")
Back to Journals » Journal of Inflammation Research » Volume 7
Systemic sensitization with the protein allergen ovalbumin augments local sensitization in atopic dermatitis
Authors Yoo J, Manicone A, McGuire J, Wang Y, Parks W
Received 9 October 2013
Accepted for publication 30 October 2013
Published 20 February 2014 Volume 2014:7 Pages 29—38
DOI https://doi.org/10.2147/JIR.S55672
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jane Yoo,1 Anne M Manicone,2 John K McGuire,3 Ying Wang,2 William C Parks2
1Center for Lung Biology, Department of Medicine, Division of Dermatology, 2Division of Pulmonary and Critical Care Medicine, 3Department of Pediatrics, Division Critical Care Medicine, University of Washington, Seattle, WA, USA
Abstract: Mouse models of atopic dermatitis based on epicutaneous sensitization have shed light on the role of epicutaneous allergen entry in the development of respiratory and gastrointestinal allergy. However, the contribution of non-cutaneous modes of sensitization to skin diseases has not been evaluated. We assessed if systemic ovalbumin administration, in conjunction with local sensitization, could prime for a robust inflammatory response. Furthermore, we attempted to elucidate important aspects of disease pathogenesis previously unaddressed in mouse models. Mice that underwent intraperitoneal ovalbumin sensitization prior to epicutaneous challenge demonstrated an acute (Th2-polarized) atopic dermatitis-like phenotype upon local challenge. The inflammatory response was strikingly more robust than in mice that underwent epicutaneous sensitization alone. The lesional infiltrate contained a dendritic cell population that corresponded phenotypically with inflammatory dendritic epidermal cells of significance in human disease. Finally, in accordance with observations in human atopic dermatitis, there was an increase in cluster of differentiation (CD) 103 (αE subunit)-expressing CD4+ T lymphocytes. However, the absence of CD103 on approximately 50% of infiltrating cells argues against a primary role for the αEβ7 integrin in tissue homing. In conclusion, we present a mouse model of atopic dermatitis that reveals novel insights into the pathogenesis of this complex disease.
Keywords: atopic dermatitis, mouse model, ovalbumin, sensitization, Th2, dendritic cells
Introduction
Atopic dermatitis (AD) is a common skin condition defined clinically by pruritus and scaly, erythematous plaques in a characteristic distribution.1 Despite the fact that allergic culprits are uncommonly identified in specific lesion development, AD is frequently depicted as resulting from repeated sensitization to allergens through the skin. Patients with AD are indeed often sensitized to numerous environmental allergens.2 Barrier defects have become increasingly emphasized as a primary factor in disease pathogenesis, allowing for allergen entry and subsequent cutaneous sensitization. Sensitization to aeroallergens is then thought to contribute to the development of respiratory atopy, a phenomenon that has been termed the atopic march.3
Allergen entry through a defective barrier is the basis of mouse models based on epicutaneous (EC) sensitization and elicitation. The most common protocol involves repeated EC sensitization with the protein allergen ovalbumin (OVA).4 The ingestion of food allergens and inhalation of aeroallergens are able to aggravate AD and non-cutaneous modes of sensitization may also play a role. Indeed, other groups have demonstrated that intraperitoneal (IP) administration of allergens serves as sufficient sensitization for Th2-mediated allergic responses in the respiratory and gastrointestinal tracts in mouse models.5,6 Li et al found that AD-like lesions followed intragastric sensitization with cow’s milk or peanuts.7 Systemic sensitization has not otherwise been demonstrated to effectively prime for the development of AD.
Acute AD is characterized by a Th2-dominant inflammatory response, which is central to activated interleukin (IL)-4-producing cluster of differentiation (CD) 4+ T cells.8 αEβ7 is an integrin found on T cells almost exclusively at epithelial surfaces and is thought to play an important role in tissue homing and/or activation via interaction with its ligand, E-cadherin ectodomain, on epithelial cells.9,10 αEβ7 is upregulated upon antigen presentation as well as by proinflammatory signals within the inflammatory milieu.11–15 Significantly, an increase in its expression by lesional T cells is seen in human AD;16 however, its expression and function have not yet been evaluated in mouse AD models.
