")
Back to Journals » OncoTargets and Therapy » Volume 10
Targeted regulation of MiR-98 on E2F1 increases chemosensitivity of leukemia cells K562/A02
Authors Huang YD, Hong XL, Hu J, Lu Q
Received 5 November 2016
Accepted for publication 15 May 2017
Published 29 June 2017 Volume 2017:10 Pages 3233—3239
DOI https://doi.org/10.2147/OTT.S126819
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ingrid Espinoza
Yingdan Huang,1,2 Xiuli Hong,2 Jiasheng Hu,2 Quanyi Lu2
1Department of Blood Transfusion, The First Affiliated Hospital of Xiamen University, 2Department of Hematology, Zhongshan Hospital of Xiamen University, Xiamen, People’s Republic of China
Background: miRNA is a microRNA that negatively regulates protein expression at post-transcriptional or translational level. It is widely involved in the pathogenesis of tumors. miR-98 belongs to the let-7 family, and its overexpression can increase the sensitivity to drugs in solid cancer cells. However, the function of miR-98 in leukemia is still unclear. In this study, the effect of miR-98 on drug resistance and proliferation of leukemia cells were investigated.
Methods: Real-time quantitative polymerase chain reaction analyzed the expression difference between miR-98 and E2F1 in leukemia cell lines, K562 and K562/A02. The downstream target gene of miR-98 was predicted by TargetScan; K562/A02 was transiently transfected with miR-98 mimic to upregulate the expression of miR-98; real-time quantitative polymerase chain reaction and Western blot were used to analyze the expression alterations of E2F1; cell counting kit-8 was used to evaluate the influence on K562/A02 proliferation and sensitivity to chemotherapeutic drugs; meanwhile, Western blot was used to analyze the expression of p21, Bax, matrix metalloproteinase 9 and ABCG2 proteins.
Results: E2F1 is one of the target genes of miR-98 proved by bioinformatics. Compared with the K562, the level of miRNA-98 expression was decreased in K562/A02, but the level of E2F1 expression was upregulated. Leukemia cell line K562/A02 was transfected with miR-98 mimic to upregulate the expression of miR-98, the expression of E2F1 was significantly decreased. After upregulating the miR-98 expression in K562/A02, the proliferation was weakened, and the sensitivity to chemotherapy was increased. Western blot showed that upregulated miR-98 expression increased the levels of p21 and BAX proteins in K562/A02 cells, and decreased the levels of matrix metalloprotease 9 and ABCG2 proteins, which were significantly different compared with those before miR-98 mimic transfection.
Conclusion: In the leukemia drug-resistant cell line K562/A02, the targeted upregulated expression of miR-98 could decrease the proliferation of leukemia cells and improve the sensitivity to chemotherapeutics by inhibiting E2F1 expression. miR-98 might be a potential target for overcoming leukemia multidrug resistance.
Keywords: miRNA-98, leukemia cells, E2F1, drug resistance, K562/A02
Background
Acute myeloid leukemia is one of the common malignant hematologic diseases, and combination chemotherapy is the primary method of treatment.1,2 Recently, due to the application of new targeting therapeutics and development of hematopoietic stem cell transplantation, the patients’ prognosis has been greatly improved. However, most of the patients succumb to drug resistance and relapse.3 That is, even if the patients with drug sensitivity are treated chemotherapeutically, after several rounds, they start showing resistance to one or multiple drugs.4,5 Therefore, the reversibility of multidrug resistance becomes an urgent issue in tumor therapy.
