")
Back to Journals » International Journal of Nanomedicine » Volume 14
Targeted anticancer potential against glioma cells of thymoquinone delivered by mesoporous silica core-shell nanoformulations with pH-dependent release
Authors Shahein SA, Aboul-Enein AM, Higazy IM , Abou-Elella F, Lojkowski W , Ahmed ER, Mousa SA , AbouAitah K
Received 28 February 2019
Accepted for publication 10 May 2019
Published 19 July 2019 Volume 2019:14 Pages 5503—5526
DOI https://doi.org/10.2147/IJN.S206899
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Samar A Shahein1,* Ahmed M Aboul-Enein,1 Iman M Higazy,2 Faten Abou-Elella,1 Witold Lojkowski,3 Esam R Ahmed,4 Shaker A Mousa,5 Khaled AbouAitah3,6,*
1Biochemistry Department, Faculty of Agriculture, Cairo University, Giza, Egypt; 2Department of Pharmaceutical Technology, Pharmaceutical and Drug Industries Research Division, National Research Centre (NRC), Giza, Egypt; 3Laboratory of Nanostructures, Institute of High Pressure Physics, Polish Academy of Sciences, Warsaw, Poland; 4Confirmatory Diagnostic Unit, Egyptian Organization for Vaccine, Sera and Biological Products (VACSERA), Giza, Egypt; 5The Pharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, New York, NY, USA; 6Medicinal and Aromatic Plants Research Department, Pharmaceutical and Drug Industries Research Division, National Research Centre (NRC), Giza, Egypt
*These authors contributed equally to this work
Background and purpose: Glioma is one of the most aggressive primary brain tumors and is incurable. Surgical resection, radiation, and chemotherapies have been the standard treatments for brain tumors, however, they damage healthy tissue. Therefore, there is a need for safe anticancer drug delivery systems. This is particularly true for natural prodrugs such as thymoquinone (TQ), which has a high therapeutic potential for cancers but has poor water solubility and insufficient targeting capacity. We have tailored novel core-shell nanoformulations for TQ delivery against glioma cells using mesoporous silica nanoparticles (MSNs) as a carrier.
Methods: The core-shell nanoformulations were prepared with a core of MSNs loaded with TQ (MSNTQ), and the shell consisted of whey protein and gum Arabic (MSNTQ-WA), or chitosan and stearic acid (MSNTQ-CS). Nanoformulations were characterized, studied for release kinetics and evaluated for anticancer activity on brain cancer cells (SW1088 and A172) and cortical neuronal cells-2 (HCN2) as normal cells. Furthermore, they were evaluated for caspase-3, cytochrome c, cell cycle arrest, and apoptosis to understand the possible anticancer mechanism.
Results: TQ release was pH-dependent and different for core and core-shell nanoformulations. A high TQ release from MSNTQ was detected at neutral pH 7.4, while a high TQ release from MSNTQ-WA and MSNTQ-CS was obtained at acidic pH 5.5 and 6.8, respectively; thus, TQ release in acidic tumor environment was enhanced. The release kinetics fitted with the Korsmeyer–Peppas kinetic model corresponding to diffusion-controlled release. Comparative in vitro tests with cancer and normal cells indicated a high anticancer efficiency for MSNTQ-WA compared to free TQ, and low cytotoxicity in the case of normal cells. The core-shell nanoformulations significantly improved caspase-3 activation, cytochrome c triggers, cell cycle arrest at G2/M, and apoptosis induction compared to TQ.
Conclusion: Use of MSNs loaded with TQ permit improved cancer targeting and opens the door to translating TQ into clinical application. Particularly good results were obtained for MSNTQ-WA.
Keywords: brain cancer targeting, drug delivery system, thymoquinone core-shell nanoformulation, mesoporous silica nanoparticles, pH-dependent release kinetics
Introduction
Glioma is one of the most aggressive primary brain tumors and is incurable.1,2 It accounts for 28% of the brain tumors, 80% of them malignant.1 Median survival is less than 2 years because of malignant cell proliferation, invasion, and migration of postoperative tumor residue, as well as a high rate of recurrence.3,4
During the last decade, surgical resection, radiation, and chemotherapies have been the standard treatments for brain tumors. However, the therapeutic approach of combining radiotherapy and chemotherapy remains unsatisfactory, with low survival rates of 12–24 months.5 Drug resistance and failure to cross the blood–brain barrier (BBB) render chemotherapy ineffective.6 Consequently, increasing the chemotherapeutic drug intake has been considered as an alternative solution; however, they have negative side effects on healthy tissue.7,8
Phytochemicals as a source of prodrugs have been extensively investigated for their role as novel anticancer compounds. Thymoquinone (TQ), the main active compound of the essential oil of Nigella sativa L. (medicinal plant known as black seed or black cumin), was first isolated in 1963.9 Its biological activity has been evaluated in vitro and in animal models for anticancer, antioxidant, anti-inflammatory, antimicrobial, antidiabetic, and other properties. TQ’s chemical composition is 2-methyl-5-isopropyl-1,4-benzoquinone. It is a monoterpene diketone, multitargeted molecule exhibiting a versatile potential for modulating numerous major molecular signaling pathways in several diseases.10
TQ has anticancer potential in several cancer types, including colorectal,11 lung,12 leukemia,13 breast,14 and others.15 However, few studies have investigated its role in brain cancer.16–20 In glioblastoma cancer cells, several molecular mechanisms were observed during tests on in vitro and in vivo models treated with TQ,21 indicating its superior anticancer efficacy. Additionally, it inhibits toxin-induced neuroinflammation and neurotoxicity in animal models.22 Interestingly, the antiproliferative effect may relate in part to the sensitivity of glioblastoma cancer cells to TQ as compared to normal cells, as Gurung et al, reported in a cytotoxicity evaluation.16
Although TQ has promise, its potential in clinical application as yet to be fulfilled because of limitations related to its pure form, including low solubility in water and bioavailability, nonspecific delivery to tumor sites, and lack of selectivity for cancer cells over normal cells. Therefore, new formulation strategies are urgently required to overcome such roadblocks.
Drug delivery systems (DDSs) are a major research field of nanomedicine.6,23,24 DDSs can be used to target cancer and enhance the therapeutic efficiency with decreasing the expected toxicity for normal cells distributed around the tumor site. Thus, several cancer’s targeting strategies have been developed. Tumor-targeting can be achieved by a specific active targeting via receptor-mediated pathways (with different ligands molecules such as antibodies, small molecules as folic acid, and others), or by stimuli drug’s release from its nanocarriers to tumor specific. Among other stimuli release systems for cancer’s targeting, the pH-controlled drug release, which is considered a general developed approach because the fact that tumor site is low acidic microenvironment compared to healthy cells.25,26 Therefore, DDS that induces pH-trigger drug release, could be importantly required for cancer treatment. Various strategies for developing DDS for TQ have been reported, such as chemical derivatives (thymoquinone-4-a-linolenoylhydrazone and thymoquinone-4-palmitoylhydrazone),27 liposomes,28 solid lipid nanoparticles,29 and chitosan nanogels.30 Among the numerous nanostructured materials that can be used for designing DDSs, mesoporous silica nanoparticles (MSNs),31 attract interest because of their chemical and mechanical stability, large surface area, high volume fraction of nanosized pores, and good biocompatibility. Furthermore, a wide range of surface functionalization of MSNs may ensure controlled drug release together with the delivery of drug molecules to specific sites.32–34 MSNs are considered promising for developing efficient anticancer DDSs34 and enhanced the BBB permeability for brain cancer targeting. In this regard, Baghirov et al,35 demonstrated that intravenous injection of MSNs had no damage to the BBB and can be potentially used to deliver drugs into the brain tissue through transcellular transport. Also, in recent in vivo study Tamba et al,36 showed that MSNs modified with glucose and glucose-poly (ethylene glycol) methyl ether amine cross the BBB to reach the brain tissues via specific or nonspecific mechanisms. An example for using of MSNs as a DDSs to target glioma cells was reported by Shi et al.37 The MSNs were loaded with doxorubicin anticancer drug and coated with peptide. In another example, Zhang et al,38 fabricated a dual-targeted system composed of MSNs with valproic acid as radiosensitizer and folic acid as targeting molecule. A high rate of cell death mediated through apoptosis was observed. Several other DDSs based on MSNs have been designed as drug nanocarriers able to cross the BBB and target cancer cells.39–41
MSNs with a hydrophilic shell can be used for delivery of hydrophobic TQ and in this way limitations of low TQ solubility and stability can be overcomed.42 Only one study of the application of MSNs for TQ DDS has been reported, indicating cancer cell apoptosis with sustained release potential.43 This result offers motivation to optimize MSNs as TQ carriers. To the best of our knowledge, no optimum formulation of TQ as an anticancer agent has been reported.
In the current study, we sought to develop DDS in the form of TQ-loaded MSN core-polymer shell structures for pH-controlled release and brain cancer targeting. Its physicochemical properties were characterized and followed by kinetic release studies and evaluation of the anticancer activity and the possible molecular mechanism.
Materials and Methods
Materials
TQ, tetraethyl orthosilicate (TEOS), octadecene, and cetyltrimethylammonium ammonium chloride solution 25 wt% in H2O (CTAC), cyclohexane, ammonium nitrate were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, USA). Ethanol was obtained from Thermo Fisher Scientific, USA. Dimethyl sulfoxide (DMSO) was obtained from Tedia (Tedia, Fairfield OH, USA). 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), gum Arabic, stearic acid (MW:284.48), and chitosan (MW:100,000–300,000 Da) were obtained from Acros Organics (Acros Organics, Geel, Belgium). Whey protein concentration was obtained from Alfasol, Istanbul, Turkey. Triethanol amine (TEA) from Molekula GmbH, Munich, Germany. Phosphate-buffered saline (PBS), DMEM (Dulbecco’s modified Eagle’s medium), RPMI 1640 Medium, and fetal bovine serum (FBS) were obtained from (Gibco/life technologies, Thermo Fisher Scientific – Langenselbold, Germany). Insulin (Novo Nordisk, Bagsvaerd, Denmark). Penicillin G, streptomycin, and MTT assay kit (Sigma-Aldrich, St. Louis, USA). Caspase-3 (active) Human ELISA kit from Invitrogen (Cat. KHO1091, Camarillo, CA, USA). Annexin V-FITC Apoptosis Detection Kit was from BioVision (Cat. K101-25, −100, −400, BioVision, CA, USA), and the Script One-Step RT-PCR Kit with SYBR® Green was obtained from BIO-RAD, Hercules, CA, USA. Ultrapure water (18.2 MΩ; Milli-Q® system, Millipore, Darmstadt, Germany) used in all prepared solutions. All other reagents used were of analytical grade.
Methods
Synthesis of MSNs
The MSNs were prepared using a diphase stratification method recently reported by Shen et al,44 with minor modifications. In a typical preparation, 24 mL CTAC (25% solution) and 0.5 mL TEA were added to 36 mL water and stirred for 1 hr at 60°C. Then, 20 mL of 20 v/v% of TEOS in octadecene was carefully added to the mixture drop by drop and stirred for another 12 hrs. After cooling, the upper layer formed (containing octadecene) and was totally removed and then replaced with 20 mL of 5 v/v% of TEOS in cyclohexane and stirred for another 12 hrs at 80°C to obtain the second generation. Finally, the solution was collected by centrifugation, washed several times with ethanol, and centrifuged again. The obtained product was dried in an oven at 60°C to remove the used template, then the powder material was dispersed and extracted in ethanol containing ammonium nitrate (0.6 wt.%) for 6 hrs at 60°C. This step was repeated two times to remove the CTAC template. The resulting product patch of the material was dried at 60°C and named as MSN.
TQ loading into MSNs and preparation of core-shell designed nanoformulations
Loading of TQ into MSNs was based on a solvent evaporation method with a loading ratio of TQ: MSN of 1:4. The following procedure was used: 1 g of MSN was dissolved in ethanol (HPLC grade) containing 250 mg TQ, then the solution was stirred for 24 hrs at room temperature. Subsequently, the solvent was evaporated at 50°C using the Rotavap (Büchi, Flawil, Switzerland), resuspended in ultrapure water (repeated several times) to remove unloaded molecules, and dried at 60°C for 12 hrs in an oven. The resulting product was labeled MSNTQ. This material was intended as the core to be used in designed nanoformulations with a shell of polymer mixtures to finally obtain core-shell delivery designs.
Two shell designs were prepared: a shell layer of chitosan–stearic acid (CS) and a shell layer of whey protein–gum Arabic (WA).
For CS, the following procedure was performed based on a coacervate and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) coupling reaction. It permitted to obtain complex polymers through the reaction of amino groups of chitosan and carboxyl groups in stearic acid. The EDC coupling reaction was reported in previous studies by Hu et al,45 Yuan et al,46 and Yang et al,47 for chitosan-stearic acid conjugation. We followed this path during the present preparations. In typical method, in a glass bottle, 0.750 g of chitosan (used as received without any further purifications) was dissolved in 25 mL ultrapure water (1.5% acetic acid) and sonicated for 10 mins, then stirred for 1.5 hrs with vigorous stirring at 65°C adjusted with a temperature sensor to obtain the chitosan solution. In a separate glass bottle, 0.375 g of stearic acid and 0.375 g EDC (1:1) were dissolved in 25 mL of acetone/ethanol by sonication for 10 mins, then also stirred for 1.5 hrs under the same conditions as for the chitosan solution to obtain the stearic acid-EDC solution. Afterwards, the stearic acid-EDC solution was dropped slowly into the chitosan solution over 5 mins to prevent aggregation (an additional 10 mins under sonication is recommended). The solution was left to stir for 24 hrs at room temperature.
Next, the solution was filtered and washed with water several times to remove excess EDS molecules, and the resulting material consisting of CS was dried in an oven at 60°C. The dried CS was resuspended again in 50 mL ultrapure water and kept at 5°C until use. To prepare the shell layer on MSNTQ, we suspended 0.5 g of dried MSNTQ in CS solution and stirred for 48 hrs at room temperature, followed by collection with centrifugation (cooling Sigma 16K, Laborzentrifugen GmbH, Osterode am Harz, Germany), several washes with ultrapure water, and oven drying at 60°C. The resulting material was labeled MSNTQ-CS core-shell nanoformulation.
