education statutory grant 02-0058/07/262 to JMW; by the Agency for Science Technology and Research (A*STAR) to AL, SR is supported by Ikerbasque (The Basque Foundation for Science), by GV-AI-HEALTH, the Basque Government through the BERC 2018-2021 program, by the Spanish State Research Agency through BCAM Severo Ochoa excellence accreditation SEV-2017-0718 and through project RTI2018-093860B-C21 funded by (AEI/FEDER, UE) with acronym “MathNEURO”. MD and SR acknowledge the support of Inria via the Associated Team “NeuroTransSF”. AEB acknowledges financial support from the National Institutes of Health, National Institute on Aging, research grant # 5DP1AG072438 (NIH Director’s Pioneer Award).
Disclosure
Prof. Dr Tamas Fulop reports grants from CIHR, during the conduct of the study; personal fees from Pfizer and Sanofi, outside the submitted work. Dr Ton Bunt is a share holder of Izumi Biosciences INC, outside the submitted work. In addition, Dr Ton Bunt is a co-inventor for patent US-2014235631-A1 pending and an inventor for a patent WO/2019/183403. Professor Annelise E Barron reports grant (# 5DP1AG072438) from NIH/NIA, during the conduct of the study. In addition, Professor Annelise E Barron has a patent US20190015361A1 pending to Stanford University not related to this study. The authors report no other conflicts of interest in this work.
References
1. Brown RC, Lockwood AH, Sonawane BR. Neurodegenerative diseases: an overview of environmental risk factors. Environ Health Perspect. 2005;113(9):1250–1256. doi:10.1289/ehp.7567
2. Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology. 2014;76(Pt A):27–50. doi:10.1016/j.neuropharm.2013.07.004
3. Jessen F, Amariglio RE, Buckley RF, et al. The characterisation of subjective cognitive decline. Lancet Neurol. 2020;19(3):271–278. doi:10.1016/S1474-4422(19)30368-0
4. Jack CR
5. Iqbal UH, Zeng E, Pasinetti GM. The use of antimicrobial and antiviral drugs in Alzheimer’s disease. Int J Mol Sci. 2020;21(14):4920. doi:10.3390/ijms21144920
6. Villemagne VL, Burnham S, Bourgeat P, et al.; Australian Imaging Biomarkers and Lifestyle (AIBL) Research Group. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a Prospective Cohort Study. Lancet Neurol. 2013;12(4):357–367. doi:10.1016/S1474-4422(13)70044-9
7. Fulop T, Witkowski JM, Bourgade K, et al. Can an infection hypothesis explain the beta amyloid hypothesis of Alzheimer’s disease? Front Aging Neurosci. 2018;10:224. doi:10.3389/fnagi.2018.00224
8. Rosario D, Boren J, Uhlen M, et al. Systems biology approaches to understand the host-microbiome interactions in neurodegenerative diseases. Front Neurosci. 2020;14:716. doi:10.3389/fnins.2020.00716
9. Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol. 2018;9:697. doi:10.3389/fimmu.2018.00697
10. Solleiro-Villavicencio H, Rivas-Arancibia S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4+T cells in neurodegenerative diseases. Front Cell Neurosci. 2018;12:114. doi:10.3389/fncel.2018.00114
11. Rothhammer V, Borucki DM, Tjon EC, et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018;557(7707):724–728. doi:10.1038/s41586-018-0119-x
12. Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener. 2020;15(1):40. doi:10.1186/s13024-020-00391-7
13. Le Page A, Dupuis G, Frost EH, et al. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp Gerontol. 2018;107:59–66. doi:10.1016/j.exger.2017.12.019
14. Fulop T, Witkowski JM, Olivieri F, Larbi A. The integration of inflammaging in age-related diseases. Semin Immunol. 2018;40:17–35. doi:10.1016/j.smim.2018.09.003
15. Fülöp T, Dupuis G, Witkowski JM, Larbi A. The role of immunosenescence in the development of age-related diseases. Rev Invest Clin. 2016;68(2):84–91.
16. Pawelec G. Immunosenescence and cancer. Biogerontology. 2017;18(4):717–721. doi:10.1007/s10522-017-9682-z
17. Appay V, Sauce D. Naive T cells: the crux of cellular immune aging? Exp Gerontol. 2014;54:90–93. doi:10.1016/j.exger.2014.01.003
18. Nguyen THO, Sant S, Bird NL, et al. Perturbed CD8+ T cell immunity across universal influenza epitopes in the elderly. J Leukoc Biol. 2018;103(2):321–339. doi:10.1189/jlb.5MA0517-207R
19. Pawelec G. Age and immunity: what is “immunosenescence”? Exp Gerontol. 2018;105:4–9. doi:10.1016/j.exger.2017.10.024
20. Yanes RE, Gustafson CE, Weyand CM, Goronzy JJ. Lymphocyte generation and population homeostasis throughout life. Semin Hematol. 2017;54(1):33–38. doi:10.1053/j.seminhematol.2016.10.003
21. Xu W, Larbi A. Markers of T cell senescence in humans. Int J Mol Sci. 2017;18(8):1742. doi:10.3390/ijms18081742
22. Pawelec G. Hallmarks of human “immunosenescence”: adaptation or dysregulation? Immun Ageing. 2012;9(1):15. doi:10.1186/1742-4933-9-15
23. Larbi A, Fulop T. From “truly naïve” to “exhausted senescent” T cells: when markers predict functionality. Cytometry A. 2014;85(1):25–35. doi:10.1002/cyto.a.22351
24. Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol. 2012;24(5):331–341. doi:10.1016/j.smim.2012.04.008
25. Montgomery RR, Shaw AC. Paradoxical changes in innate immunity in aging: recent progress and new directions. J Leukoc Biol. 2015;98(6):937–943. doi:10.1189/jlb.5MR0315-104R
26. Oh SJ, Lee JK, Shin OS. Aging and the immune system: the impact of immunosenescence on viral infection, immunity and vaccine immunogenicity. Immune Netw. 2019;19(6):e37. doi:10.4110/in.2019.19.e37
27. Fülöp T, Larbi A, Witkowski JM. Human inflammaging. Gerontology. 2019;65(5):495–504. doi:10.1159/000497375
28. Fulop T, Larbi A, Dupuis G, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. 2018;8:1960. doi:10.3389/fimmu.2017.01960
29. Bektas A, Schurman SH, Sen R, Ferrucci L. Aging, inflammation and the environment. Exp Gerontol. 2018;105:10–18. doi:10.1016/j.exger.2017.12.015
30. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9:586. doi:10.3389/fimmu.2018.00586
31. Pawelec G. Immune signatures associated with mortality differ in elderly populations from different birth cohorts and countries even within northern Europe. Mech Ageing Dev. 2019;177:182–185. doi:10.1016/j.mad.2018.04.005
32. Müller L, Fülöp T, Pawelec G. Immunosenescence in vertebrates and invertebrates. Immun Ageing. 2013;10(1):12. doi:10.1186/1742-4933-10-12
33. Tieri P, Grignolio A, Zaikin A, et al. Network, degeneracy and bow tie. Integrating paradigms and architectures to grasp the complexity of the immune system. Theor Biol Med Model. 2010;7(1):32. doi:10.1186/1742-4682-7-32
34. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. doi:10.1038/nri3399
35. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510. doi:10.1038/ni1582
36. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404. doi:10.1038/nri3671
37. Porcheray F, Viaud S, Rimaniol AC, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142(3):481–489. doi:10.1111/j.1365-2249.2005.02934.x
38. Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 2017;38(6):395–406. doi:10.1016/j.it.2017.03.001
39. Chou C, Li MO. Tissue-resident lymphocytes across innate and adaptive lineages. Front Immunol. 2018;9:2104. doi:10.3389/fimmu.2018.02104
40. Hannaway RF, Wang X, Schneider M, et al. Mucosal-associated invariant T cells and Vδ2+ γδ T cells in community acquired pneumonia: association of abundance in sputum with clinical severity and outcome. Clin Exp Immunol. 2020;199(2):201–215. doi:10.1111/cei.13377
41. Godfrey DI, Koay HF, McCluskey J, Gherardin NA. The biology and functional importance of MAIT cells. Nat Immunol. 2019;20(9):1110–1128. doi:10.1038/s41590-019-0444-8
42. Rivera A, Siracusa MC, Yap GS, Gause WC. Innate cell communication kick-starts pathogen-specific immunity. Nat Immunol. 2016;17(4):356–363. doi:10.1038/ni.3375
43. Kaufmann SH, Dorhoi A. Molecular determinants in phagocyte-bacteria interactions. Immunity. 2016;44(3):476–491. doi:10.1016/j.immuni.2016.02.014
44. Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-like receptors: significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol. 2018;37(1):20–36. doi:10.1080/08830185.2017.1380200
45. Kufer TA, Nigro G, Sansonetti PJ. Multifaceted functions of NOD-like receptor proteins in myeloid cells at the intersection of innate and adaptive immunity. Microbiol Spectr. 2016;4(4). doi:10.1128/microbiolspec.MCHD-0021-2015
46. Barik S. What really rigs up RIG-I? J Innate Immun. 2016;8(5):429–436. doi:10.1159/000447947
47. Fulop T, Le Page A, Fortin C, Witkowski JM, Dupuis G, Larbi A. Cellular signaling in the aging immune system. Curr Opin Immunol. 2014;29:105–111. doi:10.1016/j.coi.2014.05.007