MicroRNA (miRNA) is an endogenous non-coding single-stranded ribonucleic acid (RNA) molecule with 19–25 nt in eukaryotes. Although miRNA does not encode a protein, it degrades or inhibits the translation by interacting with the 3′ untranslated regions of specific target mRNA at the post-transcriptional level and regulates the expression of the target gene.6 It is further involved in tumor development, including oncobiology, invasion, metastasis, and chemotherapeutic drug resistance.7–10 An increasing number of studies found that the imbalance of miRNA in different tumors was closely related to the drug resistance of tumor cells. For instance, To et al studied the drug-resistant colon cancer cell line S1MI80 and found that the miR-519c expression level was abnormal. The miR-519c downregulated the tolerance of cells to drugs by influencing the expression of ABCG2.11 Zhu et al reported that miR-181a/b was involved in the drug resistance regulation of leukemic cells by regulating multi-antiapoptotic genes.12
miR-98 is one of the let-7 family numbers. Wendler et al discovered that in the ovarian cancer cell line, miR-98 could regulate PGRMG1 expression, thereby influencing the proliferation and chemotherapy resistance of the tumor cells.13 Xiang et al reported that miR-98 increased the sensitivity of human lung adenocarcinoma cells A549/DDP to cisplatin by upregulating HMGA2,14 indicating that miR-98 could be associated with the drug resistance of tumor cells. E2F family is a group of genes coding transcriptional regulatory factors that are crucial for cell cycle progression. Among them, E2F1 is an important positive regulatory factor in the cell cycle, playing a pivotal role in the progression from G0/G1 to S phase.15 It has been established that E2F1 is the target gene of miR-98, which is involved in the augmentation of drug resistance by regulating proliferation and apoptosis.16,17
Currently, whether the abnormal expression of miR-98 is involved in leukemia chemotherapy drug resistance and if the pathway influences the leukemia cell resistance is yet ill-understood. In our study, the mechanism of miR-98 involved in drug resistance of leukemia cells was confirmed, providing a novel pathway for overcoming the chemotherapeutic resistance in leukemic cells.
Methods
Cell line and cell culture
The human chronic myeloid leukemia cell line K562 and its multidrug-resistance counterpart K562/A02 were purchased from Tianjin Institute of Hematology (Shanghai, People’s Republic of China). K562 cells were cultured in Roswell Park Memorial Institute 1640 medium (Gibco BRL, Grand Island, NY, USA) containing 10% fetal bovine serum and penicillin–streptomycin (100 U/mL each) and at 37°C and 5% CO2 with saturated humidity. In order to continually maintain the multidrug resistance phenotype, 1 μg/mL of Adriamycin® (ADR) was added to the culture medium of K562/A02. Before the experiment, the cells were cultured in the drug-free medium for 2 weeks.
Real-time fluorescent quantitative polymerase chain reaction (RT-PCR)
The total RNA was extracted by TRIzol reagent (Invitrogen, Carlsbad, CA, USA). miRNA was extracted with the kit (Tiangen, Beijing, People’s Republic of China) and quantitated at 260 nm. RT-PCR was employed to analyze the expression level of mature miRNA-98.18 Reverse transcription kit was purchased from Thermo Fisher Scientific (Waltham, MA, USA), and fluorescent quantitation kit was purchased from SYBR Green (Bioneer, Daejeon, South Korea). U6 snRNA served as an endogenous control to normalize and estimate the relative expression of miR-98 by 2−ΔΔCt (19). ΔΔCt = ΔCtK562/A02 − ΔCtK562, ΔCt = CtmiRNA − CtU6 snRNA.
The total RNA was reverse transcribed into cDNA, according to the manufacturer’s instructions. E2F1 gene expression was quantitated by real-time fluorescent quantitation PCR. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression level was used as a reference control. The reaction condition was as follows: denaturation at 95°C for 10 min, followed by denaturation at 95°C for 10 s, and annealing and extension at 60°C for 60 s, for 40 cycles. The gene-specific primers are shown in Table 1.
 | Table 1 The primers used for RT-PCR |
miRNA transfection assay
miR-98 mimic and negative control (5′-3′ UGAGGUAGUAAGUUGUAUUGUU) were purchased from RiBoBio (Guangzhou, People’s Republic of China). K562/A02 cells were plated in a 6-well plate at 6×105/well. The miR-98 mimic and negative control were transfected into K562/A02, according to the manufacturer’s instructions (riboFECT™ CP Transfection Kit; RiBoBio, Guangzhou, People’s Republic of China).