For WA, the following procedure was performed to obtain WA-conjugated polymer material with a 3:1 ratio of whey protein to gum Arabic, according to a method previously reported by Klein et al48 in a glass bottle, 1.5 g whey protein, and 0.5 g gum Arabic were dissolved in 50 mL ultrapure water and sonicated for 10 mins, then the mixture solution was adjusted to pH 7.5 and stirred for 5 hrs. The mixture solution was filtered and kept at 5°C until further use. To prepare the shell layer on MSNTQ, 0.5 g of dried MSNTQ was suspended in WA solution (25 mL) and stirred for 24 hrs at room temperature. This step was repeated once again to ensure a good layer, then the material was collected with centrifugation and washed several times with water and dried in an oven at 60°C. The resulting material was labeled MSNTQ-WA core-shell nanoformulation design.
Characterization techniques
The nanostructures during different stages of preparation were observed by means of a high-resolution transmission electron microscope (HR-TEM, JEM 2100, JEOL Ltd., Tokyo, Japan) and field emission scanning electron microscope (FE-SEM, Ultra Plus, Zeiss, Jena, Germany). The Brunauer, Emmett, and Teller (BET) surface area analysis was used to characterize the mesoporosity characteristics using NOVA automated gas sorption system (NOVA, Quanta Chrome Instruments, Florida, USA). All materials were degassed for 24 hrs at 50°C before analysis. Fourier transformed infrared (FTIR) spectroscopy was used to distinguish the change in the surface functional group before and after loading (Bruker Optics Tensor 27, Bruker Corporation, Billerica, MA, USA). The thermal gravimetric analysis (TGA)-coupled to differential scanning calorimetry (DSC) (Shimadzu TGA-DSC 50, Shimadzu, Kyoto, Japan) analysis was performed for two reasons: (1) to determine the drug loading content with TGA, and (2) to confirm the crystalline state by with DSC analysis. The experimental condition for thermal analysis was programmed to reach 800°C with a heating rate of 10°C/min under nitrogen.
In vitro drug release studies
We followed the Franz Diffusion Cell method49 and modified the setup described by Salama et al,50 under perfect sink conditions in triplicate under different pH conditions (pH 7.4, 6.8, and 5.5). The dissolution medium reservoir of suitable PBS and cellulose membrane (molecular weight cutoff 12,000 g/mole) was incubated at 37°C and placed into a holder with a calculated amount for each nanoformulation, facing up. This face was brought into contact with dissolution medium, and the stirring speed was adjusted to 150 rpm (Shaking incubator, GFL 3032, Germany). Sampling was automated according to a defined time schedule. A 2-mL aliquot was withdrawn periodically (up to 12 hrs in the first day, then single sampling every day for 72 hrs) and refilled with fresh pre-warmed PBS buffer to maintain constant dissolution medium volume of 50 mL. The amount of TQ released was quantified spectrophotometrically, at corresponding λmax of TQ with regard to each pH at 7.4, 6.8, and 5.5.
The TQ release data were kinetically analyzed using Kinet DS3 software (Department of Pharmaceutical Technology and Biopharmaceutics, Faculty of Pharmacy, Jagiellonian University), according to various model-dependent orders. Linear regression equations were employed, and coefficient of determination (R2) was determined. Data were processed for regression analysis using MS excel statistical function (Office365, 2013, USA), and release efficiency (RE), mean dissolution time (MDT), and release rate (RR) were also computed.51
Cell cultures
We investigated two brain cancer cell lines: human glioma cells (A172) and human astrocytoma (SW1088). Human cortical neuronal cells-2 (HCN2) were used as the normal cell reference. Cells were obtained from the American Type Culture Collection (ATCC, Manassas, Virginia, USA), and all cell-based evaluations were done at the Confirmatory Diagnostic Unit, VACSERA, Dokki, Giza, Egypt. Cells were cultured in DMEM supplemented with 10% FBS, penicillin G (100 U/mL), and streptomycin (100 μg/mL), maintained at 37°C in a humidified 5% CO2 atmosphere.
In vitro cytotoxicity assessment
To assess cytotoxicity and anticancer activity of the synthesized nanoformulations and TQ, the MTT assay was used as described previously by Mosmann.52 Cells were seeded in 96-well tissue culture plates at a density of 1.2–1.8×10,000 cells/per well and kept for 24 hrs at 37°C in a humidified 5% CO2 atmosphere, until cell monolayers were confluent. Subsequently, medium was removed, washed, and fresh DMEM added containing 100 µL of tested compounds with different concentrations. For MSNs, concentrations of 12.3, 37, 111, 333, and 1,000 µg/mL were used for biocompatibility evaluation. For TQ, MSNTQ, MSNTQ-CS, and MSNTQ-WA, concentrations of 1.2, 3.7, 11, 33, and 100 µg/mL were used (in the case of nanoformulations, their concentration was designed to obtain an equivalent amount of TQ). As a control, 100 µL of medium was used. To calculate the equivalent amount of TQ in each nanoformulations we proposed simple an equation based on the weight loss data (Table 1 and Supplementary materials). After treatment, the samples were incubated for 48 and for 72 hrs under the same conditions. Thereafter, the medium was removed, fresh DMEM containing 50 µL of MTT (1 mg/mL) solution was added, and cells were further incubated for 4 hrs at 37°C. Then, the medium containing MTT solution was discarded, and 100 µL of DMSO was added into each well (to dissolve MTT formazan crystals). Afterwards, the plates were gently shaken for 5 mins to ensure that the crystals were completely dissolved. Finally, the absorbance was measured at 540 nm using a Robonik P2000 ELISA reader (Robonik India PVT LTD, Thane, India). This assay was done in triplicate, and the data are expressed as mean ± standard deviation (SD). The inhibition concentration of 50% cells (IC50) was calculated using Origin Pro. 8.5 software (OriginLab, USA).
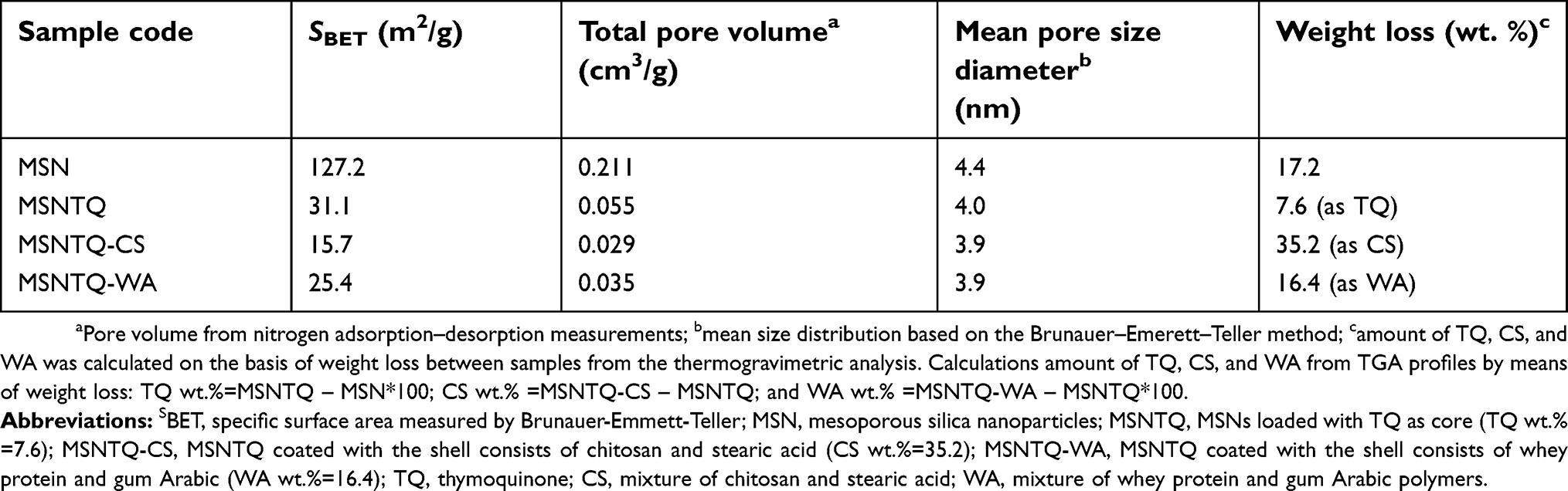 |
Table 1 Physicochemical properties of mesoporous silica nanoparticles before and after loading of thymoquinone |
Apoptosis detection and cell cycle analysis with flow cytometry
To detect apoptosis, we used the Annexin V-FITC Apoptosis Detection Kit according to the manufacturer’s instruction. Briefly, SW1088 cells were seeded (5×105 cells/well) onto six-well plates and left to adhere overnight, and the cells were treated with different samples (TQ, MSN, MSNTQ-CS, and MSNTQ-WA) at their IC50 (µg/mL, as listed in Table 2) in 100 μL sample volume per well and incubated for 48 hrs. In the case of the nanoformulations, an equivalent amount of TQ-producing IC50 concentrations was used. The IC50 concentration is universally used, and every drug or anticancer agent may have a different IC50 value depending on the type of cancer cell line. For control, cells without any treatment were used. The cells were collected by centrifugation, and the pellet was resuspended in 500 μL of binding buffer solution. Afterward, 5 μL of Annexin V-FITC and 5 μL of propidium iodide were added and incubated for 5 mins in the dark and immediately analyzed with flow cytometry (FACSCalibur, Becton Dickinson, NJ, USA). To analyze the cell cycle, after SW1088 cells were treated with the different samples mentioned above, compared to control, and incubated for 48 hrs, they were washed, collected by centrifugation, fixed in cold 70% ethanol, and labeled with propidium iodide staining. Then, the samples were analyzed by flow cytometry. During analysis, cell cycle analysis was performed with an FL2-A histogram of single cells.
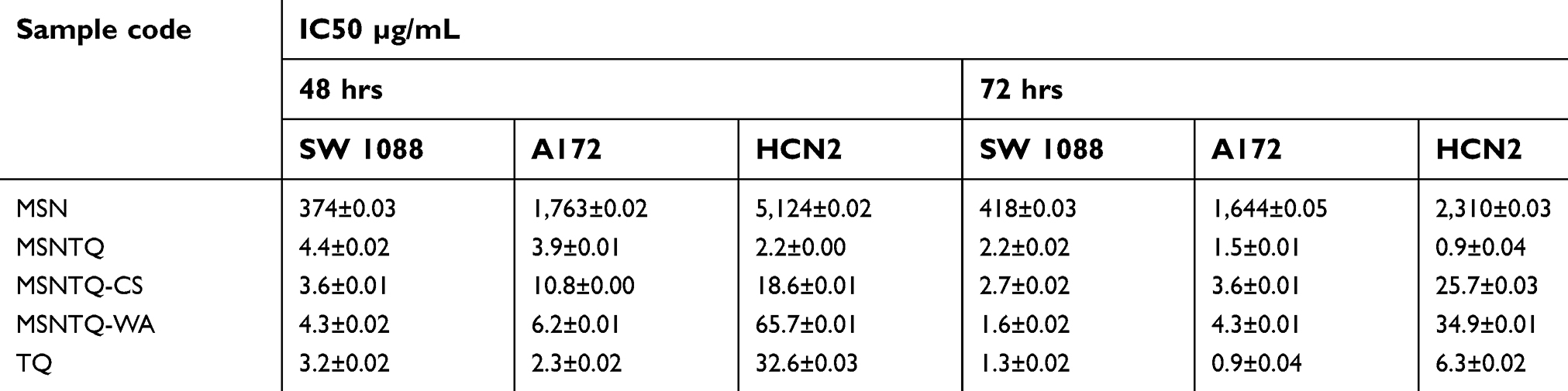 |
Table 2 The calculated IC50 expressed as µg/mL for all samples |
Caspase-3 activity assay
To assess caspase-3 activity, we followed the manufacturer’s instructions for the human active caspase-3 content assay kit. In brief outlined, SW1088, A172, and HCN2 cells were cultured onto 96-well plates to obtain a density of 1.2–1.8×10,000 cells/well in a 100 µL volume of complete growth medium (RPMI 1640 containing 10% FBS at 37°C). The cells were treated with different samples (TQ, MSN, MSNTQ-CS, and MSNTQ-WA) at their IC50 (µg/mL, as listed in Table 2) in 100 μL sample volume per well and incubated for 48 and 72 hrs prior to the assay. Cells were lysed with the cell extraction buffer; the lysates were then diluted by the standard dilution buffer of the range for the assay and assessed for human active caspase-3 content. The absorbance was measured at 450 nm using the Robonik P2000 ELISA reader. The assay was performed in triplicate, and the data are expressed as mean ± SD.
Cytochrome c quantitative analysis by RT-PCR
The cytochrome c quantitative determination by RT-PCR was done using the BIORAD iScriptTM One-Step RT-PCR Kit with SYBR® Green (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. In brief, total mRNA was isolated for all cell lines under investigation using the RNeasy extraction kit (Qiagen, Germany), and the cells (1×106). Then, cells were disrupted in buffer RLT and homogenized and disrupted, followed by addition of ethanol to the lysates to create conditions that promoted selective binding of RNA to the RNeasy membrane. The different samples (TQ, MSN, MSNTQ-CS, and MSNTQ-WA) at their IC50 (µg/mL, as listed in Table 2) in 100 μL sample volume were applied to a RNeasy Mini spin column. With total RNA bound to the membrane and contaminants efficiently filtered, high-quality RNA could be eluted in RNase-free water. Washing and elution followed by centrifugation in a microcentrifuge. For the RT-PCR, the primers and PCR conditions used were the iScript one-step RT-PCR kit with SYBR Green on a Rotor-Gene 3000 apparatus. The mRNA extract prepared above was used in the reactions, and DNA amplification was performed according to the manufacturer’s protocols and Yergeau et al.53 The iScript one-step RT-PCR kit with SYBR Green kit contents, reagent description, master mix preparation, and amplification protocol were provided for more details in Supplementary materials (RT-PCR protocol). Samples were assessed in triplicates.
Statistical analysis
Data for biological evaluations are expressed as mean ± SD. Significance differences throughout this study were calculated using analysis of variance and mean comparisons by least significant difference (LSD) at p<0.05. All statistical analysis was done using the software IRRE STST (2005; a computer program for data management and statistical analysis of experimental data, version S, biometrics and bioinformatics unit, ERRI Institute). Only, cytochrome c data analyzed with Statistix 9.0 software by LSD at p<0.05.
