")
Back to Journals » OncoTargets and Therapy » Volume 12
Targeted therapy and personalized medicine in gastrointestinal stromal tumors: drug resistance, mechanisms, and treatment strategies
Received 19 February 2019
Accepted for publication 30 May 2019
Published 1 July 2019 Volume 2019:12 Pages 5123—5133
DOI https://doi.org/10.2147/OTT.S180763
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
George Z Li,1 Chandrajit P Raut2
1Department of Surgery, Brigham and Women’s Hospital, Boston, MA, USA; 2Center for Sarcoma and Bone Oncology, Dana Farber Cancer Center, Boston, MA, USA
Abstract: Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract. Since the discovery that the KIT and PDGFRA receptor tyrosine kinases are the primary oncogenic drivers in the vast majority of GISTs, targeted therapy with tyrosine kinase inhibitors has been the mainstay of treatment for this disease. Using molecular profiling of tumor specimens, researchers also discovered that KIT and PDGFRA mutations are non-random and occur in specific regions of the receptors, and furthermore, that particular genotypes predicted response or resistance to targeted therapy. Imatinib, the first tyrosine kinase inhibitor used to treat GIST, remains the first-line therapy in advanced GIST and the only therapy confirmed through clinical trials in the adjuvant or neoadjuvant setting for resectable disease. Resistance to imatinib is well described and is either primary or secondary. Primary resistance is associated with specific tumor genotypes, so genotyping of individual patient tumors helps guide decision-making into whether to offer imatinib and at what dose. Secondary resistance occurs due to the acquisition of secondary mutations during therapy. Currently, the main strategy to combat imatinib resistance is to switch to another tyrosine kinase inhibitor, because imatinib-resistant GIST is usually still oncogenically addicted to KIT/PDGFRA signaling. Surgery can also be used to combat resistant disease in select settings. Unfortunately, progression-free and overall survival remains dismal for patients who develop imatinib-resistant disease, and further research into alternative strategies is still needed.
Keywords: GIST, targeted therapy, tyrosine kinase inhibitor, imatinib
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasm of the gastrointestinal tract, with an incidence of 3,000–5,000 cases per year in the United States.1 GISTs arise from the interstitial cells of Cajal (ICCs), which are intestinal pacemaker cells. ICCs require the KIT receptor tyrosine kinase for normal development,2 but constitutively active KIT signaling has been shown in multiple studies to be the central driving force behind GIST tumorigenesis.3–5 Indeed, based on this biological framework, most current-targeted therapy strategies against GIST are aimed against KIT and its related receptor tyrosine kinases. In this review, we will discuss the molecular basis of targeted therapy and personalized medicine in GIST, current treatment paradigms, mechanisms of resistance, strategies to overcome resistance, and novel research into alternative approaches.
Molecular characteristics of GIST
Over 90% of GISTs stain positive for KIT on immunohistochemistry, and approximately 70–85% of tumors with sequencing data have an identifiable gain-of-function KIT mutation.3,6–8 KIT is a 145 kDa transmembrane glycoprotein in the receptor tyrosine kinase family that consists of an extracellular domain (encoded by exons 1–9), a transmembrane domain (exon 10), a juxtamembrane domain (exon 11), and a tyrosine kinase domain (exons 13–21).9 Its most well-described ligand is stem cell factor (SCF),10,11 which causes receptor dimerization and activation. Following KIT activation, subsequent downstream signaling via phosphoinositide 3-kinase (PI3K), Src kinase, and mitogen-activated protein kinase result in cellular differentiation, proliferation, and survival (Figure 1).12,13 While normal KIT signaling is required for ICC differentiation and survival, constitutive ligand-independent KIT activation leads to GIST tumorigenesis.3–5,14
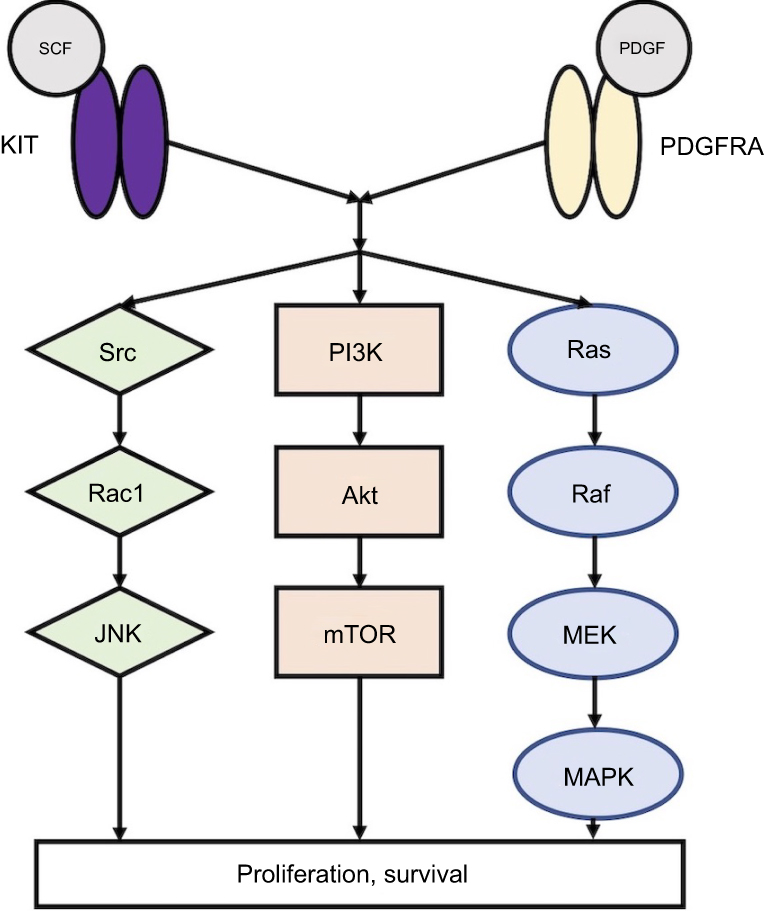 |
Figure 1 KIT and PDGFRA downstream signaling pathways. Abbreviations: SCF, stem cell factor; PDGF, platelet-derived growth factor, JNK, c-Jun N-terminal kinase; PI3K, phosphoinositide 3-kinase; mTOR, mammalian target of rapamycin; MEK, mitogen-activated protein kinase; MAPK, mitogen-activated protein kinase. |
Over 60% of mutations in GIST are located in KIT exon 11,7 which encodes the juxtamembrane domain responsible for inhibiting receptor dimerization and activation in the absence of SCF ligand (Figure 2). Another 20% of mutations are in KIT exon 9 in the extracellular domain,7 which also cause ligand-independent receptor dimerization. Less common mutations have also been described in other regions of KIT, although these are usually seen as secondary mutations in tumors exposed to tyrosine kinase inhibitors as opposed to treatment-naïve tumors, as will be discussed later.15
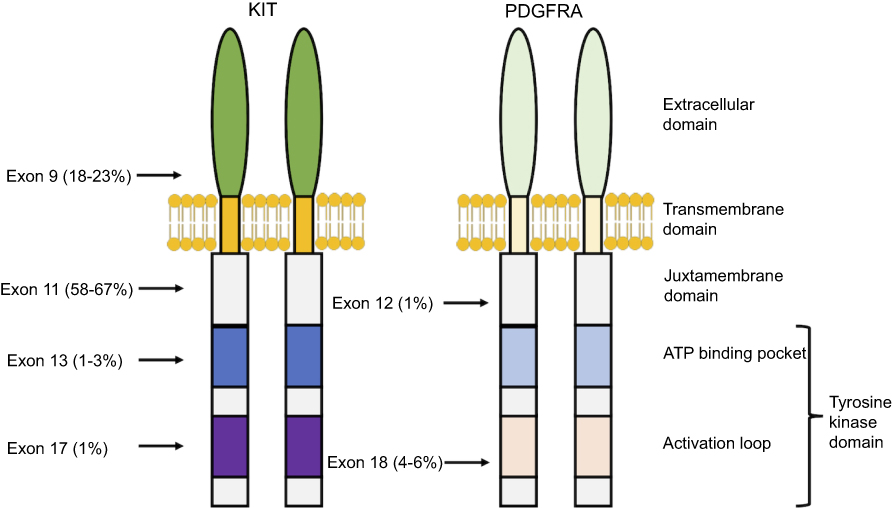 |
