")
Back to Journals » International Journal of Nanomedicine » Volume 12
Targeted multidrug delivery system to overcome chemoresistance in breast cancer
Authors Tang Y , Soroush F, Tong Z, Kiani MF, Wang B
Received 15 October 2016
Accepted for publication 14 December 2016
Published 21 January 2017 Volume 2017:12 Pages 671—681
DOI https://doi.org/10.2147/IJN.S124770
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Yuan Tang,1 Fariborz Soroush,1 Zhaohui Tong,2 Mohammad F Kiani,1 Bin Wang1,3
1Department of Mechanical Engineering, Temple University, Philadelphia, PA, 2Department of Agricultural and Biological Engineering, University of Florida, Gainesville, FL, 3Department of Biomedical Engineering, Widener University, Chester, PA, USA
Abstract: Chemotherapy has been widely used in breast cancer patients to reduce tumor size. However, most anticancer agents cannot differentiate between cancerous and normal cells, resulting in severe systemic toxicity. In addition, acquired drug resistance during the chemotherapy treatment further decreases treatment efficacy. With the proper treatment strategy, nanodrug carriers, such as liposomes/immunoliposomes, may be able to reduce undesired side effects of chemotherapy, to overcome the acquired multidrug resistance, and to further improve the treatment efficacy. In this study, a novel combinational targeted drug delivery system was developed by encapsulating antiangiogenesis drug bevacizumab into liposomes and encapsulating chemotherapy drug doxorubicin (DOX) into immunoliposomes where the human epidermal growth factor receptor 2 (HER2) antibody was used as a targeting ligand. This novel combinational system was tested in vitro using a HER2 positive and multidrug resistant breast cancer cell line (BT-474/MDR), and in vivo using a xenograft mouse tumor model. In vitro cell culture experiments show that immunoliposome delivery led to a high cell nucleus accumulation of DOX, whereas free DOX was observed mostly near the cell membrane and in cytoplasm due to the action of P-gp. Combining liposomal bevacizumab with immunoliposomal DOX achieved the best tumor growth inhibition and the lowest toxicity. Tumor size decreased steadily within a 60-day observation period indicating a potential synergistic effect between DOX and bevacizumab through the targeted delivery. Our findings clearly indicate that tumor growth was significantly delayed in the combinational liposomal drug delivery group. This novel combinational therapy has great potential for the treatment of patients with HER2/MDR double positive breast cancer.
Keywords: immunoliposome, targeted drug delivery, xenograft mouse tumor model, combination therapy, multidrug resistance
Introduction
Breast cancer is the most common invasive cancer in women worldwide, and in 2012 >1.7 million new cases were diagnosed in the US alone.1 In the US, it was estimated that ~12.3% of women will experience breast cancer at some point during their lifetime.2 According to different hormone receptor status, breast cancer can be classified into 3 categories: endocrine receptor (estrogen or progesterone receptor) positive, human epidermal growth factor receptor 2 (HER2) positive, and triple negative (negative for estrogen, progesterone, and HER2 receptor).3 Among them, the HER2 positive tumors tend to grow and spread faster than HER2 negative ones due to the fact that HER2 is a growth hormone receptor. Meanwhile, HER2 positive tumors also tend to recur more often than others.
Approximately 30% of the early-stage breast cancers progress to metastatic breast cancers (MBCs).4 To date, there are very few treatment options available for MBC management, and the response rates range from 30% to 70% after anthracycline or taxane-based chemotherapy.5 However, these responses are often not durable due to the fact that the breast cancer may develop multidrug resistance (MDR). MDR has become a major obstacle in the treatment of breast cancer, where prolonged exposure to one type of chemotherapeutic agent can lead to the resistance to not only that drug but also many other anticancer compounds of different structure and mechanism of action.6 Furthermore, most anticancer compounds have severe side effects to healthy tissues if delivered systemically.
One solution to both overcome drug resistance and concurrently minimize adverse side effects might be encapsulating anticancer compounds into drug carriers to achieve targeted delivery. Tumor tissue is usually characterized by the enhanced permeation and retention (EPR) effect, which consists of loose interconnections and intercellular openings ranging between 100 and 780 nm in size. Drug carriers at the same scale can easily exploit this unique feature and extravasate from the systemic circulation,7,8 therefore enhancing drug delivery to the targeted cancer. This method of localizing drugs to the tumor tissue is a type of passive targeting.9 However, trapping drugs into carriers also prevents them from being recognized by cellular efflux pumps, such as P-glycoprotein (P-gp), and hence helps overcome MDR in resistant tumors.10
With proper design, the therapeutic potential of drug carriers can be further enhanced to achieve active targeting capability. By tagging them with appropriate ligands such as monoclonal antibodies (mAbs), such drug carriers can specifically interact with cancer cell membrane receptors,11 thus allowing the drug to be released specifically inside the tumor by receptor-mediated endocytosis.9 Among all targeting moieties, mAbs are the most widely investigated for tumor targeting.12 Initially, mAbs were conjugated directly to anticancer compounds,13,14 although direct conjugation allows few antibodies to be linked to the drug15 and usually adversely affect the drug’s pharmacological potency.16 Conjugation of mAbs to the drug carrier surface, however, overcomes these two disadvantages while providing greater selectivity compared with direct drug-mAb conjugation.17
The heterogeneity of the breast cancer also restricts the efficacy of the cancer treatment. For this reason, targeted delivery of drugs from different classes to multiple targets in the tumor may overcome drug resistance, improve efficacy, and reduce side effects. Angiogenesis has been shown to play an important role in breast cancer progression.18,19 Highly vascularized breast cancer has a poorer prognosis and greater metastatic potential than other types with low vascularity,20–22 therefore, breast cancer has been considered as an angiogenesis-dependent cancer and a unique target for antiangiogenic therapy.23,24 Therapeutic disruption of angiogenesis is effective in mediating tumor regression.25–27 Bevacizumab (rhuMAbVEGF, Avastin; Genentech, South San Francisco, CA, USA), a recombinant humanized antibody, inhibits angiogenesis by specifically binding to vascular endothelial growth factor A and preventing it from binding to the receptor. The combination of chemotherapeutic and antiangiogenic compounds has been shown to increase the survival rate in metastatic cancer patients.28
Liposomes are US Food and Drug Administration approved spherical lipid bilayer drug delivery vehicles that are biocompatible and biodegradable. It has been studied and used widely as a nanoscale drug carrier due to its versatility in carrying both hydrophobic and hydrophilic drugs and long blood circulation time after pegylation treatment on the surface.10,29 In this study, we tested the feasibility of combining bevacizumab and chemotherapy for the treatment of HER2/MDR double positive breast cancer cells using a liposomal drug delivery system. An extracellular agent (bevacizumab) was selectively delivered to the interstitial space of the breast cancer tissue by long circulating liposomes (liposomal bevacizumab). A commonly used anthracycline drug, doxorubicin (DOX), was selectively delivered into breast tumor cells by immunoliposomes (immunoliposomal DOX) to increase efficacy whereas minimizing side effects. The HER2 antibody was used as a targeting ligand to facilitate the internalization of the immunoliposomal DOX by ligand–receptor interaction, because the HER2 receptor is overexpressed on the surface of the breast tumor. The characteristics of the drug delivery systems, their targeting potential, and treatment efficacy were verified in both in vitro cell culture and in vivo mouse tumor xenograft models.