Results and discussion
Synthesis and fabrication of TQ delivery systems
To design a suitable delivery system for TQ, we synthesized 3D dendritic MSNs-mesoporous silica nanospheres according to a previously reported method44 with minor modification. This type of MSN was selected because of good biocompatibility and advantageous 3D-dendritic structure permitting high drug loading and controlled release.44 Figure 1A shows the steps to obtaining the designed nanoformulations for the TQ delivery system.
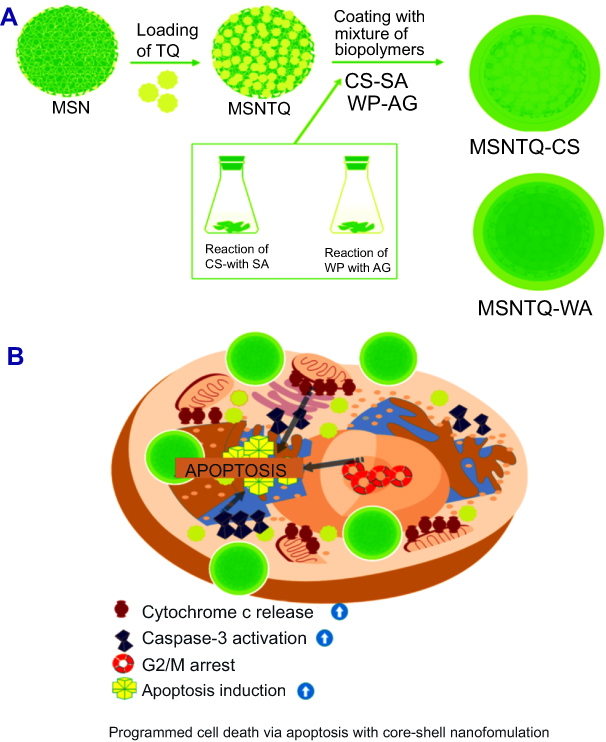 |
Figure 1 Schematic representation of preparation steps and proposed anticancer mechanism. Notes: Synthesis of MSNs and nanoformulations for delivery of TQ (A) and proposed anticancer mechanism (B) for brain cell cancer treated with nanoformulations. Abbreviations: MSN, mesoporous silica nanoparticles; MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic; TQ, thymoquinone; CS-SA, mixture solution of chitosan and stearic acid polymers; WP-AG, mixture solution of whey protein and gum Arabic. |
Characterization of MSNs and nanoformulations
HR-TEM images (Figure 2A and B) showed that MSNs have an almost sphere-like shape and a size ranging from 50 to 140 nm. Their dendritic mesoporous structure was clearly visualized. For MSNTQ, the pores of MSNs were filled with TQ molecules, as indicated by the blue arrow (Figure 2D) compared to MSNs before loading (Figure 2C). After being coated with polymer shells made of CS and WA, the MSNs were totally covered (Figure 2E and F) compared to MSNTQ. The coating may be formed by self-assembly, as seen in different layers by different darker and lighter areas on each nanoparticle (blue arrows). FE-SEM images (Figure 3A) show similar results, as seen in the HR-TEM images as far as particle size. The images show the polymer shell visible on MSNTQ-WA and MSNTQ-CS (Figure 3C and D) comparing to MSNTQ (Figure 3B) core. TEM images gave a direct demonstration of drug loading, and both TEM and SEM images gave a direct demonstration of polymer coating, indicating the successful formation of core-shell structures.
 |
Figure 2 HR-TEM images of synthesized MSNs before and after drug loading and coating with polymer shells. Notes: MSNs at 100 nm scale (A) and at 200 nm scale (B). The morphological structure differences at all stages of preparations are seen MSNs (C), MSNTQ (D), MSNTQ-CS (E), and MSNTQ-WA (F). Abbreviations: HR-TEM, high-resolution transmission electron microscopy; MSN, mesoporous silica nanoparticles; MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic. |
 |
Figure 3 FE-SEM images of MSNs before and after drug loading and coating with polymer shells. Notes: MSN (A), core MSNTQ (B), core-shell MSNTQ-CS (C), and core-shell MSNTQ-WA (D). Abbreviations: FE-SEM, field emission scanning electron microscopy; MSN, mesoporous silica nanoparticles; MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic. |
Figure 4 shows the results of specific surface characterization of the mesoporous structures before and after coating by means of a nitrogen adsorption–desorption technique. As presented in Figure 4A, a type-IV isotherm pattern and a small hysteresis loop were observed. It can be also seen that the MSNs displayed a wide range of pore size distribution with mean pore size 6.6 nm, calculated based on Brunauer–Joyner–Halenda (BJH) method (Figure 4B in MSN line). Additionally, both the surface area (BET) and total pore volume (BJH) were calculated and found to be 127 m2g−1 and 0.211 cm3g−1, respectively (Table 1). After loading with TQ and polymer coating (Figure 4A), the BET surface area decreased compared to MSN (127 m2/g) in the following order: MSNTQ (31.1 m2/g), MSNTQ-WA (25.4 m2/g), and MSNTQ-CS (15.7 m2/g). As seen in Figure 4B, pore size decreased following TQ loading and polymer coating compared to MSNTQ. In addition, the total pore volume decreased from 0.211 cm3/g in MSN to 0.055, 0.029, and 0.035 cm3/g in MSNTQ, MSNTQ-CS, and MSNTQ-WA, respectively. All these results confirmed successful loading of the pores of MSN particles with TQ and formation of core-shell structures. Our results concerning surface area, pore size, and pore volume properties are in line with previous reports involving MSNs.54–56
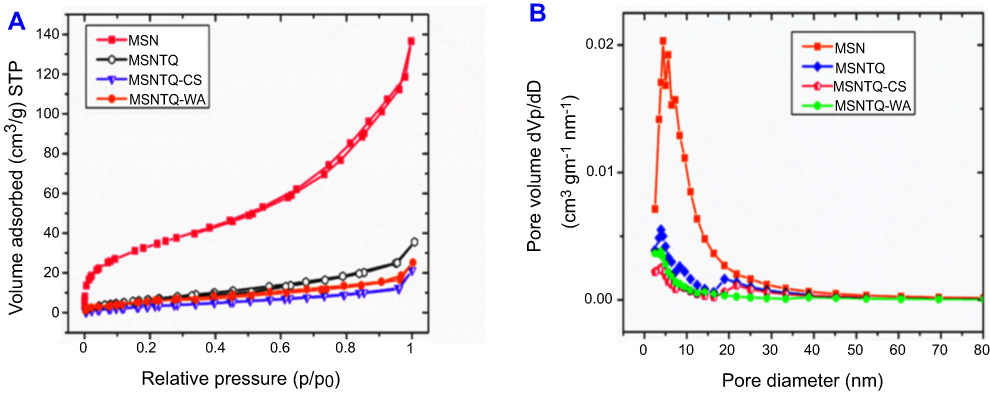 |
Figure 4 Nitrogen adsorption–desorption isotherms and pore size distributions measurements of all materials. Notes: The N2 adsorption-desorption isotherms of MSN, MSNTQ, MSNTQ-CS, and MSNTQ-WA (A). The pore diameter distribution for materials before and after TQ loading and coating with shells for nanoformulations (B). Abbreviations: MSN, mesoporous silica nanoparticles; MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic; TQ, thymoquinone; dVp/dD, pore volume distribtion; STP, standard temperature and pressure. |
We used TGA analysis to determine TQ content in MSNs and the polymer coating mass fraction on the basis of weight loss percent when the organic molecules were thermally decomposed (wt. %) (Figure 5A and B and Table 1). MSN lost about 17% of its initial weight, which could be ascribed to some water content and organic material used during synthesis. By loading of TQ to MSN to obtain MSNTQ, the weight loss increased compared to MSN, and the calculated amount of TQ-loaded to MSN was 7.62 wt. %. This observation confirms the successful loading of TQ into the pores of silica nanoparticles. Further attachment of polymer materials for each of the two coating layers to obtain the shells resulted in more relative weight loss because of decomposition of polymers: 35.2 wt. % in MSNTQN-CS nanoformulation and 16.4 wt. % in MSNTQ-WA nanoformulation. This result further confirms the successful coating process on MSNTQ core nanoformulation. Figure 5C and D shows the DSC profiles of materials. It is seen that no peaks are presented in MSNTQ, MSNTQ-CS, and MSNTQ-WA comparing to TQ with its two melting point peaks. This could attribute to TQ molecules are entrapped into pores of MSNs even though shell layer was constructed.
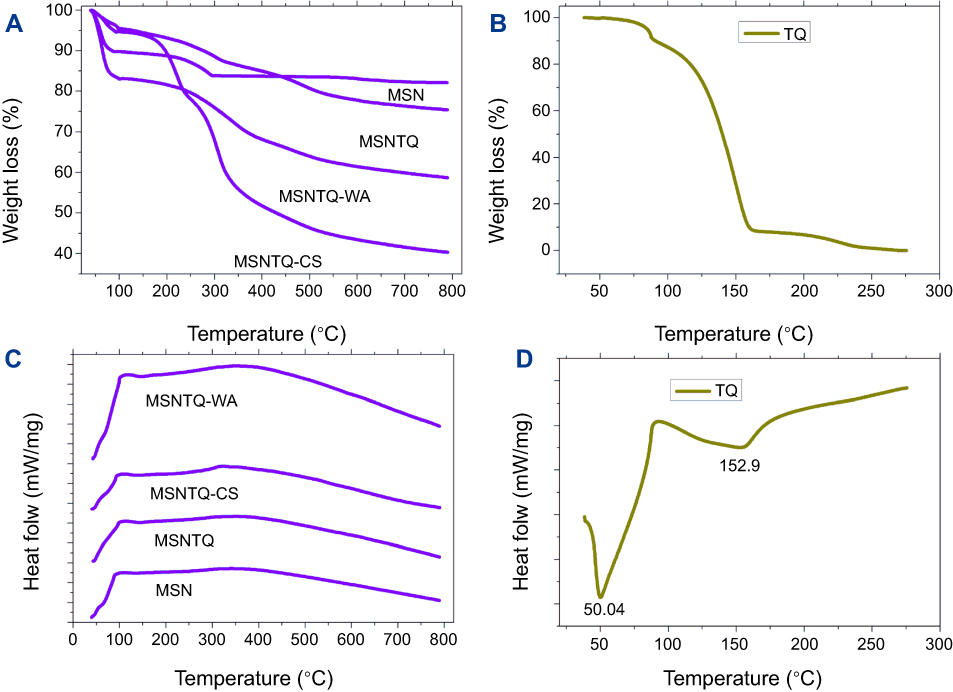 |
Figure 5 TGA and DSC measurements. Notes: TGA analysis of MSNs, TQ-loaded, and coated polymer nanoformulations (A). TQ (B). DSC profiles of MSNs, TQ-loaded, and coated polymer nanoformulations (C). TQ (D). Abbreviations: TGA, thermal gravimetric analysis; DSC, differential scanning calorimetry analysis; MSN, mesoporous silica nanoparticles; MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic; TQ, thymoquinone. |
The results of FTIR characterization are shown in Figure 6. In the MSN spectrum, two characteristic bands appeared to be related to starching vibrations for the Si–O–Si at 805 cm−1 (symmetric) and 1,090 cm−1 (asymmetric), which confirm the mesoporous silica framework formation. In addition, a broad prominent band appeared at 3,446 cm−1, corresponding to hydroxyl groups on the surface of the nanoparticles.57 The almost complete disappearance of peaks at 2,800–3,200 cm−1 and 1,470 cm−1 confirmed the successful removal of CTAC template from MSNs.58 The template can be also removed using sodium chloride-methanolic solution procedure.59 The FTIR spectrum of MSNTQ displayed two new bands at 1,380 and 1,641cm−1, corresponding to TQ, and the intensity bands at 2,934 and 3,340 cm−1 increased. The FTIR spectrum of MSNTQ-WA showed that the bands at 1,380, 1,641, and 2,934 cm−1, corresponding to TQ, slightly diminished in comparison with MSNTQ (as indicated by black arrows). This result can be explained by attachment WP and AG polymers to MSNTQ covering the loaded TQ molecules corresponded to 1,645 and 2,920 cm−1. Thus, the functional groups on the surface are strongly related to WA. In the FTIR spectrum of MSNTQ-CS, there were two small bands at 1,690, 2,870, and 2,925 cm−1 corresponding to CS and SA pure polymers, and one broadband centered at 1,430 cm−1, ascribed to chitosan (Cs) (blue dashed ring). Additionally, the intensity in the region centered at 3,440 cm−1 (indicated by black arrows) for MSNTQ-WA and MSNTQ-CS is smaller than for MSNTQ. This is presumably a result of polymer coating. The differences in the FTIR spectra for materials before, after drug loading, and after polymer coating are in line with results previously reported by Sun et al,60 for MSNs loaded doxorubicin drug and coated with multiple polyelectrolyte layers.
