")
Back to Journals » Journal of Pain Research » Volume 14
Tapentadol for the Treatment of Moderate-to-Severe Acute Pain in Children Under the Age of Two Years
Authors Eissa A, Tarau E, Beuter C , Radic T , Watson E , Sohns M, Lefeber C , Hammer GB
Received 26 June 2020
Accepted for publication 5 November 2020
Published 29 January 2021 Volume 2021:14 Pages 229—248
DOI https://doi.org/10.2147/JPR.S269530
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Michael Schatman
Ayman Eissa,1 Eva Tarau,2 Christoph Beuter,3 Tatjana Radic,3 Estelle Watson,3 Melanie Sohns,3 Claudia Lefeber,3 Gregory B Hammer4
1Anaesthetic Department, Sheffield Children’s Hospital, Western Bank, Sheffield, UK; 2Grünenthal USA Inc., Overland Park, KS, USA; 3Grünenthal GmbH, Aachen, Germany; 4Department of Pediatrics and Anesthesiology, Perioperative and Pain Management, Stanford University School of Medicine, Stanford, CA, USA
Correspondence: Christoph Beuter
Grünenthal GmbH, Zieglerstraße 6, 52078 Aachen, Germany
Tel +49 241 569 0
Email [email protected]
Background: Pharmacokinetics (PK), efficacy, and safety of the opioid analgesic tapentadol in the treatment of moderate-to-severe acute pain have so far not been investigated in pediatric patients < 2 years of age.
Patients and Methods: Two multicenter, open-label trials assessed the pharmacokinetic profile, safety, tolerability, and efficacy of single doses of tapentadol oral solution (OS; NCT02221674; n=19) or intravenous infusion (IV, EudraCT 2014-002259-24; n=38) in children from birth to < 2 years of age. Of these, 8 preterm neonates were included in the IV trial. A third randomized, double-blind, placebo-controlled trial (NCT02081391) investigated the efficacy and safety of multiple tapentadol OS doses in patients from birth to < 2 years (placebo n=4, tapentadol n=11) using an immediate rescue trial design. Patients in all three trials underwent surgery that, in the investigator’s opinion, reliably produced moderate-to-severe pain requiring opioid treatment.
Results: Administration of single tapentadol doses resulted in tapentadol serum concentrations within the targeted range known to be safe and efficacious in adults and compared well to the range observed for children aged 2 to < 18 years. Pain intensity already improved 15 min after administration. In the multiple dose trial, amounts of supplemental opioid analgesic medication within the first 24 h after start of trial medication were low (placebo 0.02 mg/kg, tapentadol 0.05 mg/kg). All patients stopped treatment with the trial medication because opioid analgesics were no longer required. Treatment-emergent adverse events occurred in 42.1% (tapentadol OS single dose), 28.9% (tapentadol IV), and 75% of placebo and 54.5% of tapentadol patients (tapentadol OS multiple doses), none of them serious.
Conclusion: Tapentadol showed a favorable PK and safety profile in children < 2 years of age. Multiple tapentadol OS dosing is efficacious and generally well tolerated in children ≥ 2 years and might also be a useful treatment option for children < 2 years in need of strong analgesics.
Keywords: infants, intravenous formulation, neonates, oral formulation, pain management, tapentadol
Introduction
Acute pain is common in children of all age groups, including preterm and term neonates and young infants; treatment is often inadequate.1,2 In the very young in particular, under- or untreated pain might have serious, adverse, long-lasting physical, cognitive, and behavioral consequences.3–5 In this vulnerable patient population and in a hospital setting, primarily major procedures (eg, surgeries), injuries such as fractures or burns, and medical conditions (with painful episodes) are associated with acute pain. These procedures/conditions can provoke pain severe enough to require opioid analgesics as recommended by the relevant guidelines.6–8 Healthcare professionals treating pain in these young patients are faced with a dilemma: while there is an ethical obligation to relieve the pain and a clinical necessity to avoid long-term consequences of untreated pain, only very few medications are labelled for pediatric use. Moreover, the younger the infant or child, the fewer labelled medications are available. In the United States (US), only acetaminophen (from neonates onwards) and ibuprofen (from 6 months onwards) are approved for the treatment of infants <2 years of age9 and some medications such as codeine even carry a black box warning for pediatric patients including the very young.10 In Europe, the situation is more complicated owing to differing pediatric drug labelling in individual countries; in addition to acetaminophen and ibuprofen, opioids are sometimes included in the pediatric armamentarium for the very young.9 The lack of labelled analgesics leaves the healthcare professional with no approved analgesic medication at hand and having to rely on using medications off-label. This may be one of the reasons for the difficulty in providing adequate pain management in this most vulnerable population. In the US, 37% of pediatric hospitalizations were associated with the use of off-label prescription analgesics.11 In an attempt to improve the situation, pediatric programs for medications in development (including analgesics) were made legally binding in both the US and Europe more than 10 years ago.12,13
The opioid analgesic tapentadol was the first opioid medication to undergo a formal pediatric development program designed to meet regulatory requirements of the US Food and Drug Administration and the Pediatric Committee of the European Medicines Agency. Tapentadol exhibits two synergistic mechanisms of action as a µ-opioid receptor agonist and a norepinephrine reuptake inhibitor.14 This leads to analgesia comparable with pure µ-opioid agonists (despite a lower µ-load15) and contributes to lower rates of opioid-typical side effects.16,17 In addition, tapentadol has a predictable pharmacokinetic (PK) profile in adults18 with no active metabolites contributing to the analgesic effect19 and a low potential for drug interactions.20,21 For these reasons, tapentadol was considered a good candidate for treating pain in very young pediatric patients. The program was set up to determine pharmacokinetics (PK), efficacy, and safety and tolerability of the medication across the entire pediatric age range from preterm neonates to adolescents <18 years of age.9 Six clinical trials were carried out in a global setting and included PK/dose-finding trials for the determination of appropriate doses for different pediatric populations and investigations of efficacy and safety of these doses. The findings led to the approval of tapentadol oral solution (OS) in the European Union for children 2 to <18 years of age with a dose recommendation of 1.25 mg/kg body weight every 4 hours.22 Detailed information about the program and the trials can be found in the thematic pediatric tapentadol series in this journal.9,23–28
Whereas efficacy and safety of tapentadol OS have been demonstrated for older pediatric patients,26 the PK profile, efficacy, and safety in patients <2 years have so far not been published. In order to investigate efficacy and safety of tapentadol OS for the treatment of acute moderate-to-severe postsurgical pain in children <2 years, a phase 3, randomized, double-blind, placebo-controlled, multiple dose efficacy and safety trial was required. However, dose selection for this trial was a multiple step process requiring input from a single dose PK trial in the same aged population. Figure 1 shows the approach taken to select appropriate tapentadol doses. Doses for the single dose PK trial were obtained from population (POP)-PK modelling of older subjects25 and from incorporation of data into a physiologically based PK (PBPK) model29 to understand maturational effects. The single dose PK trial was then conducted at the doses selected. Due to the limitation on blood sampling in subjects <2 years of age, only 2 or 3 PK samples could be drawn from each subject. Hence, to determine appropriate doses for the phase 3 trial, the PK data from the single dose trial were incorporated into a POP-PK model.27 Simple PK analysis using for example a noncompartmental approach is not appropriate for sparsely sampled trials.
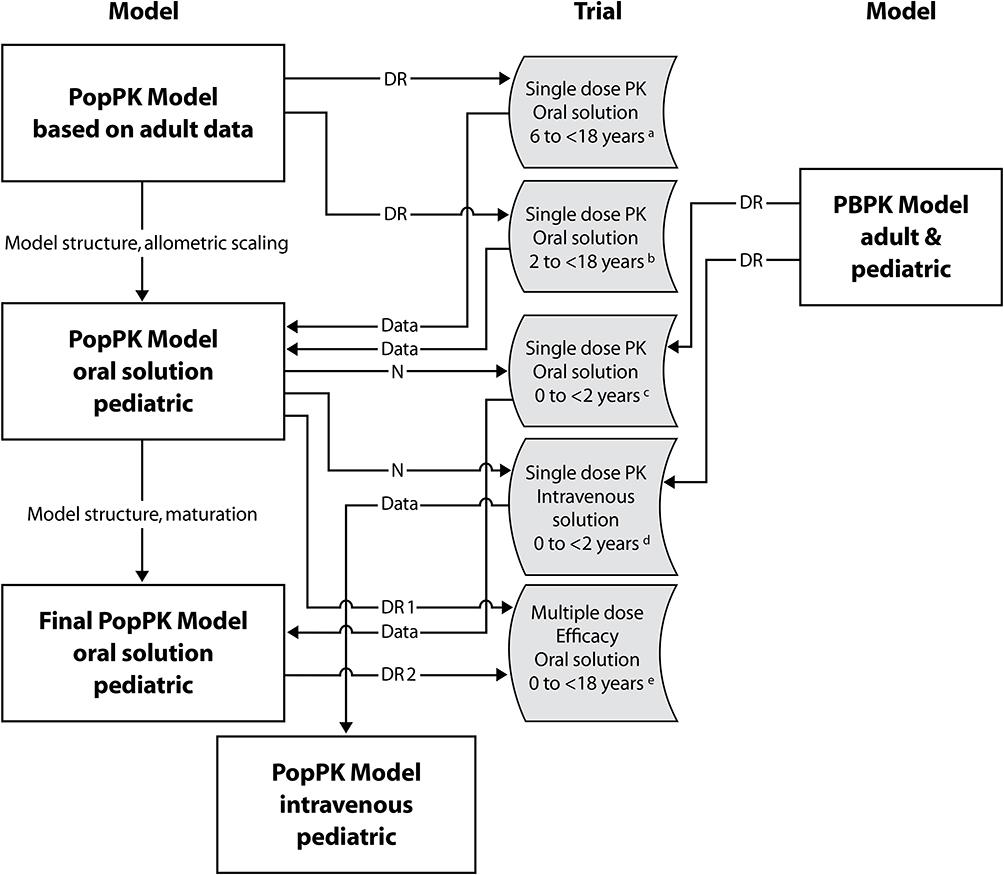 |
Figure 1 Dose finding process for tapentadol oral solution and intravenous infusion for the age range preterm neonates to children <18 years of age. Abbreviations: DR, dose recommendation; N, number; PB, physiologically based; PK, pharmacokinetic; Pop, population. Notes: aNCT01134536, bNCT01729728, cNCT02221674, dEudraCT2014-002259-24, eEudraCT2012-004359-35, NCT02081391 |
Although an oral formulation generally offers ease of use and accurate body weight dosing across a wide age range, an intravenous (IV) drug formulation is an additional useful formulation for seriously ill neonates and children unable to take oral medications.30,31 Therefore, an additional open-label, phase 2 trial was carried out which provided the first PK data in preterm neonates and infants <2 years following a single IV tapentadol infusion for surgical pain treatment. The findings of all three trials are presented in this article.
