")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Tardive dyskinesia in a South Asian population with first episode psychosis treated with antipsychotics
Authors Adam U, Husain N, Haddad P, Munshi T, Tariq F, Naeem F, Chaudhry I
Received 23 May 2014
Accepted for publication 16 July 2014
Published 14 October 2014 Volume 2014:10 Pages 1953—1959
DOI https://doi.org/10.2147/NDT.S68297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Usman Adam, Nusrat Husain, Peter M Haddad, Tariq Munshi, Fauzia Tariq, Farooq Naeem, Imran B Chaudhry
Background: Tardive dyskinesia (TD) is a side effect of antipsychotic treatment that often only appears after months or years of treatment. A systematic review of randomized controlled trials lasting more than 1 year showed that second-generation antipsychotics (SGAs) were associated with an approximately fivefold lower risk of TD compared to haloperidol in patients with chronic schizophrenia. In contrast, there is little research on the risk of TD with other first-generation antipsychotics (FGAs), and this applies especially to their use in the treatment of patients with first episode psychosis (FEP).
Objectives: To determine the severity and point prevalence of TD in a naturalistic sample of patients with FEP in Pakistan treated with FGAs or SGAs.
Methods: This was an observational study. TD was assessed by trained clinicians using the Abnormal Involuntary Movement Scale.
Results: In the total sample (number =86) the mean age of patients was 26 years and the prevalence of TD (Schooler Kane criteria) was 29% with no significant difference between those treated with FGAs and SGAs (31% FGAs versus 26% SGAs; P=0.805). The Abnormal Involuntary Movement Scale total score (items 1–7), a measure of the severity of TD, was significantly higher for patients treated with FGAs versus those treated with SGAs (P=0.033). Scores on specific items showed that this reflected higher scores for dyskinesia affecting the muscles of facial expression, as well as of the upper and lower limb, whereas scores did not differ significantly in other body areas.
Conclusion: FGAs were associated with greater severity, though not prevalence, of TD than SGAs. The study highlights the relatively high rate of TD in Asian FEP patients and the need for clinicians to monitor for this and other potential antipsychotic side effects during treatment.
Keywords: first-generation antipsychotic, second-generation antipsychotic, Abnormal Involuntary Movement Scale
Introduction
First-generation antipsychotics (FGAs) were first introduced in the mid-20th century and they remain an important treatment option for many patients suffering from schizophrenia and other psychotic disorders, particularly in developing countries.1 Despite their efficacy in treating psychotic symptoms, FGAs are perceived by many clinicians to be associated with a relatively high risk of extrapyramidal side effects (EPS). Second-generation antipsychotics (SGAs) were specifically developed and marketed as having a lower risk of EPS.2 However, the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study3 showed no significant difference in the incidence of Parkinsonism, dystonia, akathisia, or tardive dyskinesia (TD) between patients treated with SGAs and the FGA perphenazine. This finding has been supported by other recent large trials, including the Cost Utility of the Latest Antipsychotics in Schizophrenia Study Band 1 (CUtLASS-1),4 which demonstrated no significant difference between the rates of emergent Parkinsonism, akathisia, or TD in patients treated with FGAs versus those treated with SGAs. It seems that the marketing of SGAs as medications with a relatively low EPS risk reflected over-generalization of the results of industry-sponsored randomized controlled trials (RCTs) that used haloperidol, often at a relatively high dose, as the FGA comparator.5 Haloperidol is a high-potency dopamine D2 receptor antagonist and has a high risk of causing EPS.6 In contrast, low-potency FGAs, such as chlorpromazine, have a much lower propensity to cause EPS.7 It has been argued that the division of antipsychotics into FGAs and SGAs is an unhelpful dichotomy, as these are “pseudo-classes”, each containing individual drugs that differ markedly in the relative risk of a wide range of side effects.8,9 Despite this, “FGA” and “SGA” remain widely used terms.