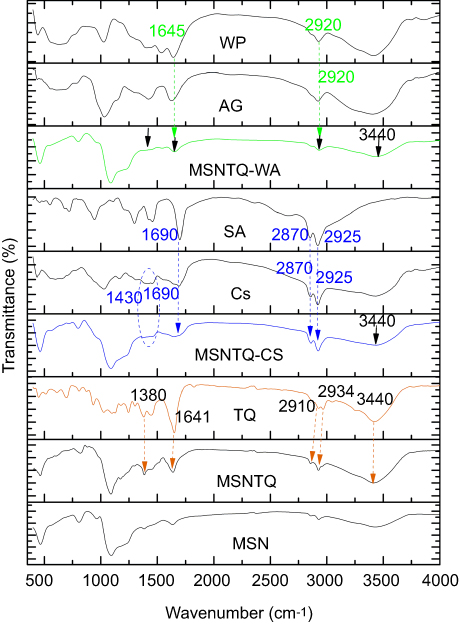 |
Figure 6 FTIR spectra of MSNs before and after loading, used polymers, and TQ. Notes: Orange arrows indicate the corresponding peaks to TQ for MSNTQ. Blue arrows specify peaks for chitosan. Green arrows specify peaks for whey protein and stearic acid. Black arrows indicate decreasing intensity of peaks corresponding to MSNTQ due to the shell for MSNTQ-WA due to used polymers and only the peak centered at 3,440 cm−1 MSNTQ-CS due to used polymers. Abbreviations: FTIR, Fourier transform infrared spectroscopy; MSN, mesoporous silica nanoparticles; TQ, thymoquinone; Cs, chitosan; AG, gum Arabic; SA, stearic acid; WP, whey protein; MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic. |
In vitro release studies
The Supplementary materials show the pre-release studies in detail. To perform the solubility studies, the linearity and strong correlation between “TQ concentration” and “absorbance” was proved through R2 values approaching one, and straight-line equation, as shown in Table SI1 and Figure SI1. The favorable pH zone for TQ solubility in PBS lies between pH 5.5 and 7.4, increasing with increasing acidity (Table SI2). To further confirm that TQ is still entrapped in nanoformulations after the coating process, the total TQ content, TQ loading efficiency/capacity, and its entrapment efficiency were investigated by UV-vis spectrophotometry method. The detailed results are shown in Table SI3. To analyze the values obtained from the release study quantitatively, we used “KinetDS3.0” software, employing different mathematical formulae to find the best fitting model. RE is defined as the area under the release curve up to time (t), expressed as a percentage of the released drug,61 while MDT characterizes drug RR and thus depends on dose/solubility ratio.62
The in vitro TQ release profiles at pH 7.4 are presented graphically in Figure 7A, and data are listed in Table SI4. It was carried out for 12 hrs only because of TQ instability at this pH. At pH 5.5 and 6.8, release was extended to 72 hrs.42 The maximum (p<0.05) TQ release of 38.9±2.2% occurred after 12 hrs for MSNTQ, followed by MSNTQ-CS (30.9±3.8%) and then MSNTQ-WA (26.7±4.1%). The nanoformulation of MSNTQ showed a significantly different release profile (p<0.05) compared to MSNTQ-CS and MSNTQ-WA. The release profiles of the two nanoformulations were not significantly different. The RR reached 3.25±0.42%/h for MSNTQ, 2.57±0.26%/h for MSNTQ-CS, and 2.22±0.32%/h for MSNTQ-WA, respectively. The RE was found to be 25.7±2.5% with MSNTQ, 19.3±1.7% with MSNTQ-CS, and 17.5±2.1% with MSNTQ-WA, respectively. As for MDT values, MSNTQ showed the lowest value (4.07±0.08 h), followed by MSNTQ-CS (4.51±0.13 hrs) and MSNTQ-WA (4.11±0.03 hrs). This result implied an effect of polymeric hindering of drug release, with the highest hindering for the MSNTQ-CS.
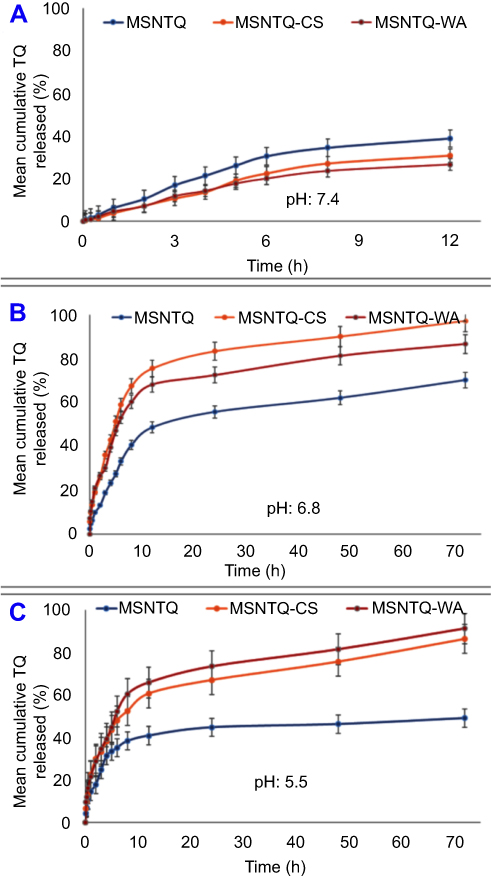 |
Figure 7 Release kinetic profiles of core nanoformulation (MSNTQ) and core-shell nanoformulations (MSNTQ-CS and MSNTQ-WA) at different pH conditions with PBS media. Notes: In vitro release profiles at pH 7.4 (A). In vitro release profiles at pH 6.8 (B). In vitro release profiles at pH 5.5 (C). The curves are expressed as mean ± SD (n=3), and error bars were created based on SD values. Abbreviations: MSNTQ, MSNs loaded with thymoquinone; MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic; PBS, phosphate-buffered saline; SD, standard deviation; n, number of replicates. |
The release profiles of TQ at pH 6.8 (Figure 7B, Table SI5) showed a sustained release pattern, reaching a maximum after 72 hrs. The TQ released for MSNTQ was 70.3±5.5%; for MSNTQ-CS it was 97.1±5.1%, and for MSNTQ-WA, it was 86.7±4.2%. A relatively rapid burst at the beginning of release for MSNTQ-CS and MSNTQ-WA was observed, followed by characteristic slow release. The MSNTQ-CS demonstrated the maximum TQ release, with a significantly high (p<0.05) mean cumulative TQ released after 72 hrs. The highest RE (82.0±5.7%) and RR (1.35±0.77%/h) were found for MSNTQ-CS compared to other nanoformulations; RE (55.5±3.3%) and RR (0.98±0.01%/h) for MSNTQ; and RE (73.2±4.0%) and RR (1.20±0.05%/h) for MSNTQ-WA. As for MDT, MSNTQ-CS and MSNTQ-WA showed insignificantly higher values: 11.19±4.99 and 11.2±4.03 hrs, respectively, but it significantly differed from MSNTQ release (1.52±0.02 h).
Release profiles of TQ at pH 5.5 (Figure 7C, Table SI6) indicated a maximum TQ release from MSNTQ-WA (91.3±5.0%), followed by MSNTQ-CS (86.3±4.8%), and MSNTQ (49.1±5.3%). We detected no significant difference between MSNTQ-CS and MSNTQ-WA. Worth noting, pH – rather than the type of polymeric shell – governs the release parameters of TQ for these core-shell nanoformulations. This relationship was reflected through the insignificance difference (p>0.05) in RE, MDT, and RR of both MSNTQ-CS and MSNTQ-WA. At the same time, the polymer shell effect was evident through the significant difference (p<0.05) for MSNTQ-CS and MSNTQ-WA release parameters compared to those of MSNTQ.
In general, the rate of TQ release into the dissolution media may depend on binding between TQ and MSN, permeability of the polymer shell, and pH of the medium. Coating of MSNTQ-CS and MSNTQ-WA hinders TQ release compared to uncoated MSNTQ. The initial burst release could be caused by dissolution of some TQ adsorbed to the outer shell.
All TQ release profiles followed the Korsmeyer–Peppas63 kinetic model, which is characteristic for drug release from polymeric systems.64 This model corresponds to Fickian diffusion of TQ into release media65 and is an anomalous form of transport based on the diffusion-controlled release, as proposed by Ritger et al.66 Factors affecting TQ release from the nanoformulations in different pHs are as follows:
- Solubility of TQ: Its maximum solubility occurs at pH 5.5 (Figure SI2), as confirmed by the MDT at pH 5.5.
- Presence of a polymeric shield: This shield slows TQ release, as seen in the RR and RE values (Table SI6).
- Dependence on composition of the shell layer: In case of a shell with chitosan/stearic acid in MSNTQ-CS, the slow release of TQ may result from ionic interactions between chitosan and stearic acid in the shell. The ionic gelation interaction between them depends on the pH condition,67 so that pH may control TQ release.68 In the case of the shell in MSNTQ-WA, TQ release may depend on pH condition because the two polymers interact to produce a polyelectrolyte complex.69 This interaction depends on pH,70 so that pH level would either hinder or facilitate TQ release. In addition, whey protein swelling is minimum at its isoelectric point (pI at 5.1).71 Thus, TQ release for core-shell nanoformulations was higher at pH 5.5 and pH 6.8 compared to pH 7.4.
In vitro biocompatibility of MSNs
A significant effect on cell viability (p<0.05), which depends on concentrations, cell line, and incubation times, was observed (Figure 8). A high dose significantly reduced cell viability. For SW1088 cancer cells, the MSNs at a high concentration of 1,000 µg/mL reduced cell viability to 44.8±1.3% (48 hrs) and 45.7±1.3% (72 hrs) compared to other concentrations used. The difference between different incubation times was insignificant (except for 12.3 µg/mL). The calculated IC50 was 374 µg/mL at 48 hrs and 418 µg/mL at 72 hrs (Figure 8A). For A172 cancer cells, an increasing concentration decreased cell viability. High reduction percentages were obtained when cells were treated with 1,000 µg/mL compared to others, and the cell viability was 54.2±0.8% (48 hrs) and 52.5±0.7% (72 hrs).
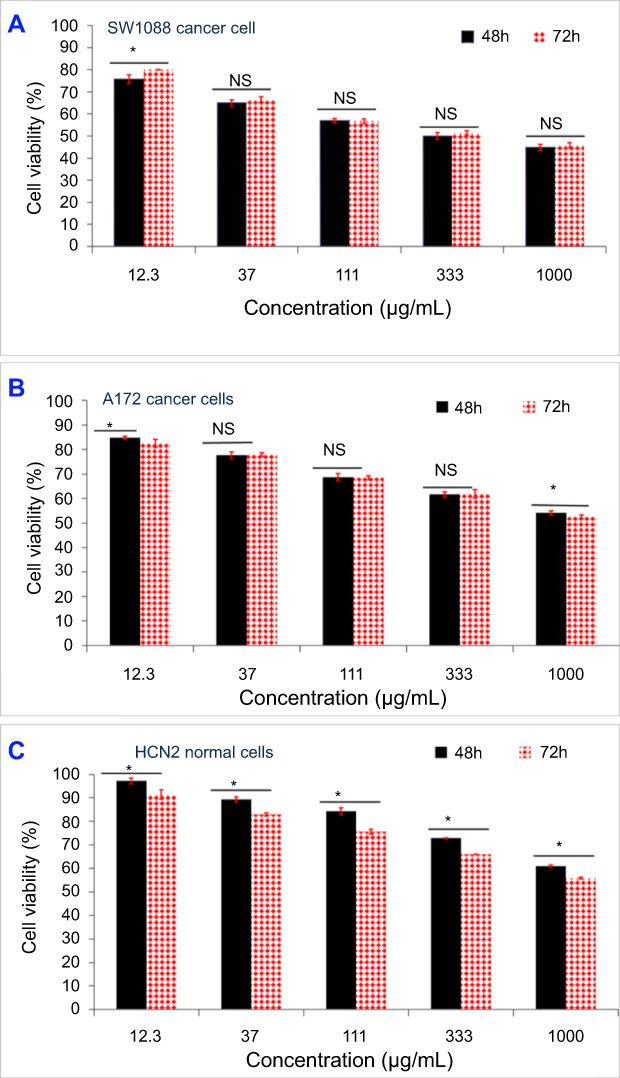 |
Figure 8 In vitro cytotoxicity of MSNs for biocompatibility evaluations on brain cancer cells (SW1088 and A172) and normal brain cells (HCN2) after 48 and 72 hrs of incubation with cells. Notes: Biocompatibility of MSN on SW1088 cancer cells with different concentrations from 12.3 to 1,000 µg/mL (A). Biocompatibility of MSN on A172 cancer cells with different concentrations from 12.3 to 1,000 µg/mL (B). Biocompatibility of MSN on HCN2 normal cells with different concentrations from 12.3 to 1,000 µg/mL (C); all data are expressed as mean ± SD. The differences are labeled with * (between the groups/samples) at p<0.05 based on the least significant difference (LSD values). Non-significant differences are marked as NS, it indicated by line linked the NS groups (for two samples).Abbreviations: MSN, mesoporous silica nanoparticles; SW1088, human astrocytoma brain cancer cells; A172, human glioma cells; HCN2, human cortical neuronal cells-2 normal cells; SD, standard deviation. |
The time effect was significant with concentrations of 12.3 and 1,000 µg/mL, respectively, compared to others. Also, the IC50 was 1,763 µg/mL at 48 hrs and 1,644 µg/mL at 72 hrs (Figure 8B). For HCN2 normal cells, a relatively high viability was obtained compared to cancer cell lines: for cells treated with 1,000 µg/mL, the cell viability reached for 61.0±0.5% and 55.9±0.5%. The time effect significantly affected viability at 48 and 72 hrs at all concentrations. Moreover, the IC50 was 5,124 µg/mL at 48 hrs and 2,310 µg/mL at 72 hrs (Figure 8C). These results demonstrate that the MSNs were less toxic for HCN2 brain normal cells compared to brain cancer cells, showing relatively higher biocompatibility with normal brain cells. This finding led us to calculate the selectivity index (SI) by means of IC50 values (Table 2), dividing the IC50 value of normal cells by that of cancer cells (Table 3). The SI values were as follows: 13.7 (with SW1088 cells) and 2.9 (with A172 cells) at 48 hrs; and 5.5 (with SW1088 cells) and 1.4 (with A172 cells) at 72 hrs. We infer that SI depends on the cancer cell line and time of incubation. The greater SI value indicates more selectivity, thus SW1088 extremely much sensitive to MSNs than A172 cells after 48 hrs and after 72 hrs.
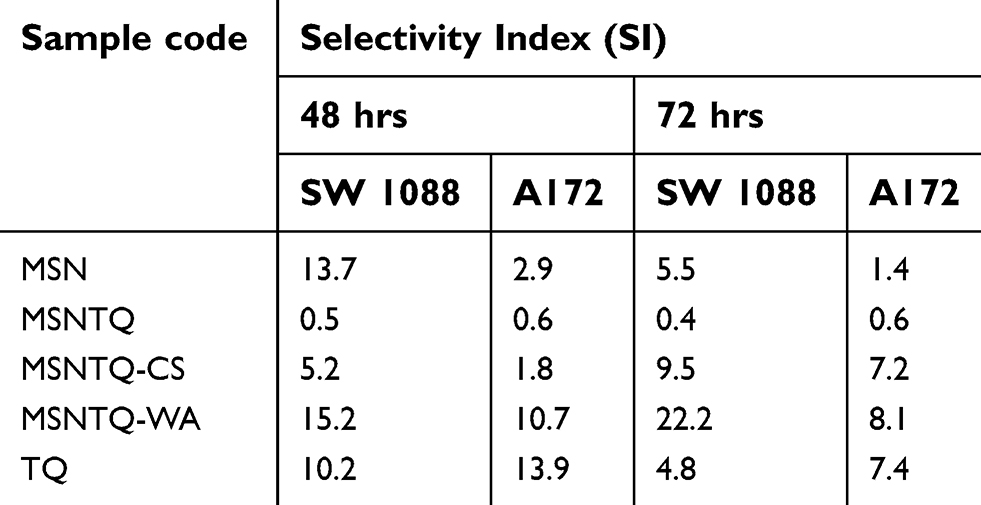 |
Table 3 The calculated selectivity index for all samples |
In vitro anticancer activity of fabricated nanoformulations for brain cancer cells
The anticancer activity was evaluated for nanoformulations and TQ by means of cell viability with the MTT assay. Cancer and normal cells were subjected to different concentrations at two incubation periods of 48 and 72 hrs (Figure 9). The data demonstrated that viability depended on cell line, TQ form, and time.