Dendritic cells (DCs) serve in the recognition and processing of antigens and subsequent polarization and activation of T cells.8 Although relatively little research has evaluated the phenotype and function of DCs in AD, both epidermal (Langerhans cells; LCs) and dermal DCs have been implicated. LCs have been shown to be critical for the acute AD response.17,18 In addition, a unique dermal inflammatory DC population, termed inflammatory dendritic epidermal cells (IDECs), appears in human AD lesions.19 IDECs were originally described as CD1a-expressing epidermal DCs demonstrating high expression of CD11b, FCεRI, HLA-DR, and CD36.19 They have since been shown to predominate in the dermis and to express CD1b, CD80, CD86, CD206, and histamine H4 receptor.20–23 They are thought to be more important in the perpetuation of inflammation,24,25 although studies have yet to identify their precise function.
In this study, we present a model of AD based on systemic, in combination with local, sensitization with the protein allergen OVA. We demonstrate that systemic exposure to allergens augments the effects of local sensitization alone, suggesting that non-cutaneous routes of allergen entry may be an important mechanism of sensitization in human disease. In addition, we present several novel insights into the phenotype and trafficking of T cells and DCs in AD that have not previously been examined in mouse models.
Materials and methods
Ovalbumin sensitization and challenge protocol
Six to eight week old male BALB/c mice from The Jackson Laboratory (Bar Harbor, ME, USA) were housed in a specific pathogen free facility. All animal experiments were approved by the University of Washington Institutional Animal Care and Use Committee. Figure 1 depicts the OVA sensitization and challenge protocol. Briefly, mice were sensitized at day −28 by IP administration of 1 μg of OVA (grade V; Sigma, St Louis, MO, USA) at 1 μg/μL in normal saline, mixed 1:1 with the adjuvant Imject Alum (Thermo Scientific Pierce, Rockford, IL, USA). Mice were resensitized by IP route on day −15, followed by EC route on day −14. For EC sensitization, mice were anesthetized with isoflurane and shaven. The mid-back was gently scratched ten times, and a 1.5 cm × 1.5 cm gauze patch impregnated with 1 mg of OVA at 10 mg/mL in dH2O was affixed to the scratched site under Tegaderm (3M Health Care, St Paul, MN, USA) and Elastikon (Johnson & Johnson Consumer Co, Inc, Skillman, NJ, USA). The patch was left intact for 7 days. Mice were challenged on day 0 by repeated daily EC application until harvest. Sham-treated mice received normal saline by IP route and dH2O by EC route.
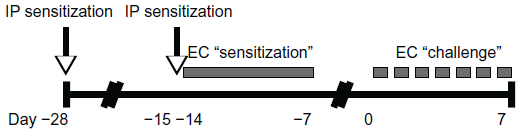 | Figure 1 Ovalbumin sensitization and challenge protocol. |
Histology and immunofluorescence staining
Samples of patched skin were fixed in 10% formalin, dehydrated by serial incubation in 30%, 50%, and 70% ethanol, and embedded in paraffin. 5 μm sections were stained with hematoxylin and eosin (H&E). For immunofluorescence, skin samples were stored in Tissue-Tek® O.C.T. compound (Sakura, Torrence, CA, USA) at −80°C until cryostat sectioning. Six to eight μm sections were stained with the following affinity purified primary anti-mouse monoclonal antibodies (eBioscience, San Diego, CA, USA, except as indicated): CD4 (L3T4, clone GK1.5), CD8a (Ly-2, clone 53–6.7), CD11c (clone N418), langerin/CD207 (eBioRMUL.2), CD103 (M290; BD Biosciences, San Diego, CA, USA), CD103 (2E7), CD11b (M1/70), CD86 (GL1), CD36 (No 72-1), and CD206 (macrophage mannose receptor, clone MR5D3; AbD Serotec, Oxford, UK). Alexa Fluor® 488-conjugated anti-hamster and Cy3-conjugated anti-rat secondary antibodies were used for detection. Images were taken using an Olympus DP70 digital microscope camera and software (Olympus America, Center Valley, PA, USA).
Serum OVA-specific IgE enzyme linked immunosorbent assay
Costar 96-well plates (Corning Inc, Corning, NY, USA) were coated and incubated overnight with 2 ng of rat monoclonal anti-mouse IgE (clone 23G3; eBioscience). Plates were blocked with 1× phosphate buffered saline (PBS)/5% bovine serum albumin (BSA), then coated with serum samples (1:4 starting dilution) or mouse monoclonal anti-OVA IgE standard (AbD Serotec; 0.5 μg/μL starting concentration), in duplicate and serially diluted 1:2. Plates were sequentially incubated with 2.5 μg of OVA (grade V; Sigma), 0.125 μg of rabbit anti-OVA horseradish peroxidase (HRP; Rockland Immunochemicals, Gilbertsville, PA), and 3-ethylbenzthiazoline-6-sulfonic acid substrate buffer (ABTS; Sigma, 0.1 M citric acid, pH 4.35). Peroxidase activity was measured at 405 nm using a Synergy 4 Multi-Mode Microplate Reader and Gen5 software (BioTek Instruments, Winooski, VT, USA).