Surveys show that a clinician’s choice of antipsychotic still strongly favors SGAs, and this is supported by prescribing data.10,11 In addition, there have been criticisms of the newer research, including CATIE and CUtLASS, and it is possible that some clinicians may regard the evidence from these studies as unrepresentative, just as authors of these studies did of earlier research that was dominated by studies comparing SGAs to haloperidol.12,13
Much of the data concerning the side effect profiles of FGAs and SGAs stem from research conducted on patients with chronic schizophrenia with relatively little data available from first episode psychosis (FEP) patients.14,15 A comprehensive meta-analysis by Leucht et al15 of 150 RCTs that compared FGAs to SGAs included only five trials of FEP patients. Sanger et al16 found that FEP patients are more likely to develop EPS, even though they are treated with lower mean doses of antipsychotics. A recent systematic review highlighted the vulnerability of patients with FEP to develop EPS when treated with haloperidol, even at a low dose, and reaffirmed guidelines that suggest that FEP patients should commence treatment with lower doses of antipsychotics than are used in patients with recurrent psychotic disorders.17 Some data suggest that Asian and Afro–Caribbean patients require lower doses of psychotropic drugs and are more prone to develop side effects than Caucasians.18,19 Both genetic and environmental factors may contribute. A review by Chaudhry et al20 suggested that although there was a strong reasoning towards genotype derived pharmacy, there are also known environmental factors that play a role in drug metabolism. These authors suggested that further research was needed to understand how treatments varied across ethnicities.20
In this paper, we report a secondary analysis of baseline data from a previously reported RCT that recruited patients within 5 years of a diagnosis of schizophrenia, who were then randomized to placebo or minocycline in addition to continuing their existing antipsychotic, irrespective of whether it was an FGA or SGA.21 The primary study, funded by Stanley Medical Research Institute, recruited inpatients and outpatients from Brazil and Pakistan and followed them for up to 3 years. This secondary analysis only considered the baseline data from the participants recruited in Pakistan. The aim of the study was to determine the prevalence and severity of TD in a representative cohort of Asian patients with FEP. To the best of our knowledge, there are few comparable studies.
Methods
Inclusion and exclusion criteria
The inclusion and exclusion criteria are reflective of the primary RCT by Chaudhry et al21 (Table 1). Full details of the study design and methodology are given in the primary manuscript. In addition, for this secondary analysis, patients who were treated with a combination of an FGA and an SGA were excluded.
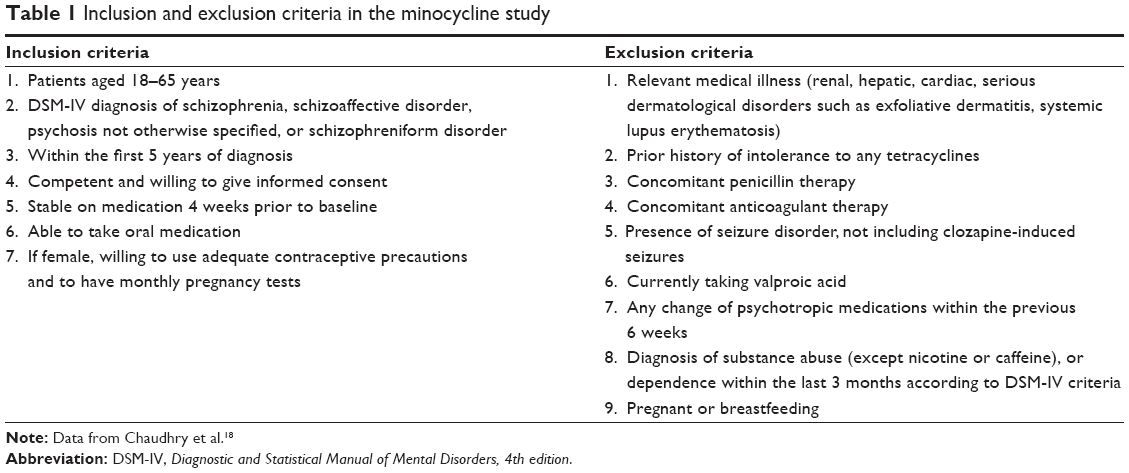 | Table 1 Inclusion and exclusion criteria in the minocycline study |
Participants
Patients were recruited from government- and charity-funded hospitals in two cities in Pakistan, Karachi and Rawalpindi. Patients were within their first 5 years of a diagnosis of schizophrenia, diagnosed using the structured clinical interview for the Diagnostic and Statistical Manual of Mental Disorders, 4th edition with schizophrenia, schizoaffective disorder, schizophreniform disorder, or psychosis not otherwise specified. The antipsychotics prescribed at baseline included haloperidol, flupentixol, fluphenazine, trifluoperazine, aripiprazole, risperidone, and olanzapine.