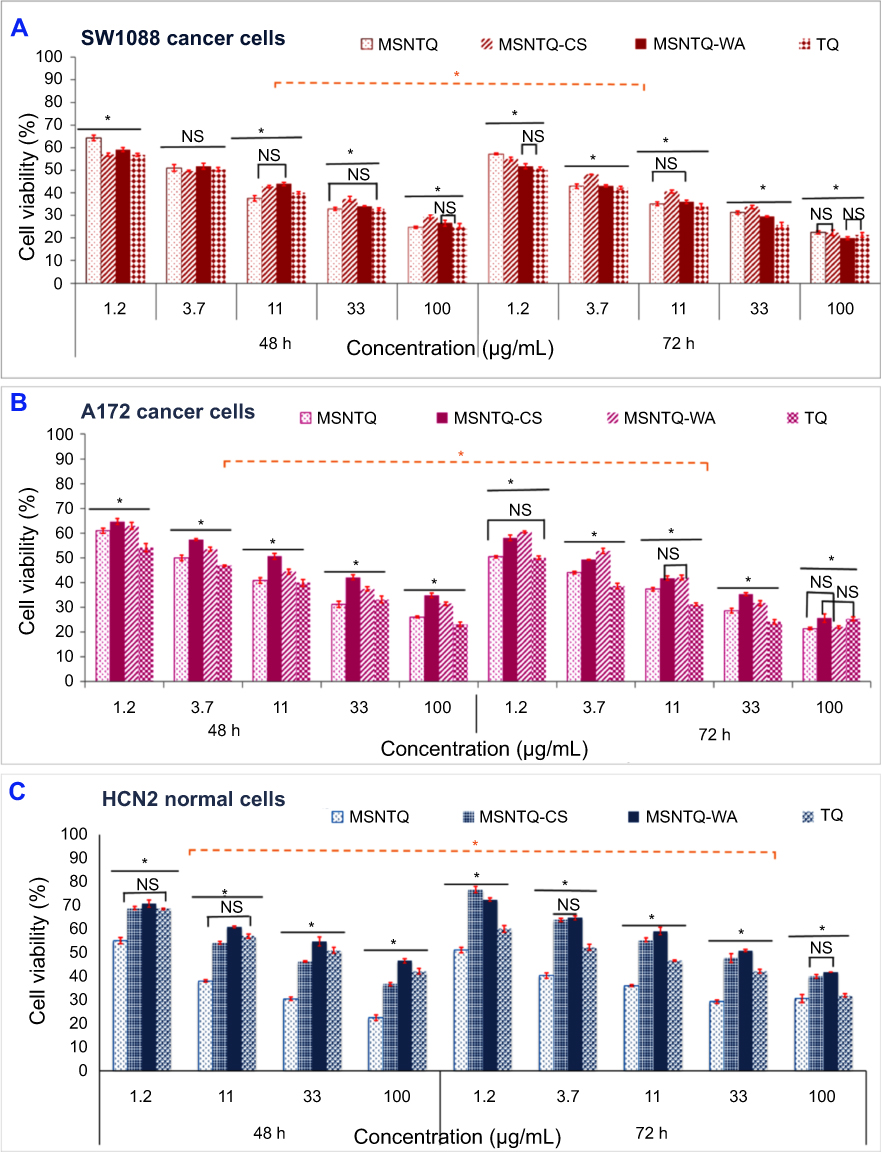 |
Figure 9 In vitro cytotoxicity of core nanoformulation (MSNTQ) and core-shell nanoformulations (MSNTQ-CS and MSNTQ-WA), and TQ in free form on brain cancer cells (SW1088 and A172), and normal brain cells (HCN2) after 48 and 72 hrs of incubation with cells. Notes: Cytotoxicity on SW1088 cancer cells with different concentrations from 1.2 to 100 µg/mL (A). Cytotoxicity on A172 cancer cells with different concentrations from 1.2 to 100 µg/mL (B). Cytotoxicity on HCN2 normal cells with different concentrations from 1.2 to 100 µg/mL (C). All data are expressed as mean ± SD. The differences are labeled with * (between the samples) at p<0.05 based on the least significant difference (LSD values). The orange line indicated the significant differences between incubation times. Non-significant differences are marked as NS, it indicated by line linked the NS (for two samples). For nanoformulations, the concentration was calculated as an equivalent amount of TQ in MSNTQ, MSNTQ-CS, and MSNTQ-WA. Abbreviations: MSNTQ, MSNs loaded with thymoquinone; MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic; TQ, thymoquinone; SW1088, human astrocytoma brain cancer cells; A172, human glioma cells; HCN2, human cortical neuronal cells-2 employed as normal cells; SD, standard deviation. |
As shown in Figure 9A for SW1088 cancer cells, a significant difference (p<0.05) was observed when incubation time was considered. After 48 hrs, low cell viability was detected when cells were treated with 100 µg/mL and reached 24.7±0.4% for MSNTQ, 25.3±1.0% for TQ, 26.5±1.3% for MSNTQ-WA, and 29.2±0.9% for MSNTQ-CS. At 72 hrs, slight changes were observed, with the following reduction percentages: MSNTQ-WA (19.9±0.9%), TQ (21.3±1.0%), MSNTQ-CS (22.3±1.2%), and MSNTQ (22.4±0.7%).
As indicated in Figure 9B, for A172 cancer cells, a significant difference (p<0.05) was found between incubation times. After 48 hrs, the lowest viability values were found when cells were treated with 100 µg/mL, ranked as follows: 23.1±0.9% (TQ), 26.1±0.3% (MSNTQ), 31.4±0.7% (MSNTQ-WA), and 34.8±1.0% (MSNTQ-CS). On further incubation to 72 hrs, some change was observed, with lower viabilities when cells were treated with 100 µg/mL, ranked as follows: MSNTQ < MSNTQ-WA < TQ < MSNTQ-CS. These results indicated that MSNTQ is more toxic on A172 cells than is TQ or other nanoformulations.
Interestingly, treating HCN2 normal cells with the present nanoformulations and TQ resulted in a less toxic effect on these cells compared to cancer cells (Figure 9C). After 48 hrs, the minimum cell viability values were recorded with 100 µg/mL as follows: MSNTQ < MSNTQ-CS < TQ < MSNTQ-WA. Also, MSNTQ significantly decreased viability as compared to other samples. Further incubation for 72 hrs resulted in less toxic effect, and MSNTQ significantly decreased the cell viability compared to other samples used.
For a better understanding of the above complex effects, we calculated the IC50 values (Table 2). IC50 calculations also confirmed that the anticancer effect in terms of cell viability depends on the cell line, TQ form, and time. For cancer cells after 48 and 72 hrs, TQ had a little more anticancer activity compared to other nanoformulations. We did not expect such an effect after 72 hrs; this action is strongly related to the kinetic release properties of TQ from nanoformulations. In this context, only about 80–85% of TQ from core-shell nanoformulations was released into PBS media after about 72 hrs as a result of its polymeric shell coating.
In contrast to cancer cell lines, a higher toxic effect was obtained when HCN2 normal cells were treated with MSN-TQN, followed by TQ, then MSNTQ-CS, and MSNTQ-WA. This pattern was seen at both incubation times, indicating that MSNTQ-WA and MSNTQ-CS showed better compatibility with normal cells than did TQ with long-time incubation at 72 hrs, which is important.
With these significant differences concerning toxicity, we calculated the SI (Table 3). The SI calculation verified that MSNTQ-WA showed better selectivity for cancer than for normal cells compared to TQ and other nanoformulations. Furthermore, combining the information from IC50 and SI demonstrated that MSNTQ-WA had a more efficient selectivity effect (22.15) and IC50 value (1.57 µg/mL) for SW1088 cancer cells than A172 cancer cells. Showing the importance of the balance between toxicity and selectivity is important. Thus, it seems that core-shell nanoformulations are required for targeted killing of brain cancer cells.
Few studies have demonstrated such an effect for other cancer cells for TQ-nanoparticles made from polymeric materials.72,73 The TQ-nanoformulations designed here are promising not only in improving TQ therapeutic efficiency but also in producing a targeted anticancer effect.
Nanoformulations improve caspase-3 activation in brain cancer cells
As Figure 10A shows, caspase activity (in fold change, FLD) significantly increased (p<0.05) with increasing time from 48 to 72 hrs. Activity significantly varied among the three cell lines. After 48 hrs of treatment, a remarkable activation was detected for A172 compared to SW1088. Caspase-3 activity expression was ranked in the following order: MSNTQ > MSNTQ-WA > MSNTQ-CS > TQ > MSN. In contrast, for HCN2 normal cells, as expected, caspase-3 activity was lower; however, there still were significant differences among the nanoformulations, pure TQ, and MSNs alone. Most important, after 72 hrs, MSNTQ-WA displayed an effectively enhanced activity in cancer cells over normal cells compared to other nanoformulations and TQ or MSN. When cells treated with MSNTQ-WA, caspase activation reached 7.13±0.05 FLD (SW1088) and 9.4±0.2 FLD (A172), compared to HCN2 with 1.35±0.01 FLD.
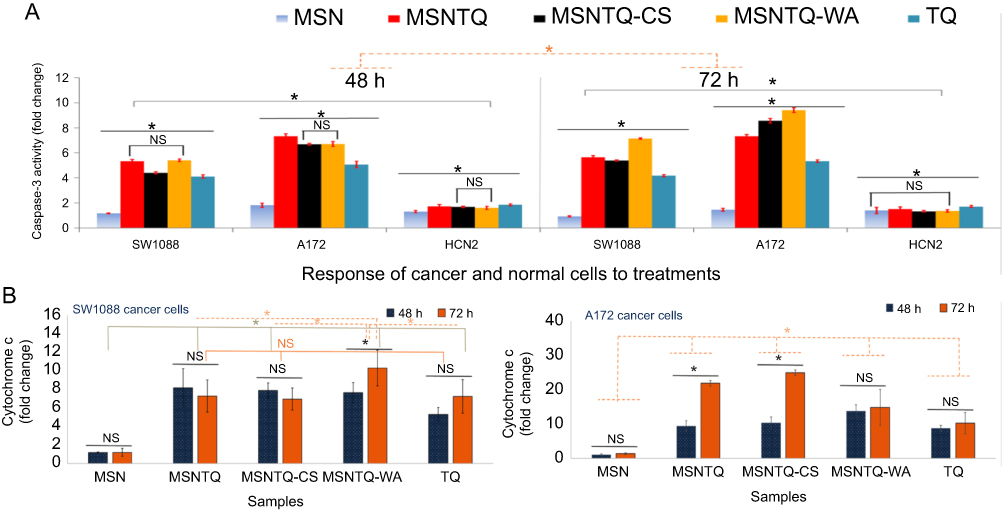 |
Figure 10 Molecular mechanism of targets of core nanoformulation (MSNTQ), and core-shell nanoformulations (MSNTQ-CS and MSNTQ-WA), and TQ in free form on brain cancer cells (SW1088 and A172), and normal brain cells (HCN2) after 48 and 72 hrs of incubation with cells. Notes: caspase-3 activation in fold change measured by ELISA for all samples after 48 and 72 hrs of incubation with SW1088, A172, and HCN2 at IC50 concentration for each sample (A). Cytochrome c intracellular release in fold change measured by RT-PCR for all samples after 48and 72 hrs of incubation with SW1088, A172, and HCN2 at IC50 concentration for each sample (B). All data are expressed as mean ± standard deviation. The differences are labeled with * (between the samples or time effect) at p<0.05 based on the least significant difference (LSD values). Non-significant differences marked as NS (between the samples or time effect). In case of caspase-3: solid-gray line indicates the significance between cell lines; the dashed-orange line indicates differences between incubation times. In case of cytochrome c: solid-orange line indicates NS between some linked samples together, the solid-olive line indicates significant differences between some linked samples together, and the dashed-orange line indicates the differences between some samples. The cells treated with IC50 concentrations of TQ, MSNTQ, MSNTQ-CS, and MSNTQ-WA. A significant difference was obtained between cell lines (SW1088 and A172, with LSD of 0.655 regarding the obtained mean values of the two groups). Abbreviations: MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic; TQ, thymoquinone; SW1088, human astrocytoma brain cancer cells; A172, human glioma cells; HCN2, human cortical neuronal cells-2 employed as normal cells; ELISA, enzyme-linked immunosorbent assays; IC50, the half maximal inhibitory concentration; SD, standard deviation. |
As expected, nanoformulation triggered caspase-3 compared to TQ, indicating that they not only inhibit proliferation of cancer cells but also promote caspase-3 activation and subsequent efficient cell death. The ability of prodrug-nanoformulations to induce caspase-3 activation better than free prodrug was reported by AbouAitah et al, reporting on curcumin-loaded MSNs on cancer cells.74 Also, Choi et al, pointed out that co-delivery of celastrol loaded in MSNs and axitinib in PEGylated lipidic bilayers increase caspsase-3 and induce apoptosis in tumor xenograft models.75 To the best of our knowledge, no data have been published on caspase-3 activation by TQ nanoformulations in brain cancer.