Materials and methods
BT474/MDR subline development and cell culture
To study the therapeutic efficacy of our combination therapy on MDR breast cancer, we have developed a breast cancer MDR cell line BT474/MDR, which overexpresses MDR transporter P-gp.30 Briefly, HER2 overexpressing human breast carcinoma cell line, BT474 cells (American Type Culture Collection, Manassas, VA, USA) were transfected with MDR1 gene packaged in a plasmid vector (EX-E2266-M02, Genecopoeia, Rockville, MD, USA) using Lipofectamin 2000 tranfection reagent (Life Technologies, Grand Island, NY, USA) according to manufacturer’s instructions. After transfection, cells were maintained in the culture media containing 60 ng/mL of colchicine (Sigma-Aldrich, St Louis, MO, USA) to select P-gp positive cells.
Cells were cultured as monolayers in Dulbecco’s Modified Eagle’s Medium/F12 (Life Technologies) medium supplemented with 10% fetal bovine serum (Life Technologies) and 1% penicillin/streptomycin (Life Technologies) in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Cells were subcultured twice weekly. For experiments, the cells were grown in tissue culture flasks and used when in the exponential growth phase.
Preparation of liposomal bevacizumab and immunoliposomal DOX
Liposomal bevacizumab preparation
Liposomes were composed of 50% hydrogenated soy L-α-phosphatidylcholine, 45% cholesterol, 3% 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[(polyethylene glycol)2000]11 (DSPE-PEG2000), and 2% DSPE-PEG-maleimide and were prepared by means of solvent evaporation and film formation.31 Briefly, all four lipid components were dissolved in chloroform (Sigma-Aldrich), which was then removed by nitrogen gas purging at room temperature, followed by overnight evaporation in a Labconco Freezone 1 freeze-dryer (Kansas City, MO, USA). The resulting lipid film was hydrated with a 250 mM ammonium sulfate (Sigma-Aldrich) solution by gentle mixing, resulting in spontaneously organized multilamellar vesicles (MLVs). The MLVs were extruded 11 times through 200 and 100 nm pore sized Whatman polycarbonate membranes (GE Healthcare Biosciences, Pittsburgh, PA, USA) using a Lipex™ Extruder (Northern Lipids Inc., Burnaby, BC, Canada) to form small unilamellar vesicles. All lipids were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL, USA).
Bevacizumab (Genentech) was loaded into liposomes by a passive loading method. Briefly, lipids dissolved in chloroform in appropriate amounts; the solvent was evaporated by first purging in nitrogen gas and then dried in a lyophilizer overnight. Afterward, the thin dry film of lipid was rehydrated in a buffer containing bevacizumab (20 mM Tris–HCl, 135 mM NaCl, 200 mM bevacizumab, pH 7.2) preheated at 50°C and vortexed for 3 minutes. Then the suspension was extruded 11 times at 50°C through a membrane of 200 nm pore size.
Immunoliposomal DOX preparation
Liposomes were prepared according to the previous section. DOX (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was loaded into liposome lipid bilayer by a remote loading method via ammonium sulfate gradient, which was created by replacing ammonium sulfate outside the liposome by phosphate-buffered saline (PBS; HyClone, South Logan, UT, USA) using a MicroKros hollow fiber diafiltration unit (Spectrum Labs, Rancho Dominguez, CA, USA). The molar ratio of DOX/lipid was 1:5. Unencapsulated DOX was removed by passing the solution through a Sepharose 4B gravity column (GE Healthcare Biosciences). After DOX encapsulation, the DOX-loaded liposomes were conjugated with anti-HER2 antibodies. Briefly, mouse monoclonal anti-human HER2 antibody (Santa Cruz Biotechnology) was first thiolated with 2-iminothiolane (Sigma-Aldrich) at pH 8.0. Then the introduced thiol groups on the antibody were coupled with maleimide groups on DSPE-PEG2000 component of the liposomes at pH 6.5 to form immunoliposomes. Unconjugated antibodies were removed by Sepharose 4B gravity column.
Characterization of liposomal bevacizumab and immunoliposomal DOX
Liposome size distribution and surface potential
Liposome hydrodynamic diameter was determined by dynamic light scattering technique using a Nano ZS Zetasizer (Malvern Instruments, Westborough, MA, USA), operating at a wavelength of 633 nm employing a nominal 5 mW He–Ne laser. Liposome surface potential was also measured by the Zetasizer.
DOX loading efficiency and release kinetics
The detection of DOX was based on its light absorption property (maximum absorption at 480 nm).32 Following the separation of free drug, immunoliposome solution was flash frozen and freeze-dried overnight. The resulting powder was reconstituted in PBS containing 1% Trition X-100 (Sigma-Aldrich) in order to break down the liposomes. DOX concentration was calculated based on its absorption measured in a spectrofluorometer (PerkinElmer, Waltham, MA, USA) against a precreated calibration curve.