Outcome measures and statistical analysis
Research assistants rated TD using the Abnormal Involuntary Movement Scale (AIMS) at baseline before patients commenced minocycline or placebo. All the research assistants were provided with training in the use of the AIMS at a university department. The AIMS is a widely used observed completed scale that allows the severity of TD to be assessed.22 Various criteria can be applied to the AIMS to allow a diagnosis of TD, including the Schooler and Kane criteria,23 which were used in this study. Thus, participants who had one score of 3 (moderate) or two scores of 2 (mild) on AIMS items 1–7, covering observed movements, were deemed to have TD.
Mean scores and standard deviations for each of the seven individual bodily areas on the AIMS (items 1–7) plus the total score of items 1–7 (items that represent the overall severity of TD) were calculated for those treated with FGAs versus SGAs. Means and standard deviations were not the most appropriate measures for these ordinal data, but they are presented here because they provide a more sensitive descriptive statistic compared with the median, which is the more usual measure for ordinal data. However because the data were ordinal, the Mann–Whitney U nonparametric test of the difference between the two groups was conducted. The nonparametric test provides the mean rank, and it is the mean rank that was compared in the test. Calculations were carried out using IBM SPSS 20.0 (IBM Corporation, Armonk, NY, USA).
Results
Patients were recruited from May 2006–April 2007. From the 114 Pakistani patients who consented to take part in the main RCT,21 86 patients completed the AIMS – of whom, 55 were prescribed an FGA and 31 were prescribed an SGA (Figure 1). Although there was little difference in the two groups in terms of their age and years of education, there was a very relatively low number of female patients taking SGAs compared with FGAs (Table 2). A total of 94.7% of patients were from a middle-class or lower background.
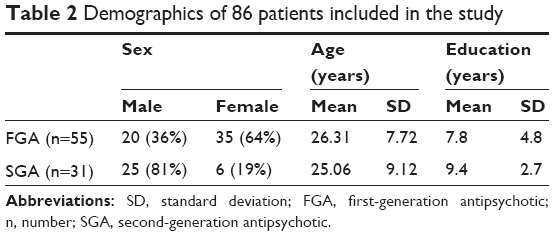 | Table 2 Demographics of 86 patients included in the study |