Nanoformulations improve intracellular release of cytochrome c in brain cancer cells
Exposure of SW1088 cells to different samples resulted in significant differences of cytochrome c (in FLD) for nanoformulations compared to MSN and TQ after various periods of incubation (Figure 10B). Statistical analysis data indicate that there was a significant (p<0.05) difference between samples, incubation times, and cell lines. For SW1088 cells, MSNTQ-WA showed a significant effect between 48 and 72 hrs compared to others. Significant differences were observed among the most of samples (except: MSNTQ, MSNTQ-CS, and TQ). Importantly, MSNTQ-WA significantly induced maximum production of cytochrome c in SW1088 cells to 10.2 FLD ±1.9 FLD after 72 hrs compared to other nanoformulations and TQ. For A172 cells, it was observed that there was a significant difference between both times for MSNTQ and MSNTQ-CS, while no significant was obtained for MSN, MSNTQ-WA, and TQ. After 48 or 72 hrs, nanoformulations significantly increased cytochrome c compared to TQ and MSN, in the following order: MSNTQ-CS > MSNTQ > MSNTQ-WA, where the maximum cytochrome c level obtained was 24.9 FLD ±0.8 FLD with MSNTQ-CS after 72 hrs. The statistical analysis also demonstrated that the cytochrome c release significantly increased in A172 cells compared to SW1088 cells.
The results confirm that cytochrome c release into cancer cells depends on cell line, TQ delivery method, and time. MSNTQ-WA induced a higher release of cytochrome c in SW1088 cells, whereas MSNTQ-CS induced a higher effect in A172 cells. They both led to increased cytochrome c compared to TQ even though they showed a slightly higher anticancer effect. This result further supports the need to develop DDSs to deliver natural prodrugs. Additionally, such enhancement observed for nanoformulations might arise from a synergistic effect because cytochrome c was higher than for MSN and TQ alone.
TQ in its free form was previously reported to induce cytochrome c release in different cancer types, in particular in cytoplasm, highlighting its essential role in apoptosis cell death.76,77 However, we could find no data on brain tumor cells regarding SW1088 or A172 cells. Thus, our results provide new confirmation that TQ increases cytochrome c level in SW1088 cancer cells, especially when administered in the form of nanoformulations. Because the release of cytochrome c is likely to inhibit cancer via an apoptosis pathway, these nanoformulations are promising as anticancer nanoformulations.
Nanoformulations improve cell cycle arrest at the G2/M phase in brain cancer cells
The growth process of cells involves subsequent phases of the cell cycle. Therefore, we investigated the cell cycle distribution after treating the SW1088 cancer cells with TQ and nanoformulations. This evaluation allowed us to explore the mechanism by which nanoformulations and TQ exert their toxic effect on cancer cells. We examined selected MSNTQ-WA, MSNTQ-CS, and TQ to assess cell cycle distribution for SW1088. Cells were exposed for 72 hrs to the concentration (µg/mL) corresponding to their respective IC50 values for TQ or an equivalent amount in the formulations and compared to controls (cells without any treatments, PBS only). The results are illustrated in Figure 11A–D. At the G0/G1 phase, the MSNTQ-CS had 61.8% of the cell distributions, similar to controls. Greater changes followed treatment with MSNTQ-WA and TQ, which reduced the cell cycle at G0/G1 phase to 55.3% and 55.8%, respectively. At S phase, following all treatments, cell division was inhibited in comparison to controls in the order MSNTQ-CS > MSNTQ-WA > TQ. For cell distribution at G0/G1 and S phases, cell populations were highly accumulated at G2/M. Increasing cell cycle distribution at G2/M ranked as follows compared to controls (4.6%): MSNTQ-WA (12.8%) > TQ (11.8%) > MSNTQ-CS (9.9%). This pattern indicated the promotion of SW1088 cancer cell arrest at the G2/M phase.
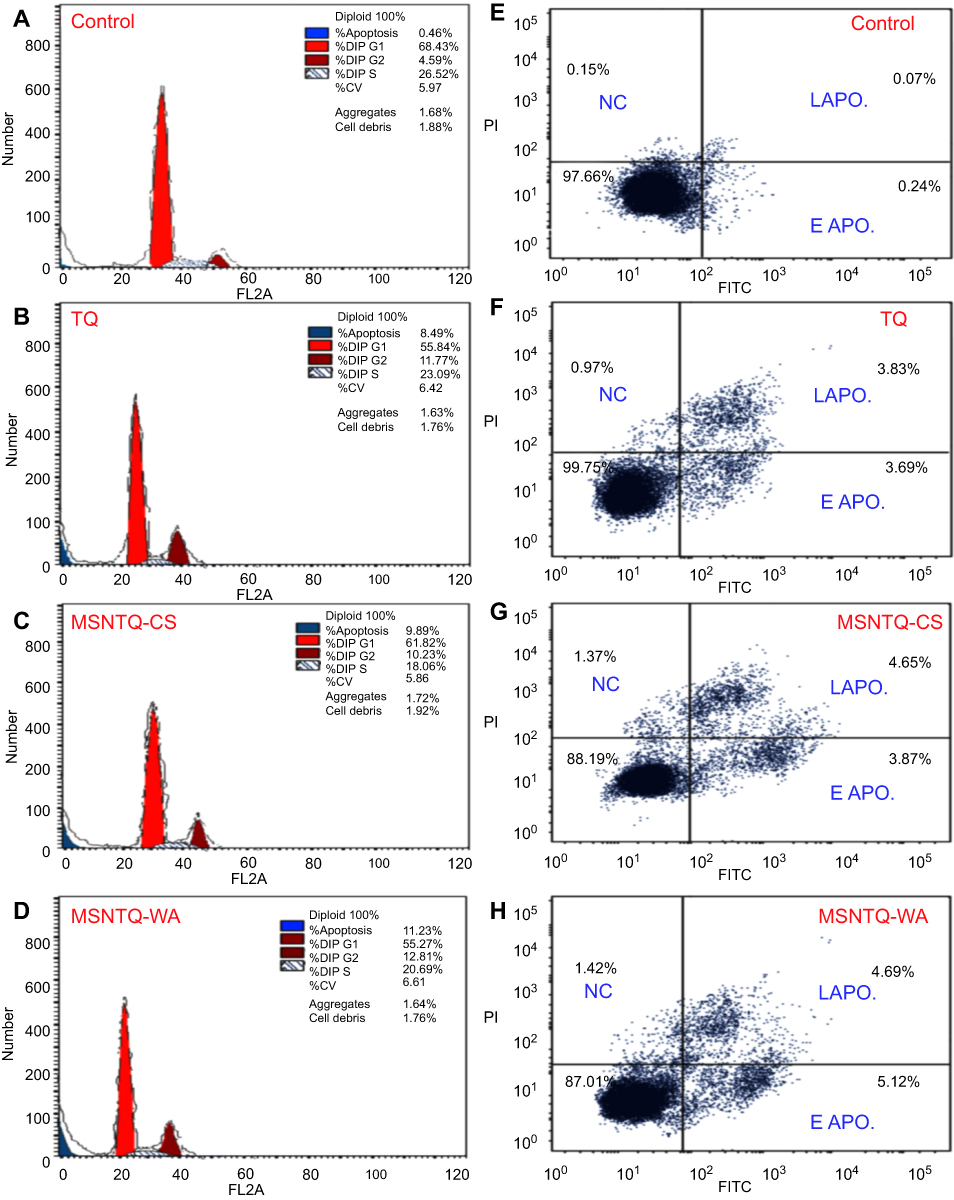 |
Figure 11 The cell cycle and apoptosis analysis by flow cytometry measurements after treating SW1088 cancer cells with core-shell nanoformulations and free TQ for 72 hrs compared without any treatments (control, only SW1088 cells). Notes: Cell cycle arrest evaluations in SW1088 investigated by means of by propidium iodide staining (A–D). Apoptosis evaluation in SW1088 cells was done through Annexin-V/FITC staining (E–H). The cells treated with IC50 concentrations. Abbreviations: MSNTQ, MSNs loaded with TQ as core; MSNTQ-CS, MSNTQ coated with the shell consists of chitosan and stearic acid; MSNTQ-WA, MSNTQ coated with the shell consists of whey protein and gum Arabic; TQ, thymoquinone; SW1088, human astrocytoma brain cancer cells; A172, human glioma cells; HCN2, human cortical neuronal cells-2 employed as normal cells; IC50, the half maximal inhibitory concentration; EAPO, early apoptosis; LAPO, late apoptosis; NC, necrosis; SD, standard deviation. |
Several studies have demonstrated the interference of TQ on cell cycle at different phases in different types of cancer, where it can arrest cell cycle at G1 and 2/M phases. Such differences could be attributed to the cancer cell line and to various up or down-regulating molecular signaling pathways. For instance, TQ arrests the G1 phase in HCT 116 human colorectal cancer cells78 and at G2/M for human cholangiocarcinomas.79 Collectively, TQ can alter the expression of multiple proteins that are responsible for cell cycle progression such as p21 and p53, resulting in cell cycle arrest at different phases.9 Furthermore, TQ was suggested to be a useful anticancer agent for brain tumors because it induces DNA damage, which is attributed to cell cycle arrest, and inhibits cell death in glioblastoma cells.16 Thus, we expect that MSNTQ-WA would enhance DNA damage in SW1088 cells, finally leading to the induction of G2/M cell cycle arrest followed by apoptosis.
Nanoformulations improve apoptosis induction in brain cancer cells
To further identify the apoptotic (late apoptosis: LAPO, and early apoptosis: EAPO) or necrotic effects (necrosis: NC), we performed the annexin V-FITC/PI assay and quantified fluorescence by flow cytometry (Figure 11E–H). The apoptotic effect appeared to increase following treatment with TQ and its nanoformulations compared to control. The effects on apoptosis were in this order: MSNTQ-WA (11.2%) > MSNTQ-CS (9.9%) > TQ (8.9%). Distribution of apoptosis varied among the tested samples, where a high late apoptosis stage was found for TQ and MSNTQ-CS, whereas MSTQ-WA induced early apoptosis. By combining various apoptosis results, we note that MSNTQ-WA was more likely to modulate the apoptotic effect in SW1088 cancer cells.
In fact, TQ is a multiple-targeting anticancer agent through its ability to interfere with various singling pathways in a wide range of glioblastoma brain cancers.21 Its apoptosis activation can be through manipulation of p53-dependent and p53-independent mechanisms.14,80 TQ also strongly modulates regulatory proteins by increasing pro-apoptotic and reducing anti-apoptotic proteins responsible for apoptosis. For instance, TQ shows direct activation of caspase-3 and subsequent cleavage of poly(ADP-ribose) polymerase, as well as upregulation of Bax and downregulation of anti-apoptotic Bcl-2, leading to an increased Bax/Bcl-2 ratio via downregulation of Bcl-2.81 Moreover, TQ increases caspase-3 and cytochrome c in cytoplasm, along with reduction of Bcl-xl and surviving proteins in breast cancer cells.82 In addition, it can activate tumor suppressor genes in cancer that effectively lead to apoptosis.10 The proposed full anticancer mechanisms in our study are presented in Figure 1B, indicating the programmed cell death of brain cancer cells after treatments with nanoformulations. Thus, the present findings are consistent with apoptosis caused by TQ activity when the prodrug TQ was delivered by targeting cancer cells using core-shell nanoformulations. Table 4 shows a schematic of the results of the present study.
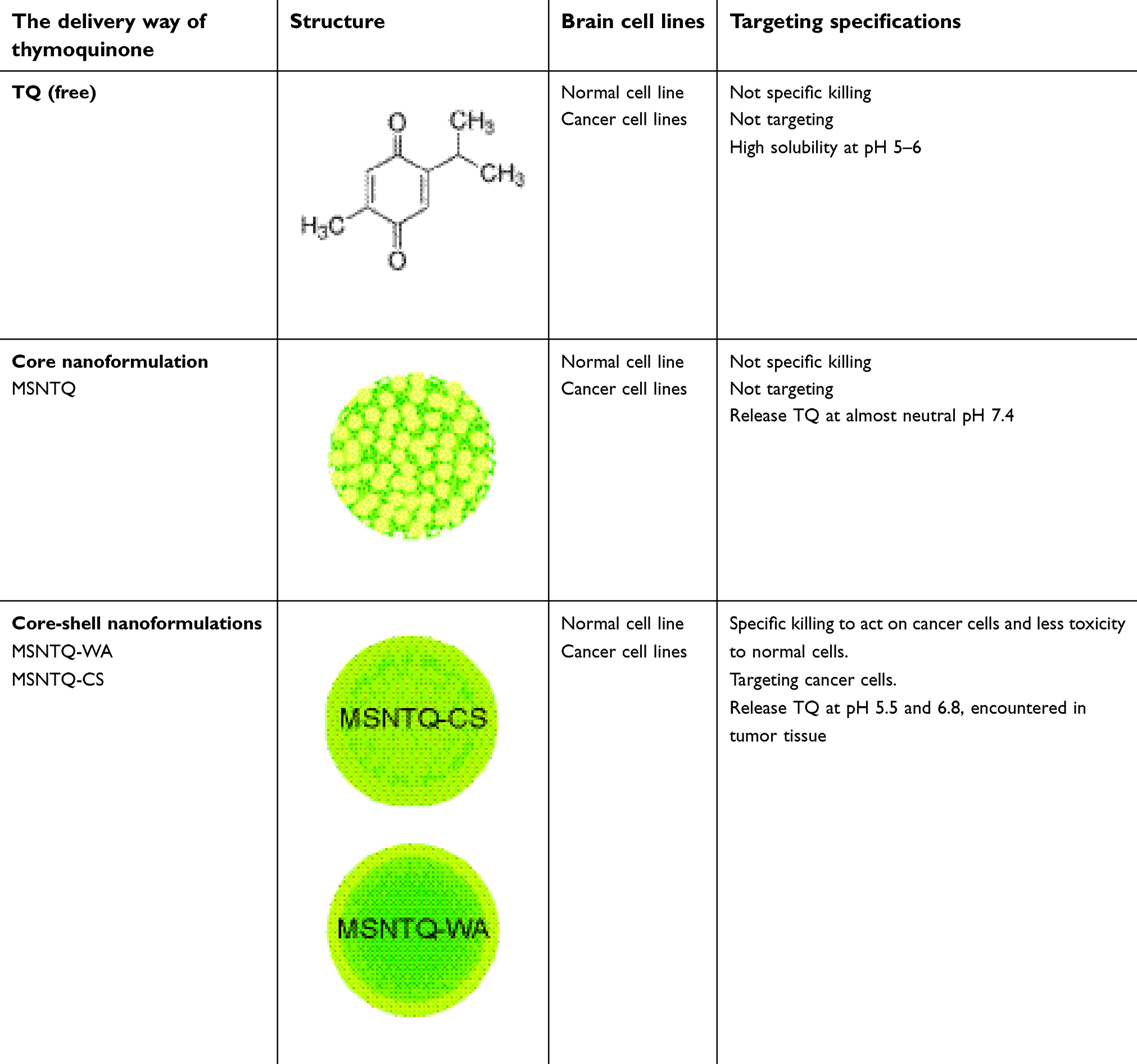 |
Table 4 Summary of the obtained results for delivery methods of thymoquinone from anticancer and kinetic release studies |
In summary, the main findings of the present study in terms of cytotoxicity and molecular mechanisms are that the MSNTQ-WA and MSNTQ-CS core-shell drug delivery nanoformulations show a high ability to target glioma brain cancer cell lines. They deliver TQ to cancer cells preferentially while delivering less TQ to normal cells. Three reasons can converge to explain the high targeting ability of prepared core-shell DDS developed here:
- The core-shell nanoformulations efficiently release TQ into an acidic environment. Acidic pH is characteristic of the tumor environment. At neutral pH, corresponding to healthy normal cells, release is less efficient. In keeping with this pattern, the core nanoformulation MSNTQ displayed more cytotoxic effects on HCN2 normal cells compared to cancer cells because in that case, release at a neutral pH was already extensive. Such particles are not optimal for targeting cancer cells, where the environment is acidic. Therefore, the pH-dependent release effect may improve selective and targeting of cancer cells.