In vitro release kinetics of DOX from the immunoliposomes was carried out at pH 7.4 using a spectrofluorometric method. Briefly, immunoliposomal DOX solution was equally distributed into microcentrifuge vials at a volume of 1 mL/vial and incubated at 37°C in the cell culture incubator. At specific time points, those vials were taken out and passed through Sepharose 4B column to separate out free DOX; the fractions where immunoliposomes were located, collected, and homogenized in 1% Trition X-100 and measured in spectrofluometer.
Bevacizumab loading efficiency and release kinetics
In order to determine the bevacizumab concentration, an enzyme-linked immunosorbent assay (ELISA) method was developed. First, a serial dilution of bevacizumab solution with known concentration was made using 50 mM carbonate buffer, containing 15 mM Na2CO3 and 35 mM NaHCO3 (Sigma-Aldrich) at pH 9.6. Second, 100 μL of bevacizumab at each concentration was added to an ELISA plate (FisherSci, Pittsburg, PA, USA). The plate was placed in a 4°C refrigerator overnight to allow for bevacizumab attachment. Unbound bevacizumab was then removed with 100 mM NaHCO3 wash at pH 9.2 (5 minutes each, on shaker, repeat 3 times). Remaining uncovered plate area was occupied with blocking buffer (3% bovine serum albumin [BSA] +0.1% Tween 20 in PBS) for 2 hours. Excessive blocking buffer was then removed by washing 3 times with a washing buffer (1% BSA +0.05% Tween 20 in PBS). After applying antihuman IgG (Fc specific)-peroxidase detection antibody (1.5 μg/mL, 100 μL/well, Thermo Scientific, Rockford, IL, USA) for 1.5 hours at room temperature, the plate was ready for color development. After 3 times wash, color develop was archived by adding peroxidase substrate 3,3,5,5′-tetramethylethylenediamine (Thermo Scientific) to each well and let it develop at room temperature for 30 minutes. Finally, color development was stopped by adding 50 μL of 1 N sulfuric acid (Sigma-Aldrich) to each well. The resulting yellow color was read at 450 nm in a plate reader.
Bevacizumab was detected with the ELISA described above. Following the separation of free bevacizumab, liposome solution was flash frozen and freeze-dried overnight. The resulting powder was reconstituted in PBS containing 1% Trition X-100 in order to break down the liposomes. In vitro release kinetics of bevacizumab from the liposomes was carried out at pH 7.4. Briefly, liposomal bevacizumab solution was equally distributed into microcentrifuge vials at a volume of 1 mL/vial and incubated at 37°C in the cell culture incubator. At specific time points, those vials were taken out and passed through Sepharose 4B column to separate out free bevacizumab; the fractions where liposomes were located, collected, and homogenized in PBS containing 1% Trition X-100 and measured by the ELISA.
Immunoliposome antibody conjugation efficiency
As lipids interfered strongly with common protein assays,33–35 an indirect ELISA method was employed in this study in order to quantify antibody conjugation efficiency. After conjugation, the immunoliposomes/antibody mixture was passed through a Sepharose CL-4B column, and fractions were collected and analyzed for the lipid content with a phosphate assay kit to first locate and then quantify the amount of liposomes. The fractions that were lipids free were first collected and then measured for unbound antibody using an HER2 ELISA kit. The amount of conjugated antibody was determined by subtracting the amount of unbound antibody from the initial amount of antibody that was added to the liposomes at the beginning of conjugation process.
In vitro immunoliposomal DOX cellular uptake
DOX cellular uptake studies were carried out for both free DOX and immunoliposomal DOX in BT474/MDR cells. On the first day, the cells were seeded into 24-well plates at cell density of 100,000–200,000 cells per well. On the second day, the cell medium was removed, and the growth medium with free DOX, or immunoliposomal DOX was added to the plates at a normalized DOX concentration of 10 μM (5.8 μg/mL) and placed in a cell incubator at 37°C for 24 hours. The control group added the growth medium without drug. After 24 hours, the cell medium was removed, and the cells were washed with ice-cold PBS (pH 7.0) 4 times and then lysed with 1 mL of 1% Triton X-100. After centrifugation, the supernatants were collected and DOX fluorescence signal was measured in a spectrofluorometer at λex=480 nm, λem=580 nm to determine DOX concentration.
Human breast cancer xenograft mouse model
As BT-474/MDR is a human cell line, BALB/c nude mice (CAnN.Cg-Foxn1nu/Crl, Charls River Laboratories, Malvern, PA, USA) were used for tumor xenograft. Six-week-old female mice were transplanted with 17-β-Estradiol hormone pellet (0.72 mg, 60-day release, Innovative Research of America, Sarasota, FL, USA) 24 hours before tumor inoculation to ensure proper tumor growth rate.36,37 Ten million cells in 200 μL of 50:50 serum free medium/Matrigel (Corning Life Sciences, Tewksbury, MA, USA) were injected under the skin in the right flank region. All the treatments were preformed once the tumor reaches 100 cm3 (measured by a caliper and tumor volume calculated by equation: V = π/6*Length*Height*Width). All experiments involving animals were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All efforts were made to minimize suffering. The protocol was approved by the Institutional Animal Care and Use Committee of Temple University (Permit Number: 3390).
Immunoliposomal DOX in vivo organ distribution
After the xenograft tumor reached 100 cm3, 200 μL of immunoliposomal DOX (8 mg/kg concentration) was infused into the mouse blood stream via tail vein injection. In order to test the immunoliposome distribution, fluorescein isothiocyanate fluorophore-labeled lipid was used as a component when forming liposomes. After 24 hours of blood circulation, the blood was flushed from the animal by injecting saline with a syringe into the left ventricle of the heart and making a small opening with microscissors in the right ventricle. The tumor along with major organs (heart, liver, kidney, brain, spleen, and lung) and a sample of blood (taken with a heparinized syringe from the heart immediately before flushing the blood) were collected for further analysis. In order to identify the immunoliposome and DOX organ distributions, collected organs were sliced, weighed, and dissolved in a 1% Triton X-100 to extract either the lipid or DOX content.