Cell viability assay
Detect median inhibitory concentration (IC50) and resistance index using cell counting kit-8 (CCK8) cell assay
We had 2 cell lines: K562 and K562/A02. The cells were seeded in 96-well plates at the rate of 5,000 cells per well. ADR was added into each well at 6 concentration gradients. Six parallel holes were set up at each concentration. ADR was added to each well according to the following concentration gradients: k562–0.02, 0.04, 0.08, 0.16, 0.32, 0.64 μg/mL; K562/A02–2, 4, 8, 16, 32, 64 μg/mL, and the cells were cultured for 48 h. After that, 10 μL CCK8 was added into each well and shaken, and then put into the incubator. Two hours later, the 96-well plate was put into a pre-heated microplate reader. The absorbance values optical density (OD) was measured at a wavelength of 450 nm in each well.
Cell growth inhibition ratio = (OD in control group-OD in experiment group)/OD in control group × 100%. The median inhibitory concentration (IC50) of ADR in the cells was calculated by SPSS12.0 (SPSS Inc., Chicago, IL, USA). IC50 of miR-98 mimic, miR-negative (NC), and untreated (UN) group in K562/A02 after 48 h post transfection were detected by the similar methods.
In K562/A02 cells, miR-98 mimic and miR-NC were transfected, respectively, and the untransfected cells were used as control (UN). After 24 h, the cells were seeded into a 96-well plate at 2×103/well (200 μL), and only medium wells in triplicates were considered as baseline. CCK8 (20 μL) was added to each well on days 1, 2, 3 and 4. OD at 450 nm was measured after 2.5 h incubation, and the average of triplicates was calculated.
Western blot analysis
After transfection with miR-98 mimic for 48 h, the cells were washed, harvested and lysed to extract the total protein. The concentration was estimated by bicinchoninic acid assay, and 50 μg protein was resolved by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. GAPDH (Cell Signaling Technology Inc, Boston, MA, USA) was used as a loading control to assess the differences in the expression of E2F1 and ABCG2 in K562 and K562/A02. The Western blot membrane was probed with E2F1 (Abcam Inc, Wuhan, People’s Republic of China), ABCG2 (Cell Signaling Technology), Bax (Abcam), p21 (Abcam), matrix metalloproteinase 9 (MMP9; Cell Signaling Technology) primary antibodies at 1:1,000. Subsequently, goat anti-rabbit and goat anti-rat secondary antibodies were added. ImageJ (National Institutes of Health, Bethesda, MD, USA) was used to analyze the images.
Data analysis
All the experiments were independently repeated 3 times, and SPSS12.0 was used to analyze the data expressed as mean ± SD. The comparison between groups was analyzed by Student’s t-test, and comparison among several groups was analyzed by one-way analysis of variance. P≤0.05 was termed as statistically significant.
Results
Expression of miR-98 and E2F1 in K562/A02 cells
The toxicity of ADR on K562 and K562/A02 was assessed by CCK8. The IC50 values of K562 and K562/A02 were 0.33±0.11 μg/mL and 24.12±1.7 μg/mL, respectively, and the drug resistance of the latter was ~73-fold of the former (Figure 1A). PCR was used to analyze the expression of miR-98 in different leukemia cell lines, and it was found that it was significantly lower in K562/A02 cells than K562 cells (Figure 1B). RT-PCR showed varied expression level of E2F1 in different cell lines, thereby indicating E2F1 was significantly increased in K562/A02 cells (Figure 1C).