48. Pinti M, Appay V, Campisi J, et al. Aging of the immune system: focus on inflammation and vaccination. Eur J Immunol. 2016;46(10):2286–2301. doi:10.1002/eji.201546178
49. Albright JM, Dunn RC, Shults JA, Boe DM, Afshar M, Kovacs EJ. Advanced age alters monocyte and macrophage responses. Antioxid Redox Signal. 2016;25(15):805–815. doi:10.1089/ars.2016.6691
50. Molony RD, Malawista A, Montgomery RR. Reduced dynamic range of antiviral innate immune responses in aging. Exp Gerontol. 2018;107:130–135. doi:10.1016/j.exger.2017.08.019
51. Metcalf TU, Wilkinson PA, Cameron MJ, et al. Human monocyte subsets are transcriptionally and functionally altered in aging in response to pattern recognition receptor agonists. J Immunol. 2017;199(4):1405–1417. doi:10.4049/jimmunol.1700148
52. Fulop T, Larbi A, Douziech N, et al. Signal transduction and functional changes in neutrophils with aging. Aging Cell. 2004;3(4):217–226. doi:10.1111/j.1474-9728.2004.00110.x
53. Fulop T, Dupuis G, Baehl S, et al. From inflamm-aging to immune-paralysis: a slippery slope during aging for immune-adaptation. Biogerontology. 2016;17(1):147–157. doi:10.1007/s10522-015-9615-7
54. Kleinnijenhuis J, Quintin J, Preijers F, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A. 2012;109(43):17537–17542. doi:10.1073/pnas.1202870109
55. van der Heijden CDCC, Noz MP, Joosten LAB, Netea MG, Riksen NP, Keating ST. Epigenetics and trained immunity. Antioxid Redox Signal. 2018;29(11):1023–1040. doi:10.1089/ars.2017.7310
56. Arts RJ, Joosten LA, Netea MG. Immunometabolic circuits in trained immunity. Semin Immunol. 2016;28(5):425–430. doi:10.1016/j.smim.2016.09.002
57. Netea MG, Joosten LAB. Trained immunity and local innate immune memory in the lung. Cell. 2018;175(6):1463–1465. doi:10.1016/j.cell.2018.11.007
58. Domínguez-Andrés J, Fanucchi S, Joosten LAB, Mhlanga MM, Netea MG. Advances in understanding molecular regulation of innate immune memory. Curr Opin Cell Biol. 2020;63:68–75. doi:10.1016/j.ceb.2019.12.006
59. Ciarlo E, Heinonen T, Théroude C, et al. Trained immunity confers broad-spectrum protection against bacterial infections. J Infect Dis. 2020;222(11):1869–1881. doi:10.1093/infdis/jiz692
60. Franceschi C, Salvioli S, Garagnani P, de Eguileor M, Monti D, Capri M. Immunobiography and the heterogeneity of immune responses in the elderly: a focus on inflammaging and trained immunity. Front Immunol. 2017;8:982. doi:10.3389/fimmu.2017.00982
61. Franceschi C, Bonafè M, Valensin S, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908(1):244–254. doi:10.1111/j.1749-6632.2000.tb06651.x
62. Franceschi C, Capri M, Monti D, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128(1):92–105. doi:10.1016/j.mad.2006.11.016
63. Franceschi C, Zaikin A, Gordleeva S, et al. Inflammaging 2018: an update and a model. Semin Immunol. 2018;40:1–5. doi:10.1016/j.smim.2018.10.008
64. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14(10):576–590. doi:10.1038/s41574-018-0059-4
65. Zhao Y, Zhan JK, Liu YA. Perspective on roles played by immunosenescence in the pathobiology of Alzheimer’s disease. Aging Dis. 2020;11(6):1594–1607. doi:10.14336/AD.2020.0205
66. Giunta B, Fernandez F, Nikolic WV, et al. Inflammaging as a prodrome to Alzheimer’s disease. J Neuroinflammation. 2008;5(1):51. doi:10.1186/1742-2094-5-51
67. Sanada F, Taniyama Y, Muratsu J, et al. Source of chronic inflammation in aging. Front Cardiovasc Med. 2018;5:12. doi:10.3389/fcvm.2018.00012
68. Frasca D, Blomberg BB, Paganelli R. Aging, obesity, and inflammatory age-related diseases. Front Immunol. 2017;8:1745. doi:10.3389/fimmu.2017.01745
69. Rubino G, Bulati M, Aiello A, et al. Sicilian centenarian offspring are more resistant to immune ageing. Aging Clin Exp Res. 2019;31(1):125–133. doi:10.1007/s40520-018-0936-7
70. Bauer ME, Fuente Mde MDL. The role of oxidative and inflammatory stress and persistent viral infections in immunosenescence. Mech Ageing Dev. 2016;158:27–37. doi:10.1016/j.mad.2016.01.001
71. Biagi E, Candela M, Fairweather-Tait S, Franceschi C, Brigidi P. Aging of the human metaorganism: the microbial counterpart. Age (Dordr). 2012;34(1):247–267. doi:10.1007/s11357-011-9217-5
72. Biagi E, Candela M, Turroni S, Garagnani P, Franceschi C, Brigidi P. Ageing and gut microbes: perspectives for health maintenance and longevity. Pharmacol Res. 2013;69(1):11–20. doi:10.1016/j.phrs.2012.10.005
73. Thevaranjan N, Puchta A, Schulz C, et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. 2017;21(4):455–466.e4. doi:10.1016/j.chom.2017.03.002
74. Coppé JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi:10.1371/journal.pbio.0060301
75. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–972. doi:10.1172/JCI64098
76. Birch J, Passos JF. Targeting the SASP to combat ageing: mitochondria as possible intracellular allies? Bioessays. 2017;39(5):1600235. doi:10.1002/bies.201600235
77. Campisi J. Cellular senescence and lung function during aging. Yin and Yang. Ann Am Thorac Soc. 2016;13 Suppl 5(Suppl5):S402–S406. doi:10.1513/AnnalsATS.201609-703AW
78. Giuliani A, Prattichizzo F, Micolucci L, Ceriello A, Procopio AD, Rippo MR. Mitochondrial (Dys) function in inflammaging: do mitomirs influence the energetic, oxidative, and inflammatory status of senescent cells? Mediators Inflamm. 2017;2017:2309034. doi:10.1155/2017/2309034
79. Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine. 2017;21:7–13. doi:10.1016/j.ebiom.2017.03.020
80. Terlecki-Zaniewicz L, Lämmermann I, Latreille J, et al. Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype. Aging (Albany NY). 2018;10(5):1103–1132. doi:10.18632/aging.101452
81. Prattichizzo F, Giuliani A, Ceka A, et al. Epigenetic mechanisms of endothelial dysfunction in type 2 diabetes. Clin Epigenetics. 2015;7(1):56. doi:10.1186/s13148-015-0090-4
82. Krämer-Albers EM. Exosomes deliver ROS for regeneration. Nat Cell Biol. 2018;20(3):225–226. doi:10.1038/s41556-018-0048-9
83. Hervera A, De Virgiliis F, Palmisano I, et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat Cell Biol. 2018;20(3):307–319. doi:10.1038/s41556-018-0039-x
84. Jiang F, Zhang Y, Dusting GJ, Sibley DR. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63(1):218–242. doi:10.1124/pr.110.002980
85. Gupta A, Pulliam L. Exosomes as mediators of neuroinflammation. J Neuroinflammation. 2014;11(1):68. doi:10.1186/1742-2094-11-68
86. Howitt J, Hill AF. Exosomes in the pathology of neurodegenerative diseases. J Biol Chem. 2016;291(52):26589–26597. doi:10.1074/jbc.R116.757955
87. Haddad M, Perrotte M, Landri S, Lepage A, Fülöp T, Ramassamy C. Circulating and extracellular vesicles levels of N-(1-carboxymethyl)-L-lysine (CML) differentiate early to moderate Alzheimer’s disease. J Alzheimers Dis. 2019;69(3):751–762. doi:10.3233/JAD-181272
88. Haddad M, Perrotte M, Khedher MRB, et al. Methylglyoxal and glyoxal as potential peripheral markers for MCI diagnosis and their effects on the expression of neurotrophic, inflammatory and neurodegenerative factors in neurons and in neuronal derived-extracellular vesicles. Int J Mol Sci. 2019;20(19):4906. doi:10.3390/ijms20194906
89. Perrotte M, Le Page A, Fournet M, et al. Blood-based redox-signature and their association to the cognitive scores in MCI and Alzheimer’s disease patients. Free Radic Biol Med. 2019;130:499–511. doi:10.1016/j.freeradbiomed.2018.10.452
90. Bourgade K, Garneau H, Giroux G, et al. β-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology. 2015;16(1):85–98. doi:10.1007/s10522-014-9538-8
91. Bourgade K, Le Page A, Bocti C, et al. Protective effect of amyloid-β peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J Alzheimers Dis. 2016;50(4):1227–1241. doi:10.3233/JAD-150652
92. Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. doi:10.1371/journal.pone.0009505
93. White MR, Kandel R, Tripathi S, et al. Alzheimer’s associated beta-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One. 2014;9(7):e101364. doi:10.1371/journal.pone.0101364
94. Kumar DK, Eimer WA, Tanzi RE, Moir RD. Alzheimer’s disease: the potential therapeutic role of the natural antibiotic amyloid-β peptide. Neurodegener Dis Manag. 2016;6(5):345–348. doi:10.2217/nmt-2016-0035
95. Kumar DK, Choi SH, Washicosky KJ, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8(340):340ra72. doi:10.1126/scitranslmed.aaf1059
96. McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126(4):479–497. doi:10.1007/s00401-013-1177-7
97. Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–388. doi:10.1016/0165-6147(91)90609-v
98. Beyreuther K, Masters CL. Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1991;1(4):241–251. doi:10.1111/j.1750-3639.1991.tb00667.x
99. Sacks CA, Avorn J, Kesselheim AS. The failure of solanezumab - how the FDA saved taxpayers billions. N Engl J Med. 2017;376(18):1706–1708. doi:10.1056/NEJMp1701047
100. Cummings J, Ritter A, Zhong K. Clinical trials for disease-modifying therapies in Alzheimer’s disease: a primer, lessons learned, and a blueprint for the future. J Alzheimers Dis. 2018;64(s1):S3–S22. doi:10.3233/JAD-179901
101. Mehta D, Jackson R, Paul G, Shi J, Sabbagh M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs. 2017;26(6):735–739. doi:10.1080/13543784.2017.1323868
102. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312–339. doi:10.1016/j.cell.2019.09.001
103. Itzhaki RF, Lathe R, Balin BJ, et al. Microbes and Alzheimer’s disease. J Alzheimers Dis. 2016;51(4):979–984. doi:10.3233/JAD-160152
104. Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18(6):794–799. doi:10.1038/nn.4017
105. Ricciarelli R, Fedele E. The amyloid cascade hypothesis in Alzheimer’s disease: it’s time to change our mind. Curr Neuropharmacol. 2017;15(6):926–935. doi:10.2174/1570159X15666170116143743
106. Osorio C, Kanukuntla T, Diaz E, Jafri N, Cummings M, Sfera A. The post-amyloid era in Alzheimer’s disease: trust your gut feeling. Front Aging Neurosci. 2019;11:143. doi:10.3389/fnagi.2019.00143
107. Tam JH, Pasternak SH. Amyloid and Alzheimer’s disease: inside and out. Can J Neurol Sci. 2012;39(3):286–298. doi:10.1017/s0317167100013408
108. Kern A, Behl C. The unsolved relationship of brain aging and late-onset Alzheimer disease. Biochim Biophys Acta. 2009;1790(10):1124–1132. doi:10.1016/j.bbagen.2009.07.016
109. Castellani RJ, Rolston RK, Smith MA. Alzheimer disease. Dis Mon. 2010;56(9):484–546. doi:10.1016/j.disamonth.2010.06.001
110. Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett. 2007;429(2–3):95–100. doi:10.1016/j.neulet.2007.09.077
111. Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol. 2009;217(1):131–138. doi:10.1002/path.2449
112. Wozniak MA, Frost AL, Preston CM, Itzhaki RF. Antivirals reduce the formation of key Alzheimer’s disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS One. 2011;6(10):e25152. doi:10.1371/journal.pone.0025152
113. Miklossy J. Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med. 2011;13:e30. doi:10.1017/S1462399411002006
114. Miklossy J. Bacterial amyloid and DNA are important constituents of senile plaques: further evidence of the spirochetal and biofilm nature of senile plaques. J Alzheimers Dis. 2016;53(4):1459–1473. doi:10.3233/JAD-160451
115. Miklossy J, McGeer PL. Common mechanisms involved in Alzheimer’s disease and type 2 diabetes: a key role of chronic bacterial infection and inflammation. Aging (Albany NY). 2016;8(4):575–588. doi:10.18632/aging.100921
116. Miklossy J. Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation. 2011;8(1):90. doi:10.1186/1742-2094-8-90
117. Pritchard AB, Crean S, Olsen I, Singhrao SK. Periodontitis, microbiomes and their role in Alzheimer’s disease. Front Aging Neurosci. 2017;9:336. doi:10.3389/fnagi.2017.00336
118. Singhrao SK, Harding A, Poole S, Kesavalu L, Crean S. Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer’s disease. Mediators Inflamm. 2015;2015:137357. doi:10.1155/2015/137357
119. Sadrameli M, Bathini P, Alberi L. Linking mechanisms of periodontitis to Alzheimer’s disease. Curr Opin Neurol. 2020;33(2):230–238. doi:10.1097/WCO.0000000000000797
120. Dioguardi M, Crincoli V, Laino L, et al. The role of periodontitis and periodontal bacteria in the onset and progression of Alzheimer’s disease: a systematic review. J Clin Med. 2020;9(2):495. doi:10.3390/jcm9020495
121. Sun YQ, Richmond RC, Chen Y, Mai XM. Mixed evidence for the relationship between periodontitis and Alzheimer’s disease: a Bidirectional Mendelian Randomization Study. PLoS One. 2020;15(1):e0228206. doi:10.1371/journal.pone.0228206
122. Dominy SS, Lynch C, Ermini F, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eaau3333. doi:10.1126/sciadv.aau3333
123. Singhrao SK, Harding A. Is Alzheimer’s disease a polymicrobial host microbiome dysbiosis? Expert Rev Anti Infect Ther. 2020;18(4):275–277. doi:10.1080/14787210.2020.1729741
124. Bourgade K, Dupuis G, Frost EH, Fülöp T. Anti-viral properties of amyloid-β peptides. J Alzheimers Dis. 2016;54(3):859–878. doi:10.3233/JAD-160517
125. Bu XL, Yao XQ, Jiao SS, et al. A study on the association between infectious burden and Alzheimer’s disease. Eur J Neurol. 2015;22(12):1519–1525. doi:10.1111/ene.12477
126. Carbone I, Lazzarotto T, Ianni M, et al. Herpes virus in Alzheimer’s disease: relation to progression of the disease. Neurobiol Aging. 2014;35(1):122–129. doi:10.1016/j.neurobiolaging.2013.06.024
127. Licastro F, Carbone I, Ianni M, Porcellini E. Gene signature in Alzheimer’s disease and environmental factors: the virus chronicle. J Alzheimers Dis. 2011;27(4):809–817. doi:10.3233/JAD-2011-110755
128. McManus RM, Heneka MT. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther. 2017;9(1):14. doi:10.1186/s13195-017-0241-2
129. Bolós M, Perea JR, Avila J. Alzheimer’s disease as an inflammatory disease. Biomol Concepts. 2017;8(1):37–43. doi:10.1515/bmc-2016-0029
130. Yang SH. Cellular and molecular mediators of neuroinflammation in Alzheimer disease. Int Neurourol J. 2019;23(Suppl 2):S54–62. doi:10.5213/inj.1938184.092
131. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. doi:10.1016/S1474-4422(15)70016-5
132. Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des. 2010;16(25):2766–2778. doi:10.2174/138161210793176572
133. Su F, Bai F, Zhou H, Zhang Z. Microglial toll-like receptors and Alzheimer’s disease. Brain Behav Immun. 2016;52:187–198. doi:10.1016/j.bbi.2015.10.010
134. Venegas C, Heneka MT. Danger-associated molecular patterns in Alzheimer’s disease. J Leukoc Biol. 2017;101(1):87–98. doi:10.1189/jlb.3MR0416-204R
135. Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):594–604. doi:10.1016/j.bcp.2014.01.008
136. Fulop T, Dupuis G, Baehl S, et al. From inflamm-aging to immune-paralysis: a slippery slope during aging for immune-adaptation. Biogerontology. 2016;17(1):147. (voir plus haut). doi:10.1007/s10522-015-9615-7
137. Kritsilis M, Rizou SV, Koutsoudaki PN, Evangelou K, Gorgoulis VG, Papadopoulos D. Ageing, cellular senescence and neurodegenerative disease. Int J Mol Sci. 2018;19(10):2937. doi:10.3390/ijms19102937
138. Nakamura K, Kawakami T, Yamamoto N, et al. Activation of the NLRP3 inflammasome by cellular labile iron. Exp Hematol. 2016;44(2):116–124. doi:10.1016/j.exphem.2015.11.002
139. Calvani R, Picca A, Lo Monaco MR, Landi F, Bernabei R, Marzetti E. Of microbes and minds: a narrative review on the second brain aging. Front Med (Lausanne). 2018;5:53. doi:10.3389/fmed.2018.00053
140. Seo DO, Boros BD, Holtzman DM. The microbiome: a target for Alzheimer disease?. Cell Res. 2019;29(10):779–780. doi:10.1038/s41422-019-0227-7
141. Friedland RP, Chapman MR. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017;13(12):e1006654. doi:10.1371/journal.ppat.1006654
142. Festoff BW, Sajja RK, van Dreden P, Cucullo L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J Neuroinflammation. 2016;13(1):194. doi:10.1186/s12974-016-0670-z
143. Blach-Olszewska Z, Zaczynska E, Gustaw-Rothenberg K, et al. The innate immunity in Alzheimer disease- relevance to pathogenesis and therapy. Curr Pharm Des. 2015;21(25):3582–3588. doi:10.2174/1381612821666150710144829
144. Busse M, Michler E, von Hoff F, et al. Alterations in the peripheral immune system in dementia. J Alzheimers Dis. 2017;58(4):1303–1313. doi:10.3233/JAD-161304
145. Bulgart HR, Neczypor EW, Wold LE, Mackos AR. Microbial involvement in Alzheimer disease development and progression. Mol Neurodegener. 2020;15(1):42. doi:10.1186/s13024-020-00378-4
146. Naughton SX, Raval U, Pasinetti GM. The viral hypothesis in Alzheimer’s disease: novel insights and pathogen-based biomarkers. J Pers Med. 2020;10(3):74. doi:10.3390/jpm10030074
147. Li H, Liu CC, Zhen H, Huang TY. Amyloid, tau, pathogen infection and antimicrobial protection in Alzheimer’s disease -conformist, nonconformist, and realistic prospects for AD pathogenesis. Transl Neurodegener. 2018;7:34. doi:10.1186/s40035-018-0139-3