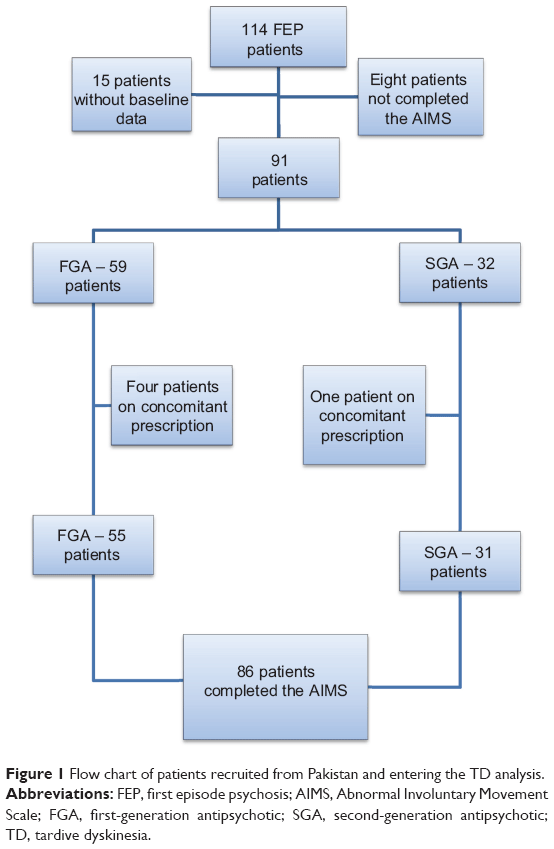 | Figure 1 Flow chart of patients recruited from Pakistan and entering the TD analysis. |
All 86 patients included in the analysis were prescribed antipsychotics within the range recommended by the British National Formulary.24 In the FGA group, 26 patients were prescribed fluphenazine deaconate (mean dose: 56.5 mg/week), 23 patients were prescribed haloperidol (mean dose: 11.2 mg/day), 13 patients were prescribed trifluoperazine (mean dose: 9.6 mg/day), and six patients received flupenthixol deaconate (mean dose: 84 mg/week). Thirteen patients in the FGA group were prescribed more than one FGA simultaneously. In the SGA group, 18 patients were prescribed risperidone (mean dose: 4.2 mg/day), ten were prescribed olanzapine (mean dose: 15.6 mg/day), and three patients were prescribed aripiprazole (mean dose: 11.6 mg/day). A number of patients were taking other psychotropic medications including procyclidine (number [n] =43), benzhexol (n=13), carbamazepine (n=7), lithium (n=6), sodium valproate (n=5), clomipramine (n=3), and various benzodiazepines (n=21).
There was no statistically significant difference in the proportion of patients diagnosed with TD according to the Schooler and Kane criteria, depending on the antipsychotic prescribed at baseline (31% FGAs versus 26% SGAs; P=0.805). However, patients prescribed SGAs scored lower on the sum of the first seven items on the AIMS, and they also had lower scores for abnormal movements in the muscles of facial expression and those of the upper and lower extremities (Table 3). When the sexes were separated, the results showed that the movements of facial expression were significantly worse (P=0.022) in male patients, but all other results showed no statistical difference between individual items.
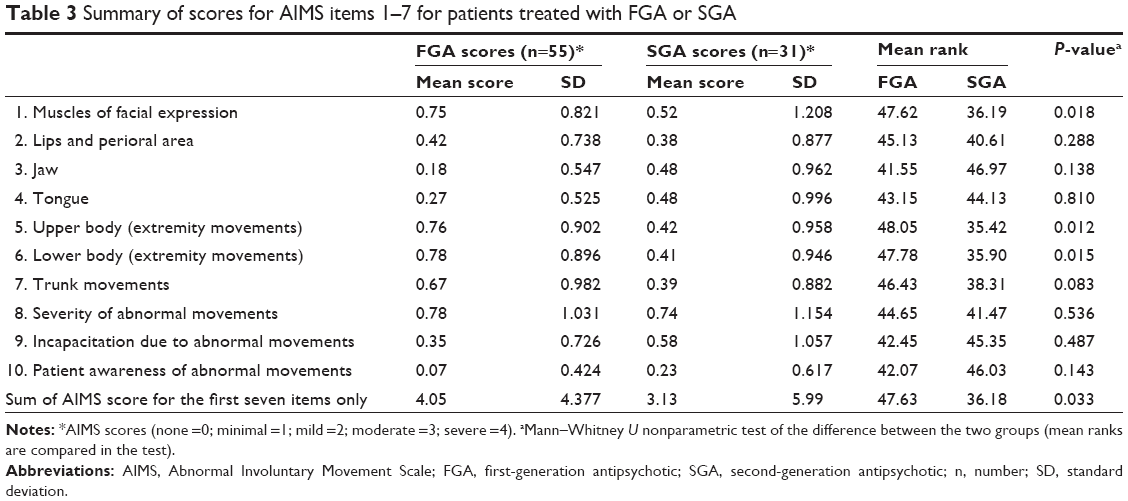 | Table 3 Summary of scores for AIMS items 1–7 for patients treated with FGA or SGA |
At baseline, 37 patients also completed the Udvalg for Kliniske Undersøgelser side effect scale (UKU).25 The individual scores showed that patients on FGAs experienced statistically worse inner unrest/tension (P=0.013), whereas SGA patients had a significantly greater tendency to experience sweating (P=0.042).
