")
Back to Journals » International Journal of Nanomedicine » Volume 14
Tailoring of PEGylated bilosomes for promoting the transdermal delivery of olmesartan medoxomil: in-vitro characterization, ex-vivo permeation and in-vivo assessment
Authors Albash R, El-Nabarawi MA, Refai H , Abdelbary AA
Received 27 April 2019
Accepted for publication 10 July 2019
Published 15 August 2019 Volume 2019:14 Pages 6555—6574
DOI https://doi.org/10.2147/IJN.S213613
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Rofida Albash,1 Mohamed A El-Nabarawi,2 Hanan Refai,1 Aly A Abdelbary2,3
1Department of Pharmaceutics, Faculty of Pharmaceutical Sciences and Drug Manufacturing, Misr University for Science and Technology, Giza, Egypt; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Cairo, Egypt; 3Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, October 6 University, Giza, Egypt
Introduction: The intention of this work was to load olmesartan medoxomil (OLM), a sparsely water soluble antihypertensive bioactive with low oral bioavailability (26%), into PEGylated bilosomes (PBs) for augmenting its transdermal delivery. PBs contain PEGylated single chain edge activator besides the components of traditional bilosomes (Span 60, cholesterol and bile salts). The PEG gives further resilience to vesicle membrane and is speculated to augment both permeability and bioavailability of OLM.
Methods: A 24 factorial experiment was constructed to inspect the impact of diverse variables on vesicles’ features and sort out the optimal formula adopting Design Expert® software utilizing thin film hydration technique. Vesicles’ evaluation was done by finding out entrapment efficiency percent (EE%), particle size (PS), polydispersity index (PDI), zeta potential (ZP) and amount of drug released after 6 hrs (Q6h). The optimal formula was selected and characterized for further investigations.
Results: The optimal formula (PB15) showed spherical vesicles with EE% of 72.49±0.38%, PS of 559.30±10.70 nm, PDI of 0.57±0.15, ZP of −38.35±0.65 mV and Q6h of 59.60±0.24%. PB15 showed higher deformability index (28.39±5.71 g) compared to traditional bilosomes (5.88±0.90 g) and transethosomes (14.94±0.63 g). Further, PB15 showed superior skin permeation from rat’s skin relative to the drug suspension. Moreover, confocal laser scanning microscopy examination revealed efficient penetration of the fluoro-labeled PB15 through skin. Histopathological study ensured the safety of PB15. In addition, in-vivo skin deposition studies showed higher OLM deposition in rat’s skin from PB15 compared to transethosomes and OLM suspension. Furthermore, pharmacodynamic and pharmacokinetic studies performed using male Wistar rats and male Albino rabbits, respectively, showed the superiority of PB15 over oral tablets. PB15 was found to have significantly higher AUC0–48 and AUC0–∞ relative to the oral tablets. As well, the relative bioavailability of PB15 was found to be 235.04%.
Conclusion: Overall, the obtained results confirmed the creditable effect of PB15 for transdermal delivery.
Keywords: bilosomes, confocal laser scanning microscopy, ex-vivo permeation, olmesartan medoxomil, pharmacodynamic study, pharmacokinetic study
Introduction
Hypertension seems to be a disorder that needs consideration for developing a transdermal drug delivery system (TDDS) because it requires long and continuing therapy.1 For hypertension management, olmesartan medoxomil (OLM) is considered a promising antihypertensive drug classified as BCS class II with low bioavailability of 26% as a result of its inadequate solubility and first-pass effect after oral administration.2 The first-pass metabolism of OLM and the considerations of hypertension disease could make transdermal delivery a recommended suggestion. TDDS could avoid hepatic first-pass effect and as a result the bioavailability is speculated to be boosted. Only small number of drugs exists as TDDS although the several merits of the skin as a drug delivery site which could be justified by the restriction of transdermal drug entry that is opposed by the stratum corneum (SC).3
Administering bioactive through a vesicular system is a successful mean for delivering drugs through transdermal route due to its deformable nature. Vesicles such as liposomes and niosomes are common carriers to encapsulate drugs. Unfortunately, they suffer from limited stability, encapsulation and scaling up problems which arouse the need for developing new vesicular systems.4 Bile salts (BS) enclosing vesicles or bilosomes have been produced as a modification for liposomes and niosomes which provide more stable vesicles by addition of BS to the lipid bilayers.5
Bilosomes have been fabricated for the orally administrated sparsely water soluble drugs and vaccines by the virtue of its high stability in opposition to pH, enzymes and BS existing in the gastro-intestinal tract.5 A recent study confirmed the tolerability of utilizing bilosomes in ocular drug delivery.6 Furthermore, bilosomes have been inspected for transdermal delivery. The studies proved the dominance of bilosomes over other vesicles as a TDDS as a result of the membrane destabilizing activity of the utilized BS.3,7
In the present work, Brij® was utilized as additional edge activator (EA) to prepare the PEGylated bilosomes (PBs). Brij® consists of PEGylated single chain EA with various PEG chain lengths and acyl chain entities.8 The presence of the hydrophilic PEG moieties in EAs could increase the water uptake in SC which leads to SC swelling and it might also cause structural alterations in SC lipids and/or corneocytes which lead to broadening of the intercellular junctions in the barrier. Hence, the SC permeation is enhanced allowing sufficiently deformed PBs to pass the skin along with hydration gradient.9 Besides the improved transport of OLM loaded PBs across the skin, it might also be possible that PEG moieties facilitate their access to the lymph nodes. In this sense, it was reported that PEG improved the drainage of vesicles and enhanced their lymph node localization and subsequently resulted in augmentation of vesicles’ ability for circumvention of the first-pass effect.10 The work in this study was planned to assess the prospective of PBs to augment both transdermal permeability and bioavailability of OLM. To accomplish this aim, a 24 full factorial experiment employing Design Expert® software was used to obtain the optimal formula. Further, ex-vivo permeation studies using dorsal rat’s skin for the optimal formula in comparison with OLM suspension was performed to estimate OLM permeation from the vesicles. Confocal laser scanning microscopy was applied to track the intensity of penetration of the fluoro-labeled optimal PB through skin layers. To assess vesicles’ membrane elasticity and the role played by Brij®, the deformability index of optimal PB was compared with transethosomes prepared in our previous work11 and traditional bilosomes. Moreover, the safety of the prepared PBs was assured using histopathology. Furthermore, to determine the amount of OLM deposited in skin of male Wistar rats, the optimal PB was compared with transethosomes and OLM suspension. In addition, pharmacodynamic and pharmacokinetic studies for the optimal PB in comparison with the oral tablets were conducted in experimental animals.
Materials
OLM was obtained from FAP Pharmaceutical Co. (Cairo, Egypt). L-ɑ phosphotidylcholine (PC) from egg yolk, cholesterol, Span 60, sodium deoxycholate (SDC), sodium taurocholate (STC), cellulose membrane (12,000–14,000 molecular weight cutoff) and fluorescein diacetate (FDA) were obtained from Sigma Aldrich Chemical Co. (St. Louis, MO, USA). Brijo20 (polyoxyethylene (20) oleyl ether) and Brij52 (polyoxyethylene (2) cetyl ether) were provided by BASF Co. (New Jersy, NY, USA). Methylprednisolone acetate was obtained from Pfizer, Cairo, Egypt. Chloroform, methanol, ethanol 95%, acetonitrile, triethylamine, ethyl acetate and ammonium formate (each HPLC grade) were obtained from Merck (Darmstadt, Germany). Angiosartan® 10 mg oral tablets were obtained from Chemipharm Co. (Cairo, Egypt) and expressed in the manuscript as the oral tablets.
Methods
Formulation of OLM PBs
PBs were assembled by applying thin film hydration method by altering both type and amount of BS and EA (Table 1). Firstly, OLM (25 mg), Span 60 (50 mg), cholesterol (25 mg), BS and EA were completely dissolved in 10 mL chloroform: methanol mixture (2:1) (v/v) in 250 mL round bottom flask and the organic phase was then removed by the aid of rotary-evaporator (Rotavapor, Heidolph VV 2000, Burladingen, Germany) in a 60°C water bath under vacuum.3 The film was hydrated with 10 mL ultra-pure water and stirred for 45 mins. To augment the yield of the obtained vesicles, glass spheres were utilized throughout the hydration stage. After that, the vesicles were sonicated using bath sonicator (Ultra Sonicator, Model LC 60/H Elma, Germany) for 10 mins for further particle size (PS) reduction. For comparative appraisal, OLM loaded traditional bilosomes and OLM loaded transethosomes were prepared by thin film hydration technique. For preparing traditional bilosomes, Span 60 (50 mg), cholesterol (25 mg) and STC (15 mg) were added in 10 mL chloroform: methanol mixture (2:1) (v/v) in 250 mL round bottom flask and the organic phase was then removed and the film was hydrated by 10 mL distilled water and then bilosomes were sonicated for 10 mins. In addition, transethosomes composed of SDC (15 mg) and phospholipid (85 mg) were developed also by thin film hydration technique and the film was hydrated using 10 mL distilled water containing 4% v/v ethanol and the preparation was continued as mentioned in our previous work.11
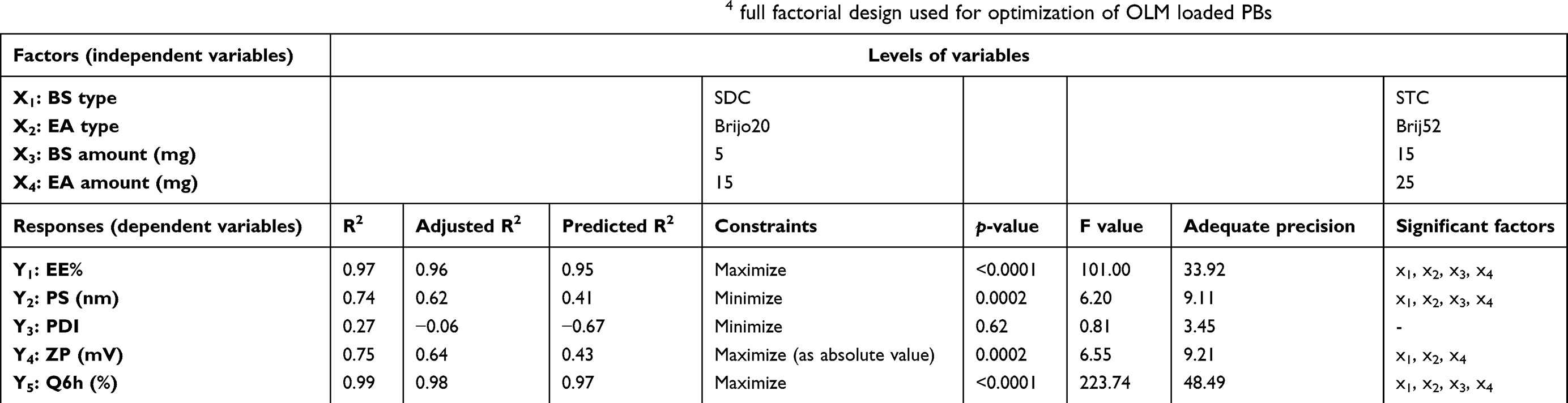 |
Table 1 The levels of independent variables and the model summary statistics of 24 full factorial design used for optimization of OLM loaded PBs |
In-vitro characterization and optimization of OLM PBs
Drug entrapment efficiency percent (EE%) analysis
The EE% of OLM from the produced formulae was determined by applying the direct method.12 The dispersions were centrifuged at 20,000 rpm for 1 hr at 4°C using a cooling centrifuge and the sediment was lysed using methanol and analyzed using UV-Vis spectrophotometer. EE% was calculated using:11
(1)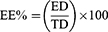
where EE% is the entrapment efficiency percentage, ED is the concentration of entrapped drug and TD is the total drug concentration. The experiments were performed three times and shown as mean±SD.