Figure 2 Location and frequency of primary mutations in KIT and PDGFRA. |
Approximately 10–15% of GISTs have an activating mutation in PDGFRA,8,16 another receptor tyrosine kinase closely related in both structure and chromosomal gene location to KIT (Figure 2).17 The most common PDGFRA mutations are located in exon 18 (the activation loop of the tyrosine kinase domain) and exon 12 (the juxtamembrane domain).7,15 There is significant overlap in signaling pathways downstream of KIT and PDGFRA in GIST, suggesting that these two receptors provide alternative entries into a common tumorigenic pathway. In fact, activating mutations of KIT and PDGFRA appear to be mutually exclusive.7 However, fundamental biological differences do seem to exist between these two types of GISTs, as gene expression profiles of KIT-mutant GISTs differ from those of PDGFRA-mutant GISTs,18 and PDGFRA-mutant GISTs occur almost exclusively in the stomach.19
Finally, there remains a small group of GISTs that do not have any detectable mutations in either KIT or PDGFRA, previously termed “wild-type” GIST, which constitutes approximately 10% of adult GISTs but up to 85% of pediatric GISTs.20 Most of these tumors are now recognized to have a deficiency in succinyl dehydrogenase (SDH) activity. A study from the National Institutes of Health found that 66% of wild-type GIST had loss-of-function SDH mutations, and 22% had methylation of the SDHC promotor which led to epigenetic silencing of SDH expression. Most of the remaining 12% of patients with SDH-competent tumors either had NF1 (usually patients with type 1 neurofibromatosis also called von Recklinghausen neurofibromatosis) or BRAF V600E mutations, with only a small minority having an idiopathic tumorigenic mechanism.21
Targeted therapy and mechanisms of drug resistance
Imatinib
Given the central role of KIT and its related receptor tyrosine kinase PDGFRA in GIST tumorigenesis, current targeted therapies are tyrosine kinase inhibitors primarily aimed at one or both of these receptors. The first anti-KIT therapy for GIST was imatinib, a tyrosine kinase inhibitor used initially for chronic myelogenous leukemia that was subsequently found to also have anti-KIT and anti-PDGFRA activity.5 Demetri and colleagues assessed the efficacy of imatinib in 147 patients with unresectable or metastatic GIST, and found a remarkable partial response rate of over 50%, with an additional 28% of patients achieving stable disease.22 Further Phase II and Phase III studies confirmed the long-term efficacy of imatinib in patients with advanced GIST, with reported median overall survivals of over 4 years.23–25
This success in patients led to studies of adjuvant imatinib in patients with resectable primary GIST. The American College of Surgeons Oncology Group (ACOSOG) Z9001 trial enrolled 713 patients with completely resected GIST at least 3 cm in diameter with KIT positivity on immunohistochemistry, and randomized them to either adjuvant imatinib therapy or placebo for 1 year.26 Patients in the imatinib group had a significantly improved 1-year recurrence-free survival (RFS) of 98% compared to 83% in the control group. A follow-up analysis of a subgroup of 645 patients with available tumor specimens to assess mitotic rate and mutational profiles found that large tumor size, small bowel location, and high mitotic rate were associated with lower RFS within both the placebo and imatinib arms. In addition, patients with KIT exon 11 deletions had improved RFS with adjuvant imatinib compared to placebo.27
Subsequent trials have examined whether longer durations of adjuvant therapy provide additional benefit. The Scandinavian Sarcoma Group (SSG) XVIII trial randomized 400 patients with resected GIST with high-risk features to 3 years versus 1 year of adjuvant imatinib. They found that patients who had 3 years of adjuvant imatinib had significantly better RFS and importantly overall survival (OS) than patients who had 1 year of adjuvant imatinib.28 A subsequent analysis of data from this trial of 341 patients with mutation data found that most of the survival benefit was realized by patients with KIT exon 11 mutations and that patients with wild-type GIST or other KIT or PDGFRA mutations did not derive benefit from the longer duration of adjuvant imatinib.29
Based on SSG XVIII, the current standard-of-care duration for adjuvant imatinib in resected high-risk GIST is at least 3 years (Figure 3), but it is possible that patients may derive additional benefit from even longer therapy provided they do not have a known mutation conferring imatinib resistance. A recent single-arm Phase II trial, PERSIST-5, examined tolerability and efficacy of 5 years of adjuvant imatinib in patients with resected GIST’s with intermediate-risk or high-risk features. The authors reported a 5-year RFS of 90% and 5-year OS of 95%.30 The only patient who recurred on adjuvant imatinib had a PDGFRA D842V mutation, which, as will be discussed, is associated with imatinib resistance. All other recurrences occurred after discontinuation of imatinib.