Discussion
Although Pakistan is the sixth largest country by population, very few studies of antipsychotic use in FEP patients have been conducted in this country, and none have compared the side effects of FGAs and SGAs. The strengths of the current study included the rating of TD using a standardized rating scale (AIMS), the diagnosis of TD using accepted operational criteria, and the assessment of a representative group of patients with FEP. The use of the AIMS allowed us to make comparisons to other studies that have used this instrument.26,27 In our sample, the mean age for those prescribed FGAs and SGAs was approximately 26 years and 25 years, respectively, and all patients were within 5 years of diagnosis of schizophrenia. The main weakness of the study is that the data were observational (ie, allocation to an FGA or SGA was not randomized). Consequently, we can only report an association between the baseline antipsychotic and TD; we cannot assume that the prevalence and/or severity of TD in those treated with FGAs versus SGAs is solely due to the antipsychotic “class” prescribed. Other factors that could influence our results include differences between the FGA and SGA groups in terms of patient sociodemographic variables, mean antipsychotic dose, the use of anticholinergic medication and benzodiazepines, the total duration of antipsychotic exposure, and the differences in prevalence of TD prior to the commencement of antipsychotic treatment. The dose ranges of all antipsychotics prescribed at baseline were within the ranges recommended by the British National Formulary.24
Our 29% prevalence rate for TD is lower than the 10.4% prevalence rate seen in the observational European SOHO (Schizophrenia Outpatient Health Outcome) study at baseline.28 The SOHO figure was based on the treating psychiatrist’s rating of TD being present, irrespective of its effect on patient functioning or quality of life.28 Variation between studies in the methodology used to detect and rate side effects can be a major problem when attempting to make comparisons about the rates of adverse effects between individual studies.29 The 29% prevalence rate for TD in our study is also higher than the 14% prevalence rate for TD seen at baseline in the United Kingdom CUtLASS band 1 study4 which, like our study, used the Schooler and Kane criteria. In summary, it seems that our sample of Asian FEP has a higher prevalence of TD than that seen in Caucasian patients with chronic schizophrenia.
We found that dyskinetic movements were more severe in those patients prescribed FGAs than those taking SGAs in specific body areas – namely, the upper and lower body extremities (P=0.012 and P=0.015, respectively), and the muscles of facial expression (P=0.018). In addition, the overall severity of TD, based on the total AIMS score of the first seven items, was greater in patients prescribed FGAs than in those prescribed SGAs (P=0.033). This could partly reflect the relatively higher mean dose of haloperidol (11.2 mg/day) and the fact that some patients received more than one FGA drug simultaneously. Although we observed a statistically significant difference in the severity of some individual AIMS items (Table 3), the mean AIMS scores for patients in both the FGA and SGA groups were between 0 and 1.2 on all of the individual movement items. This suggests that differences in abnormal movements, despite their statistical significance, may be of debatable clinical importance.
A total of 37 patients in our study completed the UKU at baseline. This 48-item scale encompasses a wide range of side effects. The individual scores showed that patients on FGAs experienced statistically worse inner unrest/tension (P=0.013), whereas SGA patients had a significantly greater tendency to experience sweating (P=0.042). Differences between all other symptoms on the scale were not significant when the two drug groups were compared. The interpretation of these results is unclear given that only 37 patients completed the UKU. The UKU requires a large degree of time and patience on behalf of the patient and their clinician, and the lack of assessment in our patients is likely a reflection of the time taken to complete this side effect assessment.