PS, polydispersity index (PDI) and zeta potential (ZP) analysis
Mean PS, PDI and ZP of the produced formulae were obtained through dynamic light scattering technique utilizing Zetasizer (Malvern Instruments Ltd., UK) after appropriate dilution of the samples.3 The ZP determination was evaluated by studying the electrophoretic mobility of the charged vesicles. All assessments were accomplished in triplicate±SD.
Determination of amount of drug released after 6 hrs (Q6h)
Two milliliters of PBs dispersions were added in plastic cylindrical tubes with a specific permeation area that have one end tightly enclosed with a cellulose membrane and the other connected to the shaft of the USP dissolution apparatus replacing baskets’ position for 6 hrs at 37°C.13 Formulations containing 5 mg of OLM were put in 50 mL PBS pH 7.4 and ethanol mixture (3:2, v/v) to keep sink situation. Aliquots were removed at 1, 2, 3, 4, 5 and 6 hrs. Samples were scanned by UV spectrophotometer at λmax 257 nm. Experiments were assessed in triplicates and represented as mean±SD.
Experimental design construction
A 24 factorial experiment was used to assess the impact of various factors in formulating PBs utilizing Design Expert® software version 7 (Stat Ease, Inc., Minneapolis, MN, USA). The design required performing 16 runs. Four factors were studied: BS type (X1), EA type (X2), BS amount (X3) and EA amount (X4) that were the independent variables, whereas EE% (Y1), PS (Y2), PDI (Y3), ZP (Y4) and Q6h (Y5) were chosen as dependent variables (Table 1).
Selection and validation of the optimal OLM loaded PB
The sorting out of optimal formula depended on the desirability function which permitted investigation of each and every response concurrently. The optimization step was decided to obtain a suggestion with the least PS and PDI and the highest EE%, ZP as absolute value and Q6h. Suggestion of highest desirability was chosen. For approving the model performance, optimal formula was prepared, characterized and correlated to the predicted responses through calculation of the bias percent by using the following equation:14
(2)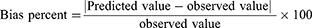
Transmission electron microscopy (TEM)
The optimal PB was morphologically identified by TEM (Joel JEM 1230, Tokyo, Japan). Stained vesicles’ dispersion was put on a carbon grid with copper coat and kept to dry to get a thin film. The sheet of copper was entered into the TEM.12
Differential scanning calorimetry (DSC)
Thermal investigation of 5 mg from each sample of OLM, cholesterol, Span 60, STC, Brij52, physical mixture of OLM with PBs constituents and the optimal PB was fulfilled by the assistance of DSC (DSC-60, Shimadzu Corp., Kyoto, Japan) in a temperature range of 10–250°C at a scanning rate of 5°C/min under inert nitrogen flow (25 mL/min) standardized with purified indium.3
Stability study
The optimal formula was retained at 4°C and 25°C for 90 days. Samples from each formulation were taken at 0, 45 and 90 days. Stability was estimated through evaluating the first measurements relative to the results observed following storing.15 The EE%, PS, PDI, ZP and Q6h of the PBs were determined as mentioned earlier. Statistical significance was analyzed by Student’s t-test using SPSS® software 22.0 (SPSS, Chicago, IL, USA). The change at p≤0.05 was regarded as significant.
Assessment of vesicles’ elasticity
Extrusion method was utilized for comparing the bilayer elasticity of the optimal PB with traditional bilosomes and transethosomes adopting the method reported by Aziz et al3 and deformability index (DI) was calculated. Statistical analysis was evaluated by one-way ANOVA adopting SPSS® software 22.0. Post-hoc test was conducted using Tukey’s HSD test.
Ex-vivo studies
Ex-vivo permeation studies
The permeation capability of OLM suspension and optimal PB via rat’s skin was evaluated. Removal of rat’s skin was done following sacrifice of animals. The setting of the study was the same as that in the in-vitro release but with putting the dorsal skin with the SC confronting the donor compartment instead of cellulose membrane. Aliquots were taken at 2, 4, 8, 10 and 12 hrs time intervals and analyzed using validated HPLC method.16 Permeation flux (Jmax) at last time point and the enhancement ratio were estimated.3 Experiments were assessed in triplicates. Statistical significance was determined by Student’s t-test using SPSS® software 22.0.
Ex-vivo tracking using confocal laser scanning microscopy study (CLSM)
To track the movement of optimal PB through deep skin tissues, the fluoro-labeled PB was formulated by thin film hydration technique. Firstly, FDA (100 mg), cholesterol (25 mg), Span 60 (50 mg), STC (15 mg) and Brij52 (15 mg) were added in 10 mL chloroform: methanol mixture (2:1) (v/v) in 250 mL round bottom flask and the organic phase was removed by the aid of rotary-evaporator in a 60°C water bath under vacuum. The film was then hydrated with 10 mL ultra-pure water and stirred for 45 mins and then sonicated for 10 mins. Dorsal rat’s skin was set with the SC confronting the donor compartment with the previously mentioned arrangement as the permeation study. FDA-packed PB was put on dorsal skin surface and remained for 6 hrs. For comparative evaluation, 1% FDA solution (control) was prepared, added on the rat’s skin and treated as mentioned for FDA-packed PB formulation. Longitudinal sections that were exposed to fluoro-labeled optimal PB and 1% FDA solution were stored in paraffin-wax and sliced using a microtome (Leica MicrosystemsSM2400, Cambridge, UK) and were examined for the existence of fluorescence in skin layers. The sample slides were inspected by invert microscope (LSM 710; Carl Zeiss, Oberkochen, Germany).11
In-vivo studies
A total of 105 male Wistar rats were enrolled in the in-vivo studies, namely histopathological, skin deposition and pharmacodynamic studies. In addition, six male Albino rabbits were involved in the pharmacokinetic study. The study design was authorized by the ethical board of the Faculty of Pharmacy, Cairo University (PI 1867). The utilization and handling of animals in all studies obeyed the EU directive 2010/63/EU. The animals were kept in cages with wide mesh wire bottoms to avoid coprophagy at a temperature of 25°C and humidity of 55%. The animals were put in a dark:light cycle of 12 hrs each and were provided with standard diet and had free access to water. Animals were kept for 7 days for adaptation before experiments.11 For in-vivo studies, lab-made containers containing drug suspension and optimal PB were used to serve as drug-pools. The containers were settled on the previously shaved dorsal animal skin. In addition, the application was performed non-occlusively into the drug-pools.3,7,11 At the end of the previously stated studies, animals were sacrificed by decapitation method following anesthesia.
In-vivo histopathological study
A total of nine rats were classified into three groups of three rats for each group and treatment duration was 1 day. Group I acted as control and group II and group III were treated with drug suspension and optimal PB, respectively. Autopsy from skin samples was fixed in 10% formol saline for 24 hrs, followed by washing and dehydration using alcohol. Specimens were cleared in xylene and fixed in paraffin wax blocks and then sectioned at 4 mm by sledge microtome (Leica MicrosystemsSM2400, Cambridge, UK). The specimens were deparaffinized, stained and histopathologically examined using light microscopy (Axiostar plus, ZEISS, Oberkochen, Germany).11
In-vivo skin deposition study
Seventy-two rats were classified into four groups of 18 rats each, where group I was control and animals in group II, group III and group IV were treated with drug suspension, transethosomes and optimal PB, respectively. A known volume (0.5 mL) of drug suspension, transethosomes and optimal PB was added non-occlusively to dorsal rat’s skin on a specific area that was shaved earlier. After various time intervals of treatment (1, 2, 4, 6, 8 and 10 hrs), three rats per group were killed and the dorsal rat’s skin was cut and analyzed.3,7,11 AUC0–10 values were calculated and compared between treatments. Statistical significance was studied using one-way ANOVA adopting SPSS® software 22.0. Post-hoc test was conducted using Tukey’s HSD test.
In-vivo pharmacodynamic study
Pharmacodynamic study was done by employing a non-invasive blood pressure arrangement (NIBP 200 A; Biopac System, Inc., Goleta, CA, USA) depending on cuff tail method. Twenty-four male Wistar rats were distributed equally into four groups (1–4). Group 1 served as normal control. Hypertension was made in the other groups (2, 3 and 4) by administration of methylprednisolone acetate for 2 weeks subcutaneously. Group 2 acted as hypertensive control. Group 3 received oral tablets (10 mg OLM) (Angiosartan® 10 mg oral tablets) that were mashed and administered by the aid of oral gavage syringe. Group 4 received topical treatment of optimal PB (10 mg OLM). The PBs dispersion was put to a shaved dorsal area of rat’s skin (4.91 cm2). The blood pressure (BP) from the tail was then taken at planned time points.1 Statistical analysis was done by one-way ANOVA using SPSS® software 22.0. Post-hoc test was performed using Tukey’s HSD test. Percent of decrease in BP from hypertensive control was determined.11
In-vivo pharmacokinetic study
Six male Albino rabbits were used in the in-vivo pharmacokinetic assessment. Animals were classified into two groups with the same number. A randomized crossover design was done on two stages separated by 1 week as washout period. Hair on the dorsal part of the first group was shaved and the optimal PB (10 mg OLM) was put on a determined area of 10 cm×5 cm. Further, oral OLM tablets equivalent to 10 mg (Angiosartan® 10 mg oral tablets) were orally given to the second group by utilizing oral gavage. Following the washout period, reverse randomization occurred. Blood samples (2 mL) were withdrawn by retro-orbital plexus puncture in heparin-coated eppendorf tubes after 0, 1, 2, 3, 4, 6, 8, 10, 12, 24 and 48 hrs of administration. Centrifugation of plasma was done at 5000 rpm for 15 mins and put at −20°C until analyzed.