- The shell layer hinders TQ release. Without a shell layer, TQ release is mainly controlled by pH level because the TQ solubility significantly increases with decreasing pH, and TQ at the nanoparticle surface has direct contact with the surrounding fluid. Therefore, for MSNTQ particles, extensive release already takes place at neutral pH, so that such particles are not optimal for targeting cancer cells. For nanoformulations with shell layers, TQ release becomes more complex and depends on the pH effect on the polymer shell. There is an ionic interaction between component polymers used for each shell at neutral or alkaline pH, leading to tightening of the shell. In contrast, there is a lack of ionic interaction between the two component polymers of each shell design in an acidic pH environment. Thus, for a longer test time of 72 hrs, a high release amount for MSNTQ-WA and MSNTQ-CS was observed at pH 5.5 and 6.8, but a slow release at pH 7.4. Hence, TQ release from the core-shell nanoformulations is favored in an acidic environment.
- Core-shell nanoformulations can target cancer cells. Regarding the CS coating, the present results are in line with a study by Ting et al,83 who reported stearic acid–grafted chitosan micelle nanoparticles with enhanced uptake by brain cancer cells. Endocytosis was the main process of internalization into cells. During an in vivo test, these particles rapidly crossed the BBB. Their study showed the ability of CS-coated nanoparticles to target brain cancer. Rapid cellular uptake enabled efficient delivery of doxorubicin (DOX), an anticancer drug, to the cells. Regarding the WA shell, presumably, whey protein probably facilitates adherence of core-shell nanoparticles to the cancer cell surface and their subsequent internalization. Whey protein has often been used as nutraceutical and pharmaceutical carrier. For example, b-Lactoglobulin protein is the main protein in whey protein concentrate, and it was formed as nanoparticles (98–192 nm) for drug delivery carrier by Ha et al.84 They found that the nanoparticles are not toxic on Caco-2 cells (human epithelial colorectal adenocarcinoma cells) but that their cellular uptake in cells increased and varied based on particle size. Such a high cellular uptake might result from receptor-mediated endocytosis in brain cells that allows molecular targeting. Also, whey protein plays a crucial role in anticancer effects; for instance, it proves an effective cysteine donor to glutathione when depleted in an immune deficiency case.85 Glutathione has an antioxidant effect against cancer cells that show resistance to oxidative stress.86
Core-shell nanoformulations, especially MSNTQ-WA, may target brain cancer cells because they include different proteins, amino acids, and growth factors of whey protein, which is needed in high amounts for cancer cell growth compared healthy normal cells. The present results may open the door to further study and development of DDSs using whey proteins as a new targeting ligand option to the currently used targeting molecules (ie, sugars, acid, antibodies, and others) for targeted anticancer DDSs. Use of novel chemistry of polymers-DDSs could lead to the development of next generations of nanomedicines for cancer therapy.87
As far as the mechanism of cancer cell killing, core-shell nanoformulations (especially MSNTQ-WA) show significantly enhanced molecular targeting of brain cancer cells through caspase-3 activation, cytochrome c triggers, cell cycle arrest at G2/M phase, and apoptosis induction comparing to application of free TQ. The mechanism of action is similar to that of TQ but is much more efficient due to targeting effect. Our study shows that the prepared optimized drug formulations not only exhibit a toxic effect on brain cells, but this effect is strongly targeted, and normal brain cells are less toxic affected. We note that these results concerning the anticancer potential of proposed core-shell nanoformulations are preliminary. Therefore, further investigations are required to understand the full mechanisms underlying brain cancer targeting.
Conclusions
We have successfully developed novel core-shell nanoformulations for targeted delivery of natural anticancer thymoquinone (TQ) against glioma cancer cells. The nanoformulation consists of MSNs loaded with TQ and with a shell made of two kinds of polymers. The prepared nanoformulations not only exhibit a highly toxic effect on brain cells but also this effect is strongly targeted, and normal brain cells are less affected.
Changing pH controls the kinetics of drug release. The core-shell nanoformulations showed favorable TQ slow release, and a higher release was detected at acidic pH (5.5 and 6.8) compared to pH 7.4. Especially MSNTQ-WA, with the shell layer made of whey protein and gum Arabic, had more toxic effects on both brain cancer cell lines (A172 and SW1088) compared to normal cells (NCN2). This finding is a favorable characteristic of the core-shell structures because enhanced TQ release is expected in cancer tissues. The nanoformulations displayed enhanced molecular targeting of cancer cells when compared to free TQ. The targeting of cancer cells could be attributed to the shell materials. The anticancer effect mechanism was similar to that expected for the general molecular mechanism of action of TQ, eg, apoptosis induction (caspase-3, apoptosis, intracellular release of cytochrome c) and cell cycle arrest at G2/M phase, but significantly improved compared to free TQ. These drug delivery designs are considered the first of their type presenting an in vitro–optimized TQ formulation. These findings are promising for further in vivo evaluations and developments.
Acknowledgments
We would like to thank the National Research Centre (NRC, Egypt) for supporting this work by means of providing use of several devices and materials available from internal funding to prepare the nanoformulations. Many thanks to the National Center for Research and Development, Poland (STRATEGMED3/306888/3/NCBR/2017, project, Method of treatment of large bone defects in oncological patients using in vivo tissue engineering approach (iTE), Poland). This research was carried out using equipment funded by the Center for Preclinical Research and Technology (CePT) project, reference: POIG.02.02.00-14-024/08, financed by the European Regional Development Fund within the Operational Programme “Innovative Economy” for 2007–2013. K AbouAitah would like to thank Adam Presz from Laboratory of Nanostructures, Institute of High Pressure Physics, Polish Academy of Sciences, for FE-SEM images. The authors thank Kelly A Keating from the Pharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, for editing and formatting our manuscript. The authors also thank professor Abdallah MA Abdullah, Agronomy Department, Faculty of Agriculture, Cairo University, for the statistical analysis of data in our manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ostrom QT, Gittleman H, Farah P, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro-Oncology. 2013;15(Suppl 2):ii1–ii56. doi:10.1093/neuonc/not151
2. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. doi:10.1056/NEJMra0708126
3. Arko L, Katsyv I, Park GE, Luan WP, Park JK. Experimental approaches for the treatment of malignant gliomas. Pharmacol Ther. 2010;128(1):1–36. doi:10.1016/j.pharmthera.2010.04.015
4. Stupp R, Mason WP, van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi:10.1056/NEJMoa043330
5. Hou LC, Veeravagu A, Hsu AR, Tse VC. Recurrent glioblastoma multiforme: a review of natural history and management options. Neurosurg Focus. 2006;20(4):E5. doi:10.3171/foc.2006.20.4.2
6. Kim SS, Harford JB, Pirollo KF, Chang EH. Effective treatment of glioblastoma requires crossing the blood-brain barrier and targeting tumors including cancer stem cells: the promise of nanomedicine. Biochem Biophys Res Commun. 2015;468(3):485–489. doi:10.1016/j.bbrc.2015.06.137
7. Nicholas MK, Lukas R, Bangalore S. Chemotherapy for malignant glioma. Expert Rev Neurother. 2005;5(6 Suppl):S41–S49. doi:10.1586/14737175.5.6.S41
8. Perry J, Laperriere N, Zuraw L, Chambers A, Spithoff K, Cairncross JG. Adjuvant chemotherapy for adults with malignant glioma: a systematic review. Can J Neurol Sci. 2007;34(4):402–410.
9. Schneider-Stock R, Fakhoury IH, Zaki AM, El-Baba CO, Gali-Muhtasib HU. Thymoquinone: fifty years of success in the battle against cancer models. Drug Discov Today. 2014;19(1):18–30. doi:10.1016/j.drudis.2013.08.021
10. Goyal SN, Prajapati CP, Gore PR, et al. Therapeutic potential and pharmaceutical development of thymoquinone: a multitargeted molecule of natural origin. Front Pharmacol. 2017;8:656. doi:10.3389/fphar.2017.00656
11. Gali-Muhtasib H, Ocker M, Kuester D, et al. Thymoquinone reduces mouse colon tumor cell invasion and inhibits tumor growth in murine colon cancer models. J Cell Mol Med. 2008;12(1):330–342. doi:10.1111/j.1582-4934.2007.00095.x
12. Jafri SH, Glass J, Shi R, Zhang S, Prince M, Kleiner-Hancock H. Thymoquinone and cisplatin as a therapeutic combination in lung cancer: in vitro and in vivo. J Exp Clin Cancer Res. 2010;29:87. doi:10.1186/1756-9966-29-87
13. El-Mahdy MA, Zhu Q, Wang QE, Wani G, Wani AA. Thymoquinone induces apoptosis through activation of caspase-8 and mitochondrial events in p53-null myeloblastic leukemia HL-60 cells. Int J Cancer. 2005;117(3):409–417. doi:10.1002/ijc.21205
14. Arafa El SA, Zhu Q, Shah ZI, et al. Thymoquinone up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat Res. 2011;706(1–2):28–35. doi:10.1016/j.mrfmmm.2010.10.007
15. Asaduzzaman Khan M, Tania M, Fu S, Fu J. Thymoquinone, as an anticancer molecule: from basic research to clinical investigation. Oncotarget. 2017;8(31):51907–51919. doi:10.18632/oncotarget.17206
16. Gurung RL, Lim SN, Khaw AK, et al. Thymoquinone induces telomere shortening, DNA damage and apoptosis in human glioblastoma cells. PLoS One. 2010;5(8):e12124. doi:10.1371/journal.pone.0012124
17. Kolli-Bouhafs K, Boukhari A, Abusnina A, et al. Thymoquinone reduces migration and invasion of human glioblastoma cells associated with FAK, MMP-2 and MMP-9 down-regulation. Invest New Drugs. 2012;30(6):2121–2131. doi:10.1007/s10637-011-9777-3
18. Racoma IO, Meisen WH, Wang QE, Kaur B, Wani AA. Thymoquinone inhibits autophagy and induces cathepsin-mediated, caspase-independent cell death in glioblastoma cells. PLoS One. 2013;8(9):e72882. doi:10.1371/journal.pone.0072882
19. Pazhouhi M, Sariri R, Rabzia A, Khazaei M. Thymoquinone synergistically potentiates temozolomide cytotoxicity through the inhibition of autophagy in U87MG cell line. Iran J Basic Med Sci. 2016;19(8):890–898.
20. Elmaci I, Altinoz MA. Thymoquinone: an edible redox-active quinone for the pharmacotherapy of neurodegenerative conditions and glial brain tumors. A short review. Biomed Pharmacother. 2016;83:635–640. doi:10.1016/j.biopha.2016.07.018
21. Chowdhury FA, Hossain MK, Mostofa AGM, Akbor MM, Bin Sayeed MS. Therapeutic potential of thymoquinone in glioblastoma treatment: targeting major gliomagenesis signaling pathways. Biomed Res Int. 2018;2018:4010629. doi:10.1155/2018/4010629
22. Jakaria M, Cho D-Y, Ezazul Haque M, et al. Neuropharmacological potential and delivery prospects of thymoquinone for neurological disorders. Oxid Med Cell Longev. 2018;2018:17. doi:10.1155/2018/1209801
23. Banerjee I, De K, Mukherjee D, et al. Paclitaxel-loaded solid lipid nanoparticles modified with Tyr-3-octreotide for enhanced anti-angiogenic and anti-glioma therapy. Acta Biomater. 2016;38:69–81. doi:10.1016/j.actbio.2016.04.026
24. Orunoğlu M, Kaffashi A, Pehlivan SB, et al. Effects of curcumin-loaded PLGA nanoparticles on the RG2 rat glioma model. Mater Sci Eng C. 2017;78:32–38. doi:10.1016/j.msec.2017.03.292
25. Lee ES, Gao Z, Bae YH. Recent progress in tumor pH targeting nanotechnology. J Controlled Release. 2008;132(3):164–170. doi:10.1016/j.jconrel.2008.05.003
26. Liu J, Huang Y, Kumar A, et al. pH-Sensitive nano-systems for drug delivery in cancer therapy. Biotechnol Adv. 2014;32(4):693–710. doi:10.1016/j.biotechadv.2013.11.009
27. Wirries A, Breyer S, Quint K, Schobert R, Ocker M. Thymoquinone hydrazone derivatives cause cell cycle arrest in p53-competent colorectal cancer cells. Exp Ther Med. 2010;1(2):369–375. doi:10.3892/etm_00000058
28. Odeh F, Ismail SI, Abu-Dahab R, Mahmoud IS, Al Bawab A. Thymoquinone in liposomes: a study of loading efficiency and biological activity towards breast cancer. Drug Deliv. 2012;19(8):371–377. doi:10.3109/10717544.2012.727500
29. Ng WK, Saiful Yazan L, Yap LH, Wan Nor Hafiza WAG, How CW, Abdullah R. Thymoquinone-loaded nanostructured lipid carrier exhibited cytotoxicity towards breast cancer cell lines (MDA-MB-231 and MCF-7) and cervical cancer cell lines (HeLa and SiHa). Biomed Res Int. 2015;2015:10. doi:10.1155/2015/263131
30. Dehghani H, Hashemi M, Entezari M, Mohsenifar A. The comparison of anticancer activity of thymoquinone and nanothymoquinone on human breast adenocarcinoma. IJPR. 2015;14(2):539–546.