148. Broxmeyer L. Are the infectious roots of Alzheimer’s buried deep in the past? J Mol Path Epidemol. 2017;3:2.
149. Jamieson GA, Maitland NJ, Craske J, Wilcock GK, Itzhaki RF. Detection of herpes simplex virus type 1 DNA sequences in normal and Alzheimer’s disease brain using polymerase chain reaction. Biochem Soc Trans. 1991;19(2):122S. doi:10.1042/bst019122s
150. Jamieson GA, Maitland NJ, Wilcock GK, Yates CM, Itzhaki RF. Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J Pathol. 1992;167(4):365–368. doi:10.1002/path.1711670403
151. Tzeng NS, Chung CH, Lin FH, et al. Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections-a nationwide, population-based cohort study in Taiwan. Neurotherapeutics. 2018;15(2):417–429. doi:10.1007/s13311-018-0611-x
152. Lopatko Lindman K, Weidung B, Olsson J, et al. A genetic signature including apolipoprotein Eε4 potentiates the risk of herpes simplex-associated Alzheimer’s disease. Alzheimers Dement. 2019;5(1):697–704. doi:10.1016/j.trci.2019.09.014
153. Baringer JR, Swoveland P. Recovery of herpes-simplex virus from human trigeminal ganglions. N Engl J Med. 1973;288(13):648–650. doi:10.1056/NEJM197303292881303
154. Romagnoli M, Porcellini E, Carbone I, Veerhuis R, Licastro F. Impaired innate immunity mechanisms in the brain of Alzheimer’s disease. Int J Mol Sci. 2020;21(3):1126. doi:10.3390/ijms21031126
155. Nicoll MP, Proença JT, Efstathiou S. The molecular basis of herpes simplex virus latency. FEMS Microbiol Rev. 2012;36(3):684–705. doi:10.1111/j.1574-6976.2011.00320.x
156. Grinde B, Causa P, Giuliani E, Nunziata A. Herpesviruses: latency and reactivation - viral strategies and host response. J Oral Microbiol. 2013;25;5(9):1436–1442. doi:10.3402/jom.v5i0.22766
157. Readhead B, Haure-Mirande JV, Funk CC, et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 2018;99(1):64–82.e7. doi:10.1016/j.neuron.2018.05.023
158. Naughton SX, Raval U, Pasinetti GM. Potential novel role of covid-19 in Alzheimer’s disease and preventative mitigation strategies. J Alzheimers Dis. 2020;76(1):21–25. doi:10.3233/JAD-200537
159. Itzhaki RF. Antivirals against SARS-CoV2: relevance to the treatment of Alzheimer’s disease. J Alzheimers Dis. 2020;78(3):905–906. doi:10.3233/JAD-200986
160. Fotuhi M, Mian A, Meysami S, Raji CA. Neurobiology of COVID-19. J Alzheimers Dis. 2020;76(1):3–19. doi:10.3233/JAD-200581
161. Balin BJ, Hammond CJ, Little CS, et al. Chlamydia pneumoniae: an etiologic agent for late-onset dementia. Front Aging Neurosci. 2018;10:302. doi:10.3389/fnagi.2018.00302
162. Dando SJ, Mackay-Sim A, Norton R, et al. Pathogens penetrating the central nervous system: infection pathways and the cellular and molecular mechanisms of invasion. Clin Microbiol Rev. 2014;27(4):691–726. doi:10.1128/CMR.00118-13
163. Leira Y, Domínguez C, Seoane J, et al. Is periodontal disease associated with Alzheimer’s disease? a systematic review with meta-analysis. Neuroepidemiology. 2017;48(1–2):21–31. doi:10.1159/000458411
164. Goyal D, Ali SA, Singh RK. Emerging role of gut microbiota in modulation of neuroinflammation and neurodegeneration with emphasis on Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2021;106:110112. doi:10.1016/j.pnpbp.2020.110112
165. Borsom EM, Lee K, Cope EK. Do the bugs in your gut eat your memories? Relationship between gut microbiota and Alzheimer’s disease. Brain Sci. 2020;10(11):814. doi:10.3390/brainsci10110814
166. Friedland RP, Chapman MR, Bliska JB. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017;13(12):e1006654. doi:10.1371/journal.ppat.1006654
167. Kelly JR, Clarke G, Cryan JF, Dinan TG. Brain-gut-microbiota axis: challenges for translation in psychiatry. Ann Epidemiol. 2016;26(5):366–372. doi:10.1016/j.annepidem.2016.02.008
168. Nwafor DC, Brichacek AL, Mohammad AS, et al. Targeting the blood-brain barrier to prevent sepsis-associated cognitive impairment. J Cent Nerv Syst Dis. 2019;11:1179573519840652. doi:10.1177/1179573519840652
169. Torres-Fuentes C, Schellekens H, Dinan TG, Cryan JF. The microbiota-gut-brain axis in obesity. Lancet Gastroenterol Hepatol. 2017;2(10):747–756. doi:10.1016/S2468-1253(17)30147-4
170. Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28(2):203–209.
171. Naseer MI, Bibi F, Alqahtani MH, et al. Role of gut microbiota in obesity, type 2 diabetes and Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2014;13(2):305–311. doi:10.2174/18715273113126660147
172. Vogt NM, Kerby RL, Dill-McFarland KA, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7(1):13537. doi:10.1038/s41598-017-13601-y
173. van de Haar HJ, Burgmans S, Jansen JF, et al. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology. 2016;281(2):527–535. doi:10.1148/radiol.2016152244
174. Bohórquez DV, Liddle RA. The gut connectome: making sense of what you eat. J Clin Invest. 2015;125(3):888–890. doi:10.1172/JCI81121
175. Zhao Y, Lukiw WJ. Bacteroidetes neurotoxins and inflammatory neurodegeneration. Mol Neurobiol. 2018;55(12):9100–9107. doi:10.1007/s12035-018-1015-y
176. Asti A, Gioglio L. Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J Alzheimers Dis. 2014;39(1):169–179. doi:10.3233/JAD-131394
177. Zhan X, Stamova B, Jin LW, DeCarli C, Phinney B, Sharp FR. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology. 2016;87(22):2324–2332. doi:10.1212/WNL.0000000000003391
178. Cerovic M, Forloni G, Balducci C. Neuroinflammation and the gut microbiota: possible alternative therapeutic targets to counteract Alzheimer’s disease? Front Aging Neurosci. 2019;18(11):284. doi:10.3389/fnagi.2019.00284
179. Tetz G, Pinho M, Pritzkow S, Mendez N, Soto C, Tetz V. Bacterial DNA promotes Tau aggregation. Sci Rep. 2020;10(1):2369. doi:10.1038/s41598-020-59364-x
180. Zhao Y, Jaber V, Lukiw WJ. Secretory products of the human GI tract microbiome and their potential impact on Alzheimer’s disease (AD): detection of lipopolysaccharide (LPS) in AD hippocampus. Front Cell Infect Microbiol. 2017;7:318. doi:10.3389/fcimb.2017.00318
181. Lee JW, Lee YK, Yuk DY, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5(1):37. doi:10.1186/1742-2094-5-37
182. Sampson TR, Mazmanian SK. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe. 2015;17(5):565–576. doi:10.1016/j.chom.2015.04.011
183. Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell. 2016;167(4):915–932. doi:10.1016/j.cell.2016.10.027
184. Erny D, Hrabě de Angelis AL, Jaitin D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965–977. doi:10.1038/nn.4030
185. Raval U, Harary JM, Zeng E, Pasinetti GM. The dichotomous role of the gut microbiome in exacerbating and ameliorating neurodegenerative disorders. Expert Rev Neurother. 2020;20(7):673–686. doi:10.1080/14737175.2020.1775585
186. Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol. 2020;98(1):28–41. doi:10.1111/imcb.12301
187. Lee CYD, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna). 2010;117(8):949–960. doi:10.1007/s00702-010-0433-4
188. Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457–470. doi:10.1038/nrneurol.2015.119
189. Nyúl-Tóth Á, Suciu M, Molnár J, et al. Differences in the molecular structure of the blood-brain barrier in the cerebral cortex and white matter: an in silico, in vitro, and ex vivo study. Am J Physiol Heart Circ Physiol. 2016;310(11):H1702–14. doi:10.1152/ajpheart.00774.2015
190. Ransohoff RM. Physiology. Good barriers make good neighbors. Science. 2014;346(6205):36–37. doi:10.1126/science.1260705
191. Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971(2):197–209. doi:10.1016/s0006-8993(03)02361-8
192. Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2(6):e167. doi:10.1038/cddis.2011.50
193. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463–477. doi:10.1038/nri3705
194. Skaper SD, Facci L, Zusso M, Giusti P. An inflammation-centric view of neurological disease: beyond the neuron. Front Cell Neurosci. 2018;12:72. doi:10.3389/fncel.2018.00072
195. Arranz AM, De Strooper B. The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol. 2019;18(4):406–414. doi:10.1016/S1474-4422(18)30490-3
196. Dionisio-Santos DA, Olschowka JA, O’Banion MK. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J Neuroinflammation. 2019;16(1):74. doi:10.1186/s12974-019-1453-0
197. Fani Maleki A, Rivest S. Innate immune cells: monocytes, monocyte-derived macrophages and microglia as therapeutic targets for alzheimer’s disease and multiple sclerosis. Front Cell Neurosci. 2019;13:355. doi:10.3389/fncel.2019.00355
198. Chan WY, Kohsaka S, Rezaie P. The origin and cell lineage of microglia: new concepts. Brain Res Rev. 2007;53(2):344–354. doi:10.1016/j.brainresrev.2006.11.002
199. Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis. 2010;37(3):503–509. doi:10.1016/j.nbd.2009.10.006
200. Mammana S, Fagone P, Cavalli E, et al. The role of macrophages in neuroinflammatory and neurodegenerative pathways of alzheimer’s disease, amyotrophic lateral sclerosis, and multiple sclerosis: pathogenetic cellular effectors and potential therapeutic targets. Int J Mol Sci. 2018;19(3):831. doi:10.3390/ijms19030831
201. Wieghofer P, Prinz M. Genetic manipulation of microglia during brain development and disease. Biochim Biophys Acta. 2016;1862(3):299–309. doi:10.1016/j.bbadis.2015.09.019
202. Ramya V, Bhuvaneshwarri R, Paddmanabhan P, Manisundar N. Alziemer’s disease and periodontal disease bidirectional interrelationships. Biosci Biotechnol Res Asia. 2014;11(1):259–261. doi:10.13005/bbra/1264
203. Ransohoff RM. The MHP36 line of murine neural stem cells expresses functional CXCR1 chemokine receptors that initiate chemotaxis in vitro. J Neuroimmunol. 2007;186(1–2):199. author reply 200. doi:10.1016/j.jneuroim.2007.03.018
204. Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Exp Neurol. 2009;215(1):5–19. doi:10.1016/j.expneurol.2008.09.003
205. Mecca C, Giambanco I, Donato R, Arcuri C. Microglia and aging: the role of the TREM2-DAP12 and CX3CL1-CX3CR1 axes. Int J Mol Sci. 2018;19(1):318. doi:10.3390/ijms19010318
206. Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):29. doi:10.1186/s13024-018-0262-8
207. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi:10.12703/P6-13
208. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19(8):987–991. doi:10.1038/nn.4338
209. Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. doi:10.1016/j.cell.2017.05.018
210. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. 2018;173(5):1073–1081. doi:10.1016/j.cell.2018.05.003
211. Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–581.e9. doi:10.1016/j.immuni.2017.08.008
212. Rangaraju S, Dammer EB, Raza SA, et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):24. doi:10.1186/s13024-018-0254-8
213. Bennett JP
214. McQuade A, Kang YJ, Hasselmann J, et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat Commun. 2020;11(1):5370. doi:10.1038/s41467-020-19227-5
215. Parvathy S, Rajadas J, Ryan H, Vaziri S, Anderson L, Murphy GM
216. Sondag CM, Dhawan G, Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation. 2009;6(1):1. doi:10.1186/1742-2094-6-1
217. El Khoury JB, Moore KJ, Means TK, et al. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197(12):1657–1666. doi:10.1084/jem.20021546
218. Weber ANR, Bittner ZA, Shankar S, et al. Recent insights into the regulatory networks of NLRP3 inflammasome activation. J Cell Sci. 2020;133(23):jcs248344. doi:10.1242/jcs.248344
219. Hemonnot AL, Hua J, Ulmann L, Hirbec H. Microglia in Alzheimer disease: well-known targets and new opportunities. Front Aging Neurosci. 2019;11:233. doi:10.3389/fnagi.2019.00233
220. Hensel N, Raker V, Förthmann B, et al. The proteome and secretome of cortical brain cells infected with herpes simplex virus. Front Neurol. 2020;11:844. doi:10.3389/fneur.2020.00844
221. Nau R, Sörgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood–brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858–883. doi:10.1128/CMR.00007-10
222. Rajendran L, Paolicelli RC. Microglia-mediated synapse loss in Alzheimer’s disease. J Neurosci. 2018;38(12):2911–2919. doi:10.1523/JNEUROSCI.1136-17.2017
223. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217(2):459–472. doi:10.1083/jcb.201709069
224. Fan H, Wu PF, Zhang L, et al. Methionine sulfoxide reductase A negatively controls microglia-mediated neuroinflammation via inhibiting ROS/MAPKs/NF-κB signaling pathways through a catalytic antioxidant function. Antioxid Redox Signal. 2015;22(10):832–847. doi:10.1089/ars.2014.6022
225. González-Sanmiguel J, Schuh CMAP, Muñoz-Montesino C, Contreras-Kallens P, Aguayo LG, Aguayo S. Complex Interaction between resident microbiota and misfolded proteins: role in neuroinflammation and neurodegeneration. Cells. 2020;9(11):2476. doi:10.3390/cells9112476
226. Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9(6):429–439. doi:10.1038/nri2565
227. Yang J, Wise L, Fukuchi KI. TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front Immunol. 2020;11:724. doi:10.3389/fimmu.2020.00724
228. Hanslik KL, Ulland TK. The role of microglia and the nlrp3 inflammasome in Alzheimer’s disease. Front Neurol. 2020;11:570711. doi:10.3389/fneur.2020.570711
229. Asiimwe N, Yeo SG, Kim MS, Jung J, Jeong NY. Nitric oxide: exploring the contextual link with Alzheimer’s disease. Oxid Med Cell Longev. 2016;2016:7205747. doi:10.1155/2016/7205747
230. Tse KH, Herrup K. DNA damage in the oligodendrocyte lineage and its role in brain aging. Mech Ageing Dev. 2017;161(Pt A):37–50. doi:10.1016/j.mad.2016.05.006
231. Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. 2017;39(1):73–82. doi:10.1080/01616412.2016.1251711
232. Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-related alterations in the dynamic behavior of microglia. Aging Cell. 2011;10(2):263–276. doi:10.1111/j.1474-9726.2010.00660.x
233. Floden AM, Combs CK. Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J Alzheimers Dis. 2011;25(2):279–293. doi:10.3233/JAD-2011-101014
234. Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW. Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain. 2016;139(Pt 3):653–661. doi:10.1093/brain/awv395
235. Ní Chasaide C, Lynch MS. The role of the immune system in driving neuroinflammation. Brain Neurosci Adv. 2020;4:2398212819901082. doi:10.1177/2398212819901082
236. Larbi A, Pawelec G, Witkowski JM, et al. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis. 2009;17(1):91–103. doi:10.3233/JAD-2009-1015
237. Jóźwik A, Landowski J, Bidzan L, Fülop T, Bryl E, Witkowski JM. Beta-amyloid peptides enhance the proliferative response of activated CD4CD28 lymphocytes from Alzheimer disease patients and from healthy elderly. PLoS One. 2012;7(3):e33276. doi:10.1371/journal.pone.0033276
238. Smolders J, Heutinck KM, Fransen NL, et al. Tissue-resident memory T cells populate the human brain. Nat Commun. 2018;9(1):4593. doi:10.1038/s41467-018-07053-9
239. Ciccocioppo F, Lanuti P, Pierdomenico L, et al. The characterization of regulatory T-cell profiles in Alzheimer’s disease and multiple sclerosis. Sci Rep. 2019;9(1):8788. doi:10.1038/s41598-019-45433-3
240. Gate D, Saligrama N, Leventhal O, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577(7790):399–404. doi:10.1038/s41586-019-1895-7
241. Merlini M, Kirabali T, Kulic L, Nitsch RM, Ferretti MT. Extravascular CD3+ T cells in brains of Alzheimer disease patients correlate with tau but not with amyloid pathology: an Immunohistochemical Study. Neurodegener Dis. 2018;18(1):49–56. doi:10.1159/000486200
242. Townsend KP, Town T, Mori T, et al. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur J Immunol. 2005;35(3):901–910. doi:10.1002/eji.200425585
243. Marsh SE, Abud EM, Lakatos A, et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A. 2016;113(9):E1316–25. doi:10.1073/pnas.1525466113
244. Wyatt-Johnson SK, Brutkiewicz RR. The complexity of microglial interactions with innate and adaptive immune cells in Alzheimer’s disease. Front Aging Neurosci. 2020;12:592359. doi:10.3389/fnagi.2020.592359
245. Mayne K, White JA, McMurran CE, Rivera FJ, de la Fuente AG. Aging and neurodegenerative disease: is the adaptive immune system a friend or foe? Front Aging Neurosci. 2020;12:572090. doi:10.3389/fnagi.2020.572090
246. Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999;10(2):119–130. doi:10.1016/s1359-6101(99)00008-8
247. Hensley K. Neuroinflammation in Alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis. 2010;21(1):1–14. doi:10.3233/JAD-2010-1414
248. Zhang Y, Zhao Y, Zhang J, Yang G. Mechanisms of NLRP3 inflammasome activation: its role in the treatment of Alzheimer’s disease. Neurochem Res. 2020;45(11):2560–2572. doi:10.1007/s11064-020-03121-z
249. Saresella M, La Rosa F, Piancone F, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener. 2016;11(1):23. doi:10.1186/s13024-016-0088-1
250. Shen H, Guan Q, Zhang X, et al. New mechanism of neuroinflammation in Alzheimer’s disease: the activation of NLRP3 inflammasome mediated by gut microbiota. Prog Neuropsychopharmacol Biol Psychiatry. 2020;100:109884. doi:10.1016/j.pnpbp.2020.109884
251. Chelbi-Alix MK, Wietzerbin J. Interferon, a growing cytokine family: 50 years of interferon research. Biochimie. 2007;89(6–7):713–718. doi:10.1016/j.biochi.2007.05.001