Pharmacokinetic assessment
Olmesartan; the active-form of OLM was determined in the samples of plasma employing a sensitive, precise and selective liquid chromatography mass spectrometry (LC/MS/MS). The concentration of olmesartan was plotted against time and analyzed by Kinetica® 5 software (Thermo Fisher scientific Inc., Waltham, MA, USA) using non-compartmental models. The Cmax and Tmax were determined. In addition, AUC0–48 and AUC0–∞ were calculated. Cmax, AUC0–48 and AUC0–∞ were compared between the treatments employing ANOVA test using SPSS® software version 22. The nonparametric Signed Rank Test (Mann–Whitney’s U test) was used to compare the medians of Tmax for the treatments. The relative bioavailability of optimal PB was calculated and compared to the oral tablets employing the following equation:
(3)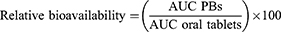
Results and discussion
Analysis of factorial design
Quality by design is an impressive statistical means where it confirms the efficacy of the procedure in term of mathematical relationships. Desirability function is an effective tool for defining the optimal levels of the variables. The model chosen was two-factor interaction. Adequate precision is used to affirm that the model might be used to navigate the design space.17 A ratio superior than four is preferred which was noticed for all dependent variables (except PDI) as illustrated in Table 1. The predicted R2 values were in a good harmony with the adjusted R2 in all dependent variables (Table 1). The negative predicted R2 value of PDI proves that the overall mean is a better interpreter of the response.18
Effect of variables on the EE%
The capability of PBs to enclose a considerable quantity of OLM is important for its potential application as a TDDS. Both cholesterol and Span 60 in PBs bilayers offered high drug entrapment; cholesterol presence provided rigidity to the bilayers19 while Span 60 with high transition temperature (Tc) offered a better adequacy to maintain a more organized gel structure and well-defined vesicles.20 EE% ranged from 40.19±0.12 to 81.21±0.44% (Table 2). ANOVA results proved that BS type (X1), EA type (X2), BS amount (X3) and EA amount (X4) significantly affected EE% (p<0.0001), which is graphically presented in 3-D surface plots (Figure 1A–C).
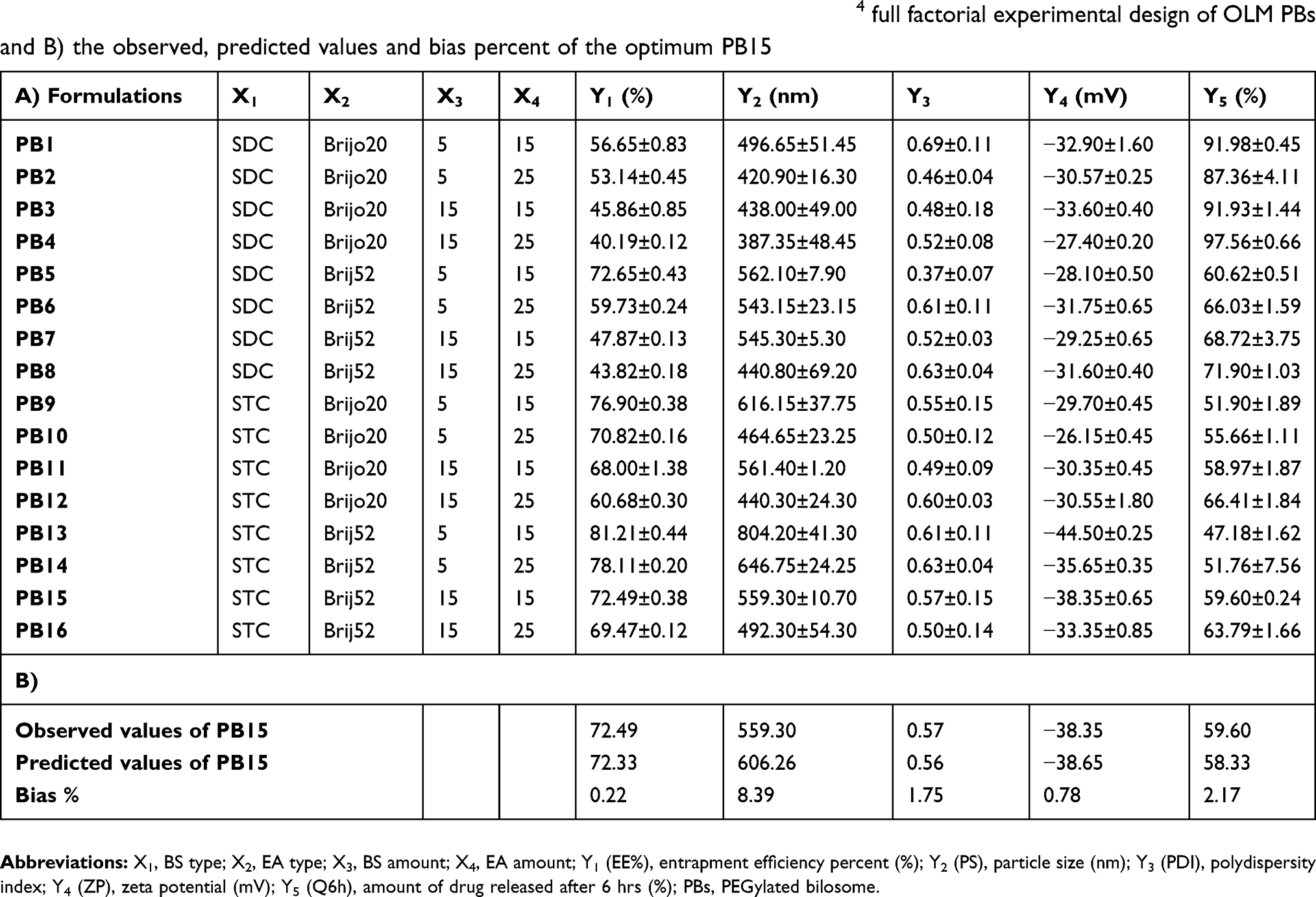 |
Table 2 A) Experimental runs, independent variables and measured responses of the 24 full factorial experimental design of OLM PBs and B) the observed, predicted values and bias percent of the optimum PB15 |
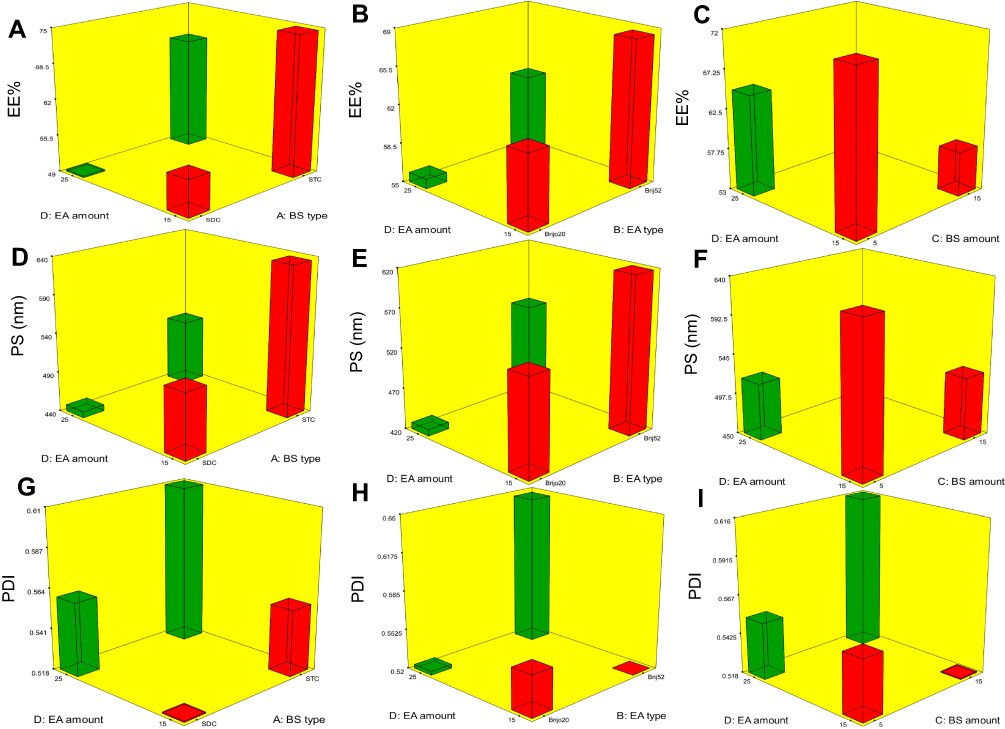 |
Figure 1 Response 3-D plots for the effect of BS type (X1), EA type (X2), BS amount (X3) and EA amount (X4) on EE%, PS and PDI of OLM loaded PBs. Abbreviations: BS, bile salt; EA, edge activator; EE%, entrapment efficiency percent; PS, particle size; PDI, polydispersity index; OLM, olmesartan medoxomil; PBs, PEGylated bilosomes. |
Taking into consideration BS type (X1), relatively high drug entrapment was obtained in formulae consisting of STC compared to SDC formulae. It is worthy to mention that the hydrophobicity of STC is higher than SDC5 which could justify that the higher EE% values observed in PBs containing STC might be related to a widely preferable hydrophobic area for the lipophilic OLM in vesicles’ bilayers.21 Regarding EA type (X2), EE% values were found to be higher in formulae containing Brij52 compared to Brijo20 which might be linked to the HLB values of both Brij52 (HLB=5.3) and Brijo20 (HLB=15.3).8,22 As previously declared, the lower HLB value of EA resulted in elevated drug EE%.23 Furthermore, Brij® consists of acyl chain with different lengths with a saturated or unsaturated carbon chain and PEG repeating units. Brijo20 contains acyl chain of C18, unsaturated double-bond and 20 PEG repeating units while Brij52 contains acyl chain of C16 and two PEG repeating units. Tagami et al8 reported that Brijo20, with an unsaturated acyl chain, did not result in formulations with good drug entrapment compared to Brij52 which contains no double-bond in its structure. Existence of the unsaturated double-bond in the chain of carbon might cause it to twist which leads to the vesicle bilayer being leakier as the packing of the molecules might not be firm.24 In addition, the lipid-phase transition temperature (Tc) of the EA could be critical factor in demonstrating EA effect on EE%. The Tc of Brij52 and Brijo20 are 36–38°C and 25–30°C, respectively.8 It was recorded that the higher the Tc of the EA, the better its capability to make a defined structure and a more ordered bilayer that might result in higher EE% values.24
ANOVA results revealed that by raising BS amount (X3) from 5 to 15 mg, a significant negative impact on EE% occurred. Hofmann and Mysels25 illustrated that BS form helical mixed micelles in the vesicles’ bilayers. This explains that at a higher BS amount (15 mg), the solubility of OLM in the aqueous phase increased due to potential presence of mixed micelles which compromised the EE% of OLM. Regarding the impact of EA amount (X4) on EE%, it was noted that increasing the EA amount from 15 to 25 mg resulted in a consequent reduction of EE%. This might be related to the formation of pores in vesicle bilayers, when the EA amount increased which decreased EE% values.26
Effect of variables on PS
PS of the vesicles has a leading impact on their in-vivo metabolism. Decreasing the PS could significantly abolish their detection by the complements in the blood and increase their circulation time which enhances the therapeutic efficiency of entrapped drugs.27 The PS of PBs was in the range of 387.40±48.45 to 804.20±41.30 nm as illustrated in Table 2. The impact of BS type (X1), EA type (X2), BS amount (X3) and EA amount (X4), on the PS of PBs is graphically represented in 3-D surface plots (Figure 1D–F).