In vivo treatment efficacy in human breast cancer xenograft mouse model
To test the therapeutic efficacy of our combination therapy in vivo, 200 μL mixture of liposomal bevacizumab (10 mg/kg) and immunoliposomal DOX (8 mg/kg) was infused into the mouse blood stream via tail vein injection after the xenograft tumor reached 100 cm3. The mice were treated in 4 different groups: liposomal bevacizumab (10 mg/kg), free DOX (8 mg/kg), liposomal DOX (8 mg/kg), and immunoliposomal DOX (8 mg/kg). Tumor bearing mice injected with 200 μL of saline or empty liposomes (10 mM lipid concentration) were used as the control. Tumor measurements were performed with calipers every day until the tumor reached 10% of the body mass, after which the animals were anesthetized with a 87/13 mg/kg Ketamine/Xylazine solution. The animals were then be euthanized (an intracardiac injection of KCl) and the tumor surgically excised.
Results
Liposome and immunoliposome size and charge
Liposomes exhibited a mean particle size ranging between 140 and 160 nm as determined using a Zetasizer. Drug-loaded liposomes exhibited diameters that were not significantly different from empty liposomes indicating that the drug loading process did not impact the particle size. The immunoliposomes showed increased diameter probably due to the added antibody layer on the surface (Table 1).
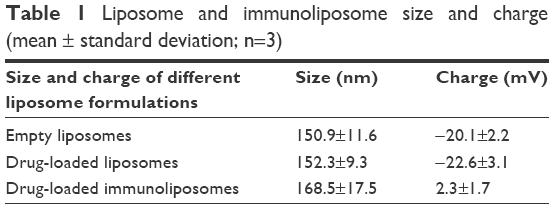 | Table 1 Liposome and immunoliposome size and charge (mean ± standard deviation; n=3) |
DOX loading, release, and antibody conjugation efficiency in immunoliposomal DOX
DOX concentrations encapsulated in immunoliposomes were measured by a spectrophotometer after disruption of immunoliposomes with the detergent. Encapsulation efficiencies of DOX were ~80%. Figure 1 shows the DOX release profile in vitro for up to 96 hours. During the first 24 hours, the DOX was released with a relatively high rate. Then, DOX was released steadily over the last 48 hours. Since the immunoliposomes have a plasma half-life of ~12 hours,38 such release profile can ensure high drug retention when the drug carrier reaches the tumor site. To quantify the amount of covalently attached antibodies, we constructed a calibration curve for the HER2 antibody, and then analyzed the different fractions collected during the separation of free antibody procedure. The amount of antibody that was covalently linked to the maleimide group on liposome surface was quantified by subtracting the amount of free antibody from the initial added amount. A 10%–15% anti-HER2 coupling efficiency was observed during various liposome preparing processes. Although conjugated antibody could not be quantified directly due to the interference from the lipids, the ELISA with a HER2 protein standard confirmed the presence of anti-HER2 in these fractions was also confirmed by ELISA with HER2 protein standard.
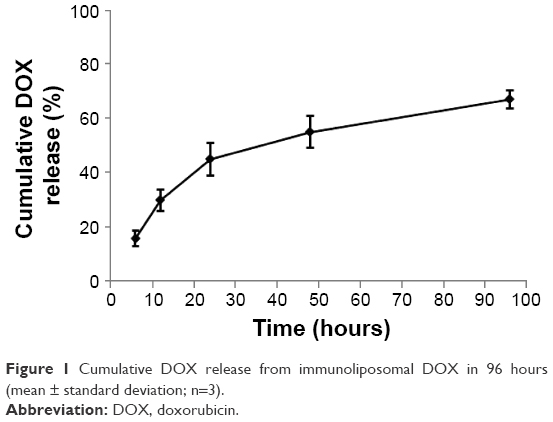 | Figure 1 Cumulative DOX release from immunoliposomal DOX in 96 hours (mean ± standard deviation; n=3). |
Bevacizumab loading and release in liposomal bevacizumab
The bevacizumab release profile was obtained by calculating the amount of bevacizumab remaining in the liposome 1, 2, 3, 4, 5, 6, 24, and 48 hours after the ultracentrifuge. The results were shown in Figure 2. According to the calibration curve (Figure S1), ~37% of the added bevacizumab was entrapped into the liposomes. Bevacizumab presented two phases of release from the liposomes. The first phase was the initial burst release; a burst release of 21.1% was estimated from the curve fitting. This fast phase was possibly due to the release of bevacizumab that was adsorbed onto the surface during the liposome formulation process. Following the burst release, further release was mediated by diffusion from the liposome core and degradation of the liposomes, which had much slower release rate compared to that of the burst release. The total bevacizumab released in 48 hours was 55.1% (Figure 2).
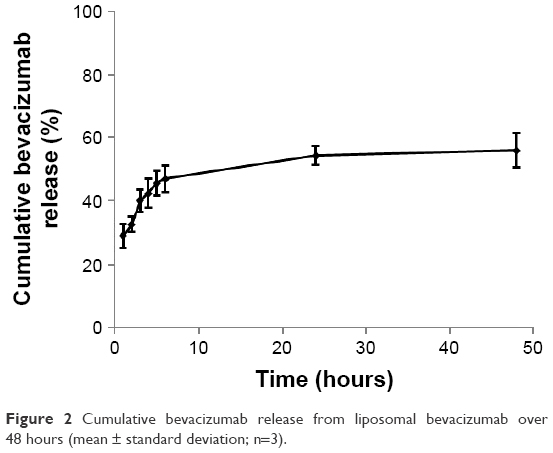 | Figure 2 Cumulative bevacizumab release from liposomal bevacizumab over 48 hours (mean ± standard deviation; n=3). |
In vitro uptake of immunoliposomal DOX
To test the ability of HER2 conjugated immunoliposomes internalizing into tumor cells in vitro, BT474/MDR cells were cultured with either free DOX or immunoliposomal DOX. As shown in Figure 3, fluorescence images were taken to visualize DOX uptake and localization. DOX was accumulated in the nucleus by the immunoliposome delivery, whereas free DOX was observed mostly near cell membrane and in cytoplasm due to the action of P-gp. As P-gp could recognize DOX and actively transport them out the cell, free DOX can no longer reach its target site of cell nucleus. Immunoliposome delivery, however, led to a high cell nucleus accumulation of DOX, probably because of the receptor-mediated endocytosis between HER2 antibody and HER2 receptor. A quantitative study (Figure 4) using lysed cells showed the highest DOX internalization by immunoliposome delivery of DOX, which confirmed our fluorescence imaging results.