252. Capobianchi MR, Uleri E, Caglioti C, Dolei A. Type I IFN family members: similarity, differences and interaction. Cytokine Growth Factor Rev. 2015;26(2):103–111. doi:10.1016/j.cytogfr.2014.10.011
253. Khorooshi R, Owens T. Injury-induced type I IFN signaling regulates inflammatory responses in the central nervous system. J Immunol. 2010;185(2):1258–1264. doi:10.4049/jimmunol.0901753
254. Licastro F, Raschi E, Carbone I, Porcellini E. Variants in antiviral genes are risk factors for cognitive decline and dementia. J Alzheimers Dis. 2015;46(3):655–663. doi:10.3233/JAD-142718
255. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity. 2019;50(4):907–923. doi:10.1016/j.immuni.2019.03.025
256. Egli A, Santer DM, O’Shea D, Tyrrell DL, Houghton M. The impact of the interferon-lambda family on the innate and adaptive immune response to viral infections. Emerg Microbes Infect. 2014;3(7):e51. doi:10.1038/emi.2014.51
257. Osterlund PI, Pietilä TE, Veckman V, Kotenko SV, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179(6):3434–3442. doi:10.4049/jimmunol.179.6.3434
258. Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12(6):399–414. doi:10.1038/gene.2011.21
259. Welling MM, Nabuurs RJ, van der Weerd L. Potential role of antimicrobial peptides in the early onset of Alzheimer’s disease. Alzheimers Dement. 2015;11(1):51–57. doi:10.1016/j.ja
260. De Lorenzi E, Chiari M, Colombo R, et al. Evidence that the human innate immune peptide LL-37 may be a binding partner of amyloid-β and inhibitor of fibril assembly. J Alzheimers Dis. 2017;59(4):1213–1226. doi:10.3233/JAD-170223
261. Lee EY, Chan LC, Wang H, et al. PACAP is a pathogen-inducible resident antimicrobial neuropeptide affording rapid and contextual molecular host defense of the brain. Proc Natl Acad Sci U S A. 2021;118(1):e1917623117. doi:10.1073/pnas.1917623117
262. Augustyniak D, Nowak J, Lundy FT. Direct and indirect antimicrobial activities of neuropeptides and their therapeutic potential. Curr Protein Pept Sci. 2012;13(8):723–738. doi:10.2174/138920312804871139
263. Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med. 2018;378(4):321–330. doi:10.1056/NEJMoa1705971
264. Egan MF, Kost J, Voss T, et al. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N Engl J Med. 2019;380(15):1408–1420. doi:10.1056/NEJMoa1812840
265. Ballard C, Aarsland D, Cummings J, et al. Drug repositioning and repurposing for Alzheimer disease. Nat Rev Neurol. 2020;16(12):661–673. doi:10.1038/s41582-020-0397-4
266. Munafò A, Burgaletto C, Di Benedetto G, et al. Repositioning of immunomodulators: a ray of hope for Alzheimer’s disease? Front Neurosci. 2020;14:614643. doi:10.3389/fnins.2020.614643
267. Lövheim H, Norman T, Weidung B, et al. Herpes simplex virus, apoeɛ4, and cognitive decline in old age: results from the Betula Cohort Study. J Alzheimers Dis. 2019;67(1):211–220. doi:10.3233/JAD-171162
268. Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. 1997;349(9047):241–244. doi:10.1016/S0140-6736(96)10149-5
269. Lathe R, Tzeng NS, Itzhaki R. Herpes infections and dementia: rebutting alternative fact. Neurotherapeutics. 2019;16(1):176–179. doi:10.1007/s13311-018-00700-5
270. Devanand DP, Andrews H, Kreisl WC, et al. Antiviral therapy: valacyclovir treatment of Alzheimer’s disease (VALAD) trial: protocol for a randomised, double-blind,placebo-controlled, treatment trial. BMJ Open. 2020;10(2):e032112. doi:10.1136/bmjopen-2019-032112
271. Molloy DW, Standish TI, Zhou Q, Guyatt G; DARAD Study Group. A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: the DARAD trial. Int J Geriatr Psychiatry. 2013;28(5):463–470. doi:10.1002/gps.3846
272. Li N, Collyer CA. Gingipains from Porphyromonas gingivalis - complex domain structures confer diverse functions. Eur J Microbiol Immunol (Bp). 2011;1(1):41–58. doi:10.1556/EuJMI.1.2011.1.7
273. Guo Y, Nguyen KA, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol. 2000;54(1):15–44. doi:10.1111/j.1600-0757.2010.00377.x
274. Arastu-Kapur S, Nguyen M, Raha D, et al. Treatment of Porphyromonas gulae infection and downstream pathology in the aged dog by lysine-gingipain inhibitor COR388. Pharmacol Res Perspect. 2020;8(1):e00562. doi:10.1002/prp2.562
275. Norins LC. Licensed anti-microbial drugs logical for clinical trials against pathogens currently suspected in Alzheimer’s disease. Antibiotics. 2021;10(3):327. doi:10.3390/antibiotics10030327
276. Rath S, Rud T, Karch A, Pieper DH, Vital M. Pathogenic functions of host microbiota. Microbiome. 2018;6(1):174. doi:10.1186/s40168-018-0542-0
277. Eiser AR. Could dietary factors reduce COVID-19 mortality rates? Moderating the inflammatory state. J Altern Complement Med. 2020;27(2):176–178. doi:10.1089/acm.2020.0441
278. Islam MA, Khandker SS, Alam F, Khalil MI, Kamal MA, Gan SH. Alzheimer’s disease and natural products: future regimens emerging from nature. Curr Top Med Chem. 2017;17(12):1408–1428. doi:10.2174/1568026617666170103163054
279. Ravi SK, Narasingappa RB, Vincent B. Neuro-nutrients as anti-Alzheimer’s disease agents: a critical review. Crit Rev Food Sci Nutr. 2019;59(18):2999–3018. doi:10.1080/10408398.2018.1481012
280. Atlante A, Amadoro G, Bobba A, Latina V. Functional foods: an approach to modulate molecular mechanisms of Alzheimer’s disease. Cells. 2020;9(11):2347. doi:10.3390/cells9112347
281. Omar SH. Mediterranean and MIND diets containing olive biophenols reduces the prevalence of Alzheimer’s disease. Int J Mol Sci. 2019;20(11):2797. doi:10.3390/ijms20112797
282. van den Brink AC, Brouwer-Brolsma EM, Berendsen AAM, van de Rest O. The mediterranean, dietary approaches to stop hypertension (DASH), and mediterranean-DASH intervention for neurodegenerative delay (MIND) diets are associated with less cognitive decline and a lower risk of Alzheimer’s disease-a review. Adv Nutr. 2019;10(6):1040–1065. doi:10.1093/advances/nmz054
283. Sanchez-Flack JC, Tussing-Humphreys L, Lamar M, et al. Building research in diet and cognition (BRIDGE): baseline characteristics of older obese African American adults in a randomized controlled trial to examine the effect of the Mediterranean diet with and without weight loss on cognitive functioning. Prev Med Rep. 2020;22:101302. doi:10.1016/j.pmedr.2020.101302
284. Wang Y, Park NY, Jang Y, Ma A, Jiang Q. Vitamin E gamma-tocotrienol inhibits cytokine-stimulated NF-kappaB activation by induction of anti-inflammatory A20 via stress adaptive response due to modulation of sphingolipids. J Immunol. 2015;195(1):126–133. doi:10.4049/jimmunol.1403149
285. Yuan L, Liu J, Ma W, et al. Dietary pattern and antioxidants in plasma and erythrocyte in patients with mild cognitive impairment from China. Nutrition. 2016;32(2):193–198. doi:10.1016/j.nut.2015.08.004
286. Chen X, Wu S, Chen C, et al. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-κB pathway following experimental traumatic brain injury. J Neuroinflammation. 2017;14(1):143. doi:10.1186/s12974-017-0917-3
287. Vafeiadou K, Vauzour D, Spencer JP. Neuroinflammation and its modulation by flavonoids. Endocr Metab Immune Disord Drug Targets. 2007;7(3):211–224. doi:10.2174/187153007781662521
288. Spencer JP, Vafeiadou K, Williams RJ, Vauzour D. Neuroinflammation: modulation by flavonoids and mechanisms of action. Mol Aspects Med. 2012;33(1):83–97. doi:10.1016/j.mam.2011.10.016
289. Spilsbury A, Vauzour D, Spencer JPE, Rattray M. Regulation of NF-κB activity in astrocytes: effects of flavonoids at dietary-relevant concentrations. Biochem Biophys Res Commun. 2012;418(3):578–583. doi:10.1016/j.bbrc.2012.01.081
290. Kennedy DO. B vitamins and the brain: mechanisms, dose and efficacy–a review. Nutrients. 2016;8(2):68. doi:10.3390/nu8020068
291. Ray B, Lahiri DK. Neuroinflammation in Alzheimer’s disease: different molecular targets and potential therapeutic agents including curcumin. Curr Opin Pharmacol. 2009;9(4):434–444. doi:10.1016/j.coph.2009.06.012
292. Bonfili L, Cecarini V, Gogoi O, et al. Microbiota modulation as preventative and therapeutic approach in Alzheimer’s disease. FEBS J. 2020. doi:10.1111/febs.15571
293. Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13(10):701–712. doi:10.1038/nrn3346
294. Borre YE, Moloney RD, Clarke G, Dinan TG, Cryan JF. The impact of microbiota on brain and behavior: mechanisms & therapeutic potential. Adv Exp Med Biol. 2014;817:373–403. doi:10.1007/978-1-4939-0897-4_17
295. Borre YE, Panagaki T, Koelink PJ, et al. Neuroprotective and cognitive enhancing effects of a multi-targeted food intervention in an animal model of neurodegeneration and depression. Neuropharmacology. 2014;79:738–749. doi:10.1016/j.neuropharm.2013.11.009