BS type (X1) showed a significant impact on PS (p=0.0009). Smaller PS was obtained for vesicles prepared with SDC compared to those containing STC. The difference in PS might be related to the difference in the structure of the used BS.6 Aburahma5 illustrated the difference in molecular weight for both SDC (414.6 gm/mol) and STC (537.68 gm/mol). Therefore, the bulkier STC might have produced larger PS of STC-PBs in comparison with SDC-PBs. Furthermore, increase in the molecular weight leads to increased viscosity of the nanodispersion and might result in aggregations and augmentation of the PS.28 Moreover, the obtained results asserted that EA type (X2) showed a significant effect on the PS of the vesicles (p=0.0007). The PS of the formulae containing Brij52 was higher compared to Brijo20 formulae. This could be associated with that the EA of higher HLB value (Brijo20) resulted in augmentation of the surface energy and thus decreased the PS. In addition, as previously mentioned, Brijo20 and Brij52 contain 20 and 2 PEG units, respectively. It is reported that the increase in PEG content might slow down the rate of vesicles’ precipitation and hence hinders vesicles’ agglomeration.29
BS amount (X3) showed a significant influence on the PS of PBs (p=0.0021). Raising BS amount from 5 to 15 mg decreased PS because of the reduction in surface tension of the vesicles.5 Another speculated theory is that BS at higher amount tend to form mixed micelles which are lesser than the original size of vesicles.30 ANOVA results confirmed that increasing the EA amount (X4) significantly decreased the PS (p=0.0010). The possible reason might be due to that EA at smaller amount (15 mg) was not satisfactory enough to effectively cover the lipids and reduce the surface tension. Hence, the PS of vesicles was higher and larger vesicles were formed. Moreover, the decrease in PS could be explained by the augmentation of the curvature of PBs at greater EA amount.15
Effect of variables on PDI
Concerning PDI, zero value demonstrates monodisperse dispersion. On the contrary, a value of 1 indicates polydisperse dispersion.7 All inspected factors; BS type (X1), EA type (X2), BS amount (X3) and EA amount (X4) revealed no significant effect with p-values of 0.37, 0.36, 0.80 and 0.33, respectively, which is graphically represented in 3-D surface plots (Figure 1G–I). PDI of the measured formulae ranged from 0.37±0.07 to 0.69±0.11 as illustrated in Table 2. This showed that the prepared PBs were polydisperse but with appropriate values.31
Effect of variables on ZP
ZP is frequently employed to assess the nanoparticle’s charge which represents a good indicator about the stability of nanoparticles.32 ZP ranged from −26.15±0.45 to −44.50±0.25 mV (Table 2). All factors were significant except BS amount (X3) (p=0.5009). The influence of BS type (X1), EA type (X2), BS amount (X3) and EA amount (X4) on the ZP of PBs is graphically depicted in 3-D surface plots (Figure 2A–C).
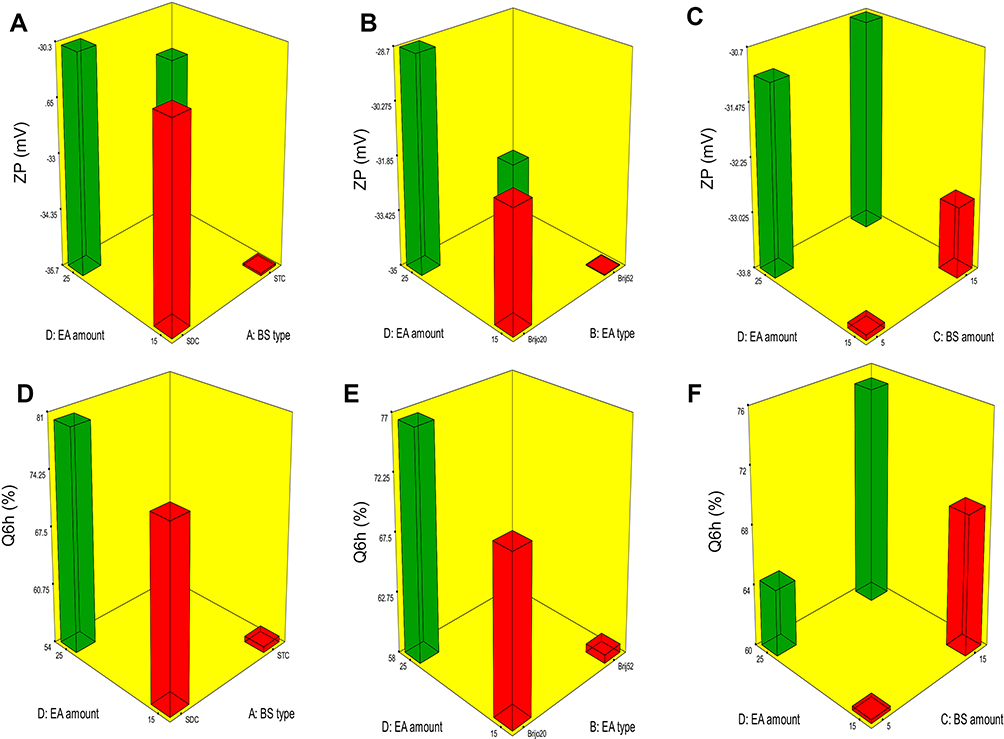 |
Figure 2 Response 3-D plots for the effect of BS type (X1), EA type (X2), BS amount (X3) and EA amount (X4) on ZP and Q6h of OLM loaded PBs. Abbreviations: BS, bile salt; EA, edge activator; ZP, zeta potential; Q6h, amount of drug released after 6 hrs; OLM, olmesartan medoxomil; PBs, PEGylated bilosomes. |
It was observed that by changing BS type (X1), ZP significantly changed (p=0.0054). STC increased ZP of vesicles compared to SDC. This difference might be due to extra OH group in STC which contains three OH groups, compared to SDC that contains two OH groups.5,33 Further, taurine BS are more acidic than unconjugated BS3 due to the presence of highly charged taurine amino-acid which increases the negative value of ZP.34 Moreover, higher molecular weight BS like STC usually have thick and malleable interfacial layers and are more reluctant to stress relative to lower molecular weight BS as SDC that are predispose to coalescence when they are present in close contact.35 It is worth mentioning that vesicles produced by using STC were significantly larger than those produced using SDC which might also be related to the greater ZP-values of the PBs containing STC. Vesicles of greater ZP-values revealed high repulsion energy among the charged vesicular bilayers with subsequent increased spaces between them resulting in production of bigger vesicles.3
Regarding EA type (X2), results displayed that it had a significant impact on ZP (p=0.0004). Higher ZP-values were obtained upon incorporation of Brij52 in vesicles in comparison to Brijo20. It was recorded that the EA with higher Tc (Brij52), as mentioned earlier, might be important in preparing more ordered stable vesicles.24 On the contrary, Brijo20 with 20 PEG repeated units had lower ZP. This could be related to the higher amount of hydrophilic PEG steric shield that slightly screened the surface charge of carboxyl groups at the surface of the vesicles and led to lower ZP-values.36 Additionally, increasing EA amount (X4) significantly decreased the ZP (p=0.0205). This could be because of the tendency of the investigated EAs to reside on the surface of the vesicular bilayers related to their hydrophilic character which might have resulted in obscuring the negative charge of the PBs.33
Effect of variables on Q6h
Amount of OLM released after 6 hrs ranged from 47.18±1.62 to 97.56±0.66% (Table 2). ANOVA results showed that BS type (X1), EA type (X2), BS amount (X3) and EA amount (X4) had a significant impact on Q6h (p<0.0001), which is graphically depicted in 3-D surface plots in Figure 2D–F. In addition, Figure 3A–B illustrates the release profiles of OLM from the prepared PBs at different time intervals.
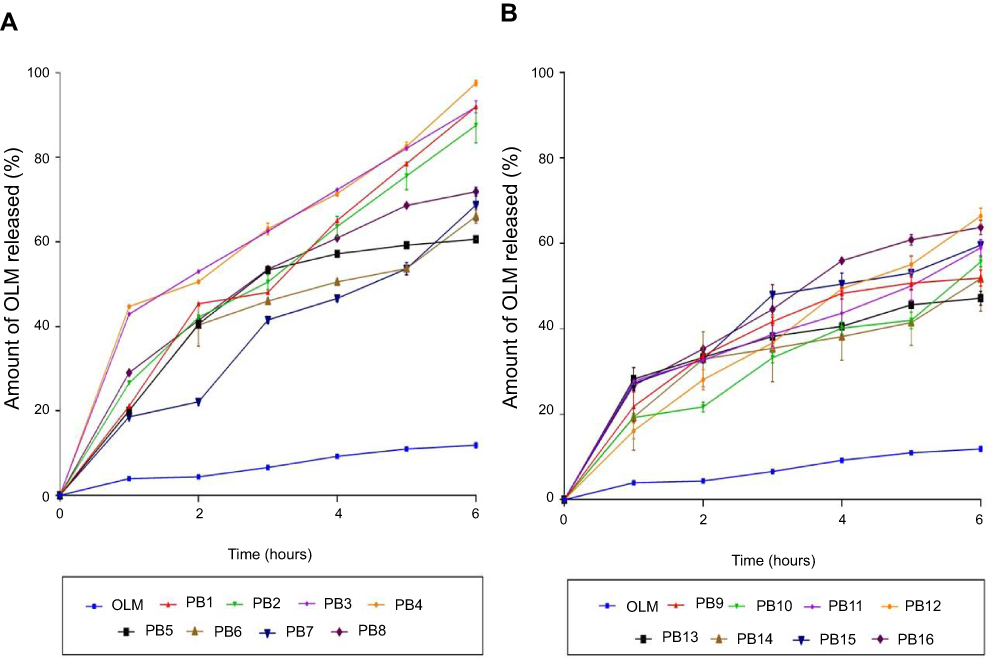 |
Figure 3 The release profiles of OLM from the prepared 16 PB formulations compared to OLM suspension (A) release profiles of OLM and PB1:PB8 and (B) release profiles of OLM and PB9:PB16. Data represented as mean±SD (n=3). Abbreviations: OLM, olmesartan medoxomil; PB, PEGylated bilosome. |
Regarding BS type (X1), it was found that PBs containing SDC produced a significantly greater amount of OLM released relative to STC containing PBs. As previously mentioned, the hydrophobicity is higher in case of STC compared to SDC while critical micelle concentration correlates inversely with hydrophobicity. The large number of methylene groups in the side chain of the BS during conjugation with taurine results in decreased critical micelle concentration values which could attribute for the decrease of OLM release from STC containing PBs.37 For EA type (X2), PBs containing Brij52 showed significantly lower amount of OLM released than the corresponding formulae containing Brijo20. Based on PEG content and HLB values, Brijo20 was found to be more hydrophilic than Brij52, as previously stated. As the hydrophilicity increases, the membrane microviscosity decreases and the rate of flip-flop and membrane penetration increase.38 In addition, it was reported that by increasing the bulk of hydrophilic portions of the EA (PEG), the flippase activity increases which could attribute to the augmentation of the amount of OLM released.39
Increase in BS amount (X3) resulted in increasing the amount of OLM released which could be related to that the incorporation of BS through the bilayer resulted in discharge of the lipophilic drug integrated. Further, BS might associate with the vesicles’ bilayers and convert them into mixed micelles which results in boosting the solubility of OLM and augmenting OLM release.5 Moreover, increasing EA amount (X4) from 15 to 25 mg resulted in an increase in the amount of OLM released. Lower OLM release at smaller EA amount might be explained because of the existence of the lipid membranes in firm and less leaky form which hinders drug release from PBs, while increasing EA amount to 25 mg resulted in pore formation in PBs bilayers which augmented OLM release.40
Selection and validation of the optimal PB
The optimal formula from experimental design was PB15 which met the criteria determined before. High correlation was noticed among the observed and predicted values of PB15 (Table 2). In addition, the average bias percent values for all the obtained responses were smaller than 10% revealing the high model’s predictability capacity.14 Subsequently, PB15 was chosen as the optimal formula for extra studies.