Flow cytometry
Single cell suspensions were prepared from skin-draining lymph nodes (SDLNs), blocked with anti-mouse CD16/CD32 (clone 2.4G2; BD Biosciences), and surface-stained with the following conjugated anti-mouse monoclonal antibodies (all eBioscience): CD3-PeCy7 (145-2C11), CD4-PE (RM4-5), CD8a-APC (53-6.7), CD103-fluorescein isothiocyanate (FITC; 2E7), CD11c-APC (N418), B220-FITC (RA3-6B2). Intracellular FoxP3 staining was performed using the PE anti-FoxP3 Staining Set (eBioscience). FACS was performed on a BD FACSCanto II multicolor flow cytometer (BD Biosciences) and analyzed using FlowJo Flow Cytometry Analysis Software (Tree Star, Inc, Ashland, OR, USA).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis
Flash-frozen tissue was stored in TRIzol Reagent at −80°C and homogenized with a Power Gen 125 tissue homogenizer (Fisher Scientific). Ribonucleic acid (RNA) isolation was performed by TRIzol method in conjunction with the Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Concentration and purity were measured with a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Complementary deoxyribonucleic acid (cDNA) was synthesized from 5 μg of total RNA using a High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). qRT-PCR was performed using primer and TaqMan probes (FAM [6-carboxyfluorescein] dye-labeled) for hypoxanthine-guanine phosphoribosyltransferase (HPRT), IL-4, and interferon (IFN)-γ on an ABI HT7900 Fast Real-Time PCR System (Life Technologies, Grand Island, NY, USA).
Results
OVA-challenged mice demonstrate characteristics of AD
Mice were harvested over a time course following initiation of EC challenge. Mice demonstrated scratching behavior at the site of OVA application throughout the challenge phase. Gross evaluation revealed erythema by day 1 and skin thickening, scale, and/or crust by day 7, as well as prominent axillary and inguinal lymphadenopathy by day 7. Characteristic histologic features included an acanthotic, spongiotic epidermis with basket-weave to compact hyperkeratosis, a predominantly mononuclear cell dermal infiltrate, and progressive dermal thickening (Figure 2A). Focal erosions and serum crust with secondary impetiginization were universal by day 7 (Figure 2B).
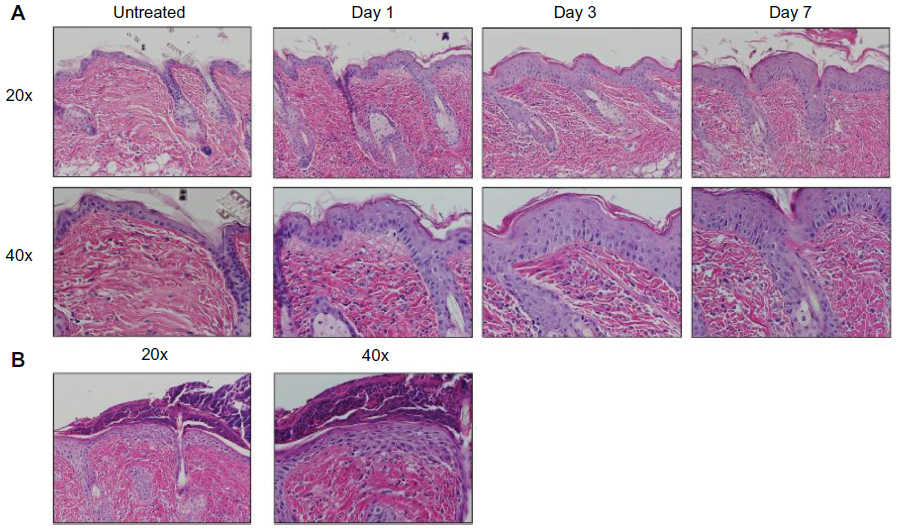 | Figure 2 AD characteristic histologic changes in OVA challenged mice. (A) Histology was analyzed at 1, 3, and 7 days of challenge and compared to skin of untreated mice, revealing a progression of characteristic histologic changes, including acanthosis, spongiosis, hyperkeratosis, dermal inflammation, and dermal thickening. Images, shown at 20× (upper) and 40× (lower) magnifications, are representative of three mice per time point. (B) Secondary changes included erosion, crust, and impetiginization. |
The dermal infiltrate consists predominantly of CD4+ T cells and CD11c+ DCs
The lesional lymphocytic infiltrate was comprised largely of CD4+ T cells in a perifollicular and perivascular distribution (Figure 3A), while CD8+ T and B220+ B cells were sparse (not shown). CD11c+ DCs comprised the majority of the remaining infiltrate (Figure 3A) and were langerin- and CD103-negative (Figure 3B), reflecting either an expansion of the majority langerin−CD103− DCs present in the steady-state dermis or a novel population derived from the periphery. To further characterize this DC population, we assessed a panel of surface markers corresponding to that of IDECs described in human AD. The majority of the dermal DCs co-expressed CD11b, CD206, CD86, and CD36 (Figure 3C).