Patients and Methods
The three trials were conducted in a global setting including several countries in accordance with the Declaration of Helsinki, Good Clinical Practice, national regulations, and applicable local laws. The protocol, amendments, patient information sheet, and applicable informed consent forms were reviewed and approved by independent ethics committees or institutional review boards at the participating trial sites (Supplemental Material). Parents/legal guardians provided written informed consent for trial participation of their children. Table 1 provides an overview of the main trial characteristics. Owing to the global setting, the medical judgment by the investigator played a central role in assessing subject eligibility and accordingly was manifested in an inclusion criterion used in all trials: patients had to be suffering from moderate-to-severe postoperative pain requiring treatment with an opioid in the investigator’s judgment. Owing to the vulnerable trial population, an independent data monitoring committee (DMC) was established to oversee patients’ safety.
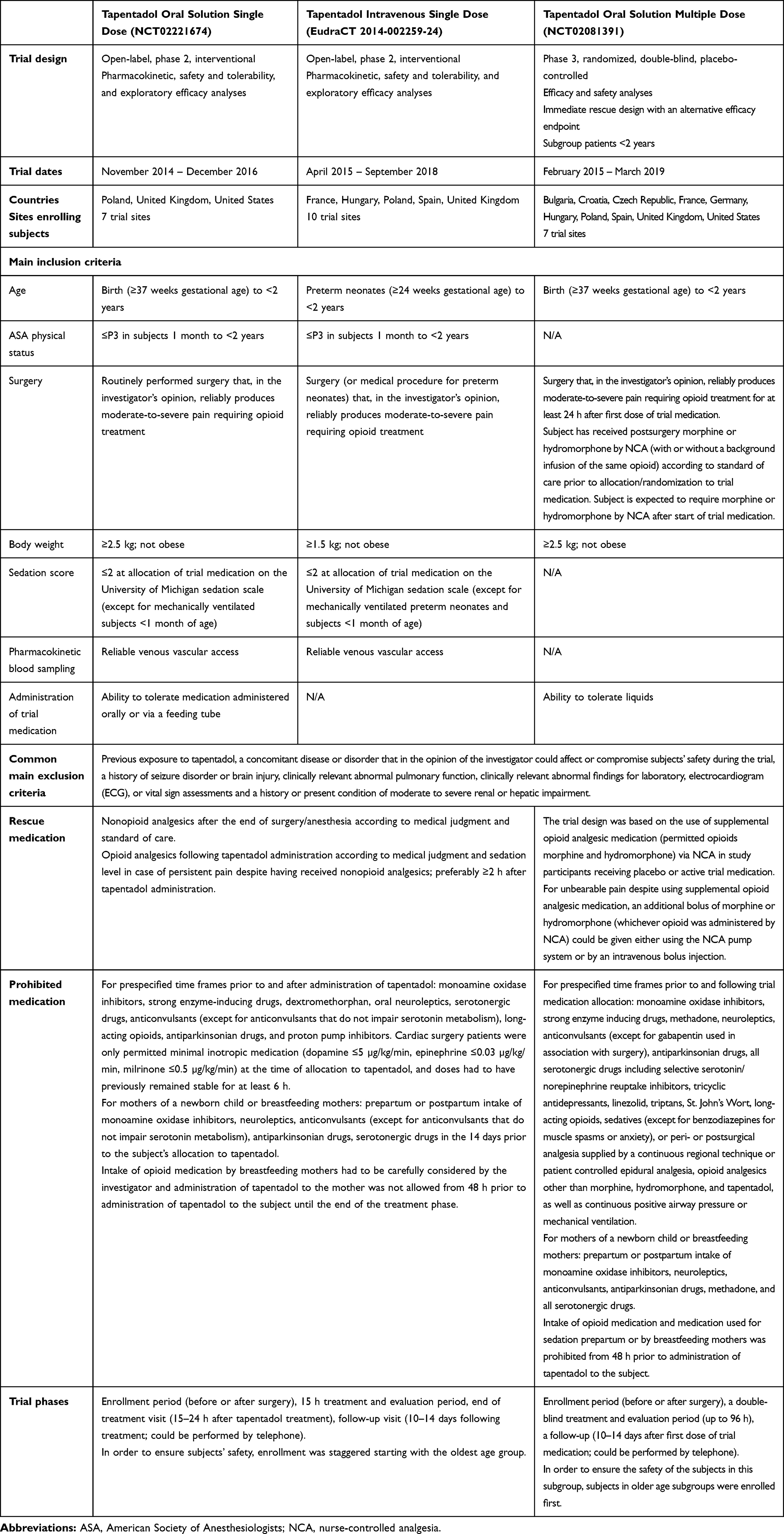 |
Table 1 Main Characteristics of the Trials |
Single Dose Trials
Two phase 2, open-label, single dose trials included patients <2 years undergoing routine surgery that, in the investigator’s opinion, would reliably produce moderate-to-severe pain requiring opioid treatment. The medication used in these trials was either tapentadol oral solution, to treat subjects from birth with a gestational age of ≥37 weeks, or tapentadol IV formulation, to include treatment of preterm neonates (≥24 weeks gestational age). These trials consisted of an enrollment period (up to 28 days before allocation to tapentadol), a 15 h treatment and evaluation period, an end-of-treatment visit (15–24 h after tapentadol treatment), and a follow-up visit (10–14 days following treatment). The trial population was stratified by age in both trials: patients aged 6 months to <2 years, patients aged 1 month to <6 months, and neonatal patients from birth to <1 month, and, for the IV trial only, preterm neonates (gestational age of 24 to <37 weeks with a postmenstrual age of ≤41 weeks).
In order to ensure patient safety, enrollment was staggered starting with the oldest age group. In the tapentadol oral solution trial, at least 2 patients from the older age group were treated with tapentadol OS to assess safety and targeted exposure prior to enrollment in the next younger age group. Enrollment of younger children was started if for the first 2 patients:
- no safety concerns were detected, or a safety concern was detected but the concern was resolved after evaluation, and
- the pharmacokinetic data were assessed as
- tapentadol blood concentrations neither exceeding the 97.5th percentile of the concentrations observed in adults administered 100 mg tapentadol IR
- nor tapentadol blood concentrations being below the 2.5th percentile of the concentrations observed in adults administered 50 mg tapentadol IR.
In the tapentadol IV trial, enrollment started with at least 4 patients in the oldest age group, moving to the next age group once (1) the targeted tapentadol exposure was verified (using the population PK model):
- tapentadol blood concentrations neither exceeding the 97.5th percentile of the concentrations observed in adults administered 100 mg tapentadol IR
- nor tapentadol blood concentrations being below the 2.5th percentile of the concentrations observed in adults administered 50 mg tapentadol IR
and (2) no safety issues such as the following were reported:
- a serious adverse event (AE) considered at least possibly related to tapentadol;
- the occurrence of any severe AE inconsistent with the known safety profile considered either by investigator or sponsor to be at least possibly related to tapentadol;
- a clinically relevant change in a safety laboratory parameter, the electrocardiogram (ECG), or a vital sign parameter which changed in a consistent direction compared with baseline in at least 2 patients.
Table 1 lists the main inclusion and exclusion criteria. Preterm and neonatal patients who were mechanically ventilated after surgery as per standard of care were eligible for inclusion if, in the investigator’s judgment, there was a need for opioid treatment. Prior to administration of tapentadol, the use of premedication, intraoperative medication, and opioid analgesics were permitted according to the usual standard of care. If possible, opioid analgesics and sedatives were not to be given within 2 h of allocation to tapentadol. Nonopioid analgesics and medications for the treatment of adverse events were permitted after the end of surgery/anesthesia according to medical judgment and standard of care. Sedatives could also be administered, but preferably not until 4 h post tapentadol dose. Rescue medication and prohibited medication details can be found in Table 1.
Study Setup
The tapentadol oral solution trial was powered to target a 95% confidence interval (CI) within 60% and 140% of the geometric mean estimate of the primary PK parameters (apparent clearance and apparent volume of distribution) for each age group.32 Simulations suggested that 15 patients (5 patients/group) with 2 PK samples each at specific timepoints would be sufficient to meet the power criterion of at least 80%.
For the tapentadol IV formulation trial, it was estimated that a sample size of 32 patients (8 in each age group) with 3 PK samples/patient at specific timepoints would be sufficient to meet the power criterion of at least 80% as described for the tapentadol OS single dose trial.
Dose Selection
Owing to the rapid and dynamic changes occurring in the neonatal period, doses for the very young children in the oral solution trial could not be reliably estimated using allometric scaling alone, and a PBPK approach29 was additionally used to define doses for the three age groups. A PBPK model was initially developed using GastroPlusTM 8.5 (Simulations Plus, Inc., Lancaster, USA) to account for the mean exposure data in adults following a variety of doses and different formulations of tapentadol. Subsequently, experimental distributions of uridine 5ʹ-diphospho-glucurono-syltransferase expression in the gastrointestinal tract, liver, and kidney were included which enabled the model to describe PK data obtained in previous pediatric trials in children aged 2 to <18 years.23,24 The model could then be used for identifying the optimal dose for children from birth to <2 years by targeting serum concentrations equivalent to the known safe and efficacious dose range of 50 to 100 mg tapentadol immediate release (IR) in adults. Underlying assumptions for this approach were that the pain mechanism and the exposure–effect curve of tapentadol would be similar in this age group compared to older children and adults.
The following doses of tapentadol OS (4 mg/mL solution) were selected for the trial population: 0.75 mg/kg body weight for patients aged 6 months to <2 years, 0.6 mg/kg for patients aged 1 month to <6 months, and 0.5 mg/kg for neonates. The route of administration was oral or could, if necessary, either be by orogastric, nasogastric, or gastric tube. Trial centers were instructed to ensure that the patient received the full dose if administered via a feeding tube.
For the IV formulation trial, the same PBPK modelling was used to identify the optimal IV tapentadol dose for the patient population of this trial as stated for the single dose tapentadol OS trial. The simulations showed that IV tapentadol infusions (1 mg/mL solution) of 0.3 mg/kg body weight for 1 h (constant rate) in patients from birth to <7 days and 0.4 mg/kg for 1 h for patients ≥7 days to <2 years old were expected to produce exposures similar to those after oral administration of tapentadol IR 75 mg to adults. Dosing was based both on the gestational age and the postnatal age of the patients as follows:
0.3 mg/kg tapentadol
- for a gestational age of ≥32 weeks and a postnatal age of <7 days
- for a gestational age of 31 weeks and a postnatal age of 7 to <14 days
- for a gestational age of 30 weeks and a postnatal age of 14 to <21 days
- for a gestational age of 26 to <30 weeks and a postnatal age of 4 to <6 weeks
- for a gestational age of 24 to <26 weeks and a postnatal age of 6 to <8 weeks
0.4 mg/kg tapentadol
- for a gestational age of ≥32 weeks and a postnatal age of ≥7 days
- for a gestational age of 31 weeks and a postnatal age of ≥14 days
- for a gestational age of 30 weeks and a postnatal age of ≥21 days
- for a gestational age of 26 to <30 weeks and a postnatal age of ≥6 weeks
- for a gestational age of 24 to <26 weeks and a postnatal age of ≥8 weeks
Pharmacokinetic Assessments
Low volume blood samples (0.1–0.2 mL) for determining serum concentrations of tapentadol and its metabolites, tapentadol-O-glucuronide and tapentadol-O-sulfate, were collected at predefined times. The sampling schemes differed for the oral solution and the IV formulation (Table 2A and B, respectively), with 2 or 3 pre-defined samples being required, respectively. Each patient was randomly assigned to one of the blood sampling schemes by an interactive response technology system.