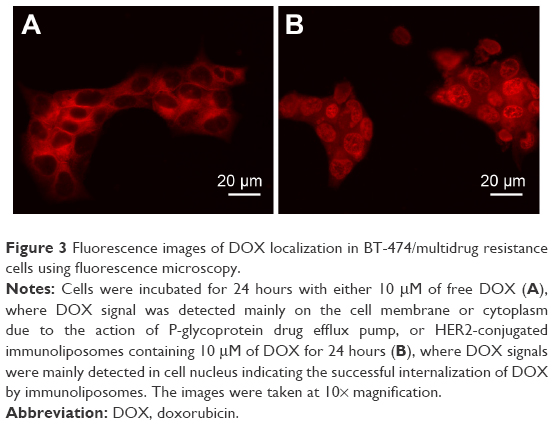 | Figure 3 Fluorescence images of DOX localization in BT-474/multidrug resistance cells using fluorescence microscopy. |
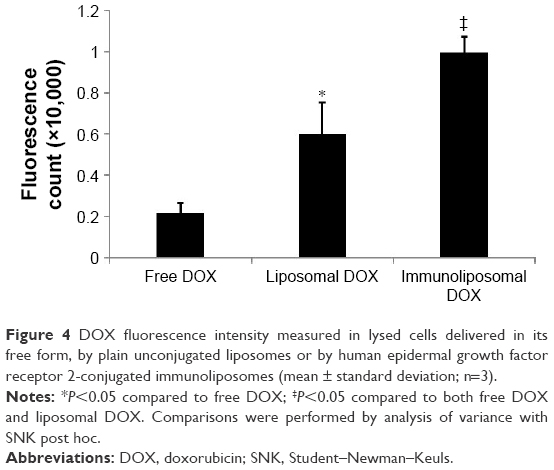 | Figure 4 DOX fluorescence intensity measured in lysed cells delivered in its free form, by plain unconjugated liposomes or by human epidermal growth factor receptor 2-conjugated immunoliposomes (mean ± standard deviation; n=3). |
Immunoliposomal DOX organ distribution
In the in vivo distribution studies, the disposition of immunoliposomal DOX or free DOX after intravenous administration into tumor xenograft was investigated (Figure 5). HER2 receptor targeting immunoliposomes had the highest concentration in tumors compared to other organs, showing its great potential as a drug delivery vehicle (Figure 5A). When comparing DOX concentration delivered by immunoliposome versus its delivery in free form, we observed significantly higher tumor DOX localization and significantly lower liver localization of immunoliposomal DOX compared to free DOX (Figure 5B). Taken together, these data demonstrated the “stealth” property of this drug delivery system.
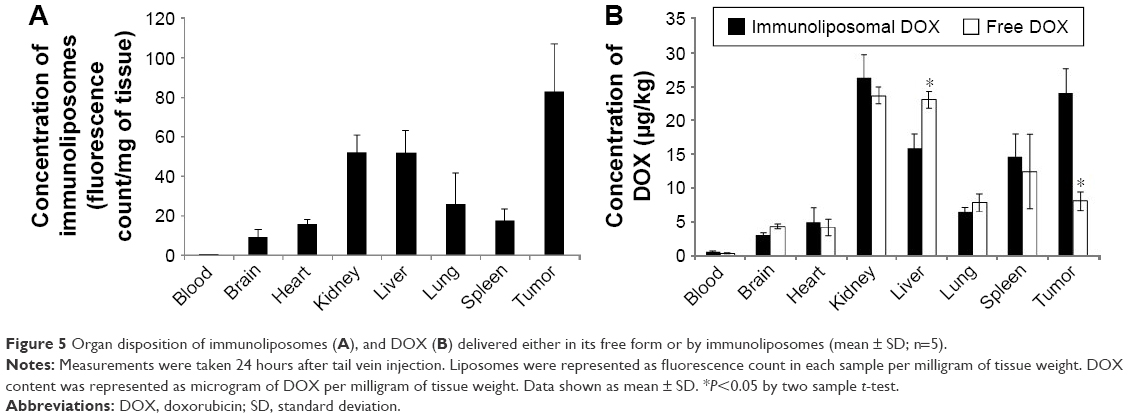 | Figure 5 Organ disposition of immunoliposomes (A), and DOX (B) delivered either in its free form or by immunoliposomes (mean ± SD; n=5). |
In vivo tumor growth
The effect of saline, empty liposome, liposomal bevacizumab, free DOX, liposomal DOX, immunoliposomal DOX, and the combination of liposomal bevacizumab and immunoliposomal DOX on tumor growth were plotted in Figure 6. Treatments started when tumor size reached 100 mm3. Tumors developed very quickly in saline and empty liposome treatment groups where animals were sacrificed when tumor volumes reached ~300 mm3. Treatment with liposomal bevacizumab delayed tumor growth significantly where the tumor size approached 300 mm3 at 60 days after transplantation.