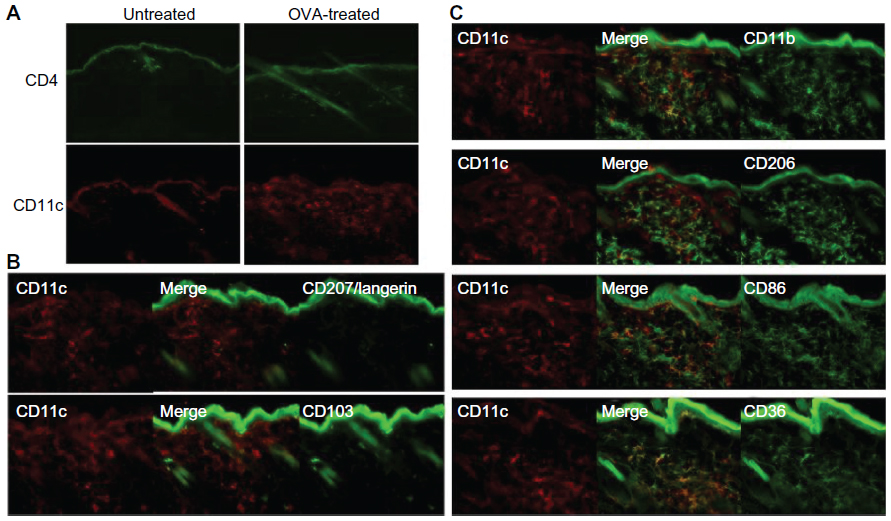 | Figure 3 CD4+ T cell and CD11c+ DC dominant inflammatory cell phenotype in OVA-challenged mice. (A) Upper panel shows a prominent, largely perivascular and perifollicular influx of CD4+ T cells. Lower panel shows a dramatic accumulation of CD11c+ DCs. (B) The CD11c+ population was CD103− and langerin negative. Rare double-positive cells presumably represent the minority steady-state DC subpopulation. (C) The majority of the inflammatory CD11c+ cells co-expressed CD11b, CD206, CD86, and CD36. |
Elevated tissue IL-4 and serum OVA-specific IgE are consistent with a Th2 allergic inflammatory profile
Th2-type allergic inflammation is characterized by increases in the prototypical cytokine IL-4 and circulating IgE. IL-4 messenger (m) RNA in lesional skin of OVA-challenged mice was approximately 8-fold higher as early as day 1 to over 40-fold by day 7, compared to unchallenged control mice (Figure 4A). Reciprocally, there was a decrease in the prototypical Th1 cytokine IFN-γ (Figure 4B). Because skin microtrauma alone induced IFN-γ (up to 4-fold higher in sham-challenged versus unchallenged mice; data not shown), IFN-γ data are presented relative to sham treatment at day 1, revealing an almost 3-fold decrease by day 7 of the challenge. Finally, OVA-specific serum IgE was measured up to 1.5 μg/μL in OVA-treated mice compared to non-detectable levels in sham-treated and untreated mice (Figure 4C).