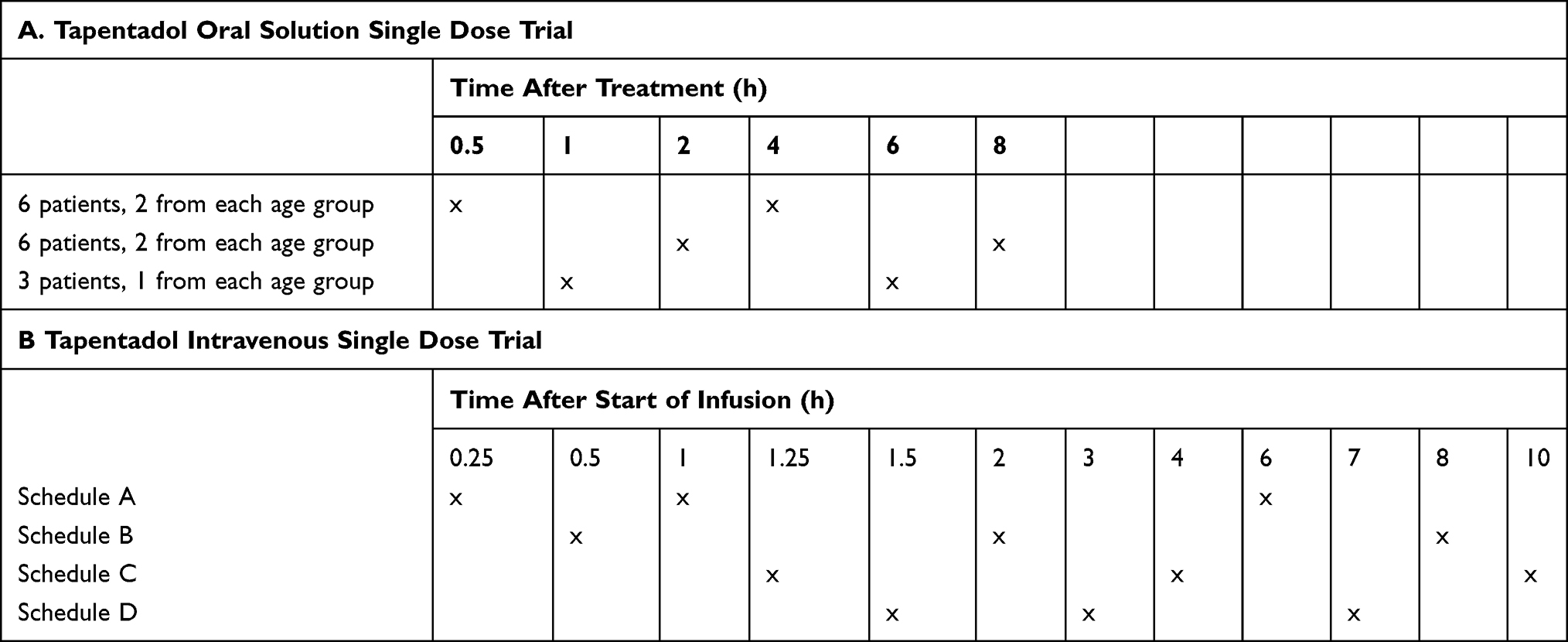 |
Table 2 Pharmacokinetic Sampling Schedule |
Serum concentrations were analyzed using a validated liquid chromatography-tandem mass spectrometry method after protein precipitation with acetonitrile. All three analytes were determined in a single analytical run. Tapentadol serum concentrations were closely monitored throughout the trial to ensure exposure remained within the expected target range and thereby enable dose corrections if required.
Efficacy Assessment
Pain intensity was evaluated using the observational Face, Legs, Activity, Cry, and Consolability (FLACC33) scale within 15 min prior to tapentadol dosing and at approximately 15 min, 30 min, 1, 2, 4, 6, 8, 12, and 15 h after dosing. Patients could be awake or asleep at the time the measurement was taken; awake/asleep status was documented for each evaluation.
Safety/Tolerability Assessments
Safety data included clinical chemistry and hematology values, 12-lead ECG assessments, physical examination, vital signs (respiratory rate, systolic and diastolic blood pressure, heart rate), and oxygen saturation by pulse oximetry (SpO2). Adverse events were recorded and assessed throughout the trial. SpO2, heart rate, and respiratory rate were continuously monitored for the first 4 h after the administration of tapentadol OS and for the first 5 h after the administration of IV tapentadol. Monitoring after these time periods was according to standard of care.
Statistical Analysis
Baseline characteristics, and efficacy and safety endpoints were analyzed with SAS versions 9.3 and 9.4 (SAS Institute Inc., Cary, NC, USA). AUC estimates were derived using R version 3.5.1., DeSolve version 1.21. The PK set in the tapentadol OS trial included all patients who had quantifiable tapentadol serum concentrations for PK analysis. Patients who did not receive the complete tapentadol dose or had fewer than 2 blood samples taken (as required by protocol) were considered non-evaluable and replaced for a sufficient sample size. Patients were also replaced if the tapentadol concentration in one of the blood samples was below the lower limit of quantification (LOQ; 0.2 ng/mL). Furthermore, replacement of a patient within the PK set was decided on a case by case basis by the sponsor if a PK sampling time deviated substantially from the planned timepoint or if the patient vomited within 3 h of tapentadol administration.
The PK set in the IV tapentadol infusion trial included all patients who had at least one quantifiable tapentadol serum concentration for subsequent pharmacometric analysis. Any patient who did not receive the complete amount of IV tapentadol solution for injection or for whom fewer than 3 blood samples for PK analysis (as required by protocol) were taken was considered non-evaluable and was replaced. Patients were also replaced if the tapentadol concentration in one of the blood samples was below the lower LOQ. Furthermore, replacement of a patient was decided on a case by case basis by the sponsor if a PK sampling time was outside the defined time window of 15 min.
Serum concentrations for tapentadol and its glucuronide and sulfate conjugates were analyzed descriptively in both trials. To ensure doses administered in the IV tapentadol trial were appropriate, the observed serum concentration-time profiles were plotted, and individual AUC0-24h values were estimated using simulation techniques which were then compared to adult exposure limits.25 As the IV infusion was given over one hour, the concentration-time profile was expected to resemble the profile obtained following a single dose of tapentadol OS. The serum concentration data of both trials were included in a POP-PK model.27 Due to the sparseness of data, formal non-compartmental analysis could not be performed.
All patients who received tapentadol as an oral solution or via IV infusion were included in the efficacy (full analysis set [FAS]) and safety/tolerability analyses that were summarized descriptively. Efficacy was investigated in an exploratory manner by the change from baseline in pain intensity. The FLACC scores of sleeping patients were included in this analysis (guidance for pain assessment in sleeping patients was given in the FLACC scale instructions). AEs were encoded using the Medical Dictionary for Regulatory Activities (MedDRA), version 19.1. Treatment-emergent AEs (TEAEs) were defined as any AE that started at the time of tapentadol OS intake or within its therapeutic reach (48 h after intake). TEAEs were considered related to treatment if the causal relationship was assessed as at least possible by the investigator.
Tapentadol OS Multiple Dose Trial
Efficacy and safety/tolerability of multiple tapentadol OS doses in the treatment of postsurgical acute pain were investigated in a phase 3, randomized, double-blind, placebo-controlled trial.26 The trial included children from birth (≥37 weeks gestational age) to <18 years. The results for the subgroup of patients aged <2 years are presented herein. The trial has been described in detail in a recent publication of the findings for older children.26 Briefly, the trial consisted of an enrollment period (up to 28 days before allocation to trial medication), a double-blind treatment and evaluation period (up to 96 h), and a follow-up (10–14 days after first dose of trial medication). An immediate rescue trial design was used where supplemental opioid analgesic medication was available to all patients. For the population aged below 2 years, supplemental medication was administered via nurse controlled analgesia pump (NCA) with morphine or hydromorphone. Efficacy was determined by comparing the use of this supplemental opioid analgesic medication between patients receiving placebo or tapentadol. Patients were included if they had undergone surgery that would reliably produce moderate-to-severe pain requiring opioid treatment for at least 24 h after the first dose of trial medication and were hospitalized until the end of treatment. Age groups included were patients aged 6 months to <2 years, patients aged 30 days to <6 months, and neonatal patients from birth to <30 days. Main inclusion and exclusion criteria as well as rescue medication and prohibited medication details are listed in Table 1.
Treatment
Patients were randomly allocated (2:1) to receive either tapentadol OS or a matching placebo OS at dosing intervals of 4 h (±15 min). Dosing for the first 24 h with tapentadol OS (4 mg/mL solution) was 1.25 mg/kg body weight for patients aged 6 months to <2 years, 0.5 mg/kg for patients aged 30 days to <6 months, and 0.1 mg/kg for neonates. If asleep at the time of the scheduled dose, the patient was awakened and dosed within a maximum time of 6 h after the previous dose. Background infusions were discontinued at administration of first trial medication. If, in the investigator’s opinion, analgesic requirements had declined after 24 h, tapentadol doses could be reduced to 1 mg/kg for patients aged 6 months to <2 years, 0.3 mg/kg for patients aged 30 days to <6 months, and 0.075 mg/kg for neonates. The tapentadol/placebo treatment period was 72 h or shorter. Treatment was stopped if opioid analgesics were no longer required. Treatment was also stopped if patients no longer needed intravenous opioids administered through NCA because their pain could be managed by exclusively administering oral opioids. Vital signs, sedation score, SpO2, and pain intensity scores were measured before each administration of trial medication.
Efficacy Assessments
Efficacy was assessed by treatment differences in the total amount of supplemental opioid analgesic medication administered via NCA within the first 12 and 24 h after first dose of trial medication, time to first administration of supplemental opioid analgesic medication, changes from baseline in pain intensity over the treatment period, and ratings regarding the patients’ overall improvement. The amount of supplemental opioid analgesic medication was expressed as morphine equivalents in mg/kg body weight, with hydromorphone considered to be 5 times more potent than morphine (eg, hydromorphone 1 mg = morphine 5 mg). Supplemental opioid analgesic medications included in the analysis were opioids given via NCA or IV bolus. Pain intensity was assessed prior to each administration of the trial medication and at end of treatment using the FLACC scale. The overall change in patients’ health was rated by investigators on the Clinical Global Impression of Change (CGIC34) scale and by parents/guardians on the Patient Global Impression of Change (PGIC34) scale from 1=very much improved to 7=very much worse after completion of double-blind treatment.