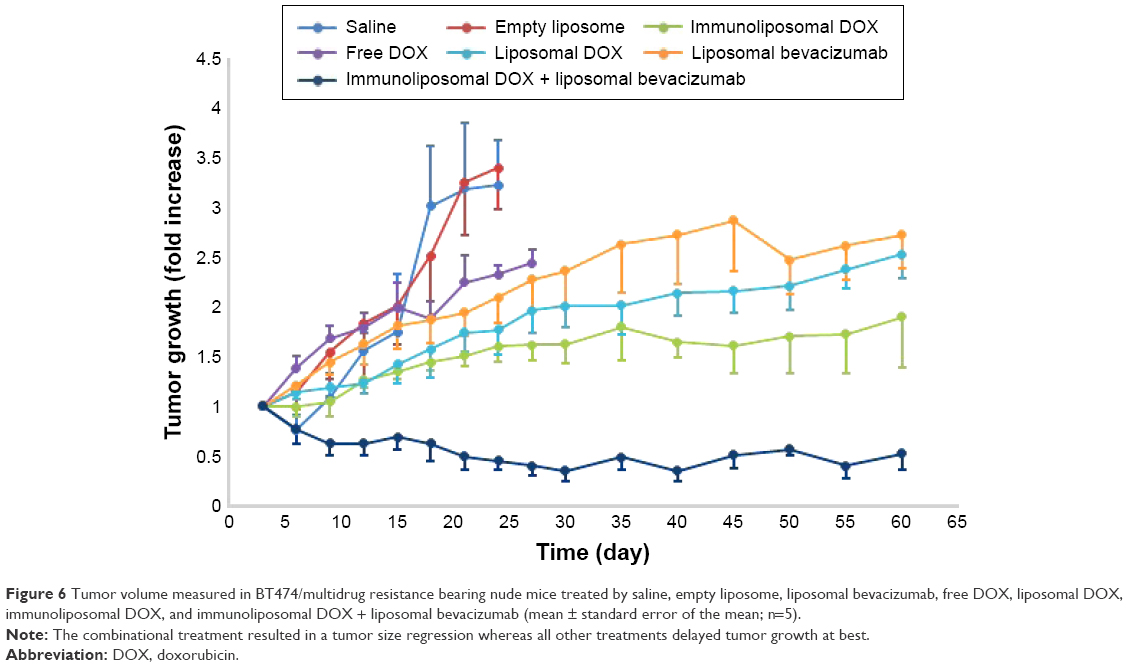 | Figure 6 Tumor volume measured in BT474/multidrug resistance bearing nude mice treated by saline, empty liposome, liposomal bevacizumab, free DOX, liposomal DOX, immunoliposomal DOX, and immunoliposomal DOX + liposomal bevacizumab (mean ± standard error of the mean; n=5). |
After applying free DOX at 8 mg/kg concentration when the tumor size reached 100 mm3, tumor growth was delayed as shown in Figure 6. However, due to its high toxicity, animals suffered significant weight loss (data not shown). Comparing to free DOX, liposomal DOX significantly decreased its toxicity. All 5 mice in this group survived beyond 60 days. However, tumor still reoccurred after an initial growth inhibition. HER2 receptor targeting immunoliposomal DOX treatment showed some improvement over liposomal DOX in treatment efficacy. Tumor size maintained at a relatively low level but still growing. Combining liposomal bevacizumab with immunoliposomal DOX achieved the best tumor growth inhibition effect. In the latter group, tumor size decreased steadily within the 60-day observation period indicating a potential synergistic effect between DOX and bevacizumab through targeted delivery.
Discussion
Breast cancer is the second leading cause of cancer deaths in women.39 Breast cancer is, generally, considered a highly heterogenic cancer type, which restricts the efficacy of the cancer treatment. DOX is one of the most widely used chemotherapeutic agent in the treatment of cancer including managing both primary and metastatic recurrent breast cancers.40 However, nonselective biodistribution and dose-limiting toxicities associated with systemic DOX chemotherapy restricted its use and therapeutic potential. Hence, approaches aiming at improving its therapeutic index while minimizing its side effects have long been sought. To tackle these challenges, in this study, we implemented a combinational therapy with targeted delivery of multiple agents from different classes to multiple targets in the tumor that enabled to overcome drug resistance, improve therapeutic efficacy, and reduce side effects. As shown in Figure 6, targeted delivery of an antiangiogenesis drug and a chemotherapy drug together greatly improved animal survival and inhibited tumor growth compared to either treatment alone, exhibiting a synergistic treatment effect between these two classes of antitumor drugs.
Clinically, HER2 positive breast cancers are treated with HER2 receptor antagonist, such as trastuzumab (Herceptin). Trastuzumab is a mAb that triggers G1 arrest in cells upon binding to the receptor. Meanwhile, trastuzumab is capable of inducing immune response to destroy the cancer cell.41 However, de novo resistance is common, occurring in ~65% of patients, furthermore, ~70% of patients will ultimately develop acquired resistance after initial response.42 To date, despite numerous preclinical studies, continued administration of HER2 targeting drug with chemotherapy is still a standard treatment method clinically for patients with recurrent HER2 positive breast cancer.43–45 In this study, we utilized an antiangiogenesis recombinant mAb bevacizumab (Avastin), to inhibit blood vessel formation in animals. Clinical trials have shown modest beneficial results in breast cancer patients treated with bevacizumab,46–48 which is in accordance with our animal experiments (Figure 6).
Conventional systemic administration of chemotherapeutic agents lacks targeting effect and can have severe side effects, resulting in suboptimal therapeutic effect. More importantly, MDR can occur after repeated exposure to chemotherapeutic agents. In the drug-resistant cells, ATP-binding cassette proteins can actively translocate a variety of structurally different substrates outside the cell membrane utilizing the energy from ATP hydrolysis,6 leading to accelerated drug efflux and decreased net drug accumulation in tumor.32 It is, generally, believed that upregulation of the drug transporter proteins in the cell membrane is responsible for the increased efflux.
Over the past decades, nanosized drug carriers have been extensively studied to achieve targeted drug delivery for cancer therapy. Nanocarriers have the properties of controlled drug release, prolonged blood circulation, and superior biocompatibility. When the surface of nanocarrier was modified with targeting moieties, such as mAbs, they can be selectively delivered to the tumor region. In this study, we utilized an immunoliposomal drug delivery system to overcome drug efflux pump. As a result, the encapsulated DOX was found to be much more concentrated in the tumor than systemic delivery of DOX (Figure 4).