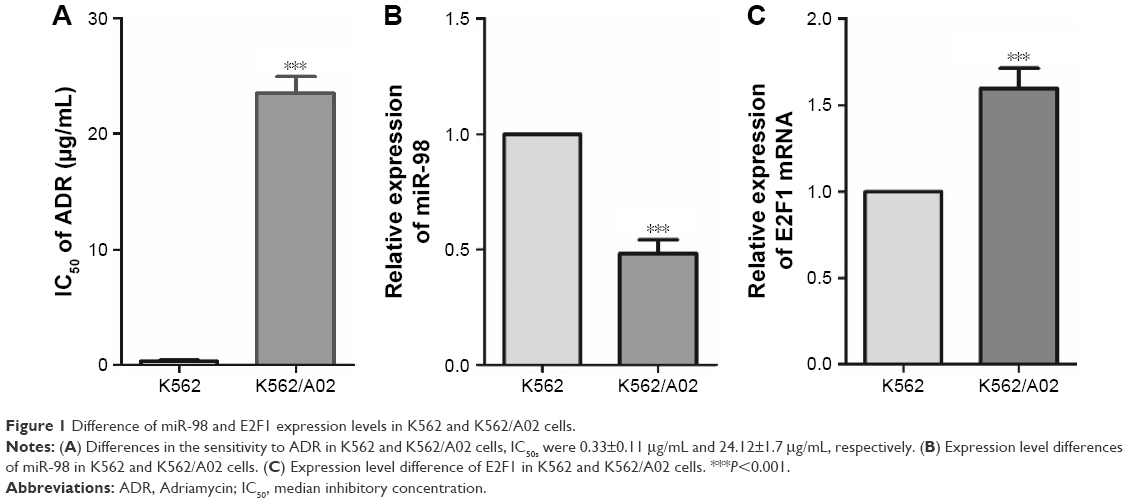 | Figure 1 Difference of miR-98 and E2F1 expression levels in K562 and K562/A02 cells. |
Influence of miR-98 expression on E2F1 in leukemia cells
The downstream target gene of miR-98 was predicted by TargetScan (http://www.Targetscan.org). The sequence comparison indicated that one of the miR-98 downstream target genes was E2F1 (Figure 2A).
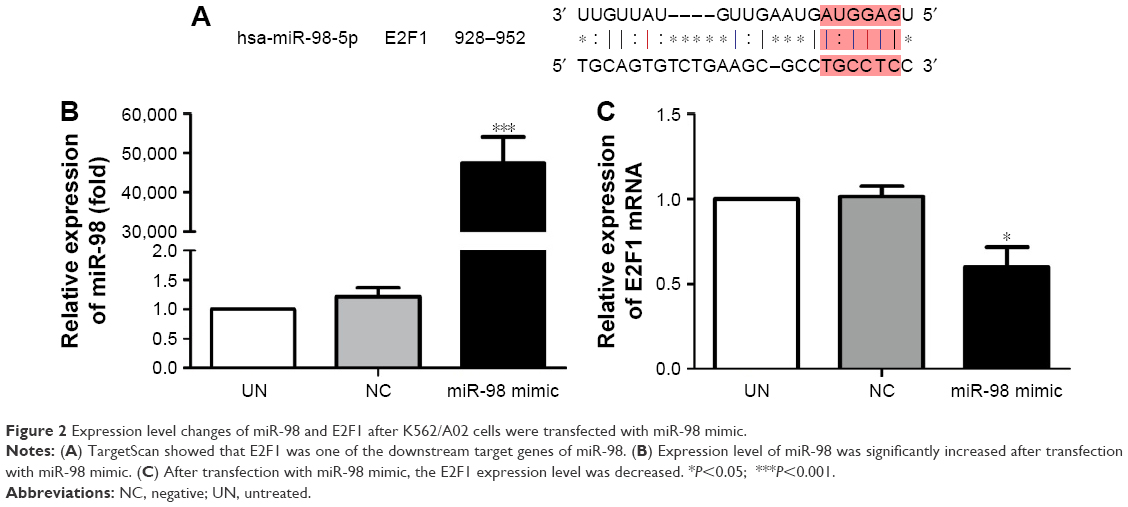 | Figure 2 Expression level changes of miR-98 and E2F1 after K562/A02 cells were transfected with miR-98 mimic. |
In order to confirm the influence of miR-98 expression on endogenous E2F1 expression in K562/A02 cells, the mature miR-98 mimic was transfected in K562/A02 cells causing the endogenous miR-98 expression level to upregulate by 5,000-fold (Figure 2B). Furthermore, RT-PCR established that post K562/A02 cells transfection with miR-98 mimic, the E2F1 expression level was significantly down-regulated, lower than that before transfection (Figure 2C). Compared with K562 cells, higher levels of ABCG2 and E2F1 proteins were expressed in K562/A02 cells, which were analyzed by Western blot (Figure 3A). After transfection with miR-98 mimic, the E2F1 protein expression level was significantly downregulated, lower than that before transfection (Figure 3B).