Safety/Tolerability Assessments
Safety assessments included clinical chemistry and hematology results, physical examination, 12-lead ECG, vital signs, SpO2, and sedation scores. SpO2 was continuously monitored for 4 h, heart rate and respiratory rate for 24 h after the first administration of trial medication. Monitoring after these time periods was according to standard of care. Patients were monitored for adverse events throughout the trial. All TEAEs, as well as the proportion of patients discontinuing treatment due to TEAEs or drug-related TEAEs were analyzed for tolerability assessment.
Statistical Analysis
All analyses were performed with SAS version 9.4 (SAS Institute Inc., Cary, USA). All randomized patients who received at least one dose of trial medication were analyzed for safety and efficacy (FAS). Because a limited sample size was expected for this young age group, it was accepted that the analysis would be of a descriptive nature and would not allow for an assessment of statistical significance. CGIC and PGIC responders were defined as patients with at least “much improved” ratings at end of treatment. Safety data were summarized descriptively. AEs were encoded using MedDRA version 19.1.
Results
Tapentadol OS Single Dose Trial
Of 40 enrolled patients, 21 were not included in the trial due to inclusion/exclusion criteria issues (16 patients), withdrawn consent (4 parents/legal guardians), and unspecified other (1 patient). Nineteen patients (8 patients aged 6 months to <2 years, 6 patients aged 1 to <6 months, 5 neonates) received tapentadol OS treatment and were included in the safety set and the FAS for efficacy analysis. One patient in the 6 months to <2 year group was lost to follow-up; all other patients completed the trial. Baseline characteristics of the trial population are listed in Table 3. All patients aged 6 months to <2 years, 50% of the patients aged 1 month to <6 months, and 80% of neonates had concomitant diseases. All patients received prior and concomitant medication, mostly relating to medication administered peri-operatively. Table 4 shows the surgeries performed in this patient population.
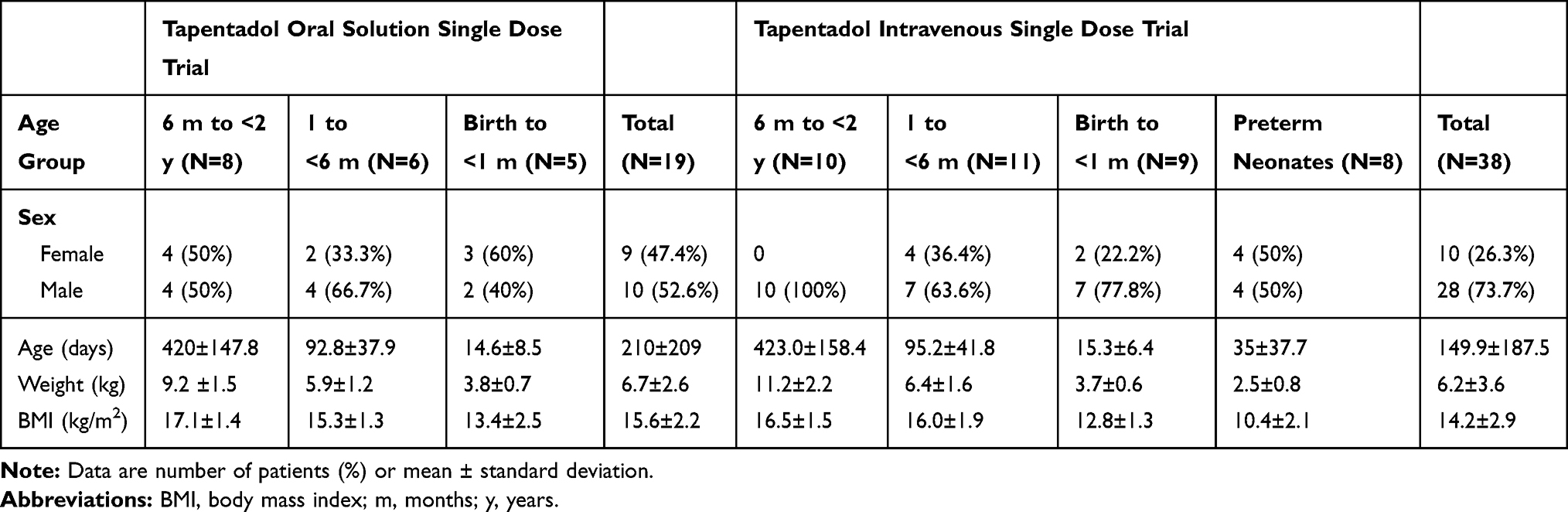 |
Table 3 Baseline Data of the Single Dose Trial Populations (Full Analysis Set) |
 |
Table 4 Type of Surgery Performed in the Three Trials |
Pharmacokinetics
Although it was planned to include 5 patients with 2 evaluable blood samples in each of the three age groups, the trial was terminated with one of the 5 neonatal patients having only one evaluable sample. By re-running the sample size calculation, POP-PK modelling indicated that sufficient information would still be available to characterize tapentadol PK in this group and further recruitment was therefore halted. Overall, the PK population comprised 18 patients.
Mean serum concentrations of tapentadol, tapentadol-O-glucuronide, and tapentadol-O-sulfate after tapentadol intake for the three age groups are summarized in the Supplemental Material (Supplemental Table 1). Figure 2 shows the observed tapentadol concentrations for each age group for the respective doses administered (0.75 mg/kg, 0.6 mg/kg, and 0.5 mg/kg, for patients aged 6 months to <2 years, patients aged 1 month to <6 months, and neonates, respectively). There was no obvious difference in serum concentrations between the groups, suggesting that the estimated doses for each group adequately compensated for the maturational changes occurring from birth up to 2 years of age. Mean tapentadol concentrations were similar to the observed range for children 2 to <18 years dosed with 1 mg/kg tapentadol OS23,24 and most of the observed tapentadol concentrations in young infants were within or at the lower end of the concentration range shown to be efficacious and safe in adults (Figure 2; 2.5th and 97.5th percentile of the simulated adult exposure following administration of 50 mg and 100 mg tapentadol IR, respectively25).
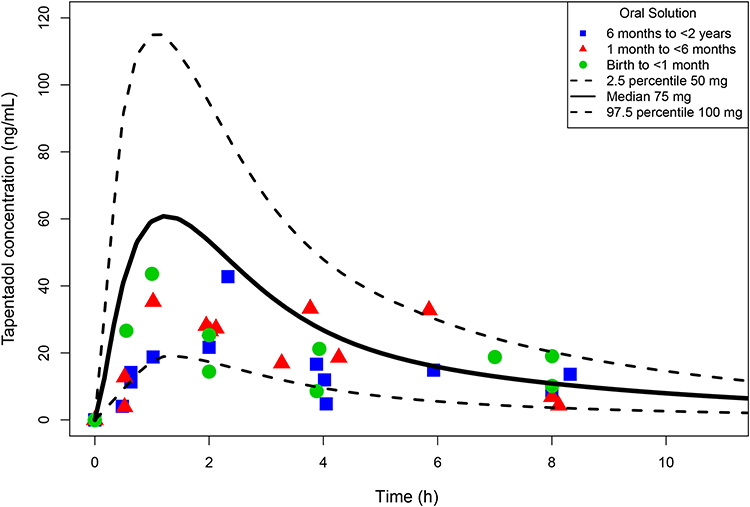 |
Figure 2 Tapentadol serum concentrations in young infants <2 years of age following a single dose of tapentadol oral solution (overlaid with adult simulated exposures). |
Mean serum concentrations for tapentadol-O-glucuronide were below or at the lower end of the range observed for older children (2 to <18 years24) and generally lower than the concentrations observed in adults (data on file). Tapentadol-O-sulfate concentrations were highest in the youngest age group. Data could not be compared with older age groups, as this metabolite was not analyzed in previous trials.
Efficacy
Pain intensity was reduced 15 min after tapentadol OS administration in all three patient groups with maximum mean decreases at 2 h for patients aged 6 months to <2 years, at 30 min for patients aged 1 to <6 months, and at 15 min for neonates (Figure 3A). Most of the neonatal patients were asleep at scoring times and variation in scores was high.
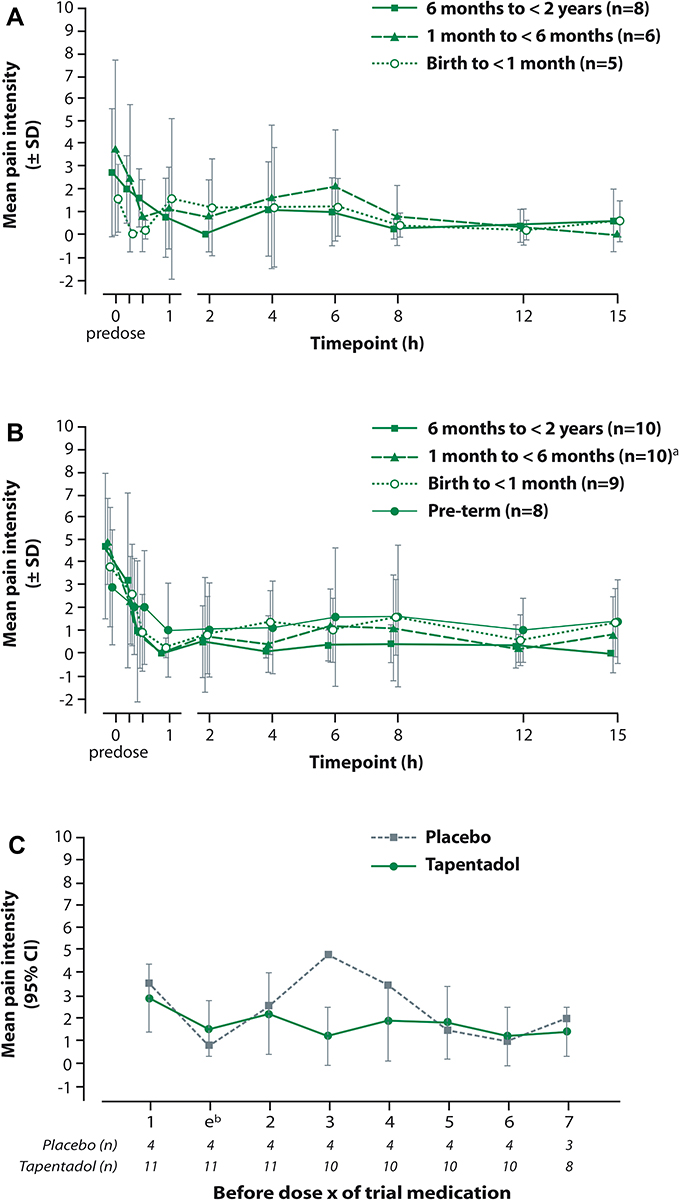 |
Figure 3 Mean pain intensity over time scored with the observational Face, Legs, Activity, Cry, and Consolability scale (A) following a single dose of tapentadol oral solution, (B) following a single dose of intravenous tapentadol, (C) prior to each intake of tapentadol oral solution (first seven intakes, multiple dose trial). Notes: (A and B) Missing data were not imputed. Predose scores are displayed at 15 min prior to dosing; data points at each observation time are slightly offset to enhance readability. The intervals show the mean value ± standard deviation; in some cases, there was a high standard deviation exceeding the mean value and this resulted in negative numbers for mean value - standard deviation. (B) aPain values were not available for one patient. (C) bAssessment was performed 30–60 mins after first intake of trial medication. |
The majority of patients (94.7%) received concomitant analgesic medication; 73.7% were given opioid analgesics, 89.5% were administered nonopioids/antipyretics. In most cases, additional analgesic medications were not administered for at least 2 h after administration of tapentadol OS.