31. Vallet-Regi M, Rámila A, Del Real RP, Pérez-Pariente J. A new property of MCM-41: drug delivery system. Chem Mater. 2001;13(2):308–311. doi:10.1021/cm0011559
32. Kwon S, Singh RK, Perez RA, Abou Neel EA, Kim H-W, Chrzanowski W. Silica-based mesoporous nanoparticles for controlled drug delivery. J Tissue Eng. 2013;4:2041731413503357. doi:10.1177/2041731413503357
33. Wang Y, Zhao Q, Han N, et al. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomedicine. 2015;11(2):313–327. doi:10.1016/j.nano.2014.09.014
34. Watermann A, Brieger J. Mesoporous silica nanoparticles as drug delivery vehicles in cancer. Nanomaterials (Basel, Switzerland). 2017;7(7). doi:10.3390/nano7120458
35. Baghirov H, Karaman D, Viitala T, et al. Feasibility study of the permeability and uptake of mesoporous silica nanoparticles across the blood-brain barrier. PLoS One. 2016;11(8):e0160705. doi:10.1371/journal.pone.0160705
36. Tamba BI, Streinu V, Foltea G, et al. Tailored surface silica nanoparticles for blood-brain barrier penetration: preparation and in vivo investigation. Arab J Chem. 2018;11(6):981–990. doi:10.1016/j.arabjc.2018.03.019
37. Shi J, Hou S, Huang J, et al. An MSN-PEG-IP drug delivery system and IL13Rα2 as targeted therapy for glioma. Nanoscale. 2017;9(26):8970–8981. doi:10.1039/c6nr08786h
38. Zhang H, Zhang W, Zhou Y, Jiang Y, Li S. Dual functional mesoporous silicon nanoparticles enhance the radiosensitivity of VPA in glioblastoma. Transl Oncol. 2017;10(2):229–240. doi:10.1016/j.tranon.2016.12.011
39. You Y, Yang L, He L, Chen T. Tailored mesoporous silica nanosystem with enhanced permeability of the blood–brain barrier to antagonize glioblastoma. J Mat Chem B. 2016;4(36):5980–5990. doi:10.1039/C6TB01329E
40. Mo J, He L, Ma B, Chen T. Tailoring particle size of mesoporous silica nanosystem to antagonize glioblastoma and overcome blood–brain barrier. ACS Appl Mater Interfaces. 2016;8(11):6811–6825. doi:10.1021/acsami.5b11730
41. Song Y, Du D, Li L, Xu J, Dutta P, Lin Y. In vitro study of receptor-mediated silica nanoparticles delivery across blood-brain barrier. ACS Appl Mater Interfaces. 2017;9(24):20410–20416. doi:10.1021/acsami.7b03504
42. Salmani JM, Asghar S, Lv H, Zhou J. Aqueous solubility and degradation kinetics of the phytochemical anticancer thymoquinone; probing the effects of solvents, pH and light. Molecules. 2014;19(5):5925–5939. doi:10.3390/molecules19055925
43. El-Toni AM, Khan A, Ibrahim MA, et al. Synthesis of double mesoporous core-shell silica spheres with tunable core porosity and their drug release and cancer cell apoptosis properties. J Colloid Interface Sci. 2012;378(1):83–92. doi:10.1016/j.jcis.2012.04.006
44. Shen D, Yang J, Li X, et al. Biphase stratification approach to three-dimensional dendritic biodegradable mesoporous silica nanospheres. Nano Lett. 2014;14(2):923–932. doi:10.1021/nl404316v
45. Hu F-Q, Zhao M-D, Yuan H, You J, Du Y-Z, Zeng S. A novel chitosan oligosaccharide–stearic acid micelles for gene delivery: properties and in vitro transfection studies. Int J Pharm. 2006;315(1):158–166. doi:10.1016/j.ijpharm.2006.02.026
46. Yuan H, Bao X, Du Y-Z, You J, Hu F-Q. Preparation and evaluation of SiO2-deposited stearic acid-g-chitosan nanoparticles for doxorubicin delivery. Int J Nanomedicine. 2012;7:5119–5128. doi:10.2147/IJN.S35575
47. Yang T-S, Liu -T-T, Lin IH. Functionalities of chitosan conjugated with stearic acid and gallic acid and application of the modified chitosan in stabilizing labile aroma compounds in an oil-in-water emulsion. Food Chem. 2017;228:541–549. doi:10.1016/j.foodchem.2017.02.035
48. Klein M, Aserin A, Ben Ishai P, Garti N. Interactions between whey protein isolate and gum Arabic. Colloids Surf B. 2010;79(2):377–383. doi:10.1016/j.colsurfb.2010.04.021
49. Franz TJ. Percutaneous absorption on the relevance of in vitro data. J Invest Dermatol. 1975;64(3):190–195.
50. Salama RO, Traini D, Chan HK, Young PM. Preparation and characterisation of controlled release co-spray dried drug-polymer microparticles for inhalation 2: evaluation of in vitro release profiling methodologies for controlled release respiratory aerosols. Eur J Pharm Biopharm. 2008;70(1):145–152. doi:10.1016/j.ejpb.2008.04.009
51. Ammar HO, Ghorab MM, Mahmoud AA, Higazy IM. Lamotrigine loaded poly-varepsilon-(d,l-lactide-co-caprolactone) nanoparticles as brain delivery system. Eur J Pharm Sci. 2018;115:77–87. doi:10.1016/j.ejps.2018.01.028
52. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63.
53. Yergeau E, Arbour M, Brousseau R, et al. Microarray and real-time PCR analyses of the responses of high-arctic soil bacteria to hydrocarbon pollution and bioremediation treatments. Appl Environ Microbiol. 2009;75(19):6258–6267. doi:10.1128/AEM.01029-09
54. Qu F, Zhu G, Huang S, et al. Controlled release of Captopril by regulating the pore size and morphology of ordered mesoporous silica. Microporous Mesoporous Mater. 2006;92(1–3):1–9. doi:10.1016/j.micromeso.2005.12.004
55. AbouAitah K, Farghali A, Swiderska-Sroda A, Lojkowski W, Razin A, Khedr M. pH-controlled release system for curcumin based on functionalized dendritic mesoporous silica nanoparticles. J Nanomed Nanotechnol. 2016;7:1. doi:10.4172/2157-7439.1000351
56. AbouAitah K, Farghali A, Swiderska-Sroda A, Lojkowski W, Razin A. Mesoporous silica materials in drug delivery system: pH/glutathione-responsive release of poorly water-soluble pro-drug quercetin from two and three-dimensional pore-structure nanoparticles. J Nanomed Nanotechnol. 2016;7:1. doi:10.4172/2157-7439.1000360
57. Sahoo B, Devi KSP, Sahu SK, et al. Facile preparation of multifunctional hollow silica nanoparticles and their cancer specific targeting effect. Biomater Sci. 2013;1(6):647–657. doi:10.1039/c3bm00007a
58. Möller K, Kobler J, Bein T. Colloidal suspensions of nanometer-sized mesoporous silica. Adv Funct Mater. 2007;17(4):605–612
59. Poudel BK, Soe ZC, Ruttala HB, et al. In situ fabrication of mesoporous silica-coated silver-gold hollow nanoshell for remotely controllable chemo-photothermal therapy via phase-change molecule as gatekeepers. Int J Pharm. 2018;548(1):92–103. doi:10.1016/j.ijpharm.2018.06.056
60. Sun Y, Sun Y-L, Wang L, Ma J, Yang Y-W, Gao H. Nanoassembles constructed from mesoporous silica nanoparticles and surface-coated multilayer polyelectrolytes for controlled drug delivery. Microporous Mesoporous Mater. 2014;185:245–253. doi:10.1016/j.micromeso.2013.11.020
61. Khan KA. The concept of dissolution efficiency. J Pharm Pharmacol. 1975;27(1):48–49.
62. Rinaki E, Dokoumetzidis A, Macheras P. The mean dissolution time depends on the dose/solubility ratio. Pharm Res. 2003;20(3):406–408.
63. Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. doi:10.1016/0378-5173(83)90064-9
64. Karasulu HY, Ertan G, Kose T. Modeling of theophylline release from different geometrical erodible tablets. Eur J Pharm Biopharm. 2000;49(2):177–182. doi:10.1016/S0939-6411(99)00082-X
65. Surekha R, Aishwarya V, Sumathi T. Thymoquinone loaded solid lipid nanoparticle: formulation, characterization and in-vitro cell viability assay. Int J Pharm Bio Sci. 2014;6:449–464.
66. Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Controlled Release. 1987;5(1):37–42. doi:10.1016/0168-3659(87)90035-6
67. Wang C, Fu X, Yang L. Water-soluble chitosan nanoparticles as a novel carrier system for protein delivery. Chin Sci Bull. 2007;52(7):883–889. doi:10.1007/s11434-007-0127-y
68. Barahuie F, Dorniani D, Saifullah B, et al. Sustained release of anticancer agent phytic acid from its chitosan-coated magnetic nanoparticles for drug-delivery system. Int J Nanomedicine. 2017;12:2361–2372. doi:10.2147/IJN.S126245
69. Williams PA, Phillips GO. 11 - Gum arabic. In: Phillips GO, Williams PA, editors. Handbook of Hydrocolloids.
70. Cornelsen PA, Quintanilha RC, Vidotti M, Gorin PA, Simas-Tosin FF, Riegel-Vidotti IC. Native and structurally modified gum arabic: exploring the effect of the gum’s microstructure in obtaining electroactive nanoparticles. Carbohydr Polym. 2015;119:35–43. doi:10.1016/j.carbpol.2014.11.020
71. Gunasekaran S, Ko S, Xiao L. Use of whey proteins for encapsulation and controlled delivery applications. J Food Eng. 2007;83(1):31–40. doi:10.1016/j.jfoodeng.2006.11.001
72. Abu-Dahab R, Odeh F, Ismail SI, Azzam H, Al Bawab A. Preparation, characterization and antiproliferative activity of thymoquinone-β-cyclodextrin self assembling nanoparticles. Die Pharm-Int J Pharm Sci. 2013;68(12):939–944.
73. Bhattacharya S, Ahir M, Patra P, et al. PEGylated-thymoquinone-nanoparticle mediated retardation of breast cancer cell migration by deregulation of cytoskeletal actin polymerization through miR-34a. Biomaterials. 2015;51:91–107. doi:10.1016/j.biomaterials.2015.01.007
74. AbouAitah K, Swiderska-Sroda A, Farghali AA, et al. Folic acid-conjugated mesoporous silica particles as nanocarriers of natural prodrugs for cancer targeting and antioxidant action. Oncotarget. 2018;9(41):26466–26490. doi:10.18632/oncotarget.25470
75. Choi JY, Ramasamy T, Kim SY, et al. PEGylated lipid bilayer-supported mesoporous silica nanoparticle composite for synergistic co-delivery of axitinib and celastrol in multi-targeted cancer therapy. Acta Biomater. 2016;39:94–105. doi:10.1016/j.actbio.2016.05.012
76. Park EJ, Chauhan AK, Min KJ, Park DC, Kwon TK. Thymoquinone induces apoptosis through downregulation of c-FLIP and Bcl-2 in renal carcinoma Caki cells. Oncol Rep. 2016;36(4):2261–2267. doi:10.3892/or.2016.5019
77. Zhang M, Du H, Huang Z, et al. Thymoquinone induces apoptosis in bladder cancer cell via endoplasmic reticulum stress-dependent mitochondrial pathway. Chem Biol Interact. 2018;292:65–75. doi:10.1016/j.cbi.2018.06.013
78. Gali-Muhtasib H, Diab-Assaf M, Boltze C, et al. Thymoquinone extracted from black seed triggers apoptotic cell death in human colorectal cancer cells via a p53-dependent mechanism. Int J Oncol. 2004;25(4):857–866.
79. Xu D, Ma Y, Zhao B, et al. Thymoquinone induces G2/M arrest, inactivates PI3K/Akt and nuclear factor-kappaB pathways in human cholangiocarcinomas both in vitro and in vivo. Oncol Rep. 2014;31(5):2063–2070. doi:10.3892/or.2014.3059
80. Alhosin M, Ibrahim A, Boukhari A, et al. Anti-neoplastic agent thymoquinone induces degradation of alpha and beta tubulin proteins in human cancer cells without affecting their level in normal human fibroblasts. Invest New Drugs. 2012;30(5):1813–1819. doi:10.1007/s10637-011-9734-1
81. Paramasivam A, Sambantham S, Shabnam J, et al. Anti-cancer effects of thymoquinone in mouse neuroblastoma (Neuro-2a) cells through caspase-3 activation with down-regulation of XIAP. Toxicol Lett. 2012;213(2):151–159. doi:10.1016/j.toxlet.2012.06.011
82. Rajput S, Kumar BN, Dey KK, Pal I, Parekh A, Mandal M. Molecular targeting of Akt by thymoquinone promotes G(1) arrest through translation inhibition of cyclin D1 and induces apoptosis in breast cancer cells. Life Sci. 2013;93(21):783–790. doi:10.1016/j.lfs.2013.09.009
83. Xie Y-T, Du Y-Z, Yuan H, Hu F-Q. Brain-targeting study of stearic acid-grafted chitosan micelle drug-delivery system. Int J Nanomedicine. 2012;7:3235–3244. doi:10.2147/IJN.S32701
84. Ha H-K, Kim JW, Lee M-R, Jun W, Lee W-J. Cellular uptake and cytotoxicity of β-lactoglobulin nanoparticles: the effects of particle size and surface charge. Asian-Aust J Anim Sci. 2015;28(3):420–427. doi:10.5713/ajas.14.0761
85. Bounous G. Whey protein concentrate (WPC) and glutathione modulation in cancer treatment. Anticancer Res. 2000;20(6c):4785–4792.
86. Traverso N, Ricciarelli R, Nitti M, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2013;2013:10. doi:10.1155/2013/972913
87. Ramasamy T, Ruttala HB, Gupta B, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Controlled Release. 2017;258:226–253. doi:10.1016/j.jconrel.2017.04.043
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.