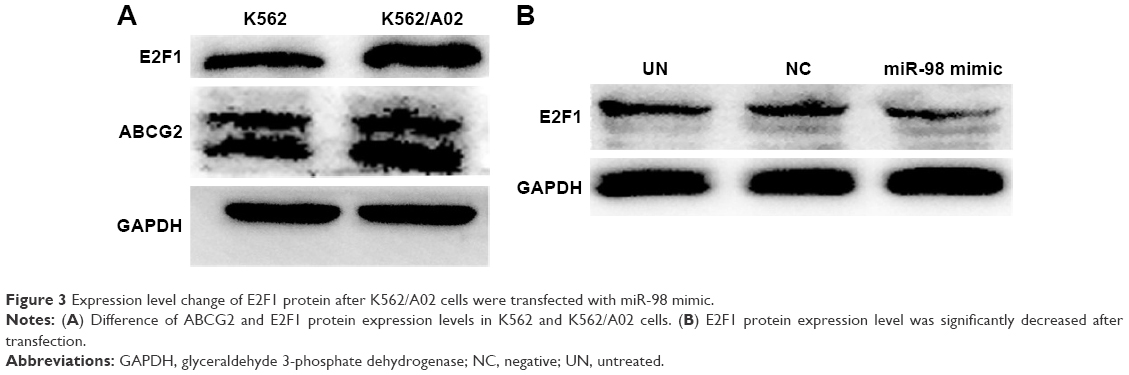 | Figure 3 Expression level change of E2F1 protein after K562/A02 cells were transfected with miR-98 mimic. |
Upregulation of miR-98 expression inhibits proliferation of leukemia cells and increases the sensitivity to ADR
The influence of miR-98 overexpression on the proliferation of K562/A02 was detected by CCK-8. It was found that the proliferation of the cells transfected by miR-98 mimic was significantly decreased compared with the control group (NC and UN). Moreover, NC group did not exhibit any statistical difference when compared with UN group (Figure 4A). It suggested that the increase in miR-98 expression level could significantly inhibit the proliferation of K562/A02 cells. On the other hand, after transfection with miR-98 mimic, IC50 of ADR was 12.19±1.78 μg/mL, which was lower than the NC (24.52±1.7 μg/mL) and UN (24.12±1.5 μg/mL) groups (P<0.05). It was suggested that upregulation of miR-98 expression improved the sensitivity of cells to ADR (Figure 4B).
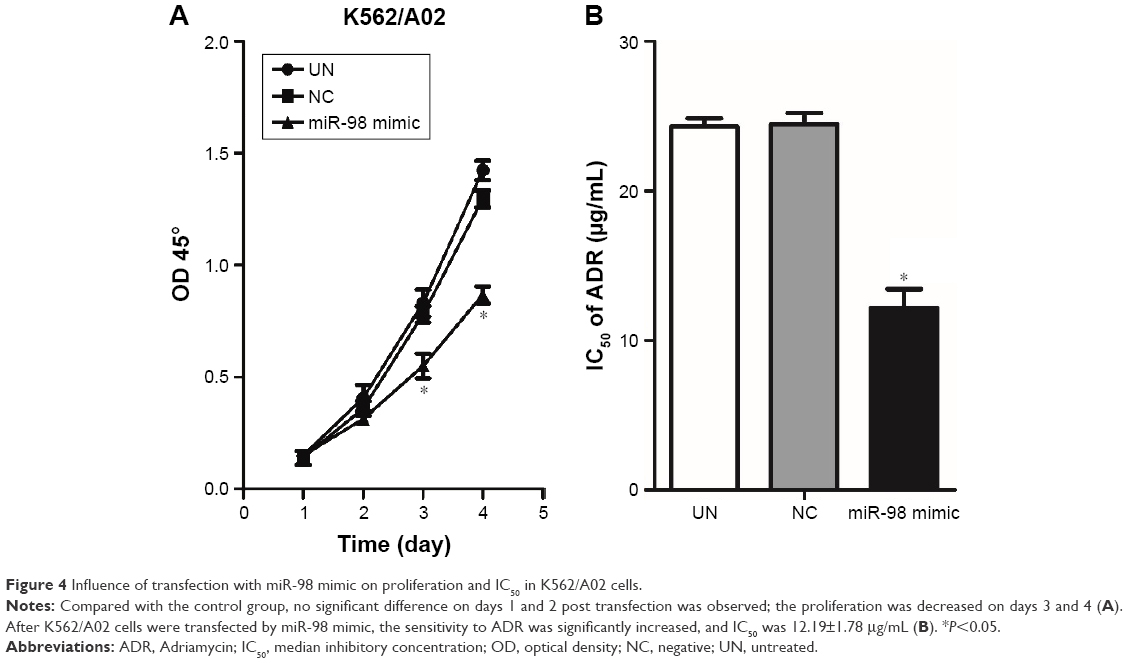 | Figure 4 Influence of transfection with miR-98 mimic on proliferation and IC50 in K562/A02 cells. |
Overexpression of miR-98 downregulated ABCG2 and MMP9 expression
The influences of miR-98 overexpression on the apoptotic proteins BAX, cell cycle regulation factor p21, drug resistance protein ABCG2, and MMP9 were analyzed by Western blot. The result showed that overexpression of miR-98 enhanced BAX and p21 expression and decreased the expression of ABCG2 and MMP9, which further increased the sensitivity of K562/A02 to ADR (Figure 5).