Safety and Tolerability
Eight patients (42.1%) experienced 16 TEAEs (3 patients [37.5%] aged 6 months to <2 years, 3 patients [50%] aged 1 to <6 months, 2 neonates [40%]; Table 5). All were mild or moderate in intensity except for one severe TEAE reported for a neonatal patient (SpO2 decreased; detailed further below). Serious TEAEs did not occur and none of the patients discontinued treatment due to a TEAE. Only one event (abdominal distension in the neonatal group) was considered possibly related to tapentadol by the investigator.
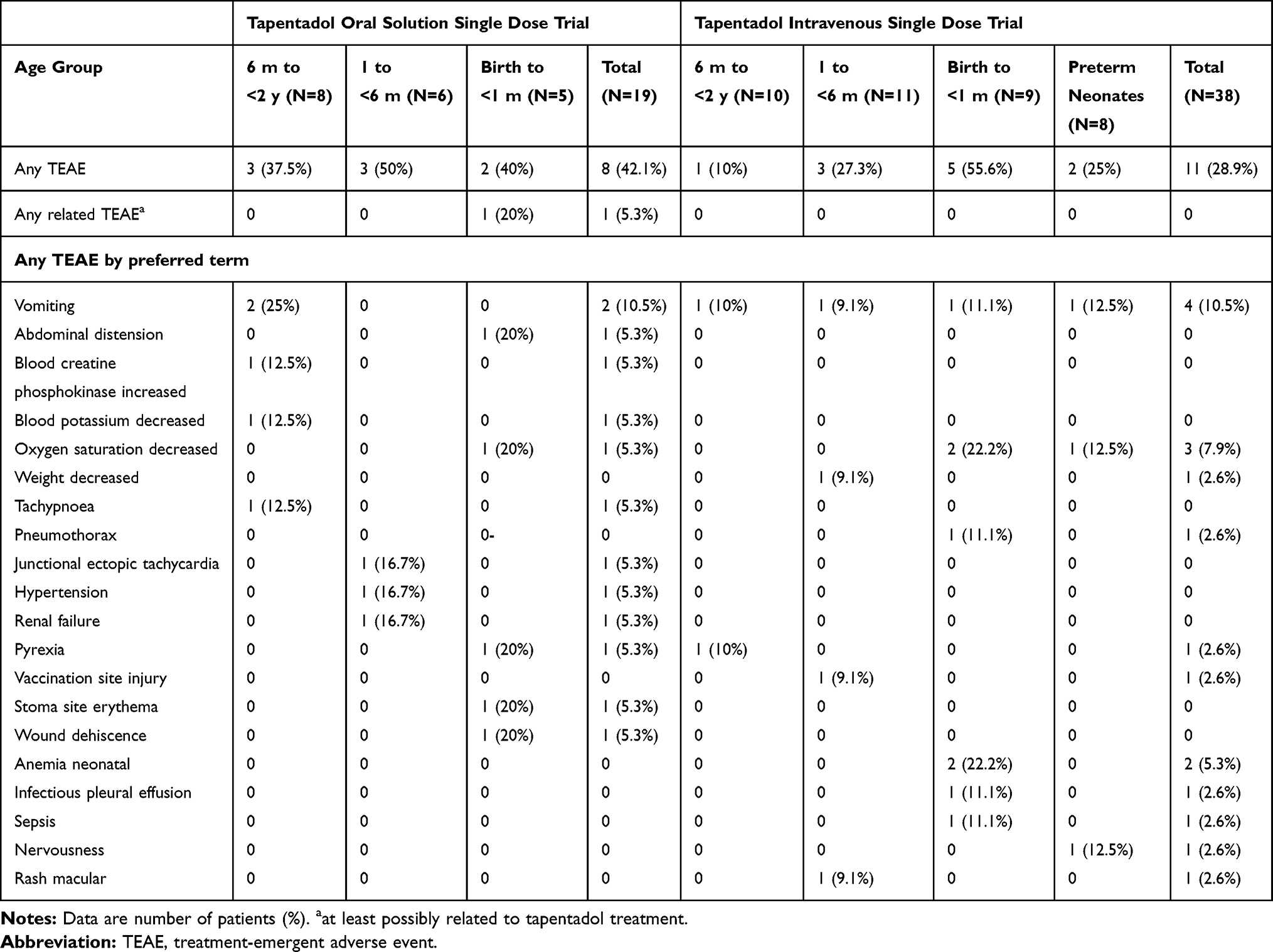 |
Table 5 Treatment-Emergent Adverse Events Profile (Single Dose Trials; Safety Set) |
As expected for a population of very young patients after surgery, fluctuation in hematological and clinical chemistry values was high. However, the changes from baseline did not suggest a particular trend in the time course of any parameter. Three changes in clinical laboratory parameters (blood creatine phosphokinase increased, blood potassium decreased, and creatinine increased reported as renal insufficiency) were clinically relevant and were reported as TEAEs; they were considered by the investigator as not related to the administration of tapentadol OS. There were no clinically relevant changes in vital signs, physical examination, or ECG assessments. SpO2 decreased below 92% for >60 sec in two neonates: one patient had an event with the lowest oxygen saturation value of 87% for 1.5 min (not reported as a TEAE), the other had a decrease on 3 occasions (lowest value of 77%; all reported as TEAEs). None of these three TEAEs was considered by the investigator to be related to tapentadol. All three resolved, the first after suction of tracheal tube (secretion), the other two without countermeasures. This second patient suffered concomitantly from cardiovascular insufficiency, atrial septal defect, pulmonary hypertension, gastrointestinal haemorrhage, diaphragmatic hernia (surgery indication), and prerenal failure.
Tapentadol IV Single Dose Trial
Thirty-eight patients (10 patients aged 6 months to <2 years, 11 patients aged 1 month to <6 months, 9 neonates, 8 preterm neonates) received an IV tapentadol infusion and were included in the safety set and the FAS. Eight patients did not participate due to inclusion/exclusion criteria issues (6 patients) and withdrawn consent (2 parents/legal guardians). All 38 patients completed the trial. Baseline characteristics of the trial population are listed in Table 3 and type of surgery is shown in Table 4. More male than female patients participated (73.7% vs 26.3%). Eighty percent of patients aged 6 months to <2 years, 18.2% of patients aged 1 to <6 months, 77.8% of neonates, and all preterm neonates had concomitant diseases. Nearly all patients (97.4%) received concomitant medication.
Pharmacokinetics
Thirty-eight patients were included in the PK analysis. Mean serum concentrations of tapentadol, tapentadol-O-glucuronide and tapentadol-O-sulfate in the four age groups after tapentadol intake are summarized in the Supplemental Material (Supplemental Table 2). Figure 4 shows the observed tapentadol concentrations. There was no obvious difference in serum concentrations between the age groups, suggesting that the estimated doses for each group adequately compensated for the maturational changes occurring in this age range. Furthermore, the estimates of AUC based on the concentrations observed in this study ranged from 75.7 to 692 ng.h/mL across the 4 age groups. The majority of the AUCs were within the expected range (130.7–706.0 ng.h/mL) simulated for adults after dosing with 50–100 mg tapentadol IR. The highest individual concentrations observed in the present trial were in the range of 57.8 ng/mL (preterm neonates) to 189 ng/mL (children aged 6 months to <2 years). This is similar to or slightly above the range of 60 ng/mL (children aged 3–6 years) to 139 ng/mL (adolescents aged 12 to <18 years) in children aged from 2 to <18 years after dosing with 1 mg/mL tapentadol OS.24
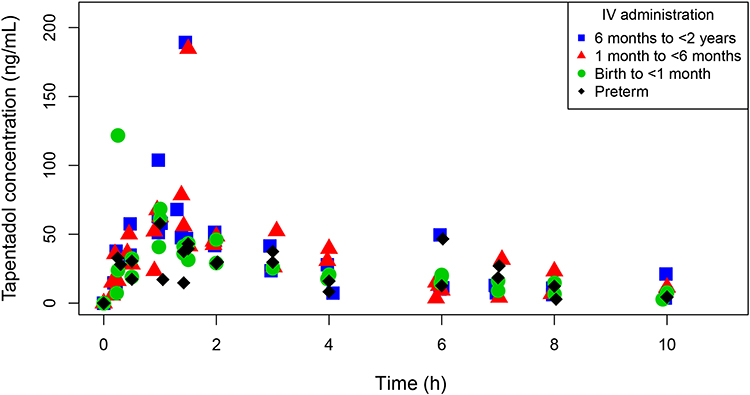 |
Figure 4 Individual tapentadol serum concentration-time profiles following a single dose of intravenous tapentadol. |
The lower doses and the infusion duration of 1 hour were chosen to generate serum concentrations similar to serum concentrations after oral dosing. This approach took the first pass metabolism of tapentadol into account. Figure 5 shows the oral solution (birth to <2 years) and IV (preterm to <2 years) serum concentration data overlaid in one graph: generally similar profiles were observed after both administrations. Furthermore, data generated in two previous PK trials using tapentadol OS in children aged 2 to <18 years23,24 are also included in Figure 5. It is apparent that similar concentrations were achieved across the age range with the dosing regimens adopted in each of these trials.