The CATIE study3 found no significant differences in the rates of emergent EPS between SGAs and perphenazine. One of the criticisms of the CATIE study was the use of perphenazine as the FGA comparator.12 Perphenazine was chosen by the CATIE investigators, not to reflect clinical practice, but because they thought it could be used effectively at modest doses with a low risk of EPS.30 Moreover, consumer groups advised that haloperidol would be an unwise drug choice, as patients could be reluctant to enter the study.13 Patients with pre-existing TD were excluded from the perphenazine arm of the study. The CATIE finding that perphenazine caused the same rates of TD as the individual SGAs assessed12 cannot be generalized to all FGAs. A secondary analysis of the CUtLASS-1 study4 found that the SGA cohort did not differ to the FGA cohort in terms of the rate of emergent and resolved EPS. The CUtLASS-1 study included only a small proportion of patients who were prescribed haloperidol. In contrast, sulpiride (probably the most atypical FGA) was the most widely used FGA in CUtLASS-1. The CATIE and CUtLASS-1 studies do not contradict evidence that lower rates of TD are found in patients taking SGAs compared to patients prescribed haloperidol.31 Our finding of no difference in the prevalence of TD between patients prescribed FGAs and SGAs is consistent with the EPS results in CATIE and CUtLASS-1. However, once again we highlight that, unlike CATIE and CUtLASS-1, our study is nonrandomized.
The severity and clinical course of TD vary markedly between individuals.32,33 TD is thought to reflect dopaminergic overactivity,34 which is paradoxical given that all effective antipsychotics are dopamine receptor antagonists. It has been suggested that this paradox is due to dopamine receptor antagonism caused by antipsychotics, resulting in a compensatory state of “super-sensitivity” in the postsynaptic dopamine receptor.35,36 Chakos et al37 additionally suggested that TD reflected a disease-related vulnerability, and that factors including neurodevelopmental structural changes may have a role to play. We cannot ascertain how many of the patients in our sample had TD prior to commencement with the antipsychotic medication. Prevalence rates as high as 53% have been reported for spontaneous TD in FEP patients.38
Antipsychotic use is not the sole risk factor for the development of TD.37,39,40 Higher antipsychotic dose; more severe negative, depressive, and Parkinsonian symptoms; older age; and poor response to the treatment of FEP have all been reported as risk factors for the development of TD. The mean age of the patients in our antipsychotics study was similar between those patients taking FGAs and those taking SGAs (Table 1) and is unlikely to have contributed to the differences found in TD between the two groups. Morgenstern and Glazer39 found “non-white patients” were at higher risk of TD. Most, but not all, previous studies report that women have a greater risk of TD than men.41
Thirteen patients in our sample were taking two FGAs simultaneously. The National Institute for Health and Clinical Excellence (2014) guidelines42 for the management of schizophrenia suggest that patients should not be prescribed regular combined antipsychotic medication, except for short periods (for example, when changing medication). The cross-sectional nature of our data means that it is not possible to comment on the reasons for the coprescribing, and it may have been clinically appropriate (for example, as part of a cross-taper during antipsychotic switching, or a small dose of oral antipsychotic may have been added temporarily to a patient on a long-acting antipsychotic injection to control breakthrough psychotic symptoms).
Conclusion
As far as we are aware, this is the first observational study conducted in Pakistan to compare the prevalence of TD in patients prescribed FGAs and SGAs. We found a high prevalence for TD (29%), but this did not differ between those treated with an FGA and an SGA, though there was some evidence of greater TD severity associated with FGA use. As this is an observational study, we cannot conclude that the results are causally related to the index antipsychotic, and the contribution of spontaneous dyskinesia is uncertain. Nevertheless, our results are consistent with those of the CATIE and CUtLASS-1 studies,3,4 which reported no difference in the incidence of TD in patients taking FGAs and SGAs. With an increasing focus on patient-centered care, it is important that psychiatrists and other members of the multidisciplinary team are familiar with the occurrence and management of medication-related side effects.