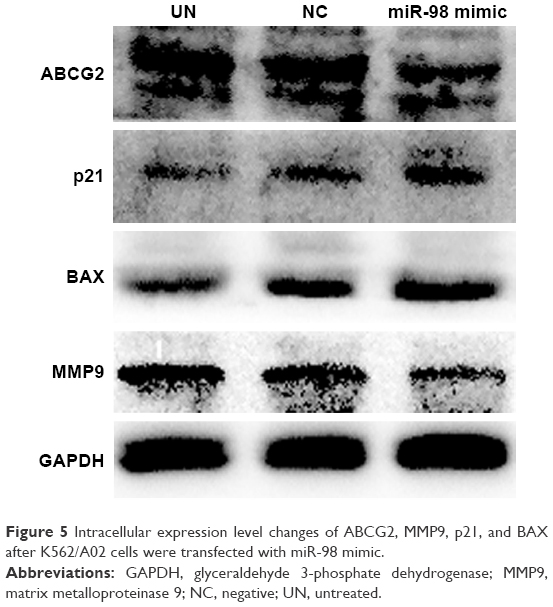 | Figure 5 Intracellular expression level changes of ABCG2, MMP9, p21, and BAX after K562/A02 cells were transfected with miR-98 mimic. |
Discussion
Currently, combination therapy is the primary mode of treatment for acute leukemia. However, the drug tolerance of leukemic cells to chemotherapeutic drugs resulting from multiple regimens is responsible for reduced efficacy.19 A deeper insight into the molecular mechanism of drug resistance is the key to resolving the issue. Several researchers have proved that expression dysregulation of miRNA in tumor cells could influence their sensitivity of the tumor to drugs by regulating target gene expression.20–22 Therefore, target-regulating miRNA is the advanced pathway for overcoming chemotherapy resistance of tumor cells.
In 1986, Kovesdi et al first discovered the cell cycle-related factor E2F1.23 Since then, a myriad of studies have demonstrated that E2F is not only involved in cell cycle regulation and apoptotic signal transduction but also closely related to growth, apoptosis, metastasis, and drug resistance of tumor.24–27 In the study of renal clear cell carcinoma, Ma et al16 reported that the overexpressed E2F1 improved metastasis of tumor cells by activating MMP2 and MMP9. Yoshihara et al also showed that28 p21 and BAX expressions could be increased and the apoptosis caused by ADR also enhanced by decreasing intracellular E2F1 expression. Rosenfeldt et al found that E2F1 could increase the resistance to chemotherapeutic drugs by regulating ABCG2 expression level in the cells.29 Altogether, these results indicated that the high expression of E2F was one of the factors improving tumor drug resistance and could be a potential target for regulating tumor cells’ drug resistance.
miR-98 has been shown to be one of the miRNA molecules possibly associated with the mechanism of tumor cells drug resistance.13,14 Siragam et al30 found that the upregulation of miR-98 expression could decrease angiogenesis and lead to the invasion of breast cancer by target inhibiting anaplastic lymphoma kinase (ALK4) and MMP11 expressions. Huang et al31 also reported that the upregulation of miR-98 expression could significantly decrease the invasion and metastasis of esophageal squamous cancer, indicating that miR-98 might be a cancer suppressor gene. TargetScan result showed that E2F1 was one of the target genes of miR-98. In our study, the miR-98 expression level was significantly downregulated in leukemia drug resistance cells. Through target regulating miR-98 expression level, the sensitivity to chemotherapy drugs could be altered, and the regulation was mediated by influencing the E2F1 expression.