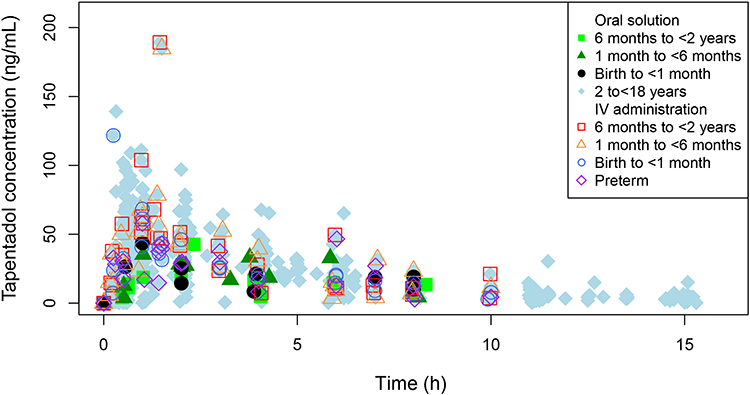 |
Figure 5 Individual tapentadol serum concentration-time profiles following single dose administrations of oral tapentadol solution or an intravenous tapentadol infusion. Figure includes the data of children ≥2 years.23,24 |
Following administration of IV tapentadol, the observed tapentadol-O-glucuronide concentrations were within the observed range for patients aged 2 to <18 years old following administration of tapentadol OS24 but were lower than following oral tapentadol administration in the <2-year trial population. Mean tapentadol-O-sulfate concentrations were within the range observed in the same age group after dosing with tapentadol oral solution.
Efficacy
Mean pain intensity was reduced in all age groups 15 min after start of IV tapentadol treatment and continued to decrease over the first hour before settling at a mean FLACC score of under 1 for the oldest age group and between 1 and 2 for the other three groups for the remainder of the evaluation period (Figure 3B). Many of the patients were asleep at scoring times and variation in scores was high. The majority of patients (86.6%) received concomitant analgesic medication (26.3% opioid analgesics, 84.2% nonopioids/antipyretics).
Safety and Tolerability
Eleven patients (28.9%) experienced 20 TEAEs (1 patient [10%] aged 6 months to <2 years, 3 patients [27.3%] aged 1 to <6 months, 5 neonatal patients [55.6%], and 2 preterm neonates [25%]; Table 5). None of the TEAEs were considered by the investigators to be related to tapentadol administration; serious TEAEs did not occur and none of the patients discontinued treatment due to a TEAE. All incidences were mild or moderate in intensity except for two severe TEAEs. One of them was the third reported episode of postsurgical decreased oxygen saturation in an intubated neonatal patient with postsurgical respiratory depression, pulmonary hypertension, and circulatory insufficiency prior to tapentadol administration. Oxygen saturation values dropped to between 45% and 47% requiring modifications to the mechanical ventilation parameters as a countermeasure. The second severe TEAE (nervousness) occurred in the preterm neonatal group; the patient was mechanically ventilated after surgery and throughout the treatment period. The event resolved with the administration of midazolam. Two neonatal patients had SpO2 below 92% during continuous pulse oximetry. One patient had three episodes (already described above), the second patient had 4 episodes between 83% and 89%; one of the events was reported as a TEAE occurring 14.5 h after the start of the tapentadol infusion. The patient underwent surgery for esophageal atresia and experienced respiratory failure and hypotension in connection with the surgery and was mechanically ventilated after surgery and during the treatment period. None of these events were considered related to tapentadol by the investigator. There was also one preterm patient with a drop in SpO2 to 87% occurring 45 min after tapentadol administration. The patient underwent surgery for gastroschisis followed by surgery for a shoulder abscess, experienced respiratory insufficiency and was mechanically ventilated after surgery during the treatment period. Apart from the above, there were no other changes in vital signs, SpO2, clinical laboratory tests, physical examination, or ECG evaluations that raised any safety concerns.
Tapentadol OS Multiple Dose Trial
Twenty-three patients were enrolled in the subgroup of patients aged <2 years reported here; eight were not randomized due to inclusion/exclusion criteria issues (6 patients) and withdrawn consent (2 parents/legal guardians). Fifteen patients were randomly allocated to placebo (4 patients) or tapentadol OS (11 patients); all were included in efficacy and safety analyses. All patients discontinued tapentadol treatment because opioid analgesics were no longer needed, one patient in the initial 12 h period after first dosing, one in the next 12 h period; the other 13 patients completed the first 24-h treatment period. Baseline characteristics of the trial population are listed in Table 6; type of surgery is shown in Table 4. Most patients (86.7%) had concomitant diseases and all received concomitant medications. The most commonly used concomitant medications were analgesics, blood substitutes and perfusion solutions, and antibacterial agents for systemic use (all used in 93.3% of patients), antidiarrheals and intestinal anti-inflammatory/anti-infective agents (each 66.7%). These medications are common in the postsurgical setting. Both treatment groups received comparable amounts of morphine or hydromorphone in the 24 h prior to first dose of trial medication (Table 6).
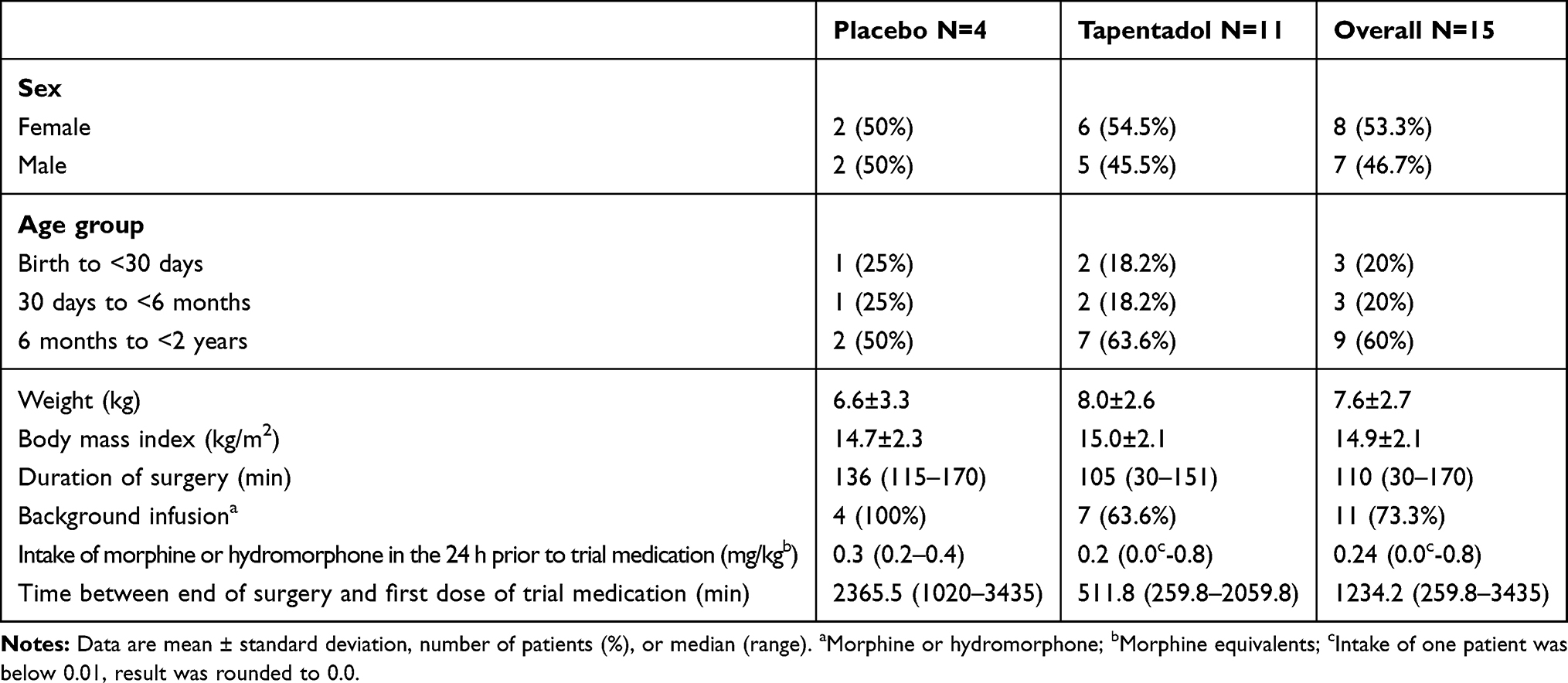 |
Table 6 Baseline Data of the Multiple Dose Trial Population (Full Analysis Set) |
Treatment
Mean exposure to trial medication was comparable between the groups with 29.4 h (SD 8.9) for placebo and 29.1 h (SD 13.3) for tapentadol patients. The 4 placebo patients all received 3 doses within the first 12 h and at least 5 doses within the first 24 h. In the tapentadol group, 54.5% and 36.4% of patients were administered 3 and 4 doses, respectively, within the first 12 h, and the majority (90.9%) received at least 5 doses within the first 24 h treatment period.
Efficacy
All subjects had received supplemental opioid analgesic medication through opioid background infusion and/or NCA after surgery before the first dose of trial medication. Three of the 4 placebo patients and 6 of the 11 tapentadol patients required supplemental opioid analgesic medication in addition to the trial medication.
Two tapentadol patients also received oral ibuprofen and IV acetaminophen, respectively, within 12 h after first trial medication intake.
The total amount of supplemental opioid analgesic medication administered in both treatment groups was generally low. It was numerically higher for tapentadol compared to placebo patients during the first 12 h after start of trial medication (0.028 mg/kg [SD 0.036] vs 0.014 mg/kg [SD 0.017]) and during the first 24 h after start of trial medication (0.054 mg/kg [SD 0.09] vs 0.016 mg/kg [SD N/A]). This was mainly due to one tapentadol patient receiving a considerably higher amount of supplemental opioid analgesic medication compared to all other placebo and tapentadol patients (0.1 mg/kg over 12 h and 0.3 mg/kg over 24 h). This patient underwent skin graft surgery for second and third-degree thermal burns whereas all other patients underwent surgeries that were not expected to produce comparable pain intensities. Time to first administration of supplemental opioid analgesic medication was numerically longer for tapentadol compared to placebo patients (median 155 min for placebo and 960 min for tapentadol, with a hazard ratio of 1.8 [standard error 1.3], p=0.4084 [Log rank test]).
Figure 3C shows the change in pain intensity during treatment. Pain intensities did not differ notably between placebo and tapentadol. This was expected, as patients in both treatment groups had access to supplemental opioid analgesic medication and were also allowed nonopioids such as acetaminophen or ibuprofen.
CGIC data were available for 4 placebo patients and 9 tapentadol patients. Investigators considered 3 placebo patients (75%) and 5 tapentadol patients (55.6%) “much” or “very much” improved (CGIC responder). Parents’ ratings on the PGIC were available for 4 placebo patients and 8 tapentadol patients. They assessed all placebo patients and 5 tapentadol patients (62.5%) as responders.