296. Wang X, Sun G, Feng T, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019;29(10):787–803. doi:10.1038/s41422-019-0216-x
297. Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255–2263. doi:10.1016/S0140-6736(15)60461-5
298. Panza F, Lozupone M, Solfrizzi V, Watling M, Imbimbo BP. Time to test antibacterial therapy in Alzheimer’s disease. Brain. 2019;142(10):2905–2929. doi:10.1093/brain/awz244
299. Ahmad MH, Fatima M, Mondal AC. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: rational insights for the therapeutic approaches. J Clin Neurosci. 2019;59:6–11. doi:10.1016/j.jocn.2018.10.034
300. Erb L, Woods LT, Khalafalla MG, Weisman GA. Purinergic signaling in Alzheimer’s disease. Brain Res Bull. 2019;151:25–37. doi:10.1016/j.brainresbull.2018.10.014
301. Kang YJ, Diep YN, Tran M, Cho H. Therapeutic targeting strategies for early- to late-staged Alzheimer’s disease. Int J Mol Sci. 2020;21(24):9591. doi:10.3390/ijms21249591
302. Sánchez-Sarasúa S, Fernández-Pérez I, Espinosa-Fernández V, Sánchez-Pérez AM, Ledesma JC. Can we treat neuroinflammation in Alzheimer’s disease? Int J Mol Sci. 2020;21(22):8751. doi:10.3390/ijms21228751
303. Townsend KP, Praticò D. Novel therapeutic opportunities for Alzheimer’s disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005;19(12):1592–1601. doi:10.1096/fj.04-3620rev
304. Minhas PS, Latif-Hernandez A, McReynolds MR, et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature. 2021;590(7844):122–128. doi:10.1038/s41586-020-03160-0
305. Kobayashi Y, Sugahara H, Shimada K, et al. Therapeutic potential of Bifidobacterium breve strain A1 for preventing cognitive impairment in Alzheimer’s disease. Sci Rep. 2017;7(1):13510. doi:10.1038/s41598-017-13368-2
306. Xiao J, Katsumata N, Bernier F, et al. Probiotic bifidobacterium breve in improving cognitive functions of older adults with suspected mild cognitive impairment: a randomized, double-blind, placebo-controlled trial. J Alzheimers Dis. 2020;77(1):139–147. doi:10.3233/JAD-200488
307. Holmes A, Finger C, Morales-Scheihing D, Lee J, McCullough LD. Gut dysbiosis and age-related neurological diseases; an innovative approach for therapeutic interventions. Transl Res. 2020;226:39–56. doi:10.1016/j.trsl.2020.07.012
308. Kesika P, Suganthy N, Sivamaruthi BS, Chaiyasut C. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci. 2021;264:118627. doi:10.1016/j.lfs.2020.118627
309. Gentile F, Doneddu PE, Riva N, Nobile-Orazio E, Quattrini A. Diet, microbiota and brain health: unraveling the network intersecting metabolism and neurodegeneration. Int J Mol Sci. 2020;21(20):7471. doi:10.3390/ijms21207471
310. Suganya K, Koo BS. Gut-brain axis: role of gut microbiota on neurological disorders and how probiotics/prebiotics beneficially modulate microbial and immune pathways to improve brain functions. Int J Mol Sci. 2020;21(20):7551. doi:10.3390/ijms21207551
311. Zhu F, Li C, Chu F, Tian X, Zhu J. Target dysbiosis of gut microbes as a future therapeutic manipulation in Alzheimer’s disease. Front Aging Neurosci. 2020;12:544235. doi:10.3389/fnagi.2020.544235
312. Liu S, Gao J, Zhu M, Liu K, Zhang HL. Gut microbiota and dysbiosis in Alzheimer’s disease: implications for pathogenesis and treatment. Mol Neurobiol. 2020;57(12):5026–5043. doi:10.1007/s12035-020-02073-3
313. Loera-Valencia R, Cedazo-Minguez A, Kenigsberg PA, et al. Current and emerging avenues for Alzheimer’s disease drug targets. J Intern Med. 2019;286(4):398–437. doi:10.1111/joim.12959
314. Obrenovich M, Jaworski H, Tadimalla T, et al. The role of the microbiota-gut-brain axis and antibiotics in ALS and neurodegenerative diseases. Microorganisms. 2020;8(5):784. doi:10.3390/microorganisms8050784
315. Akbari E, Asemi Z, Kakhaki RD, et al. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer’s disease: a randomized, double-blind and controlled trial. Front Aging Neurosci. 2016;8:256. doi:10.3389/fnagi.2016.00256
316. Leblhuber F, Steiner K, Schuetz B, Fuchs D, Gostner JM. Probiotic supplementation in patients with Alzheimer’s dementia - an explorative intervention study. Curr Alzheimer Res. 2018;15(12):1106–1113. doi:10.2174/1389200219666180813144834
317. Fortier M, Castellano CA, St-Pierre V, et al. A ketogenic drink improves cognition in mild cognitive impairment: results of a 6-month RCT. Alzheimers Dement. 2020;17(3):543–552. doi:10.1002/alz.12206
318. Rawat K, Singh N, Kumari P, Saha L. A review on preventive role of ketogenic diet (KD) in CNS disorders from the gut microbiota perspective. Rev Neurosci. 2020;32(2):143–157. /j/revneuro.ahead-of-print/revneuro-2020-0078/revneuro-2020-0078.xml. doi:10.1515/revneuro-2020-0078
319. Morrill SJ, Gibas KJ. Ketogenic diet rescues cognition in ApoE4+ patient with mild Alzheimer’s disease: a Case Study. Diabetes Metab Syndr. 2019;13(2):1187–1191. doi:10.1016/j.dsx.2019.01.035
320. Vinciguerra F, Graziano M, Hagnäs M, Frittitta L, Tumminia A. Influence of the mediterranean and ketogenic diets on cognitive status and decline: a narrative review. Nutrients. 2020;12(4):1019. doi:10.3390/nu12041019
321. Dahlgren K, Gibas KJ. Ketogenic diet, high intensity interval training (HIIT) and memory training in the treatment of mild cognitive impairment: a Case Study. Diabetes Metab Syndr. 2018;12(5):819–822. doi:10.1016/j.dsx.2018.04.031
322. Aronica L, Volek J, Poff A, D’agostino DP. Genetic variants for personalised management of very low carbohydrate ketogenic diets. BMJ Nutr Prev Health. 2020;3(2):363–373. doi:10.1136/bmjnph-2020-000167
323. Nagpal R, Neth BJ, Wang S, Craft S, Yadav H. Modified mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine. 2019;47:529–542. doi:10.1016/j.ebiom.2019.08.032
324. Libonati L, Onesti E, Gori MC, et al. Vitamin D in amyotrophic lateral sclerosis. Funct Neurol. 2017;32(1):35–40. doi:10.11138/fneur/2017.32.1.035
325. Egger F, Jakab M, Fuchs J, et al. Effect of glycine on BV-2 microglial cells treated with interferon-γ and lipopolysaccharide. Int J Mol Sci. 2020;21(3):804. doi:10.3390/ijms21030804
326. Bredesen DE. Reversal of cognitive decline: a novel therapeutic program. Aging (Albany NY). 2014;6(9):707–717. doi:10.18632/aging.100690
327. Famenini S, Rigali EA, Olivera-Perez HM, et al. Increased intermediate M1-M2 macrophage polarization and improved cognition in mild cognitive impairment patients on ω-3 supplementation. FASEB J. 2017;31(1):148–160. doi:10.1096/fj.201600677RR
328. Fiala M, Kooij G, Wagner K, Hammock B, Pellegrini M. Modulation of innate immunity of patients with Alzheimer’s disease by omega-3 fatty acids. FASEB J. 2017;31(8):3229–3239. doi:10.1096/fj.201700065R
329. Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC; Cache County Study Investigators. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. 2002;59(6):880–886. doi:10.1212/wnl.59.6.880
330. Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327(7407):128. doi:10.1136/bmj.327.7407.128
331. Guillot-Sestier MV, Doty KR, Gate D, et al. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015;85(3):534–548. doi:10.1016/j.neuron.2014.12.068
332. Cherry JD, Olschowka JA, O’Banion MK. Arginase 1+ microglia reduce Aβ plaque deposition during IL-1β-dependent neuroinflammation. J Neuroinflammation. 2015;12(1):203. doi:10.1186/s12974-015-0411-8
333. MacDonald ML, Favo D, Garver M, et al. Laser capture microdissection-targeted mass spectrometry: a method for multiplexed protein quantification within individual layers of the cerebral cortex. Neuropsychopharmacology. 2019;44(4):743–748. doi:10.1038/s41386-018-0260-0
334. Uddin MS, Kabir MT, Rahman MS, et al. Revisiting the amyloid cascade hypothesis: from anti-abeta therapeutics to auspicious new ways for Alzheimer’s disease. Int J Mol Sci. 2020;21(16):5858. doi:10.3390/ijms21165858
335. Uddin MS, Kabir MT, Mamun AA, et al. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. Int Immunopharmacol. 2020;84:106479. doi:10.1016/j.intimp.2020.106479
336. Jackson L, Eldahshan W, Fagan SC, Ergul A. Within the brain: the renin angiotensin system. Int J Mol Sci. 2018;19(3):876. doi:10.3390/ijms19030876
337. Nelson L, Gard P, Tabet N. Hypertension and inflammation in Alzheimer’s disease: close partners in disease development and progression! J Alzheimers Dis. 2014;41(2):331–343. doi:10.3233/JAD-140024
338. Anderson C, Teo K, Gao P, et al.; ONTARGET and TRANSCEND Investigators. Renin-angiotensin system blockade and cognitive function in patients at high risk of cardiovascular disease: analysis of data from the ONTARGET and TRANSCEND studies. Lancet Neurol. 2011;10(1):43–53. doi:10.1016/S1474-4422(10)70250-7