We optimized the liposome preparation process to specifically accommodate the different delivery requirements of bevacizumab and DOX. In order to facilitate the internalization of DOX, the targeted drug delivery system immunoliposomes were used. Immunoliposomes were prepared by conjugating liposomes with HER2 antibodies producing a positive charge on the liposome. The positive charge could promote binding of the immunoliposomes with negatively charged cell membrane via electrostatic interactions. Besides, the immunoliposomes can gradually release DOX to ensure a continuous effect in the tumor. Active targeting has been also used to enhance drug delivery into the tumor, and biodistribution results tell us that DOX delivered by immunoliposomes mainly distribute in transplanted tumors much more than DOX in its free form, although they still have some concentration in kidney, liver, and spleen, but much less than free DOX.
The idea of combining bevacizumab and liposomal DOX is not new. Clinical trials involving systemic delivery of bevacizumab and liposomal DOX have achieved positive response in the treatment of recurrent or refractory ovarian cancers.49 In a phase II study, where bevacizumab was combined with liposomal DOX for patients with platinum and taxane resistant ovarian cancers, researchers found an increased patient response but also increased toxicity, mainly because of the nonspecificity of bevacizumab due to systemic delivery.50 However, in the treatment of locally recurrent or MBCs, the combination of systemic bevacizumab and liposomal DOX only achieved moderate activity while higher than anticipated toxicity.51 One possible strategy to solve this problem is to add a new chemotherapy drug which belongs to a different class to minimize the dose of either DOX or bevacizumab. For example, one currently ongoing clinical trial (NCT02456857) is looking at the combinational effect of liposomal DOX, bevacizumab, and Temsirolimus in the treatment of triple-negative breast cancer.52 However, the addition of a new drug inevitably increases the system complexity. In this study, we utilized a different approach by encapsulating bevacizumab into liposomes therefore maximizing its distribution in tumor tissue through EPR effect and minimizing its distribution in healthy tissues. We also conjugated targeting antibodies to the liposome surface to maximize internalization of the DOX. The findings from this study indicate that this combination therapy is promising for treating breast cancer.
Conclusion
In the present study, we examined the treatment efficacy of a combinational therapy involving antiangiogenesis therapy and DOX chemotherapy on the treatment of HER2/MDR double positive breast cancer cells using a novel liposome drug delivery system. Both bevacizumab and DOX were successfully encapsulated into liposomes and subsequently released in a controlled fashion. Our animal studies indicate that in this targeted treatment both liposomes and DOX preferentially accumulated at the tumor site. Our findings clearly indicate that tumor growth has been significantly delayed in the combinational liposomal drug delivery group. This novel combinational therapy has great potential for treatment of patients with HER2/MDR double positive breast cancer.
Acknowledgment
This study was financially supported by Susuan G. Komen for the Cure foundation, grant no KG100492.
Disclosure
The authors report no conflicts of interest in this work.
References
International Agency for Research on Cancer (IARC) and World Health Organization (WHO). GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Lyon, France: International Agency for Research on Cancer (IARC) and World Health Organization (WHO); 2014. | ||
Rojas K, Stuckey A. Breast Cancer Epidemiology and Risk Factors. Clin Obstet Gynecol. 2016;59(4):651–672. | ||
Tang Y, Wang Y, Kiani MF, Wang B. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin Breast Cancer. 2016;16(5):335–343. | ||
O’Shaughnessy J. Extending survival with chemotherapy in metastatic breast cancer. Oncologist. 2005;10(Suppl 3):20–29. | ||
Gralow JR. Optimizing the treatment of metastatic breast cancer. Breast Cancer Res Treat. 2005;89(Suppl 1):S9–S15. | ||
Perez EA. Impact, mechanisms, and novel chemotherapy strategies for overcoming resistance to anthracyclines and taxanes in metastatic breast cancer. Breast Cancer Res Treat. 2009;114(2):195–201. | ||
Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–3756. | ||
Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–4612. | ||
Lei T, Srinivasan S, Tang Y, et al. Comparing cellular uptake and cytotoxicity of targeted drug carriers in cancer cell lines with different drug resistance mechanisms. Nanomedicine. 2011;7(3):324–332. | ||
Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55(3):329–347. | ||
Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res. 2003;42(5):439–462. | ||
Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–1157. | ||
Garnett MC. Targeted drug conjugates: principles and progress. Adv Drug Deliv Rev. 2001;53(2):171–216. | ||
Funaro A, Horenstein AL, Santoro P, Cinti C, Gregorini A, Malavasi F. Monoclonal antibodies and therapy of human cancers. Biotechnol Adv. 2000;18(5):385–401. | ||
Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13:5043–5054. | ||
Nobs L, Buchegger F, Gurny R, Allemann E. Current methods for attaching targeting ligands to liposomes and nanoparticles. J Pharm Sci. 2004;93(8):1980–1992. | ||
Dinauer N, Balthasar S, Weber C, Kreuter J, Langer K, Von Briesen H. Selective targeting of antibody-conjugated nanoparticles to leukemic cells and primary T-lymphocytes. Biomaterials. 2005;26(29):5898–5906. | ||
Boudreau N, Myers C. Breast cancer-induced angiogenesis: multiple mechanisms and the role of the microenvironment. Breast Cancer Res. 2003;5(3):140–146. | ||