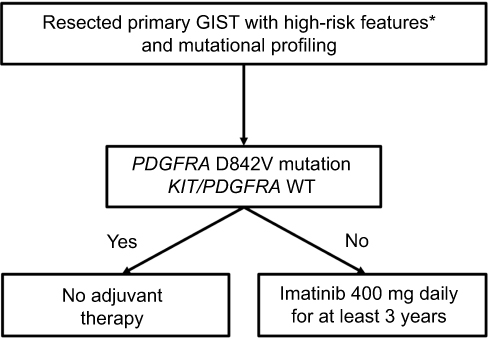 |
Figure 3 Treatment algorithm for patients with resectable gastrointestinal stromal tumor (GIST) and mutational profiling.Notes: *High-risk features include: tumor rupture; mitotic rate >10 per 50 high-powered fields (HPF); diameter >10 cm; mitotic rate >5/50 HPF and diameter >5 cm; mitotic rate >5/50 HPF and diameter >2 cm and non-gastric GIST; diameter >5 cm and non-gastric GIST. |
Imatinib therapy in the neoadjuvant setting is less well established. RTOG 0132/ACRIN 6665 was a Phase II trial that enrolled 63 patients with resectable primary GIST at least 5 cm in diameter or recurrent/metastatic GIST at least 2 cm in diameter. Patients received 8–12 weeks of neoadjuvant imatinib at 600 mg daily, followed by surgery. The study authors noted low rates of postoperative complications and a post-operative mortality rate of 2.2%, confirming the safety of imatinib use in the neoadjuvant setting.31 However, one criticism of this trial is that the neoadjuvant treatment duration was too short to see a response, and indeed only 7% of resectable primary tumors and 4.5% of recurrent/metastatic tumors had partial responses to neoadjuvant therapy. Another multicenter Phase II trial in Asia examined longer neoadjuvant imatinib therapy in 53 patients with gastric GIST’s at least 10 cm in diameter. Patients were treated with imatinib for at least 6 months, with extension to 9 months if there was a complete or partial response, unless the tumor progressed at 1 or 3 months at which point neoadjuvant treatment was stopped and the patient proceeded to surgery. Of the 53 patients, 46 received at least 6 months of neoadjuvant therapy, 33 (62%) had a partial response, and 91% (48/53) received an R0 resection.32 Of note, other than 1 patient who was excluded from the Asian trial for a PDGFRA D842V mutation, there were no other mentions of mutation status in either of the neoadjuvant trials. Overall, neoadjuvant therapy seems to be safe, and may be especially useful for esophageal, duodenal, and rectal GISTs, as pre-operative tumor shrinkage may facilitate sphincter/organ preservation and negative margins based on retrospective series.33,34 Unfortunately, there are no randomized trials that examine the effect of neoadjuvant therapy on RFS or OS.
Resistance to imatinib
Imatinib resistance is a well-described problem in GIST. Resistance is characterized as primary resistance, in which the tumor progresses through an initial imatinib challenge, or secondary resistance, in which the tumor progresses after an initial period of stabilization or response to imatinib. Several GIST genotypes are associated with primary resistance. For example, patients with KIT exon 9 mutations were much less likely to respond to imatinib than those with KIT exon 11 mutations,7 though response rates could be improved by increasing imatinib dosing from 400 mg daily to 800 mg daily.35 KIT exon 9 mutations cause receptor dimerization in the absence of ligand in a conformation that may sterically hinder imatinib binding.36,37 The PDGFRA D842V mutation, located in exon 18 in the activation loop, is also associated with imatinib resistance, although most of these tumors seem to have low mitotic rates and an indolent clinical course.38 Cassier and colleagues examined 58 patients with PDGFRA-mutant GIST’s treated with imatinib. There were no responses among patients with PDGFRA D842V, and only 32% of these patients achieved stable disease as their best response.16 Laboratory studies have shown that the D842V mutation diminishes accessibility of imatinib’s binding site to PDGFRA.39 Wild-type tumors also tend to be resistant to imatinib, but they also tend to have an indolent overall clinical course.21
The development of secondary resistance usually occurs through the acquisition of secondary mutations. Unlike primary mutations, which usually occur in exons 9 and 11, secondary mutations most often occur in exons 13 and 14 of the ATP binding pocket of the tyrosine kinase domain, and in exons 17 and 18 of the activation loop of the tyrosine kinase domain. Antonescu and colleagues examined 31 patients with GIST who were treated with imatinib prior to surgical resection, of which 15 had developed secondary resistance. Seven (46%) of these patients developed secondary mutations,15 and while the primary KIT mutations in these patients were all in either exons 9 or 11, secondary KIT mutations occurred in exon 17 (6 of 7 patients), and exons 13 and 14 (1 patient each). Another study by Heinrich and colleagues examined genetic profiles of pre- and post-imatinib treatment tumor samples from 78 patients with metastatic GIST, and found secondary mutations in 33 patients. Sixteen patients had exon 13 mutations, three patients had exon 14 mutations, 12 patients had exon 17 mutations, and two patients had exon 18 mutations, with one patient having both an exon 13 and exon 17 mutation.40 Exon 14 mutations are thought to disrupt an H-bond involved in imatinib binding,41 while other secondary mutations are thought to stabilize KIT in a conformation that imatinib cannot bind efficiently.41,42
Strategies to overcome drug resistance
Switch tyrosine kinase inhibitors
Interestingly, imatinib resistance does not correlate with KIT activity,15 and cell lines derived from imatinib-resistant GISTs still rely on KIT to activate downstream signaling,43 suggesting that these tumors may still rely on KIT activation for survival and proliferation. Thus, the primary strategy for overcoming imatinib resistance is to switch to another tyrosine kinase inhibitor (Figure 4).