339. Williamson JD, Pajewski NM, Auchus AP, et al.; SPRINT MIND Investigators for the SPRINT Research Group. Effect of intensive vs standard blood pressure control on probable dementia: a randomized clinical trial. JAMA. 2019;321(6):553–561. doi:10.1001/jama.2018.21442
340. Ono-Saito N, Niki I, Hidaka H. H-series protein kinase inhibitors and potential clinical applications. Pharmacol Ther. 1999;82(2–3):123–131. doi:10.1016/s0163-7258(98)00070-9
341. Song Y, Chen X, Wang L-Y, Gao W, Zhu M-J. Rho kinase inhibitor fasudil protects against β-amyloid-induced hippocampal neurodegeneration in rats. CNS Neurosci Ther. 2013;19(8):603–610. doi:10.1111/cns.12116
342. Becker RE, Greig NH, Lahiri DK, et al. (-)-Phenserine and inhibiting pre-programmed cell death: in pursuit of a novel intervention for Alzheimer’s disease. Curr Alzheimer Res. 2018;15(9):883–891. doi:10.2174/1567205015666180110120026
343. Lecca D, Bader M, Tweedie D, et al. (-)-Phenserine and the prevention of pre-programmed cell death and neuroinflammation in mild traumatic brain injury and Alzheimer’s disease challenged mice. Neurobiol Dis. 2019;130:104528. doi:10.1016/j.nbd.2019.104528
344. Winblad B, Giacobini E, Frölich L, et al. Phenserine efficacy in Alzheimer’s disease. J Alzheimers Dis. 2010;22(4):1201–1208. doi:10.3233/JAD-2010-101311
345. Judge A, Garriga C, Arden NK, et al. Protective effect of antirheumatic drugs on dementia in rheumatoid arthritis patients. Alzheimers Dement. 2017;3(4):612–621. doi:10.1016/j.trci.2017.10.002
346. Maragakis NJ, Rothstein JD. Glutamate transporters in neurologic disease. Arch Neurol. 2001;58(3):365–370. doi:10.1001/archneur.58.3.365
347. Kumari S, Deshmukh R. beta-lactam antibiotics to tame down molecular pathways of Alzheimer’s disease. Eur J Pharmacol. 2021;13:173877. doi:10.1016/j.ejphar.2021.173877
348. Chou RC, Kane M, Ghimire S, Gautam S, Gui J. Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: a nested case-control analysis. CNS Drugs. 2016;30(11):1111–1120. doi:10.1007/s40263-016-0374-z
349. Butchart J, Brook L, Hopkins V, et al. Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology. 2015;84(21):2161–2168. doi:10.1212/WNL.0000000000001617
350. Salvagno C, Ciampricotti M, Tuit S, et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat Cell Biol. 2019;21(4):511–521. doi:10.1038/s41556-019-0298-1
351. Oyewole-Said D, Konduri V, Vazquez-Perez J, Weldon SA, Levitt JM, Decker WK. Beyond T-cells: functional characterization of CTLA-4 expression in immune and non-immune cell types. Front Immunol. 2020;11:608024. doi:10.3389/fimmu.2020.608024
352. Baruch K, Deczkowska A, Rosenzweig N, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med. 2016;22(2):135–137. doi:10.1038/nm.4022
353. Rogers NK, Romero C, SanMartín CD, et al. Inverse relationship between Alzheimer’s disease and cancer: how immune checkpoints might explain the mechanisms underlying age-related diseases. J Alzheimers Dis. 2020;73(2):443–454. doi:10.3233/JAD-190839
354. Rosenzweig N, Dvir-Szternfeld R, Tsitsou-Kampeli A, et al. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat Commun. 2019;10(1):465. doi:10.1038/s41467-019-08352-5
355. Koronyo Y, Salumbides BC, Sheyn J, et al. Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer’s disease. Brain. 2015;138(Pt 8):2399–2422. doi:10.1093/brain/awv150
356. Kaeberlein M, Galvan V. Rapamycin and Alzheimer’s disease: time for a clinical trial? Sci Transl Med. 2019;11(476):eaar4289. doi:10.1126/scitranslmed.aar4289
357. Decourt B, Drumm-Gurnee D, Wilson J, et al. Poor safety and tolerability hamper reaching a potentially therapeutic dose in the use of thalidomide for Alzheimer’s disease: results from a double-blind, placebo-controlled trial. Curr Alzheimer Res. 2017;14(4):403–411. doi:10.2174/1567205014666170117141330
358. Schubert D, Currais A, Goldberg J, Finley K, Petrascheck M, Maher P. Geroneuroprotectors: effective geroprotectors for the brain. Trends Pharmacol Sci. 2018;39(12):1004–1007. doi:10.1016/j.tips.2018.09.008
359. Zhao Y, Qian R, Zhang J, et al. Young blood plasma reduces Alzheimer’s disease-like brain pathologies and ameliorates cognitive impairment in 3Tg-AD mice. Alzheimers Res Ther. 2020;12(1):70. doi:10.1186/s13195-020-00639-w
360. Sha SJ, Deutsch GK, Tian L, et al. Safety, tolerability, and feasibility of young plasma infusion in the plasma for Alzheimer symptom amelioration study: a randomized clinical trial. JAMA Neurol. 2019;76(1):35–40. doi:10.1001/jamaneurol.2018.3288
361. Mannick JB, Del Giudice G, Lattanzi M, et al. mTOR inhibition improves immune function in the elderly. Sci Transl Med. 2014;6(268):268ra179. doi:10.1126/scitranslmed.3009892
362. Pardon MC. Anti-inflammatory potential of thymosin β4 in the central nervous system: implications for progressive neurodegenerative diseases. Expert Opin Biol Ther. 2018;18(sup1):165–169. doi:10.1080/14712598.2018.1486817
363. Seledtsov VI, von Delwig AA. Immune memory limits human longevity: the role of memory СD4+ T cells in age-related immune abnormalities. Expert Rev Vaccines. 2020;19(3):209–215. doi:10.1080/14760584.2020.1745638
364. Ng TP, Feng L, Yap KB, Lee TS, Tan CH, Winblad B. Long-term metformin usage and cognitive function among older adults with diabetes. J Alzheimers Dis. 2014;41(1):61–68. doi:10.3233/JAD-131
365. Gurău F, Baldoni S, Prattichizzo F, et al. Anti-senescence compounds: a potential nutraceutical approach to healthy aging. Ageing Res Rev. 2018;46:14–31. doi:10.1016/j.arr.2018.05.001
366. Witherden EA, Moyesa DL, Brucea KD, Ehrlich SD, Shoaie S. Using systems biology approaches to elucidate cause and effect in host–microbiome interactions. Curr Opin Syst Biol. 2017;3:141–146. doi:10.1016/j.coisb.2017.05.003
367. Pastore A, Raimondi F, Rajendran L, Temussi PA. Why does the Aβ peptide of Alzheimer share structural similarity with antimicrobial peptides? Commun Biol. 2020;3(1):135. doi:10.1038/s42003-020-0865-9
368. Dicks LMT, Geldenhuys J, Mikkelsen LS, Brandsborg E, Marcotte H. Our gut microbiota: a long walk to homeostasis. Benef Microbes. 2018;9(1):3–20. doi:10.3920/BM2017.0066
369. Kulkarni S, Min J. Biotechnology by Ganacord Genuity, US equity research. 28 September 2020.
370. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. 2020;6(1):e12050. doi:10.1002/trc2.12050
371. Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L. Development of azeliragon, an oral small molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of cognition in mild Alzheimer’s disease. J Prev Alzheimers Dis. 2018;5(2):149–154.
372. Piedra-Quintero ZL, Wilson Z, Nava P, Guerau-de-arellano M. CD38: an immunomodulatory molecule in inflammation and autoimmunity. Front Immunol. 2020;11:597959. doi:10.3389/fimmu.2020.597959
373. Guerreiro S, Privat AL, Bressac L, Toulorge D. CD38 in neurodegeneration and neuroinflammation. Cells. 2020;9(2):471. doi:10.3390/cells9020471
374. Fülöp T, Munawara U, Larbi A, et al. Targeting infectious agents as a therapeutic strategy in Alzheimer’s disease. CNS Drugs. 2020;34:673–695. doi:10.1007/s40263-020-00737-1
375. Choi SH, Kim YH, Hebisch M, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515(7526):274–278. Epub 2014 Oct 12. PMID: 25307057; PMCID: PMC4366007. doi:10.1038/nature13800
376. Marton RM, Miura Y, Sloan SA, et al. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat Neurosci. 2019;22(3):484–491. Epub 2019 Jan 28. PMID: 30692691; PMCID: PMC6788758. doi:10.1038/s41593-018-0316-9
377. Loose C, Jensen K, Rigoutsos I, Stephanopoulos G. A linguistic model for the rational design of antimicrobial peptides. Nature. 2006;443(7113):867–869. PMID: 17051220. doi:10.1038/nature05233
378. Fülöp T, Desroches M, Cohen A, Santos FAN, Rodrigues S. Why we should use topological data analysis in ageing: towards defining the “topological shape of ageing”. Mech Ageing Dev. 2020;192:111390. Epub 2020 Oct 27. PMID: 33127442. doi:10.1016/j.mad.2020.111390
379. World Health Organization. First WHO Ministerial Conference on Global Action Against Dementia: Meeting Report. Geneva: World Health Organization; 2015.
380. Crescenzi O, Tomaselli S, Guerrini R, et al. Solution structure of the Alzheimer amyloid beta-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur J Biochem. 2002;269(22):5642–5648. doi:10.1046/j.1432-1033.2002.03271.x
381. Wang G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J Biol Chem. 2008;283(47):32637–32643. Epub 2008 Sep 25. PMID: 18818205. doi:10.1074/jbc.M805533200
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.