Safety and Tolerability
Nine patients (60%) experienced 11 TEAEs, 3 patients (75%) in the placebo and 6 patients (54.5%) in the tapentadol OS group (Table 7). All were mild or moderate in intensity; there were no serious TEAEs or discontinuations due to a TEAE. Four related TEAEs were reported in 3 patients, one placebo and two tapentadol OS patients. The placebo patient had a mild administration-related reaction: the patient liked the taste of the mediation very much and demanded to receive more investigational drug after treatment had been stopped. The event as such was assessed to be reportable by the site staff; the patient was not considered to show signs of drug dependency. In the tapentadol OS group, one patient had mild constipation (probably related); vomiting (possibly related) and “aspartate aminotransferase increased” (possibly related) occurred in a second patient. This patient had a medical history of hypospadias and urethral meatotomy, had undergone urethral repair, and had received 13 doses of tapentadol OS. The aspartate aminotransferase increased from 42 U/L at enrollment to 66 U/L at end of treatment. The latter was outside the reference range of the local laboratory (0–50 U/L). In the same time frame, alanine aminotransferase values were within the reference range. The investigator assessed the increase in aspartate aminotransferase as mild and possibly related. Morphine, electrolytes, and 5% glucose were the only concomitant treatments the patient received while being treated with tapentadol OS. The outcome of the TEAE is unknown (retests were not reported). One tapentadol OS patient experienced a mild drop in SpO2 to 82% for 1 min (reported as a TEAE unrelated to the trial medication by the investigator). The event started 26.2 h after the first tapentadol dose. It resolved without sequelae and the tapentadol dose was not changed. This patient underwent skin graft surgery due to thermal burns of chest and upper extremities.
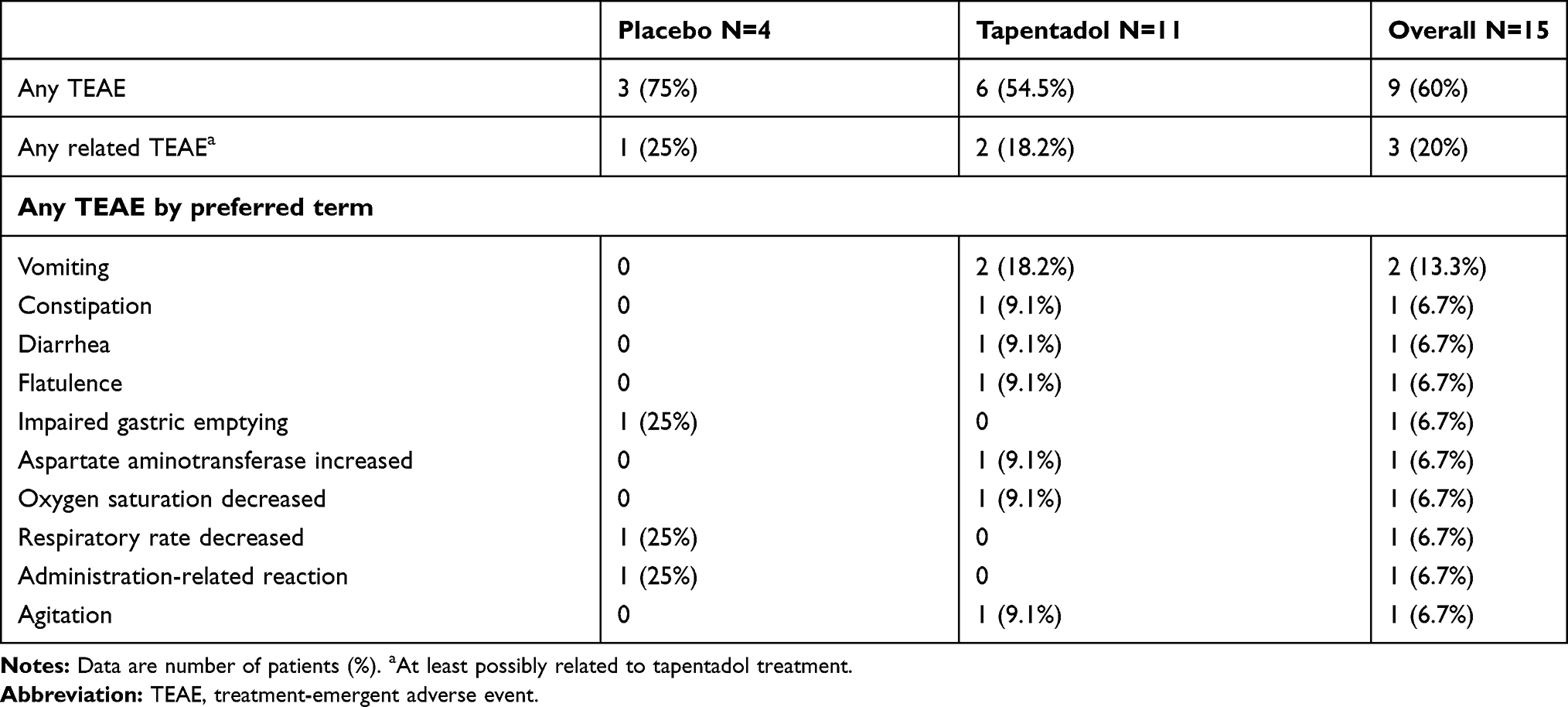 |
Table 7 Treatment-Emergent Adverse Events Profile (Multiple Dose Trial; Safety Set) |
There were no other clinically relevant changes in clinical laboratory tests, ECG, physical examination, vital signs, or SpO2.
Sedation was assessed in relation to dosing. Many patients were sedated before the first administration of trial medication (placebo 50%, tapentadol OS 63.7%); there was a slight tendency of deeper sedation in the tapentadol OS group. All patients could be aroused.
Discussion
We conducted trials of the use of tapentadol for moderate-to-severe pain in infants and children under the age of 2 years and found that a single, weight-adjusted tapentadol dose (enteral or IV) resulted in tapentadol serum concentrations within the targeted range known to be safe and efficacious in adults. Serum concentrations also compared well to the observed range for children aged 2 to <18 years receiving a single tapentadol OS dose of 1 mg/kg.23,24
The two single-dose trials additionally determined serum concentrations of two tapentadol metabolites formed by phase II conjugation, the major metabolite tapentadol-O-glucuronide, and tapentadol-O-sulfate. Although neither metabolite contributes to the analgesic activity of tapentadol, it was of interest to assess whether any alteration in metabolic pattern might occur with decreasing age. It has been reported that the contribution of glucuronidation to the elimination of acetaminophen decreases with decreasing age throughout early childhood, whereas the contribution of sulfation is independent of age.35,36 The two PK trials described herein are the first tapentadol pediatric trials in which the sulfate metabolite was quantified. Following oral tapentadol administration, the highest concentrations of tapentadol-O-sulfate were observed in the youngest age group. Although patient numbers were low, this may suggest a reduction in the glucuronide-to-sulfate ratio for tapentadol with decreasing age (<2 years) that is similar to that reported for acetaminophen. Nonetheless, the concentrations of the sulfate metabolite were still much lower than the glucuronide concentrations, indicating that in the youngest children the glucuronide was also the major conjugated metabolite. Similar metabolite concentrations were observed following the IV tapentadol infusion.
Owing to the sparse sampling approach, non-compartmental PK evaluations were neither appropriate nor intended. The data were instead incorporated into a population PK model developed in older children;25 this updated model is described in a recent publication.27
The two single dose trials were primarily designed to evaluate the PK profile of tapentadol in children <2 years but also provided the first (exploratory) efficacy and tolerability/safety data for tapentadol in this age group. Mean pain intensity improved in all age groups; reductions were already observed 15 min after start of tapentadol treatment. However, the majority of patients (94.7% on tapentadol OS, 86.6% on IV tapentadol) received concomitant analgesic medications that may have biased the findings. Treatment was generally well tolerated; TEAEs were reported for 42.1% of the patients administered tapentadol OS and 28.9% of patients on an IV tapentadol infusion. Discontinuations due to a TEAE were not reported and serious adverse events did not occur. Only one TEAE (abdominal distension) was deemed possibly related to tapentadol OS by the investigator.
The challenges faced in conducting pediatric trials in general and pediatric trials for testing new analgesics in particular have been described in detail in the first paper of the thematic pediatric tapentadol series.9 Although using a standardized pain model with comparable surgical procedures and anesthetic techniques is advantageous for an analgesic trial in the postsurgical setting, a wider selection of surgeries was permitted in the two single dose trials in order to achieve an adequate sample size. Owing to the vulnerability of this patient population, great emphasis was placed on the safety of the participating children. In case of lack of efficacy, all children had immediate access to other analgesic medication. Discomfort was minimized by limiting the number of venipunctures required and permitting the use of a local anesthetic patch or cream to reduce any associated pain. All trial center staff had to be experienced in the treatment of children. Enrollment followed a sequential age-down approach with constant surveillance of safety and PK data by the sponsor’s safety management team composed of experts from the relevant departments prior to dosing the next lower age group. In addition, safety was monitored by an external DMC throughout the trial.
Poor patient accrual has been cited as a major reason for the termination of randomized clinical pediatric trials.37 Recruitment experiences during the tapentadol OS multiple dose trial reported here highlight the major challenge of pediatric trials of finding a sufficient number of eligible trial subjects within a reasonable time frame, in particular in a young age group. Recruitment for this trial had to be paused in 2017 to identify sites able to manage patients aged <6 months. In the end, two trial sites participated in the recruitment of this very young patient population. Overall, 23 patients could be enrolled and 15 of them were dosed and included in efficacy and safety analyses. Owing to this limited sample size, only descriptive analyses were possible.
Patients <2 years received only low amounts of supplemental opioid analgesic medication within the first 24 h compared to older children (2 to <18 years) in this trial:26 placebo 0.02 mg/kg and tapentadol OS 0.05 mg/kg for the <2 group vs placebo 0.25 mg/kg and tapentadol OS 0.16 mg/kg for older children. All patients aged <2 years stopped treatment with the trial medication because opioid analgesics were no longer required.
Changes in pain intensity were similar between the groups. This was expected since patients in both treatment groups had access to supplemental opioid analgesic medication and were also allowed nonopioids such as acetaminophen or ibuprofen. It should be noted that this was an analysis of the subgroup of very young children in the multiple dose trial. Given the limited sample size it was accepted that the analysis would be of a descriptive nature and would not allow for an assessment of statistical significance.
Patients were closely monitored for safety. Multiple tapentadol doses were generally well tolerated. None of the patients discontinued treatment due to tolerability issues, none of the observed TEAEs were deemed serious, and only three events in the tapentadol OS group were considered related to tapentadol treatment by the investigator. Constipation and vomiting are known side effects of tapentadol. The increased aspartate aminotransferase values observed in one patient were unexpected and possibly associated with the intake of concomitant medications. There were no indications of higher susceptibility of this vulnerable age group to the adverse effects of the body weight-adjusted tapentadol doses in comparison to the patients aged 2 years and above.