Transmission electron microscopy (TEM)
TEM configuration as observed in Figure 4 confirmed that vesicles were spherical with narrow size distribution and no vesicles with irregular shape were visible. The PS of the vesicles determined by Zetasizer was in a perfect harmony with that observed using TEM. In addition, TEM images demonstrated that the vesicles had a smooth surface with the presence of PEG (lighter colored peripheries) around the vesicles.41
 |
Figure 4 Morphology of the selected vesicle (PB15). Abbreviation: PB, PEGylated bilosome. |
Differential scanning calorimetry (DSC)
Figure 5 illustrates the thermograms of OLM, cholesterol, Span 60, STC, Brij52, physical mixture of OLM along with PBs constituents and PB15. The DSC of OLM demonstrated a melting endothermic peak at 183.86°C.2 Thermogram of cholesterol showed a sharp melting endotherm at 146.81°C.3 Span 60 revealed a narrow melting endotherm at 55.01°C revealing its crystalline state which is attributed to its melting point.3 Furthermore, Brij52 demonstrated a Tc of 36–38°C.8 The DSC thermogram of STC showed a very wide endotherm that included the 50–120°C temperature range followed by exothermic crystallization peak at 238.30°C.3,42 The DSC of the physical mixture of OLM with PBs constituents disclosed that the OLM particular peak was existent supporting its presence in crystalline state. For PB15, the distinctive peak of OLM completely vanished which might be related to that OLM was entrapped inside the vesicles.43 These findings might also suggest a major interaction between the drug and the constituents of the bilayer structure which might be the reason for the high EE% of OLM. These interactions could account for the good vesicle shape, structure and excellent stability.44
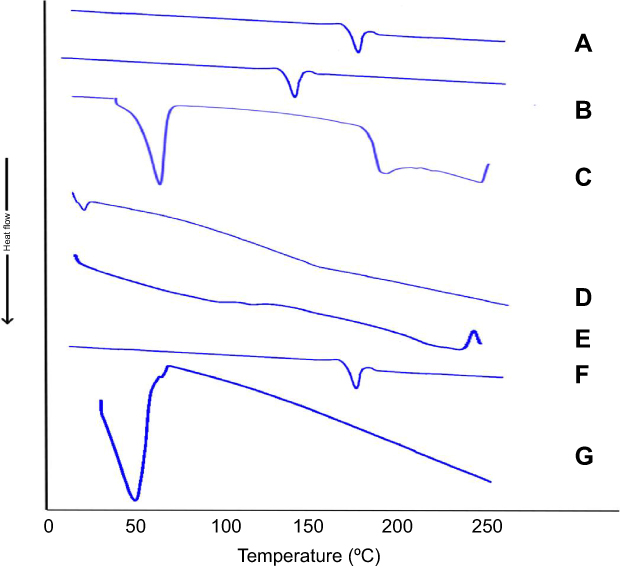 |
Figure 5 DSC thermograms of (A) OLM, (B) cholesterol, (C) Span 60, (D) Brij52, (E) STC, (F) physical mixture of PB15 components and (G) PB15. Abbreviations: STC, sodium taurocholate; OLM, olmesartan medoxomil; PB, PEGylated bilosome. |
Stability study
After 45 and 90 days in both 4°C and 25°C, stored vesicles showed no recognized aggregation or transformation in their shape. No significant change in EE%, PS, PDI, ZP and Q6h of the stored vesicles relative to the freshly prepared PBs was observed as shown in Table 3. Basically, charged vesicles are more stable against aggregation than un-charged vesicles. The prepared PBs were negatively charged because of STC that increased the ZP and prohibited the coalescence of the vesicles. Moreover, it was reported that Span 60 containing formulae were generally very stable and drug leakage from vesicles prepared using Span 60 was small and this might be related to its high Tc and its capability to form more ordered bilayers.20 In addition, the high stability of the PBs was related to the presence of the PEG in Brij52 structure which stabilized vesicle membrane by steric hindrance.36
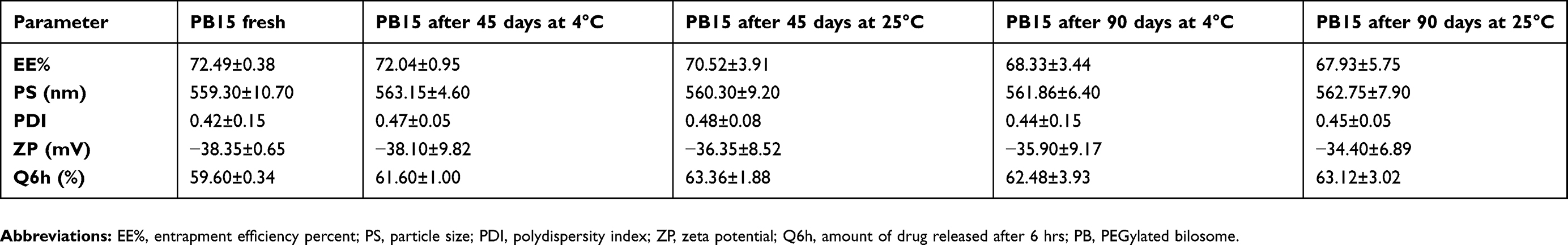 |
Table 3 Effect of storage on the physical properties of PB15 |
Elasticity measurement
BS are able of destabilizing vesicles’ bilayers and hence increase their deformable properties. The deformability index values of PB15, traditional bilosomes and transethosomes were 28.39±5.71 g, 5.88±0.90 g and 14.94±0.63 g, respectively. The significant (p<0.05) dominance of PB15 over traditional bilosomes and transethosomes could be attributed to the presence of extra PEGylated EA in PBs bilayers. In addition, the higher DI of transethosomes compared to traditional bilosomes could be justified by the presence of both BS and ethanol rather than BS alone in traditional bilosomes.11 The previous results verified the advantage of PB15 over traditional bilosomes and transethosomes in squeezing through skin with superior elasticity while preserving PS.
Ex-vivo studies
Ex-vivo permeation studies
Numerous animal models have been recommended as an alternative for human skin and have been utilized to measure percutaneous permeation of drugs. These included porcine, primates, mouse, rat, snake and guinea pig models.11,45 Rat’s skin was recognized to mimic human SC in thickness, composition and transport resistance and it consists of SC, epidermis and dermis.46 From Figure 6, it could be declared that the amount of OLM permeated from PB15 was significantly greater than that permeated from OLM suspension. The ER exceeded six folds for PB15 relative to drug suspension as presented in Table 4. The significant lower permeability of OLM suspension compared to PB15 might be due to the properties of OLM. It is suggested that the log partition coefficient value of 5.6 could be the reason of the low permeation of OLM.2 A parabolic relationship between the skin permeation ability and lipophilic character of several drugs has been formerly recorded with the highest permeability at log P of about three to four.47 For highly deformable vesicles as PBs, the condition is dissimilar as they could permeate intact through the SC and hence the permeability of the vesicle basically controls the movement of the drug.48
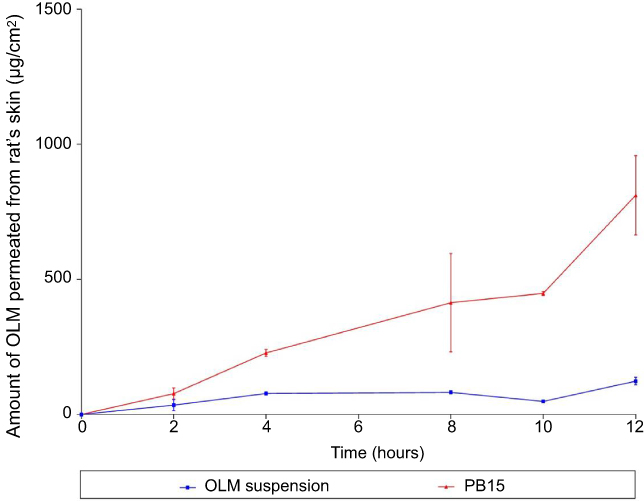 |
Figure 6 Cumulative amount of OLM permeated per unit area from PB15 relative to OLM suspension. Data represented as mean±SD (n=3). Abbreviations: OLM, olmesartan medoxomil; PB, PEGylated bilosome. |
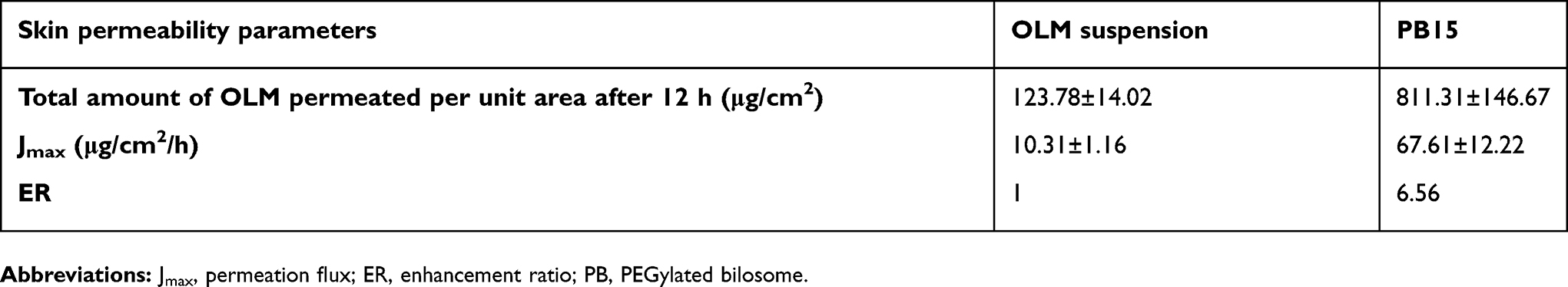 |
Table 4 Skin permeability parameters of OLM after topical application of OLM suspension and PB15 |
Ex-vivo tracking using CLSM