Disclosure
UA and FT have no competing interests to report. IBC, NH, and PMH have received support to attend conferences and/or fees for lecturing and consultancy work (including attending advisory boards) from the manufacturers of various antipsychotics. The other authors report no conflicts of interest in this work.
References
Tandon R. Antipsychotics in the treatment of schizophrenia: an overview. J Clin Psychiatry. 2011;72 Suppl 1:4–8. | ||
Margolese HC, Chouinard G, Kolivakis TT, Beauclair L, Miller R, Annable L. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 2: Incidence and management strategies in patients with schizophrenia. Can J Psychiatry. 2005;50(11):703–714. | ||
Miller DD, Caroff SN, Davis SM, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279–288. | ||
Peluso MJ, Lewis SW, Barnes TR, Jones PB. Extrapyramidal motor side-effects of first- and second-generation antipsychotic drugs. Br J Psychiatry. 2012;200(5):387–392. | ||
Kane JM, Kim E, Kan HJ, Guo Z, Bates JA, Whitehead R, Pikalov A. Comparative utility of aripiprazole and haloperidol in schizophrenia: post hoc analysis of two 52-week, randomized, controlled trials. Appl Health Econ Health Policy. 2009;7(2):109–119. | ||
Hugenholtz GW, Heerdink ER, Stolker JJ, Meijer WE, Egberts AC, Nolen WA. Haloperidol dose when used as active comparator in randomized controlled trials with atypical antipsychotics in schizophrenia: comparison with officially recommended doses. J Clin Psychiatry. 2006;67(6):897–903. | ||
Tardy M, Dold M, Engel RR, Leucht S. Flupenthixol versus low-potency first-generation antipsychotic drugs for schizophrenia. Cochrane Database Syst Rev. Epub 2014 Sep 1. | ||
Haddad PM, Sharma SG. Adverse effects of atypical antipsychotics: differential risk and clinical implications. CNS Drugs. 2007;21(11): 911–936. | ||
Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951–962. | ||
Chaudhry IB, Minhas M, Rahman R, et al. Which antipsychotics would mental health professionals from a low income country choose for themselves? Journal of Pakistan Psychiatric Society. 2011;8(2):79–83. | ||
Verdoux H, Tournier M, Bégaud B. Antipsychotic prescribing trends: a review of pharmaco-epidemiological studies. Acta Psychiatr Scand. 2010;121(1):4–10. | ||
Naber D, Lambert M. The CATIE and CUtLASS studies in schizophrenia: results and implications for clinicians. CNS Drugs. 2009;23(8): 649–659. | ||
Kraemer HC, Glick ID, Klein DF. Clinical trials design lessons from the CATIE study. Am J Psychiatry. 2009;166(11):1222–1228. | ||
Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167–177. | ||
Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373(9657):31–41. | ||
Sanger TM, Lieberman JA, Tohen M, Grundy S, Beasley C, Tollefson GD. Olanzapine versus haloperidol treatment in first-episode psychosis. Am J Psychiatry. 1999;156(1):79–87. | ||
Haddad PM, Das A, Keyhani S, Chaudhry IB. Antipsychotic drugs and extrapyramidal side effects in first episode psychosis: a systematic review of head-head comparisons. J Psychopharmacol. 2012; 26(5 Suppl):15–26. | ||
Frackiewicz EJ, Sramek JJ, Herrera JM, Kurtz NM, Cutler NR. Ethnicity and antipsychotic response. Ann Pharmacother. 1997;31(11): 1360–1369. | ||
Strickland TL, Stein R, Lin KM, Risby E, Fong R. The pharmacologic treatment of anxiety and depression in African Americans. Considerations for the general practitioner. Arch Fam Med. 1997;6(4):371–375. | ||