In our study, we used miRNA mimic transfection method to upregulate miR-98 expression in drug-resistant cells. The results showed that when miR-98 was significantly upregulated, intracellular E2F1 expression was substantially decreased along with the proliferation, whereas the sensitivity to ADR was remarkably improved. Herein, we showed for the first time that the miR-98 expression changes in leukemic cells influenced the sensitivity to chemotherapy through the expression of E2F1. miR-98 regulated the proliferation speed and sensitivity to drugs by target downregulation of E2F1 and the downstream protein expressions. We also confirmed that upregulation of miR-98 expression led to decrease in MMP9 and ABCG2 protein expression, an increase in p21 and BAX protein expression, which was in agreement with previous studies.14–16 Therefore, we established that upregulating the miR-98 level increased the sensitivity to chemotherapeutic drugs mediated by inhibiting E2F1 expression, decreasing MMP9 and ABCG2 expression, and increasing p21 and BAX expression. miR98-E2F1 could be speculated as potential machinery for overcoming leukemia cells’ drug resistance.
It is widely known that the expression changes of miRNA can influence several intracellular gene expression levels. One ordinary miRNA can regulate hundreds of downstream target genes,32 which is different from small interfering RNA silencing the expression of a single gene. Except for E2F1, miR-98 also includes many downstream genes, including E2F2,17 Hmga2,33 MYC34 and NCOA3.35 Our study proved that the upregulation of miR-98 increased the sensitivity of drug resistance cells to ADR by target decreasing E2F1. However, whether the upregulation influences other downstream genes and whether miR-98 can be used as an indicator for diagnosis and prognosis of leukemia require further studies. Due to the complicated formation mechanism of leukemia cell drug resistance, the regulatory effect of miR-98 and another miRNA expression on these cells also need further exploration.
Acknowledgment
Our work was supported by the Medical Innovate Fund of Fujian Province 2014-CXB-42.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferrara F, Schiffer CA. Acute myeloid leukaemia in adults. Lancet. 2013;381(9865):484–495. | ||
Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. | ||
Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106(4):1154–1163. | ||
Sorrentino A, Liu CG, Addario A, Peschle C, Scambia G, Ferlini C. Role of microRNAs in drug-resistant ovarian cancer cells. Gynecol Oncol. 2008;111(3):478–486. | ||
Ellis LM, Hicklin DJ. Resistance to targeted therapies. Refining anticancer therapy in the era of molecular oncology. Clin Cancer Res. 2009;15(24):7471–7478. | ||
Llave C, Xie Z, Kasschau KD, Carrington JC. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002;297(5589):2053–2056. | ||
McManus MT. MicroRNAs and cancer. Semin Cancer Biol. 2003;13(4):253–258. | ||
Gregory RI, Shiekhattar R. MicroRNA biogenesis and cancer. Cancer Res. 2005;65(9):3509–3512. | ||
Fan YH, Ye MH, Wu L, et al. Overexpression of miR-98 inhibits cell invasion in glioma cell lines via downregulation of IKKepsilon. Eur Rev Med Pharmacol Sci. 2015;19(19):3593–3604. | ||
Zhang S, Zhang C, Li Y, Wang P, Yue Z, Xie S. miR-98 regulates cisplatin-induced A549 cell death by inhibiting TP53 pathway. Biomed Pharmacother. 2011;65(6):436–442. | ||
To KK, Zhan Z, Litman T, Bates SE. Regulation of ABCG2 expression at the 3′ untranslated region of its mRNA through modulation of transcript stability and protein translation by a putative microRNA in the S1 colon cancer cell line. Mol Cell Biol. 2008;28(17):5147–5161. | ||