Conclusions
The PK of tapentadol in children <2 years of age following single oral and IV administration was well within the targeted range known to be safe and efficacious in adults. Safety and tolerability of single and multiple doses of tapentadol were in line with those observed in children ≥2 years and adults. Multiple tapentadol OS dosing was efficacious in children ≥2 years of age suffering from moderate-to-severe pain.26 Our findings give a first indication about tapentadol as a possible new treatment option for children <2 years in need of strong analgesics.
Abbreviations
AE, adverse event; CGIC, Clinical Global Impression of Change; CI, confidence interval; DMC, data monitoring committee; ECG, electrocardiogram; FAS, full analysis set; FLACC, Face, Legs, Activity, Cry, and Consolability scale; IR, immediate release; IV, intravenous; LOQ, limit of quantification; MedDRA, Medical Dictionary for Regulatory Activities; NCA, nurse controlled analgesia; OS, oral solution; PB, physiologically based; PGIC, Patient Global Impression of Change; PK, pharmacokinetic; POP, population; SD, standard deviation; SpO2, oxygen saturation; TEAE, treatment-emergent adverse event; US, United States.
Data Sharing Statement
Any request for data should be submitted via the following website: https://www.grunenthal.com/en/research-and-development/clinical-trials/data-sharing-clinical-trials.
Acknowledgments
The authors thank all patients, parents/legal guardians, investigators, and the trial site teams involved in this investigation. The trials were funded by Grünenthal GmbH. Writing assistance was provided by Elke Grosselindemann, Martin and Birgit Brett and was funded by Grünenthal GmbH.
Disclosure
AE and GBH were investigators on the clinical trials published here and were paid by Grünenthal GmbH for their contributions to these trials. ET was an employee of Grünenthal USA Inc. at the time the trials were conducted. EW was an employee of Grünenthal GmbH at the time the trials were conducted. All other authors are current employees of Grünenthal GmbH. The authors report no other potential conflicts of interest for this work.
References
1. Wilson CA, Sommerfield D, Drake-Brockman TFE, Lagrange C, Ramgolam A, von Ungern-Sternberg BS. A prospective audit of pain profiles following general and urological surgery in children. Paediatr Anaesth. 2017;27:1155–1164. doi:10.1111/pan.13256
2. Groenewald CB, Rabbitts JA, Schroeder DR, Harrison TE. Prevalence of moderate-severe pain in hospitalized children. Paediatr Anaesth. 2012;22:661–668. doi:10.1111/j.1460-9592.2012.03807.x
3. Ranger M, Grunau RE. Early repetitive pain in preterm infants in relation to the developing brain. Pain Manag. 2014;4(1):57–67. doi:10.2217/pmt.13.61
4. Fitzgerald M, Walker SM. Infant pain management: a developmental neurobiological approach. Nat Clin Pract Neurol. 2009;5(1):35–50. doi:10.1038/ncpneuro0984
5. Beggs S. Long-term consequences of neonatal injury. Can J Psychiatry. 2015;60(4):176–180. doi:10.1177/070674371506000404
6. American Academy of Pediatrics, Committee on Psychosocial Aspects of Child and Family Health, Task Force on Pain in Infants, Children, and Adolescents. The assessment and management of acute pain in infants, children, and adolescents. Pediatrics. 2001;108(3):793–797. doi:10.1542/peds.108.3.793
7. Batton DG, Barrington KJ, Wallman C. American Academy of Pediatrics Committee on Fetus and Newborn, American Academy of Pediatrics Section on Surgery, Canadian Paediatric Society Fetus and Newborn Committee. Prevention and management of pain in the neonate: an update. Pediatrics. 2006;118(5):2231–2241. doi:10.1542/peds.2006-2277
8. Association of Paediatric Anaesthetists of Great Britain and Ireland. Good practice in postoperative and procedural pain management, 2nd edition. Paediatr Anaesth. 2012;22(suppl1):1–79. doi:10.1111/j.1460-9592.2012.03838.x
9. Eerdekens M, Beuter C, Lefeber C, van den Anker J. The challenge of developing pain medications for children: therapeutic needs and future perspectives. J Pain Res. 2019;12:1649–1664. doi:10.2147/JPR.S195788
10. US Food and Drug Administration. FDA drug safety communication: FDA restricts use of prescription codeine pain and cough medicines and tramadol pain medicines in children; recommends against use in breastfeeding women. Available from: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-restricts-use-prescription-codeine-pain-and-cough-medicines-and.
11. Carmack M, Berde C, Monuteaux MC, Manzi S, Bourgeois FT. Off-label use of prescription analgesics among hospitalized children in the United States. Pharmacoepidemiol Drug Saf. 2020;29:474–481. doi:10.1002/pds.4978
12. Califf RM Best Pharmaceuticals for Children Act and Pediatric Research Equity Act Status Report to Congress; July 2016. Available from: https://www.fda.gov/downloads/scienceresearch/specialtopics/pediatrictherapeuticsresearch/ucm509815.pdf.
13. European Union. Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use, OJ L 378, 27.12.2006. Available from: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2006_1901/reg_2006_1901_en.pdf.
14. Tzschentke TM, Christoph T, Kögel BY. The mu-opioid receptor agonist/noradrenaline reuptake inhibition (MOR-NRI) concept in analgesia: the case of tapentadol. CNS Drugs. 2014;28:319–329. doi:10.1007/s40263-014-0151-9
15. Raffa RB, Elling C, Tzschentke TM. Does ‘strong analgesic’ equal ‘strong opioid’? Tapentadol and the concept of ‘µ-load’. Adv Ther. 2018;35:1471–1484. doi:10.1007/s12325-018-0778-x
16. Lange B, Kuperwasser B, Okamoto A, et al. Efficacy and safety of tapentadol prolonged release for chronic osteoarthritis pain and low back pain. Adv Ther. 2010;27:381–399. doi:10.1007/s12325-010-0036-3
17. Biondi D, Xiang J, Benson C, Etropolski M, Moskovitz B, Rauschkolb C. Tapentadol immediate release versus oxycodone immediate release for treatment of acute low back pain. Pain Physician. 2013;16:E237–E246.
18. Göhler K, Brett M, Smit JW, Rengelshausen J, Terlinden R. Comparative pharmacokinetics and bioavailability of tapentadol following oral administration of immediate- and prolonged-release formulations. Int J Clin Pharmacol Ther. 2013;51(4):338–348. doi:10.5414/CP201722
19. Terlinden R, Kogel BY, Englberger W, Tzschentke TM. In vitro and in vivo characterization of tapentadol metabolites. Methods Find Exp Clin Pharmacol. 2010;32:31–38. doi:10.1358/mf.2010.32.1.1434165
20. Kneip C, Terlinden R, Beier H, Chen G. Investigations into the drug-drug interaction potential of tapentadol in human liver microsomes and fresh human hepatocytes. Drug Metab Lett. 2008;2:67–75. doi:10.2174/187231208783478434
21. Smit JW, Oh C, Rengelshausen J, et al. Effects of acetaminophen, naproxen, and acetylsalicylic acid on tapentadol pharmacokinetics: results of two randomized, open-label, crossover, drug-drug interaction studies. Pharmacotherapy. 2010;30:25–34. doi:10.1592/phco.30.1.25
22. Electronic Medicines Compendium. Palexia oral solution 20 mg/mL. Available from: https://www.medicines.org.uk/emc/product/5346/smpc.
23. Finkel J, Goldberg J, Rosenburg R, et al. First evaluation of tapentadol oral solution for the treatment of moderate to severe acute pain in children aged 6 to <18. J Pain Res. 2019;12:1925–1936. doi:10.2147/JPR.S197348
24. Muse D, Tarau E, Lefeber C, et al. Pharmacokinetics, safety, and efficacy of tapentadol oral solution for treating moderate to severe pain in pediatric patients. J Pain Res. 2019;12:1777–1790. doi:10.2147/JPR.S197039
25. Watson E, Khandelwal A, Freijer J, van den Anker J, Lefeber C, Eerdekens M. Population pharmacokinetic modelling to facilitate dose selection of tapentadol in the pediatric population. J Pain Res. 2019;12:2835–2850. doi:10.2147/JPR.S208454
26. Beuter C, Volkers G, Radic T, Goldberg J, van den Anker J. Efficacy and safety of multiple doses of tapentadol oral solution in the treatment of moderate to severe acute pain in children aged 2 to <18 years – a randomized, double-blind, placebo-controlled trial. J Pain Res. 2019;12:3099–3112. doi:10.2147/JPR.S207010
27. Khalil F, Choi SL, Watson E, et al. Population pharmacokinetics of tapentadol in children from birth to <18 years. J Pain Res. 2020;13:3107–3123. doi:10.2147/JPR.S269549
28. Howard R, Radic T, Sohns M, Eerdekens M, Waßmuth A. Tapentadol prolonged release for long-term treatment of pain in children. J Pain Res. 2020;13:3157–3170. doi:10.2147/JPR.S272751
29. Khandelwal A, Bolger M, Brett M, Rosenburg R, Bursi R. Physiologically-based pharmacokinetic (PBPK) model of tapentadol and tapentadol-O-glucuronide in adult and pediatric population. Available from: http://www.pkuk.org.uk/Contentimages/PKUK_2014_Programme_and_Abstractsnew.pdf.
30. Nunn T, Williams J. Formulation of medicines for children. Br J Clin Pharmacol. 2005;59(6):674–676. doi:10.1111/j.1365-2125.2005.02410.x
31. European Medicines Agency. Guideline on pharmaceutical development of medicines for paediatric use. EMA/CHMP/QWP/805880/2012 Rev. 2; August 1, 2013. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-pharmaceutical-development-medicines-paediatric-use_en.pdf.
32. Wang Y, Jadhav PR, Lala M, Gobburu JV. Clarification on precision criteria to derive sample size when designing pediatric pharmacokinetic studies. J Clin Pharmacol. 2012;52(10):1601–1606. doi:10.1177/0091270011422812
33. Merkel SI, Voepel-Lewis T, Shayevitz JR, Malviya S. The FLACC: a behavioral scale for scoring postoperative pain in young children. Pediatr Nurs. 1997;23(3):293–297.
34. Guy W. ECDEU Assessment Manual for Psychopharmacology (DHEW Publication No. ADM 76-338). Washington DC: US Government Printing Office; 1976.
35. Allegaert K, de Hoon J, Verbesselt R, Vanhole C, Devlieger H, Tibboel D. Intra- and interindividual variability of glucuronidation of paracetamol during repeated administration of propacetamol in neonates. Acta Paediatr. 2005;94(9):1273–1279. doi:10.1111/j.1651-2227.2005.tb02088.x
36. van der Marel CD, Anderson BJ, van Lingen RA, et al. Paracetamol and metabolite pharmacokinetics in infants. Eur J Clin Pharmacol. 2003;59(3):243–251. doi:10.1007/s00228-003-0608-0
37. Pica N, Bourgeois F. Discontinuation and nonpublication of randomized clinical trials conducted in children. Pediatrics. 2016;138(3):e20160223. doi:10.1542/peds.2016-0223
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.