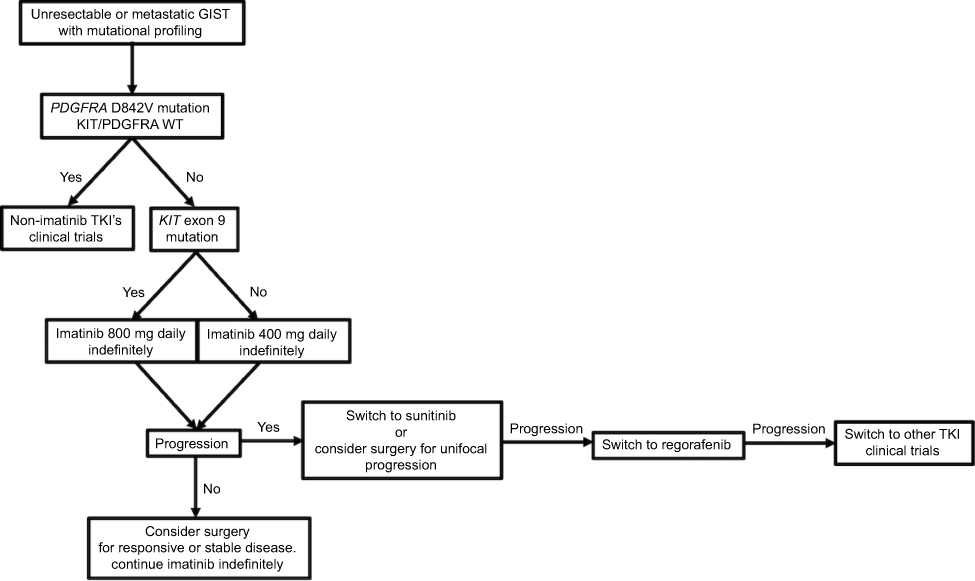 |
Figure 4 Treatment algorithm for patients with unresectable or metastatic GIST and mutational profiling. Abbreviations: TKI, tyrosine kinase inhibitor; GIST, gastrointestinal stromal tumor. |
Sunitinib is currently established as the second-line tyrosine kinase inhibitor for patients with GIST refractory to imatinib. It has activity against KIT, PDGFRA, and also against vascular endothelial growth factor receptor (VEGFR).44,45 A randomized double-blinded clinical trial by Demetri and colleagues compared sunitinib to placebo in 312 patients with metastatic or unresectable GIST who were resistant to or intolerant of previous treatment with imatinib. Patients treated with sunitinib had significantly improved median progression-free survival (PFS, 27.3 weeks vs 6.4 weeks), although the best response achieved by the vast majority of patients in the sunitinib group was stable disease, as only 7% of patients in the sunitinib group had a partial response and there were no complete responders.46 Patients with KIT exon 9 mutations had higher response rates to sunitinib than patients with KIT exon 11 mutations, and patients with secondary KIT exon 13 or 14 mutations had higher response rates compared to patients with secondary KIT exon 17 or 18 mutations.40
Regorafenib, another tyrosine kinase inhibitor with broad activity against KIT, PDGFRA, VEGFR, and BRAF,47 is third-line therapy for patients with GIST refractory to both imatinib and sunitinib. A Phase III clinical trial, also by Demetri and colleagues, of 199 GIST patients refractory to or intolerant of imatinib and sunitinib randomized to regorafenib or best supportive care showed a significantly improved PFS with regorafenib (4.8 months vs 0.9 months).48 Again, similar to sunitinib, there were no complete responders and only 4.5% of patients achieved a partial response on regorafenib.
For patients with progression on regorafenib, other tyrosine kinase inhibitors such as nilotinib,49 pazopanib,50 dasatanib,51,52 or sorafenib53 can be considered. Table 1 summarizes the current list of tyrosine kinase inhibitors used for GIST. These agents have all shown some efficacy in imatinib and sunitinib-resistant GIST in Phase I and Phase II clinical trials, although times to progression are all on the order of months, and partial response rates are uniformly low. The low patient numbers in these trials only allow for limited assessment of associations between mutation status and response to therapy. Thus, decisions to treat patients with these further line tyrosine kinase inhibitors are empiric and not necessarily genotype-based.