Moses MA, Harper J, Fernandez CA. A role for antiangiogenic therapy in breast cancer. Curr Oncol Rep. 2004;6(1):42–48. | ||
Magennis DP. Angiogenesis: a new prognostic marker for breast cancer. Br J Biomed Sci. 1998;55(3):214–220. | ||
Toi M, Bando H, Kuroi K. The predictive value of angiogenesis for adjuvant therapy in breast cancer. Breast Cancer. 2000;7:311–314. | ||
Weidner N, Folkman J, Pozza F, et al. Gasparini, Tumor angiogenesis: a new significant and independent prognostic indicator in early-stage breast carcinoma. J Natl Cancer Inst. 1992;84:1875–1887. | ||
Miller KD. Issues and challenges for antiangiogenic therapies. Breast Cancer Res Treat. 2002;75(Suppl 1):S45–S50; discussion S57–S58. | ||
Leek RD. The prognostic role of angiogenesis in breast cancer. Anticancer Res. 2001;21(6B):4325–4331. | ||
Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. | ||
Nallamothu R, Wood GC, Pattillo CB, et al. A tumor vasculature targeted liposome delivery system for combretastatin A4: design, characterization, and in vitro evaluation. AAPS PharmSciTech. 2006;7(2):E32. | ||
Pattillo CB, Sari-Sarraf F, Nallamothu R, Moore BM, Wood GC, Kiani MF. Targeting of the antivascular drug combretastatin to irradiated tumors results in tumor growth delay. Pharm Res. 2005;22(7):1117–1120. | ||
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. | ||
Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Polym Ed. 2006;17(3):247–289. | ||
Tang Y, Wang Y, Deosarkar S, Soroush F, Kiani MF, Wang B. Fast, stable induction of P-glycoprotein-mediated drug resistance in BT-474 breast cancer cells by stable transfection of ABCB1 gene. Anticancer Res. 2015;35(5):2531–2538. | ||
Tang Y, Gan X, Cheheltani R, et al. Targeted delivery of vascular endothelial growth factor improves stem cell therapy in a rat myocardial infarction model. Nanomedicine. 2014;10(8):1711–1718. | ||
Tang Y, Lei T, Manchanda R, et al. Simultaneous delivery of chemotherapeutic and thermal-optical agents to cancer cells by a polymeric (PLGA) nanocarrier: an in vitro study. Pharm Res. 2010;27(10):2242–2253. | ||
Kessler RJ, Fanestil DD. Interference by lipids in the determination of protein using bicinchoninic acid. Anal Biochem. 1986;159(1):138–142. | ||
Klegerman ME, Hamilton AJ, Huang SL, et al. Quantitative immunoblot assay for assessment of liposomal antibody conjugation efficiency. Anal Biochem. 2002;300:46–52. | ||
Danila D, Partha R, Elrod DB, Lackey M, Casscells SW, Conyers JL. Antibody-labeled liposomes for CT imaging of atherosclerotic plaques: in vitro investigation of an anti-ICAM antibody-labeled liposome containing iohexol for molecular imaging of atherosclerotic plaques via computed tomography. Tex Heart Inst J. 2009;36(5):393–403. | ||
Liang Y, Benakanakere I, Besch-Williford C, Hyder RS, Ellersieck MR, Hyder SM. Synthetic progestins induce growth and metastasis of BT-474 human breast cancer xenografts in nude mice. Menopause. 2010;17(5):1040–1047. | ||
Mafuvadze B, Liang Y, Besch-Williford C, Zhang X, Hyder SM. Apigenin induces apoptosis and blocks growth of medroxyprogesterone acetate-dependent BT-474 xenograft tumors. Horm Cancer. 2012;3:160–171. | ||
Pattillo CB, Venegas B, Donelson FJ, et al. Radiation-guided targeting of combretastatin encapsulated immunoliposomes to mammary tumors. Pharm Res. 2009;26(5):1093–1100. | ||
Tang Y, Wang Y, Kiani MF, Wang B. Classification, treatment strategy and associated drug resistance in breast cancer. Clin Breast Cancer. 2016;16(5):335–343. | ||
Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339(13):900–905. | ||
Hudis CA. Trastuzumab – mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39–51. | ||
Vu T, Claret FX. Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front Oncol. 2012;2:62. | ||
Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–2743. | ||
von Minckwitz G, Schwedler K, Schmidt M, et al. G.B.S. Group Investigators Participating. Trastuzumab beyond progression: overall survival analysis of the GBG 26/BIG 3-05 phase III study in HER2-positive breast cancer. Eur J Cancer. 2011;47:2273–2281. | ||
Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. | ||
Wedam SB, Low JA, Yang SX, et al. Antiangiogenic and antitumor effects of bevacizumab in patients with inflammatory and locally advanced breast cancer. J Clin Oncol. 2006;24(5):769–777. | ||
Ramaswamy B, Elias AD, Kelbick NT, et al. Phase II trial of bevacizumab in combination with weekly docetaxel in metastatic breast cancer patients. Clin Cancer Res. 2006;12(10):3124–3129. | ||
Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–2676. | ||
Kudoh K, Takano M, Kouta H, et al. Effects of bevacizumab and pegylated liposomal doxorubicin for the patients with recurrent or refractory ovarian cancers. Gynecol Oncol. 2011;122(2):233–237. | ||
Verschraegen CF, Czok S, Muller CY, et al. Phase II study of bevacizumab with liposomal doxorubicin for patients with platinum- and taxane-resistant ovarian cancer. Ann Oncol. 2012;23(12):3104–3110. | ||
Rochlitz C, Ruhstaller T, Lerch S, et al; Swiss Group for Clinical Cancer. Combination of bevacizumab and 2-weekly pegylated liposomal doxorubicin as first-line therapy for locally recurrent or metastatic breast cancer. A multicenter, single-arm phase II trial (SAKK 24/06). Ann Oncol. 2011;22(1):80–85. | ||
National Institutes of Health. Liposomal Doxorubicin, Bevacizumab and Temsirolimus (DAT) in Triple-Negative Breast Cancer (TNBC) Insensitive to Standard Neoadjuvant Chemotherapy. Houston, TX: National Institutes of Health; 2016. |
Supplementary material
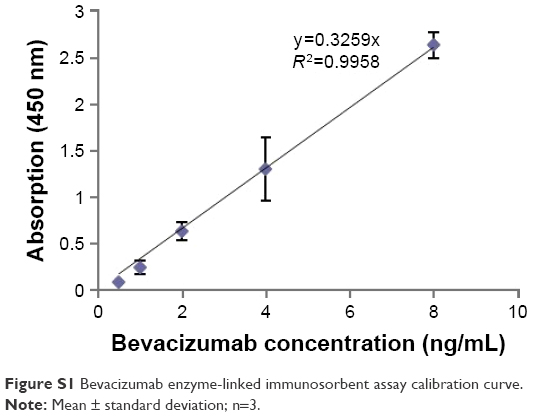 | Figure S1 Bevacizumab enzyme-linked immunosorbent assay calibration curve. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.