Chaudhry I, Neelam K, Duddu V, Husain N. Ethnicity and psychopharmacology. J Psychopharmacol. 2008;22(6):673–680. | ||
Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacol. 2012;26(9):1185–1193. | ||
Guy W. ECDEU Assessment Manual for Psychopharmacology. Washington, DC: U.S. Department of Health, Education and Welfare; 1976:534–537. | ||
Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486–487. | ||
Joint Formulary Committee. British National Formulary (BNF). London, UK: Pharmaceutical Press 2013. | ||
Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl. 1987;334:1–100. | ||
Caroff SN, Davis VG, Miller DD, et al. Treatment Outcomes of Patients With Tardive Dyskinesia and Chronic Schizophrenia. J Clin Psychiatry. 2011;72(3):10. | ||
Mathews M, Gratz S, Adetunji B, George V, Mathews M, Basil B. Antipsychotic-induced movement disorders. Psychiatry (Edgmont). 2005;2(3):36–41. | ||
Novick D, Haro JM, Bertsch J, Haddad PM. Incidence of extrapyramidal symptoms and tardive dyskinesia in schizophrenia: thirty-six-month results from the European schizophrenia outpatient health outcomes study. J Clin Psychopharmacol. 2010;30(5):531–540. | ||
Hamer S, Haddad PM. Adverse effects of antipsychotics as outcome measures. Br J Psychiatry Suppl. 2007;50:s64–s70. | ||
Morgenstern H, Glazer WM. Identifying risk factors for tardive dyskinesia among long-term outpatients maintained with neuroleptic medications. Results of the Yale Tardive Dyskinesia Study. Arch Gen Psychiatry. 1993;50(9):723–733. | ||
Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414–425. | ||
Yassa R, Nair NP. A 10-year follow-up study of tardive dyskinesia. Acta Psychiatr Scand. 1992;86(4):262–266. | ||
Caroff SN, Davis VG, Miller DD, et al; CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295–303. | ||
Marsden CD, Jenner P. The pathophysiology of extrapyramidal side-effects of neuroleptic drugs. Psychol Med. 1980;10(1):55–72. | ||
Carlsson A. Biochemical implications of dopa-induced actions on the central nervous system with particular reference to abnormal movements. In: Barbeau A, McDowell FH, editors. L-DOPA and Parkinsonism. Philadelphia, PA: Davis; 1970. | ||
Klawans HL, Rubovits R. Effect of cholinergic and anticholinergic agents on tardive dyskinesia. J Neurol Neurosurg Psychiatry. 1974; 37(8):941–947. | ||
Chakos MH, Alvir JM, Woerner MG, et al. Incidence and correlates of tardive dyskinesia in first episode of schizophrenia. Arch Gen Psychiatry. 1996;53(4):313–319. | ||
Lee J, Poon LY, Chong SA. Spontaneous dyskinesia in first-episode psychosis in a Southeast Asian population. J Clin Psychopharmacol. 2008;28(5):536–539. | ||
Morgenstern H, Glazer WM. Identifying risk factors for tardive dyskinesia among long-term outpatients maintained with neuroleptic medications. Results of the Yale Tardive Dyskinesia Study. Arch Gen Psychiatry. 1993;50(9):723–733. | ||
Oosthuizen PP, Emsley RA, Maritz JS, Turner JA, Keyter N. Incidence of tardive dyskinesia in first-episode psychosis patients treated with low-dose haloperidol. J Clin Psychiatry. 2003;64(9):1075–1080. | ||
Yassa R, Jeste DV. Gender differences in tardive dyskinesia: a critical review of the literature. Schizophr Bull. 1992;18(4):701–715. | ||
National Institute for Health and Care Excellence (NICE). Psychosis and Schizophrenia in Adults: Treatment and Management. Clinical Guideline CG178. London, UK: National Institute for Health and Care Excellence; 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.