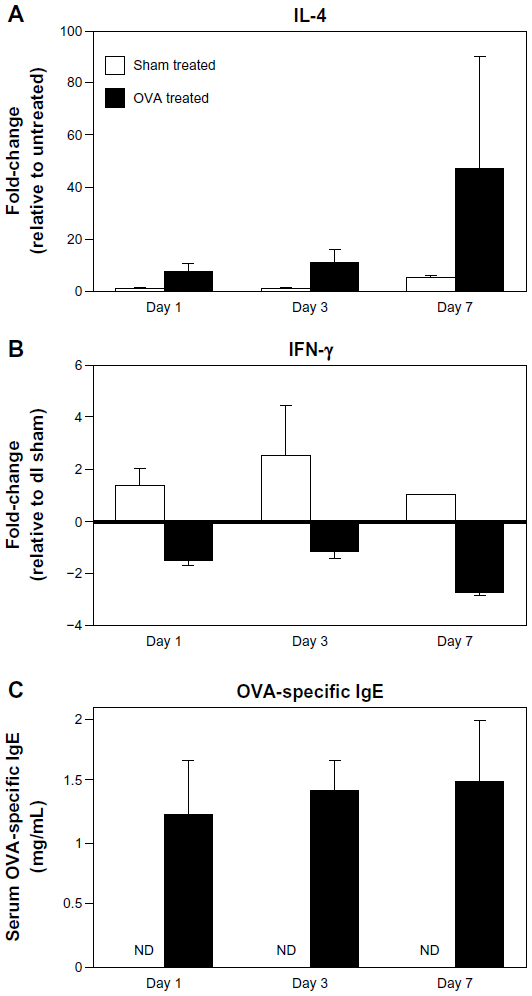 | Figure 4 Elevated tissue IL-4 expression and circulating OVA-specific IgE in OVA-challenged mice. (A and B) IL-4 and IFN-γ mRNA levels were measured by qRT-PCR. IL-4 increased over time, while IFN-γ decreased. (C) Serum OVA-specific IgE was measured by ELISA. Compared to nondetectable (ND) levels in sham-treated mice, there was a significant increase in IgE levels as early as day 1 in OVA-treated mice, with a continued increase over time. Data are presented as mean ± SEM of two (day 7) or four (days 1, 3) sham-treated mice, and five (days 1, 7) or nine (day 3) OVA-treated mice. |
T cell efflux from SDLNs coincides with the arrival of antigen-presenting DCs
In acute AD, local tissue DCs migrate to the SDLNs to present antigen to naïve T cells which in turn, upon Th2 polarization and activation, migrate back to the site of inflammation. SDLNs thus serve as important indicators of the evolution of acute inflammation in AD.
There was a significant increase in CD11c+ DCs within the SDLNs as early as day 1 with continued accumulation over the challenge period (Figure 5A). Reciprocally, there was a progressive decrease in the proportion of CD3+ T lymphocytes versus B220+ B lymphocytes over the first 3 days of the challenge (Figure 5B), concurrent with the arrival of T cells into the skin. Although this proportion stabilized by day 7, the coincident lymphadenopathy observed at this time point suggests a continued prominent egress of T lymphocytes to the skin. Finally, there was a significant increase in differentiation of CD4+FoxP3+ T regulatory cells (Tregs) in the SDLNs within the first 24 hours of challenge (Figure 5C), indicating that the early DCs carry signals to induce both effector and induced Treg (iTreg) phenotypes. The subsequent decrease in the proportion of Tregs suggests efflux to the skin and/or relatively less Treg differentiation (Figure 5C).