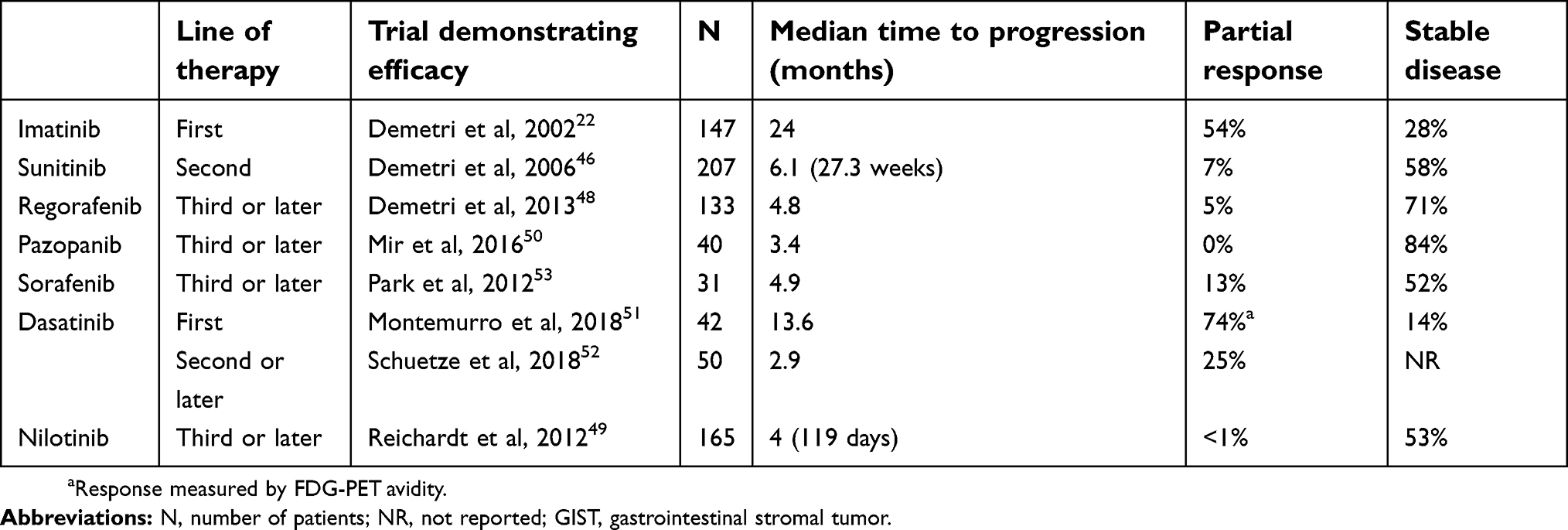 |
Table 1 Tyrosine kinase inhibitors currently used to treat GIST |
Another option for patients with GIST that progressed on imatinib and then sunitinib is to re-challenge them with imatinib. The RIGHT trial was a Phase III trial that randomized 81 patients with metastatic or unresectable GIST who progressed on at least both imatinib and sunitinib to imatinib re-challenge or placebo.54 Median PFS was statistically significantly higher in the imatinib-treated group, but was only 1.8 months, compared to 0.9 months on placebo.
Metastasectomy
As discussed previously, tumors exposed to imatinib develop secondary mutations that confer resistance. Thus, another strategy has been to surgically debulk metastatic disease with the hope of removing resistant clones, removing as much stable or responding disease as is feasible to minimize the development of secondary resistance, and prolonging time to progression. However, data on the efficacy of surgery in patients with metastatic GIST on tyrosine kinase inhibitors are limited to retrospective studies.55–61 A Phase III trial in China attempted to determine if patients undergoing metastasectomy while on imatinib therapy had better outcomes compared to those maintained on drug alone, but closed due to poor accrual (41 of a planned 210 patients).62 There was no significant difference in PFS, though there was a trend favoring surgery (2-year PFS 88.4% vs 57.7%, P=0.089). There was a difference in median OS favoring surgery (not reached vs 49 months, P=0.024), but that was not the primary endpoint. The European Organization for Research and Treatment of Cancer (EORTC) also sought to conduct a randomized controlled trial to determine whether surgery provided additional benefit to patients with metastatic GIST on imatinib (NCT00956072). Unfortunately, the trial was terminated due to low accrual.
The largest retrospective series comes from Brigham and Women’s Hospital/Dana-Farber Cancer Institute and Memorial-Sloan Kettering Cancer Center, which included 400 resections in 323 patients with metastatic GIST on imatinib, sunitinib, or third-line tyrosine kinase inhibitors.57 As expected, patients with responsive disease at the time of surgery did the best, with a median PFS of 31 months after surgery. Patients with stable disease, unifocal progression, or multifocal progression at the time of surgery had a PFS of 19, 10, and 5 months, respectively (P<0.001). Multifocal progression and a high tumor mitotic rate were independent predictors of worse PFS in a multivariable regression model. The subgroup of patients who were on imatinib at the time of surgery had a median PFS of 16 months. Within this subgroup, patients with responsive disease, stable disease, or unifocal progression at the time of surgery had a median PFS of 36, 30, and 11 months, respectively. The PFS of the patients with unifocal progression, in particular, compares favorably to a historical median PFS of <7 months (27 weeks) with sunitinib in the large Phase III trial, which would be the other option for these patients. These data suggest that it is reasonable to consider surgery on patients with advanced GIST responding to, stable on, or having unifocal progression on imatinib (Figure 4). However, only a Phase III trial can confirm whether or not these patients truly benefit from surgery plus imatinib versus imatinib alone. In contrast, in the subgroup of patients on sunitinib at the time of surgery (n=93), radiographic response at the time of surgery was not significantly associated with PFS, and overall these patients had a poor median PFS of 7 months. This is likely reflective of more aggressive tumor biology and suggests that surgery should play a more limited role in patients who are already on second-line or further tyrosine kinase inhibitor therapy.
Ongoing research into alternative strategies
Novel anti-tyrosine kinase agents
Several novel anti-tyrosine kinase agents are currently being tested in Phase I clinical trials. PLX9486 is a tyrosine kinase inhibitor that was found in pre-clinical studies to be effective against patient-derived imatinib-resistant GIST xenografts in mice with both primary exon 9 and 11 mutations as well as secondary exon 17 and 18 mutations,63 PLX9486 did not have good activity against GISTs with KIT exon 13 or 14 mutations, but since tumors with these mutations respond to sunitinib, a Phase I clinical trial was designed to examine PLX9486 activity both alone and in combination with sunitinib or pexidartinib, another tyrosine kinase inhibitor with activity against KIT exon 13 and 14 mutations. The study enrolled 36 patients with metastatic GIST on at least second-line tyrosine kinase inhibitor therapy, and the investigators reported a median PFS of >24 weeks with PLX9486 alone, and a PFS not yet reached for either combination.64
DCC-2618 is a tyrosine kinase inhibitor that inhibits the switch pocket adjacent to the ATP domain of both KIT and PGDFRA. It has been shown to broadly inhibit the activity of all KIT mutants.65 A Phase I clinical trial of DCC-2618 in patients with metastatic GIST on at least second-line therapy found that patients treated with DCC-2618 had a median PFS of 24 weeks.66 DCC-2618 is currently being investigated in the fourth-line setting in a randomized Phase III trial (INVICTUS, NCT03353753).