CLSM investigation was done to evaluate the capability of penetration and fluorescence intensity following transdermal delivery of fluoro-labeled PB15. Images obtained with CLSM (Figure 7A–B) depicted that the dye loaded PB15 exhibited accumulation of fluorescence in different skin tissues. Images were captured from longitudinal section (perpendicular to the skin surface) which provided details about the allocation in the SC and skin tissues. The vesicles demonstrated symmetric distribution on the skin surface and maximum penetration of fluoro-labeled PB15 relative to control solution which might be related to the elasticity provided by the EAs present in fluoro-labeled PB15. High intensity of fluoro-labeled PB15 was noticed near the hair-follicles. This observation agrees with the former investigations supporting that the hair-follicles serve as a depot for the transdermally applied carriers from which the liberation of the entrapped drug might happen. It is worthy to mention that the hair-follicles are surrounded by blood capillaries and thus the substances penetrating through the hair-follicles could reach the tissues surrounding the follicle and get to the blood circulation.14
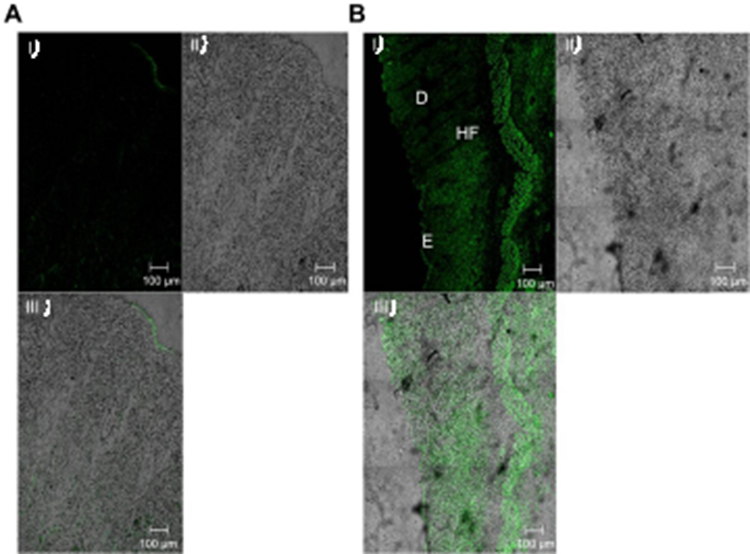 |
Figure 7 A tile scan confocal laser microscope photomicrographs of longitudinal section in rat’s skin treated with (A) FDA control solution (B) fluoro-labeled PB15. (I) fluorescence light, (II) transmitted light and (III) merge between fluorescence light and transmitted light. Abbreviations: FDA, fluorescein diacetate; E, epidermis; D, dermis; HF, hair follicles; PB, PEGylated bilosome. |
In-vivo studies
In-vivo histopathological study
Histopathological scanning (Figure 8) of group II (OLM suspension) and group III (PB15) displayed no alterations in the histopathology of different skin layers compared to untreated skin sections of group I (control). The treated skin samples showed perfect normal skin features. These findings asserted that the prepared PBs had good safety toward skin.
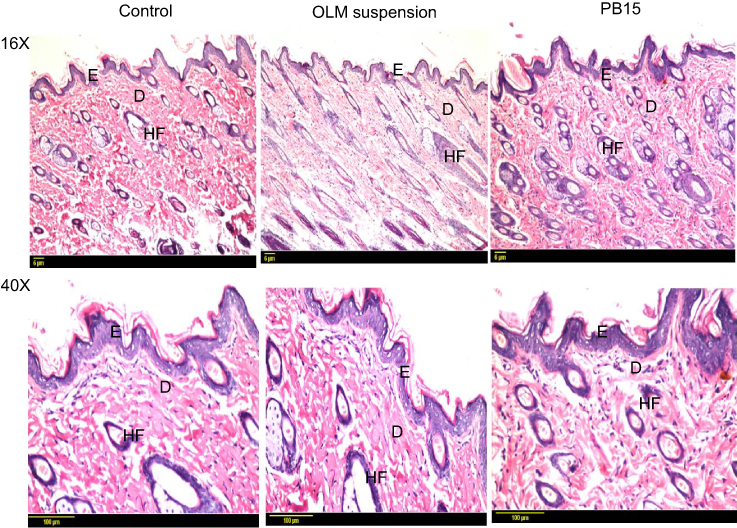 |
Figure 8 Photomicrographs showing histopathological sections (hematoxylin and eosin stained) of normal untreated rat’s skin (group I), rat’s skin treated with OLM suspension (group II) and rat’s skin treated with PB15 (group III) with magnification power of 16x and 40x, respectively. Abbreviations: E, epidermis; D, dermis; HF, hair follicles; OLM, olmesartan medoxomil; PB, PEGylated bilosome. |
In-vivo skin deposition
In-vivo skin deposition results of OLM from transethosomes and PB15 relative to OLM suspension are depicted in Figure 9. Statistical evaluation displayed that there was a significant difference (p<0.05) among the AUC0-10 of OLM suspension, transethosomes and PB15 topically treated groups. The AUC0-10 calculated for OLM loaded PBs (1115.64±110.70 μgh/cm2) and for OLM loaded transethosomes (680.46±17.99 μgh/cm2) were about 5.68 and 3.47 folds higher than that of OLM suspension (196.30±99.66 μgh/cm2), respectively. Further, multiple comparison demonstrated the superiority of PB15 over transethosomes and OLM suspension. The significantly higher skin deposition capacity of OLM from PB15 was related to the capability of PBs as highly deformable vesicles, based on elasticity results, to partition themselves via SC layers enclosing the entrapped OLM to deeper tissues. It is worth stating that adequate material transfer across the skin is not only a result of the modified skin features but rather counts on the movement of the intact and highly deformed vesicles through the SC.48 In addition, SC is constructed of proteins and fats. BS are known to solubilize membrane proteins and phospholipids effectively which could result in enhanced vesicles’ penetration.3 Moreover, the results declared that the amount of OLM deposited decreased by increasing time. This could be attributed to that the cellular uptake of skin became saturated and hence the amount of OLM transferred into the blood from the skin exceeded the quantity of drug penetrating the skin with resultant reduction of the skin deposition.49
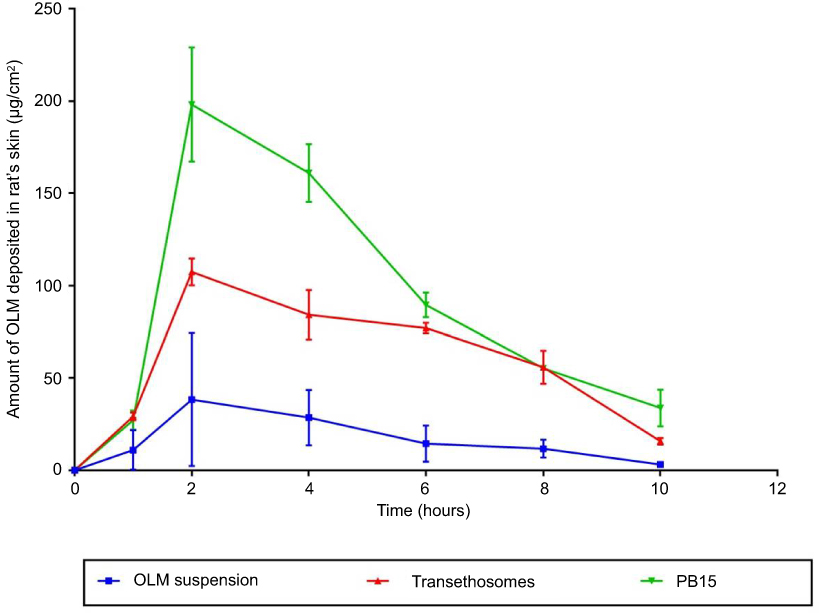 |
Figure 9 In-vivo skin deposition profiles of OLM versus time from PB15, transethosomes and OLM suspension after topical application. Data represented as mean±SD (n=3). Abbreviations: OLM, olmesartan medoxomil; PB, PEGylated bilosome. |
In-vivo pharmacodynamic study
The antihypertensive effect of transdermally applied PB15 was studied by measuring the lowering effect of BP upon applying the optimal formula to the skin of hypertensive rats. Both oral tablets and PB15 treated groups showed similar antihypertensive activity and there was no significant difference between both groups and normal control group at (1, 2, 4 and 6 hrs) after treatment. However, the BP of oral tablets treated group was maintained at normal levels up to 6 hrs only posttreatment and oral tablets were unable to revert BP to normal level at 8, 24 and 26 hrs posttreatment (Table 5). On the contrary, transdermally applied PB15 reduced the BP to normal values with the highest percent reduction of 35.00±5.00% at 24 hrs time point and BP was maintained at normal range for up to 26 hrs. Although the BP values of PB15 treated group at 28 and 48 hrs were not at normal values likewise oral tablets but there was a significant difference (p<0.05) between PB15 treated group and oral tablets treated group with superior percent reduction in BP values of 20.00±6.00% and 13.00±4.00% compared to 1.00±0.00% and 3.00±1.00% for PB15 and oral tablets treated group at 28 and 48 hrs, respectively (Table 5). Furthermore, post-hoc test showed that there was no significant change (p>0.05) between the normal control group and PB15 treated group while there was a significant difference (p<0.05) between oral tablets treated group and normal control group at 8, 24 and 26 hrs posttreatment. The obtained results could be related to the presence of PEG moieties in PBs which could inhibit effectively the in-vivo clearance of OLM by mononuclear phagocyte.50 These results asserted that the PBs were efficient in maintaining the rat’s BP for prolonged period compared to oral tablets.