Zhu DX, Zhu W, Fang C, et al. miR-181a/b significantly enhances drug sensitivity in chronic lymphocytic leukemia cells via targeting multiple anti-apoptosis genes. Carcinogenesis. 2012;33(7):1294–1301. | ||
Wendler A, Keller D, Albrecht C, Peluso JJ, Wehling M. Involvement of let-7/miR-98 microRNAs in the regulation of progesterone receptor membrane component 1 expression in ovarian cancer cells. Oncol Rep. 2011;25(1):273–279. | ||
Xiang Q, Tang H, Yu J, Yin J, Yang X, Lei X. MicroRNA-98 sensitizes cisplatin-resistant human lung adenocarcinoma cells by up-regulation of HMGA2. Pharmazie. 2013;68(4):274–281. | ||
Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002;296(5570):1132–1136. | ||
Ma X, Gao Y, Fan Y, et al. Overexpression of E2F1 promotes tumor malignancy and correlates with TNM stages in clear cell renal cell carcinoma. PLoS One. 2013;8(9):e73436. | ||
Bhat-Nakshatri P, Wang G, Collins NR, et al. Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic Acids Res. 2009;37(14):4850–4861. | ||
Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33(20):e179. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. | ||
Chen S, Chen X, Xiu YL, Sun KX, Zong ZH, Zhao Y. microRNA 490–3P enhances the drug-resistance of human ovarian cancer cells. J Ovarian Res. 2014;7:84. | ||
Yu PN, Yan MD, Lai HC, et al. Downregulation of miR-29 contributes to cisplatin resistance of ovarian cancer cells. Int J Cancer. 2014;134(3):542–551. | ||
He X, Xiao X, Dong L, et al. MiR-218 regulates cisplatin chemosensitivity in breast cancer by targeting BRCA1. Tumour Biol. 2015;36(3):2065–2075. | ||
Kovesdi I, Reichel R, Nevins JR. E1A transcription induction: enhanced binding of a factor to upstream promoter sequences. Science. 1986;231(4739):719–722. | ||
Engelmann D, Putzer BM. The dark side of E2F1: in transit beyond apoptosis. Cancer Res. 2012;72(3):571–575. | ||
Engelmann D, Putzer BM. Translating DNA damage into cancer cell death-A roadmap for E2F1 apoptotic signalling and opportunities for new drug combinations to overcome chemoresistance. Drug Resist Updat. 2010;13(4–5):119–131. | ||
Louie MC, Zou JX, Rabinovich A, Chen HW. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol. 2004;24(12):5157–5171. | ||
Andersen JB, Factor VM, Marquardt JU, et al. An integrated genomic and epigenomic approach predicts therapeutic response to zebularine in human liver cancer. Sci Transl Med. 2010;2(54):54ra77. | ||
Yoshihara Y, Wu D, Kubo N, Sang M, Nakagawara A, Ozaki T. Inhibitory role of E2F-1 in the regulation of tumor suppressor p53 during DNA damage response. Biochem Biophys Res Commun. 2012;421(1): 57–63. | ||
Rosenfeldt MT, Bell LA, Long JS, et al. E2F1 drives chemotherapeutic drug resistance via ABCG2. Oncogene. 2014;33(32):4164–4172. | ||
Siragam V, Rutnam ZJ, Yang W, et al. MicroRNA miR-98 inhibits tumor angiogenesis and invasion by targeting activin receptor-like kinase-4 and matrix metalloproteinase-11. Oncotarget. 2012;3(11):1370–1385. | ||
Huang SD, Yuan Y, Zhuang CW, et al. MicroRNA-98 and microRNA-214 post-transcriptionally regulate enhancer of zeste homolog 2 and inhibit migration and invasion in human esophageal squamous cell carcinoma. Mol Cancer. 2012;11:51. | ||
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. | ||
Hebert C, Norris K, Scheper MA, Nikitakis N, Sauk JJ. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol Cancer. 2007;6:5. | ||
Sampson VB, Rong NH, Han J, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67(20):9762–9770. | ||
Gennarino VA, Sardiello M, Avellino R, et al. MicroRNA target prediction by expression analysis of host genes. Genome Res. 2009;19(3):481–490. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.