As previously discussed, advanced GISTs with PDGFRA D842V mutations are particularly challenging to treat due to their inherent imatinib resistance. Avapratinib (BLU-285) is a novel inhibitor that specifically targets KIT exon 17 and PDGFRA D842 mutations.67 A Phase I trial (NAVIGATOR) has enrolled 231 patients with advanced GIST either with a PDGFRA D842V mutation (n=56) or progression on at least second-line tyrosine kinase inhibitor therapy. Preliminary results, which were presented at the 2018 Connective Tissue Oncology Society meeting, showed a remarkable 86% response rate in patients with PDGFRA D842V mutations, with 8 complete responses by RECIST 1.1, while the other subgroups, including patients on fourth-line therapy or further, had response rates of 20–26%.68 Currently, avapritinib is being compared to regorafenib in the third- or fourth-line setting in a Phase III randomized trial (VOYAGER, NCT03465722).
Other targeted therapy
The overall diminishing returns of switching from one tyrosine kinase inhibitor to another highlights the need for alternative therapeutic strategies against GIST. Some groups are investigating combining imatinib with inhibition of other molecules further downstream in the KIT signaling pathway. Floris and colleagues assessed the effect of imatinib plus the PI3K inhibitor GDC-0941 on the growth of human GIST xenografts in mice. They found that combination therapy induced significantly more tumor regression compared to imatinib alone, and furthermore that tumor regression was sustained after withdrawal of combination therapy, while tumors treated with imatinib alone regrew after cessation of therapy.69 Chi and colleagues showed that high levels of the transcription factor ETV1 cooperate with KIT to enhance GIST tumorigenesis from ICC precursors.70 Furthermore, in human GIST cell lines and xenograft mouse models, dual inhibition of ETV1 and KIT was more effective than inhibition of KIT alone.71
As previously discussed, wild-type GISTs tend to be imatinib-resistant. However, a small proportion of wild-type GISTs that are also SDH-proficient harbor an ETV6-NTRK3 gene fusion.72 A recent Phase II study of larotrectinib, a TRK inhibitor, enrolled 55 patients with TRK-fusion-positive cancers. Three patients had TRK-fusion-positive GIST, of whom 2 achieved a partial response and 1 achieved a complete response.73 While TRK-fusion-positive tumors represent a small proportion of GISTs, the response rates are impressive and highlight the growing importance of molecular tumor characterization in guiding GIST therapy.
Immunotherapy
There appears to be a good rationale for immunotherapy in GIST, either alone or in combination with KIT inhibition, based on several preclinical studies. The presence of high immune infiltration of GIST specimens is predictive of PFS.74 In addition, imatinib seems to increase anti-tumor T-cell responses in GIST mouse models. Balachandran and colleagues found that imatinib treatment of a mouse model that develops spontaneous GISTs increased CD8+ T cell activation within the tumor and induced regulatory T cell apoptosis. This immunogenic effect seemed to occur through imatinib inhibition of tumor cell expression of Ido, an immunosuppressive enzyme.75
Unfortunately, clinical results so far have been modest at best for GIST. In a Phase I trial by D’Angelo and colleagues, 28 patients with advanced sarcoma (of which 20 were GIST) were treated with dasatinib plus the CTLA-4 inhibitor ipilimumab. There were no partial or complete responses by RECIST, although there were 7 partial responses by Choi criteria in 13 GIST patients in which this was evaluable. Further research is needed to identify GIST patients that would be most likely to benefit from immunotherapy, and also to determine what the optimal regimen for these patients is.
Summary and conclusions
Current therapeutic strategies for GIST revolve around targeted therapy against KIT and PDGFRA. As experience with imatinib and other tyrosine kinase inhibitors has increased, clinicians have also been able to identify genotypic predictors of response and resistance to therapy to tailor targeted therapies for individual patients. The mutations with the most robust associations with response to imatinib include PDGFRA D842V (imatinib resistance), KIT/PDGFRA WT (imatinib resistant), and KIT exon 9 mutations (partial imatinib resistance, responds to higher-dose imatinib). There are other associations between mutational status and response to therapy, as summarized in Table 2, but they are often derived from smaller studies and have more limited clinical applicability at this time.
 |
Table 2 Treatment implications of KIT and PDGFRA mutations |
For patients with available mutational profiling, Figure 3 summarizes the current management of resectable GIST, and Figure 4 summarizes the management of metastatic or unresectable GIST. Currently, the outcomes for patients with advanced GIST are still quite poor, and there is an urgent need for novel therapeutic strategies. There has been some promise in novel tyrosine kinase inhibitors, non-tyrosine kinase-targeted therapy, and immunotherapy, but further research into these avenues is required.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gold JS, Dematteo RP. Combined surgical and molecular therapy: the gastrointestinal stromal tumor model. Ann Surg. 2006;244:176–184. doi:10.1097/01.sla.0000218080.94145.cf
2. Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373:347–349. doi:10.1038/373347a0
3. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580.
4. Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–8121.
5. Tuveson DA, Willis NA, Jacks T, et al. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20:5054–5058. doi:10.1038/sj.onc.1204704
6. Emile JF, Brahimi S, Coindre JM, et al. Frequencies of KIT and PDGFRA mutations in the MolecGIST prospective population-based study differ from those of advanced GISTs. Med Oncol. 2012;29:1765–1772. doi:10.1007/s12032-011-0074-y
7. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi:10.1200/JCO.2003.04.190
8. Wozniak A, Rutkowski P, Piskorz A, et al. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): polish clinical GIST registry experience. Ann Oncol. 2012;23:353–360. doi:10.1093/annonc/mdr127
9. Lev S, Blechman J, Nishikawa S, Givol D, Yarden Y. Interspecies molecular chimeras of kit help define the binding site of the stem cell factor. Mol Cell Biol. 1993;13:2224–2234. doi:10.1128/mcb.13.4.2224
10. Besmer P, Murphy JE, George PC, et al. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature. 1986;320:415–421. doi:10.1038/320415a0