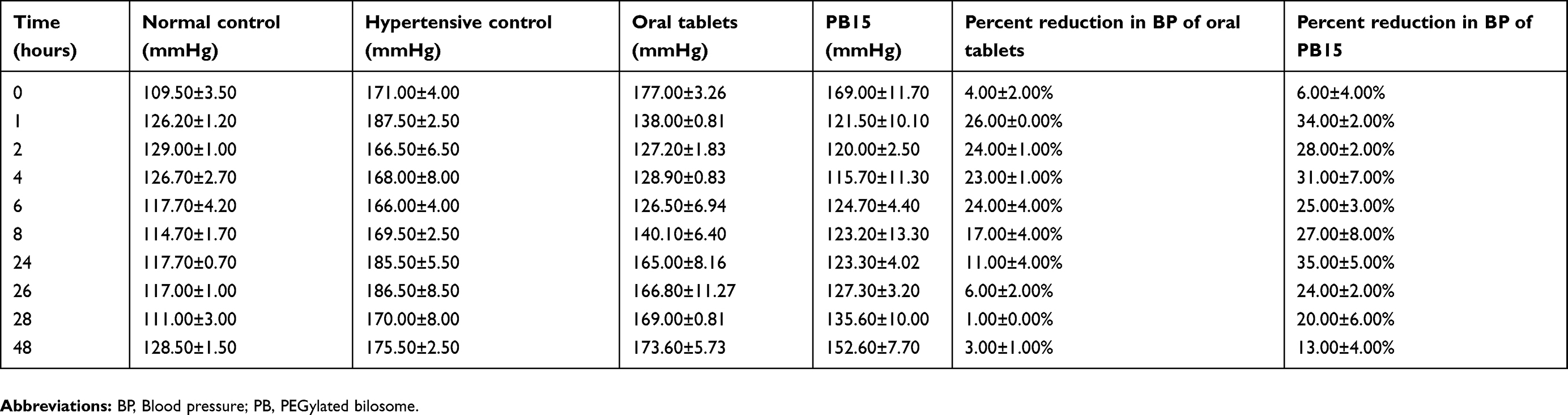 |
Table 5 Influence of oral tablets and PB15 on mean BP in methylprednisolone acetate-induced hypertensive rats |
In-vivo pharmacokinetic study
OLM is ester prodrug of olmesartan.2 Subsequently in assessing the transdermal bioavailability of OLM from PB15, it is speculated to be converted in blood circulation to olmesartan due to the presence of esterase enzymes.51 Hence, olmesartan was used as indicator for determination of the in-vivo pharmacokinetic behavior of OLM. The mean concentration time plots in male Albino rabbits after 48 hrs posttreatment are shown in Figure 10 and the main pharmacokinetic parameters are tabulated in Table 6. There was a significant difference (p<0.05) between PB15 and oral tablets in AUC0–48 and AUC0–∞ while there was no significant difference between PB15 and oral tablets in Cmax and Tmax with p-values of 0.96 and 0.10, respectively. The AUC0–48 and AUC0–∞ values of PB15 treated group were about 2.35 times higher than those of oral tablets treated group. In addition, the Tmax value of PB15 was numerically but not significantly higher compared to oral tablets. The high relative bioavailability of PB15 (235.04%) confirmed what was hypothesized at the beginning of the work and clearly defined the superiority of PB15 over oral tablets which could be associated with bypassing the liver first-pass metabolism in case of transdermal PB15 which consequently enhanced OLM bioavailability. Moreover, based on PB15 composition, the presence of PEG might have provided higher stability levels in the bloodstream preventing interactions with plasma opsonins which could have led to longer circulation time in the blood and thus prolonged the systemic efficacy. In addition, PEG moieties might have facilitated the access of PBs to the lymph nodes and subsequently resulted in augmentation of vesicles’ ability for bypassing the first-pass effect, as previously mentioned.10 Concisely, the findings verified that PB15 could act as effective transdermal delivery approach for OLM by avoiding its oral problems.
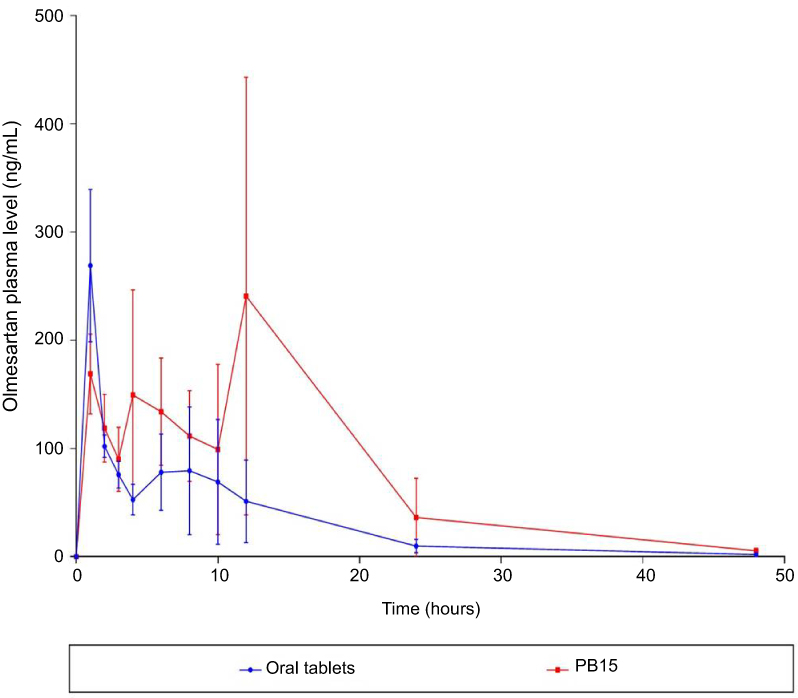 |
Figure 10 Pharmacokinetic profiles of olmesartan versus time from oral tablets and topically applied PB15. Data represented as mean±SD (n=6). Abbreviation: PB, PEGylated bilosome. |
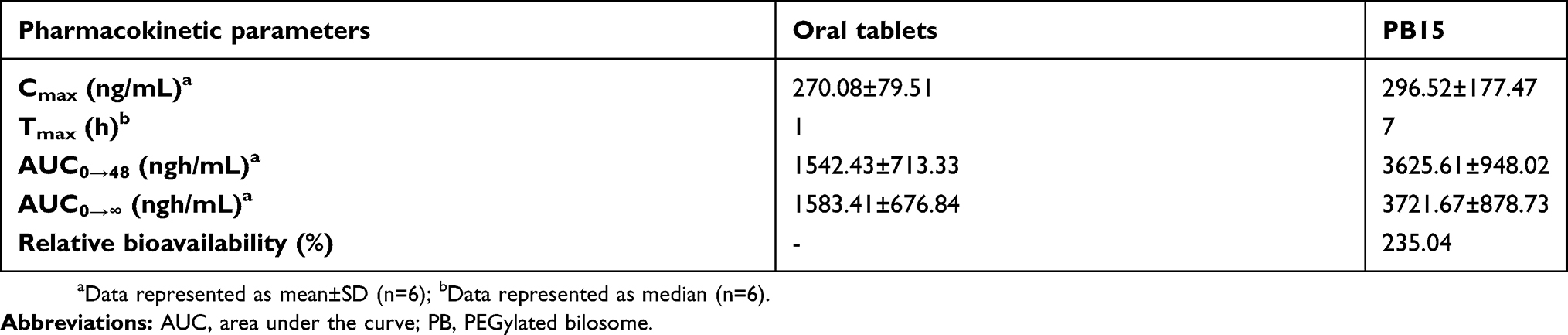 |
Table 6 Pharmacokinetic parameters of olmesartan after administration of oral tablets and PB15 |
Conclusion
In the present study, PBs were developed for transdermal delivery of OLM. A complete 24 full factorial experiment assisted in formulating the optimal formula (PB15) that showed spherical shape, high drug EE% and small PS. In addition, ex-vivo studies showed superior permeation from PB15 over OLM suspension. CLSM verified that the PBs could penetrate the deep skin layers. In addition, histopathological study confirmed the tolerability of PB15. In-vivo skin deposition study confirmed higher retention tendency of OLM in rat’s skin from PB15 compared to transethosomes and drug suspension. Further, PB15 showed a controlled effect on hypertensive rats that lasted for prolonged time. Pharmacokinetic study proved the capability of PB15 to augment the bioavailability of OLM compared to oral tablets. In the consequence, the findings proved that PB15 could be a potential TDDS for OLM by abolishing its extensive first-pass metabolism and its oral problems.
Disclosure
The authors affirm that they do not have conflicts of interest to disclose in this work.