11. Zsebo KM, Williams DA, Geissler EN, et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63:213–224.
12. Hemesath TJ, Price ER, Takemoto C, Badalian T, Fisher DE. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature. 1998;391:298–301. doi:10.1038/34681
13. Timokhina I, Kissel H, Stella G, Besmer P. Kit signaling through PI 3-kinase and Src kinase pathways: an essential role for Rac1 and JNK activation in mast cell proliferation. Embo J. 1998;17:6250–6262. doi:10.1093/emboj/17.21.6250
14. Nishida T, Hirota S, Taniguchi M, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet. 1998;19:323–324. doi:10.1038/1209
15. Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–4190. doi:10.1158/1078-0432.CCR-04-2245
16. Cassier PA, Fumagalli E, Rutkowski P, et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res. 2012;18:4458–4464. doi:10.1158/1078-0432.CCR-11-3025
17. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi:10.1126/science.1079666
18. Subramanian S, West RB, Corless CL, et al. Gastrointestinal stromal tumors (GISTs) with KIT and PDGFRA mutations have distinct gene expression profiles. Oncogene. 2004;23:7780–7790. doi:10.1038/sj.onc.1208056
19. Wasag B, Debiec-Rychter M, Pauwels P, et al. Differential expression of KIT/PDGFRA mutant isoforms in epithelioid and mixed variants of gastrointestinal stromal tumors depends predominantly on the tumor site. Mod Pathol. 2004;17:889–894. doi:10.1038/modpathol.3800136
20. Pappo AS, Janeway KA. Pediatric gastrointestinal stromal tumors. Hematol Oncol Clin North Am. 2009;23:15–34,vii. doi:10.1016/j.hoc.2008.11.005
21. Boikos SA, Pappo AS, Killian JK, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors: a report from the national institutes of health gastrointestinal stromal tumor clinic. JAMA Oncol. 2016;2:922–928. doi:10.1001/jamaoncol.2016.0256
22. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi:10.1056/NEJMoa020461
23. Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–625. doi:10.1200/JCO.2007.13.4403
24. Gastrointestinal Stromal Tumor Meta-Analysis G. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247–1253. doi:10.1200/JCO.2009.24.2099
25. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–1134. doi:10.1016/S0140-6736(04)17098-0
26. Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373:1097–1104. doi:10.1016/S0140-6736(09)60500-6
27. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563–1570. doi:10.1200/JCO.2013.51.2046
28. Joensuu H, Eriksson M, Sundby Hall K, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. Jama. 2012;307:1265–1272. doi:10.1001/jama.2012.347
29. Joensuu H, Wardelmann E, Sihto H, et al. Effect of KIT and PDGFRA mutations on survival in patients with gastrointestinal stromal tumors treated with adjuvant imatinib: an exploratory analysis of a randomized clinical trial. JAMA Oncol. 2017;3:602–609. doi:10.1001/jamaoncol.2016.5751
30. Raut CP, Espat NJ, Maki RG. et al. Efficacy and tolerability of 5-Year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: the PERSIST-5 clinical trial. JAMA Oncol;2018. e184060. doi:10.1001/jamaoncol.2018.4060
31. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42–47. doi:10.1002/jso.21160
32. Kurokawa Y, Yang HK, Cho H, et al. Phase II study of neoadjuvant imatinib in large gastrointestinal stromal tumours of the stomach. Br J Cancer. 2017;117:25–32. doi:10.1038/bjc.2017.144
33. Cavnar MJ, Wang L, Balachandran VP, et al. Rectal Gastrointestinal Stromal Tumor (GIST) in the Era of Imatinib: organ preservation and improved oncologic outcome. Ann Surg Oncol. 2017;24:3972–3980. doi:10.1245/s10434-017-6087-9
34. Tielen R, Verhoef C, van Coevorden F, et al. Surgical management of rectal gastrointestinal stromal tumors. J Surg Oncol. 2013;107:320–323. doi:10.1002/jso.23223
35. Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–1103. doi:10.1016/j.ejca.2006.01.030
36. Gounder MM, Maki RG. Molecular basis for primary and secondary tyrosine kinase inhibitor resistance in gastrointestinal stromal tumor. Cancer Chemother Pharmacol. 2011;67 Suppl 1:S25–S43. doi:10.1007/s00280-010-1526-3
37. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510–7518. doi:10.1158/1078-0432.CCR-09-0190
38. Lasota J, Dansonka-Mieszkowska A, Sobin LH, Miettinen M. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest. 2004;84:874–883. doi:10.1038/labinvest.3700122
39. Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23:5357–5364. doi:10.1200/JCO.2005.14.068
40. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–5359. doi:10.1200/JCO.2007.15.7461
41. Antonescu CR, DeMatteo RP. CCR 20th anniversary commentary: a genetic mechanism of imatinib resistance in gastrointestinal stromal tumor – where are we a decade later? Clin Cancer Res. 2015;21:3363–3365. doi:10.1158/1078-0432.CCR-14-3120
42. Tamborini E, Pricl S, Negri T, et al. Functional analyses and molecular modeling of two c-Kit mutations responsible for imatinib secondary resistance in GIST patients. Oncogene. 2006;25:6140–6146. doi:10.1038/sj.onc.1209639
43. Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–4774. doi:10.1200/JCO.2006.06.2265
44. Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2:471–478.
45. Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337.
46. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–1338. doi:10.1016/S0140-6736(06)69446-4
47. Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129:245–255. doi:10.1002/ijc.25864
48. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302. doi:10.1016/S0140-6736(12)61857-1
49. Reichardt P, Blay JY, Gelderblom H, et al. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann Oncol. 2012;23:1680–1687. doi:10.1093/annonc/mdr598
50. Mir O, Cropet C, Toulmonde M, et al. Pazopanib plus best supportive care versus best supportive care alone in advanced gastrointestinal stromal tumours resistant to imatinib and sunitinib (PAZOGIST): a randomised, multicentre, open-label phase 2 trial. Lancet Oncol. 2016;17:632–641. doi:10.1016/S1470-2045(16)00075-9
51. Montemurro M, Cioffi A, Domont J, et al. Long-term outcome of dasatinib first-line treatment in gastrointestinal stromal tumor: a multicenter, 2-stage phase 2 trial (Swiss Group for Clinical Cancer Research 56/07). Cancer. 2018;124:1449–1454. doi:10.1002/cncr.31234
52. Schuetze SM, Bolejack V, Thomas DG, et al. Association of dasatinib with progression-free survival among patients with advanced gastrointestinal stromal tumors resistant to imatinib. JAMA Oncol. 2018;4:814–820. doi:10.1001/jamaoncol.2018.0601
53. Park SH, Ryu MH, Ryoo BY, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs. 2012;30:2377–2383. doi:10.1007/s10637-012-9795-9
54. Kang YK, Ryu MH, Yoo C, et al. Resumption of imatinib to control metastatic or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib (RIGHT): a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2013;14:1175–1182. doi:10.1016/S1470-2045(13)70453-4
55. Andtbacka RH, Ng CS, Scaife CL, et al. Surgical resection of gastrointestinal stromal tumors after treatment with imatinib. Ann Surg Oncol. 2007;14:14–24. doi:10.1245/s10434-006-9034-8
56. DeMatteo RP, Maki RG, Singer S, Gonen M, Brennan MF, Antonescu CR. Results of tyrosine kinase inhibitor therapy followed by surgical resection for metastatic gastrointestinal stromal tumor. Ann Surg. 2007;245:347–352. doi:10.1097/01.sla.0000236630.93587.59
57. Fairweather M, Balachandran VP, Li GZ, et al. Cytoreductive surgery for metastatic gastrointestinal stromal tumors treated with tyrosine kinase inhibitors: a 2-institutional analysis. Ann Surg. 2018;268:296–302. doi:10.1097/SLA.0000000000002281
58. Gronchi A, Fiore M, Miselli F, et al. Surgery of residual disease following molecular-targeted therapy with imatinib mesylate in advanced/metastatic GIST. Ann Surg. 2007;245:341–346. doi:10.1097/01.sla.0000242710.36384.1b
59. Raut CP, Posner M, Desai J, et al. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol. 2006;24:2325–2331. doi:10.1200/JCO.2005.05.3439
60. Raut CP, Wang Q, Manola J, et al. Cytoreductive surgery in patients with metastatic gastrointestinal stromal tumor treated with sunitinib malate. Ann Surg Oncol. 2010;17:407–415. doi:10.1245/s10434-009-0784-y
61. Rutkowski P, Nowecki Z, Nyckowski P, et al. Surgical treatment of patients with initially inoperable and/or metastatic gastrointestinal stromal tumors (GIST) during therapy with imatinib mesylate. J Surg Oncol. 2006;93:304–311. doi:10.1002/jso.20466
62. Du CY, Zhou Y, Song C, et al. Is there a role of surgery in patients with recurrent or metastatic gastrointestinal stromal tumours responding to imatinib: a prospective randomised trial in China. Eur J Cancer. 2014;50:1772–1778. doi:10.1016/j.ejca.2014.03.280
63. Gebreyohannes YK, Burton EA, Wozniak A, et al. PLX9486 shows anti-tumor efficacy in patient-derived, tyrosine kinase inhibitor-resistant KIT-mutant xenograft models of gastrointestinal stromal tumors. Clin Exp Med. 2018;19:20.
64. Wagner AJ, Tap WD, Shields AF, et al. A phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of PLX9486 alone and in combination (combo) with the KIT inhibitors pexidartinib (pexi) or sunitinib (su) in patients (Pts) with advanced solid tumors and gastrointestinal stromal tumor (GIST). J Clin Oncol. 2018;36(15_suppl):11509.
65. Smith BD, Hood MM, Wise SC, et al. Abstract 2690: DCC-2016 is a potent inhibitor of wild-type and mutant KIT, including refractory Exon 17 D816 KIT mutations, and exhibits efficacy in refractory GIST and AML xenograft models. Cancer Res. 2015;75:2690. doi:10.1158/0008-5472.CAN-14-3569
66. George S, Heinrich M, Chi P, et al. Abstract: initial results of phase I study of DCC-2618, a broad-spectrum KIT and PDGFRa inhibitor, in patients (pts) with gastrointestinal stromal tumor (GIST) by number of prior regimens. Ann Oncol. 2018;29. doi:10.1093/annonc/mdx807
67. Evans EK, Hodous BL, Gardino AK, et al. Abstract 791: BLU-285, the first selective inhibitor of PDGFRa D842V and KIT Exon 17 mutants. Cancer Res. 2015;75. doi:10.1158/0008-5472.CAN-14-3569
68. Heinrich MC, von Mehren M, Jones RL, et al. Abstract 012: avapritinib is highly active and well-tolerated in patients with advanced GIST driven by a diverse variety of oncogenic mutations in KIT and PDGFRA. Rome, Italy:
69. Floris G, Wozniak A, Sciot R, et al. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res. 2013;19:620–630. doi:10.1158/1078-0432.CCR-12-2853
70. Chi P, Chen Y, Zhang L, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;467:849–853. doi:10.1038/nature09409
71. Ran L, Sirota I, Cao Z, et al. Combined inhibition of MAP kinase and KIT signaling synergistically destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discov. 2015;5:304–315. doi:10.1158/2159-8290.CD-14-0985
72. Brenca M, Rossi S, Polano M, et al. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J Pathol. 2016;238:543–549. doi:10.1002/path.4677
73. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731–739. doi:10.1056/NEJMoa1714448
74. Rusakiewicz S, Semeraro M, Sarabi M, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013;73:3499–3510. doi:10.1158/0008-5472.CAN-13-0371
75. Balachandran VP, Cavnar MJ, Zeng S, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17:1094–1100.
76. Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26:5360–5367. doi:10.1200/JCO.2008.17.4284
77. Ben-Ami E, Barysauskas CM, von Mehren M, et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann Oncol. 2016;27:1794–1799. doi:10.1093/annonc/mdw228
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.