References
1. Ahad A, Aqil M, Kohli K, Sultana Y, Mujeeb M. Nano vesicular lipid carriers of angiotensin II receptor blocker: anti-hypertensive and skin toxicity study in focus. Artif Cells Nanomed Biotechnol. 2016;44(3):1002–1007. doi: 10.3109/21691401.2015.1008509
2. B A, D N, Veerabrahma K. Development of olmesartan medoxomil lipid based nanoparticles and nanosuspension: preparation, characterization and comparative pharmacokinetic evaluation. Artif Cells Nanomed Biotechnol. 2018;46(1):126–137. doi:10.1080/21691401.2017.1299160
3. Aziz DE, Abdelbary AA, Elassasy AI. Investigating superiority of novel bilosomes over niosomes in the transdermal delivery of diacerein: in vitro characterization ex vivo permeation and in vivo skin deposition study. J Liposome Res. 2019;29(1):73–85. doi:10.1080/08982104.2018.1430831
4. Ammar HO, Ibrahim M, Mahmoud AA, Shamma RN, El Hoffy NM. Nonionic surfactant based in situ forming vesicles as controlled parenteral delivery systems. AAPS PharmSciTech. 2018;19(3):1001–1010. doi:10.1208/s12249-018-0981-8
5. Aburahma MH. Bile salts containing vesicles: promising pharmaceutical carriers for oral delivery of poorly water-soluble drugs and peptide/protein-based therapeutics or vaccines. Drug Deliv. 2016;23(6):1847–1867. doi: 10.3109/10717544.2014.976892
6. Abdelbary AA, Abd-Elsalam WH, Al-mahallawi AM. Fabrication of novel ultradeformable bilosomes for enhanced ocular delivery of terconazole: in vitro characterization, ex vivo permeation and in vivo safety assessment. Int J Pharm. 2016;513(1–2):688–696. doi:10.1016/j.ijpharm.2016.10.006
7. Al-mahallawi AM, Abdelbary AA, Aburahma MH. Investigating the potential of employing bilosomes as a novel vesicular carrier for transdermal delivery of tenoxicam. Int J Pharm. 2015;485(1):329–340. doi:10.1016/j.ijpharm.2015.03.033
8. Tagami T, Ernsting MJ, Li SD. Optimization of a novel and improved thermosensitive liposome formulated with DPPC and a brij surfactant using a robust in vitro system. J Control Release. 2011;154(3):290–297. doi:10.1016/j.jconrel.2011.05.020
9. Rangsimawong W, Opanasopit P, Rojanarata T, Ngawhirunpat T. Terpene containing PEGylated liposomes as transdermal carriers of a hydrophilic compound. Biol Pharm Bull. 2014;37(12):1936–1943. doi:10.1248/bpb.b14-00535
10. Jain S, Tiwary AK, Jain NK. PEGylated elastic liposomal formulation for lymphatic targeting of zidovudine. Curr Drug Deliv. 2008;5(4):275–281. doi:10.2174/156720108785915078
11. Albash R, Abdelbary AA, Refai H, El-Nabarawi MA. Use of transethosomes for enhancing the transdermal delivery of olmesartan medoxomil: in vitro, ex vivo and in vivo evaluation. Int J Nanomedicine. 2019;14:1953–1986. doi:10.2147/IJN.S196771
12. Abdellatif MM, Khalil IA, Khalil MAF. Sertaconazole nitrate loaded nanovesicular systems for targeting skin fungal infection: in-vitro, ex-vivo and in-vivo evaluation. Int J Pharm. 2017;527(1–2):1–11. doi:10.1016/j.ijpharm.2017.05.029
13. Morsi N, Ghorab D, Refai H, Teba H. Ketoroloac tromethamine loaded nanodispersion incorporated into thermosensitive in situ gel for prolonged ocular delivery. Int J Pharm. 2016;506(1–2):57–67. doi:10.1016/j.ijpharm.2016.04.021
14. Abdel-Hafez SM, Hathout RM, Sammour OA. Tracking the transdermal penetration pathways of optimized curcumin loaded chitosan nanoparticles via confocal laser scanning microscopy. Int J Biol Macromol. 2018;108:753–764. doi:10.1016/j.ijbiomac.2017.10.170
15. Zeb A, Qureshi OS, Kim HS, Cha JH, Kim HS, Kim JK. Improved skin permeation of methotrexate via nanosized ultradeformable liposomes. Int J Nanomedicine. 2016;11:3813–3824. doi:10.2147/IJN.S109565
16. Bajerski L, Rossi RC, Dias CL, Bergold AM, Frӧehlich PE. Development and validation of a discriminating in vitro dissolution method for a poorly soluble drug olmesartan medoxomil: comparison between commercial tablets. AAPS PharmSciTech. 2010;11(2):637–644. doi:10.1208/s12249-010-9421-0
17. De Lima LS, Araujo MDM, Quináia SP, Migliorine DW, Garcia JR. Adsorption modeling of Cr, Cd and Cu on activated carbon of different origins by using fractional factorial design. Chem Eng J. 2011;166(3):881–889. doi:10.1016/j.cej.2010.11.062
18. Alexander D, Tropsha A, Winkler DA. Beware of R2: simple, unambiguous assessment of the prediction accuracy of QSAR and QSPR models. J Chem Inf Model. 2015;55(7):1316–1322. doi:10.1021/acs.jcim.5b00206
19. Gaafar PM, Abdallah OY, Farid RM, Abdelkader H. Preparation characterization and evaluation of novel elastic nanosized niosomes (ethoniosomes) for ocular delivery of prednisolone. J Liposome Res. 2014;24(3):204–215. doi:10.3109/08982104.2014.881850
20. Vora B, Khopade AJ, Jain NK. Proniosome based transdermal delivery of levonorgestrel for effective contraception. J Control Release. 1998;54(2):149–165. doi:10.1016/S0168-3659(97)00100-4
21. Arzani G, Haeri A, Daeihamed M, Bakhtiari Kaboutaraki H, Dadashzadeh S. Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge. Int J Nanomedicine. 2015;10:4797–4813. doi:10.2147/IJN.S84703
22. Abdelkader H, Ismail S, Kamal A, Alany RG. Preparation of niosomes as an ocular delivery system for naltrexone hydrochloride: physicochemical characterization. Pharmazie. 2010;65(11):811–817. doi: 10.1691/ph.2010.0138
23. AbouSamra MM, Salama AH. Enhancement of the topical tolnaftate delivery for the treatment of tinea pedis via provesicular gel systems. J Liposome Res. 2017;27(4):324–334. doi:10.1080/08982104.2016.1239634
24. Bnyan R, Khan I, Ehtezazi T, et al. Surfactant effects on lipid based vesicles properties. J Pharm Sci. 2018;107(5):1237–1246. doi:10.1016/j.xphs.2018.01.005
25. Hofmann AF, Mysels KJ. Bile salts as biological surfactants. Colloids Surf. 1987;30(1):145–173. doi:10.1016/0166-6622(87)80207-X
26. Al-Mahallawi AM, Khowessah OM, Shoukri RA. Enhanced non invasive trans-tympanic delivery of ciprofloxacin through encapsulation into nanospanlastic vesicles: fabrication, in-vitro characterization and comparative ex-vivo permeation studies. Int J Pharm. 2017;522(1–2):157–164. doi:10.1016/j.ijpharm.2017.03.005
27. Abdelbary GA, Aburahma MH. Orodental mucoadhesive proniosomal gel formulation loaded with lornoxicam for management of dental pain. J Liposome Res. 2015;25(2):107–121. doi:10.3109/08982104.2014.941861
28. Yang X, Li Y, Li M, Zhang L, Feng L, Zhang N. Hyaluronic acid coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013;334(2):338–345. doi:10.1016/j.canlet.2012.07.002
29. Caliceti P, Salmaso S, Elvassore N, Bertucco A. Effective protein release from PEG/PLA nano-particles produced by compressed gas anti-solvent precipitation techniques. J Control Release. 2004;94(1):195–205. doi:10.1016/j.jconrel.2003.10.015
30. Al-Mahallawi AM, Khowessah OM, Shoukri RA. Nano-transfersomal ciprofloxacin loaded vesicles for non-invasive trans-tympanic ototopical delivery: in-vitro optimization, ex-vivo permeation studies and in-vivo assessment. Int J Pharm. 2014;472(1–2):304–314. doi:10.1016/j.ijpharm.2014.06.041
31. Stetefeld J, McKenna SA, Patel TR. Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophys Rev. 2016;8(4):409–427. doi:10.1007/s12551-016-0218-6
32. Smith MC, Crist RM, Clogston JD, McNeil SE. Zeta potential: a case study of cationic anionic and neutral liposomes. Anal Bioanal Chem. 2017;409(24):5779–5787. doi:10.1007/s00216-017-0527-z
33. Basha M, Abd El-Alim SH, Shamma RN, Awad GE. Design and optimization of surfactant based nanovesicles for ocular delivery of Clotrimazole. J Liposome Res. 2013;23(3):203–210. doi:10.3109/08982104.2013.788025
34. Gordon GS, Moses AC, Silver RD, Flier JS, Carey MC. Nasal absorption of insulin: enhancement by hydrophobic bile salts. Proc National Acad Sci USA. 1985;82(21):7419–7423. doi:10.1073/pnas.82.21.7419
35. Drapala KP, Auty MA, Mulvihill DM, O’Mahony JA. Influence of emulsifier type on the spray-drying properties of model infant formula emulsions. Food Hydrocol. 2017;69:56–66. doi:10.1016/j.foodhyd.2016.12.024
36. Hu Y, Xie J, Tong YW, Wang CH. Effect of PEG conformation and particle size on the cellular uptake efficiency of nanoparticles with the HepG2 cells. J Control Release. 2007;118(1):7–17. doi:10.1016/j.jconrel.2006.11.028
37. Roda A, Hofmann AF, Mysels KJ. The influence of bile salt structure on self association in aqueous solutions. J Biol Chem. 1983;258(10):6362–6370.
38. Demina T, Grozdova I, Krylova O, et al. Relationship between the structure of amphiphilic copolymers and their ability to disturb lipid bilayers. Biochemistry. 2005;44(10):4042–4054. doi:10.1021/bi048373q
39. Pantaler E, Kamp D, Haest CW. Acceleration of phospholipid flip flop in the erythrocyte membrane by detergents differing in polar head group and alkyl chain length. Biochim Biophys Acta. 2000;1509(1–2):397–408. doi:10.1016/S0005-2736(00)00322-9
40. Aboud HM, Ali AA, El-Menshawe SF, Elbary AA. Nanotransfersomes of carvedilol for intranasal delivery: formulation, characterization and in vivo evaluation. Drug Deliv. 2016;23(7):2471–2481. doi: 10.3109/10717544.2015.1013587
41. Abdelbary AA, Abd-Elsalam WH, Al-mahallawi AM. Fabrication of levofloxacin polyethylene glycol decorated nanoliposomes for enhanced management of acute otitis media: statistical optimization, trans-tympanic permeation and in vivo evaluation. Int J Pharm. 2019;559:201–209. doi:10.1016/j.ijpharm.2019.01.037
42. Gniado K, MacFhionnghaile P, Mcardle P, Erxleben A. The natural bile acid surfactant sodium taurocholate (NaTC) as a coformer in coamorphous systems: enhanced physical stability and dissolution behavior of coamorphous drug NaTc systems. Int J Pharm. 2018;535(1–2):132–139. doi:10.1016/j.ijpharm.2017.10.064
43. Salama HA, Mahmoud AA, Kamel AO, Abdel Hady M, Awad GA. Brain delivery of olanzapine by intranasal administration of transfersomal vesicles. J Liposome Res. 2012;22(4):336–345. doi:10.3109/08982104.2012.700460
44. El-Sayed MM, Hussein AK, Sarhan HA, Mansour HF. Flurbiprofen loaded niosomes in gel system improves the ocular bioavailability of flurbiprofen in the aqueous humor. Drug Dev Ind Pharm. 2017;43(6):902–910. doi:10.1080/03639045.2016.1272120
45. Godin B, Touitou E. Transdermal skin delivery: predictions for humans from in vivo, ex vivo and animal models. Adv Drug Deliv Rev. 2007;59(11):1152–1161. doi:10.1016/j.addr.2007.07.004
46. Harada K, Murakami T, Kawasaki E, Higashi Y, Yamamoto S, Yata N. In vitro permeability to salicylic acid of human rodent and shed snake skin. J Pharm Pharmacol. 1993;45(5):414–418. doi:10.1111/j.2042-7158.1993.tb05567.x
47. Kim MK, Lee CH, Kim DD. Skin permeation of testosterone and its ester derivatives in rats. J Pharm Pharmacol. 2000;52(4):369–375. doi: 10.1211/0022357001774101
48. Cevc G, Schätzlein A, Blume G. Transdermal drug carriers: basic properties, optimization and transfer efficiency in the case of epicutaneously applied peptides. J Control Release. 1995;36(1–2):3–16. doi:10.1016/0168-3659(95)00056-E
49. Shen LN, Zhang YT, Wang Q, Xu L, Feng NP. Enhanced in vitro and in vivo skin deposition of apigenin delivered using ethosomes. Int J Pharm. 2014;460(1–2):280–288. doi:10.1016/j.ijpharm.2013.11.017
50. Wan F, You J, Sun Y, et al. Studies on PEG modified SLNs loading vinorelbine bitartrate (I): preparation and evaluation in vitro. Int J Pharm. 2008;359(1–2):104–110. doi:10.1016/j.ijpharm.2008.04.003
51. Alsofany JM, Hamza MY, Abdelbary AA. Fabrication of nanosuspension directly loaded fast dissolving films for enhanced oral bioavailability of olmesartan medoxomil: in vitro characterization and pharmacokinetic evaluation in healthy human volunteers. AAPS PharmSciTech. 2018;19(5):2118–2132. doi:10.1208/s12249-018-0981-8
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.