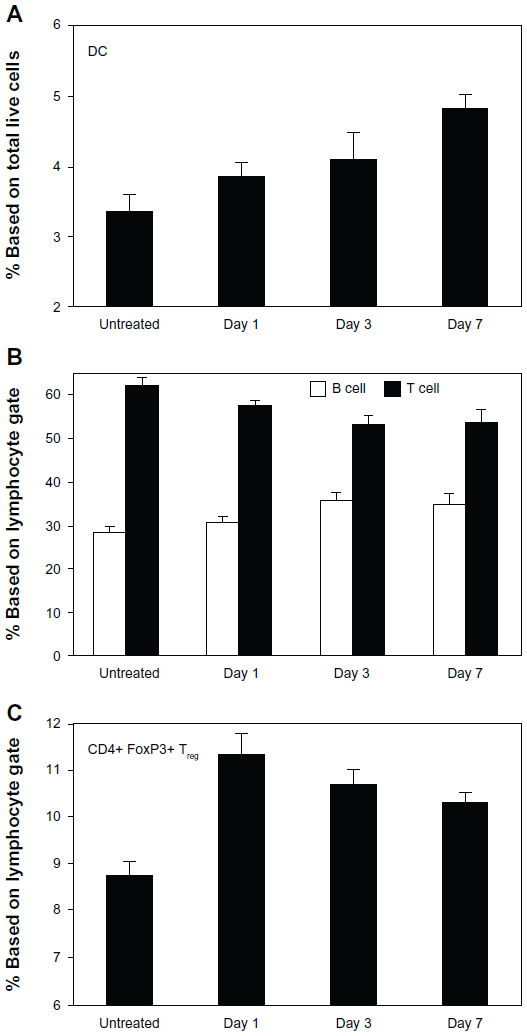 | Figure 5 DC and lymphocyte trends in SDLNs of OVA-challenged mice. (A) CD11c+ DCs were increased in SDLNs within the first 24 hours of challenge and continued to accumulate over time. (B) There was a progressive decrease in T (CD3+) versus B (B220+) cells through day 3 of challenge. (C) In addition, there was an early proliferation of CD4+CD25+ Tregs at day 1, with the proportion of Tregs decreasing thereafter. Data are presented as mean ± SEM of five mice at each time point. |
The αEβ7 integrin is not necessary for T cell homing to inflamed skin
As a potential skin-homing marker, we analyzed CD103 (αE subunit) expression on T cells both in SDLNs and inflamed skin. CD103 was upregulated over time on both effector and regulatory CD4+ T cells in SDLNs (Figure 6A), consistent with its proposed upregulation by antigen presentation and/or other inflammatory signals. CD103 was also found on infiltrating CD4+ T cells in lesional skin (Figure 6B). However, it was absent from approximately 50% of the lesional T cells, suggesting that the αEβ7 integrin is not necessary for tissue homing of these cells.
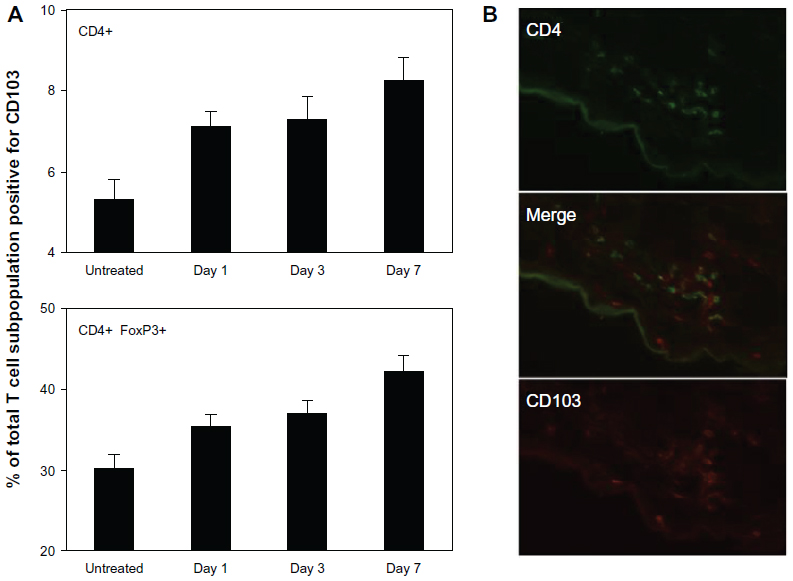 | Figure 6 CD103 (αE subunit) is upregulated in SDLNs but is not necessary for skin homing. (A) Percent of CD4+ T cells (upper) and CD4+FoxP3+ Tregs (lower) that were positive for CD103, demonstrating progressive upregulation of CD103 expression over time between both populations. Data are presented as mean ± SEM of five mice at each time point. (B) CD103 was expressed on approximately 50% of lesional CD4+ cells. CD103 was also expressed on CD8+ T cells and a small fraction of DCs (not shown), which accounts for scattered single-positive CD103+ cells. |
Systemic sensitization augments local sensitization in the development of AD
Our protocol was derived from an asthma model based on systemic sensitization with OVA and subsequent airway challenge.26 The most commonly reported OVA-based AD model is one by Spergel et al, in which mice are subjected to three 1-week sensitizations by EC route over a 7-week experimental protocol.4 However, as this model is long and tedious, we assessed if IP sensitization could serve as a more effective route of sensitization and could effectively prime for a robust AD response to EC challenge, by comparing our protocol with and without IP sensitization (Figure 7A).
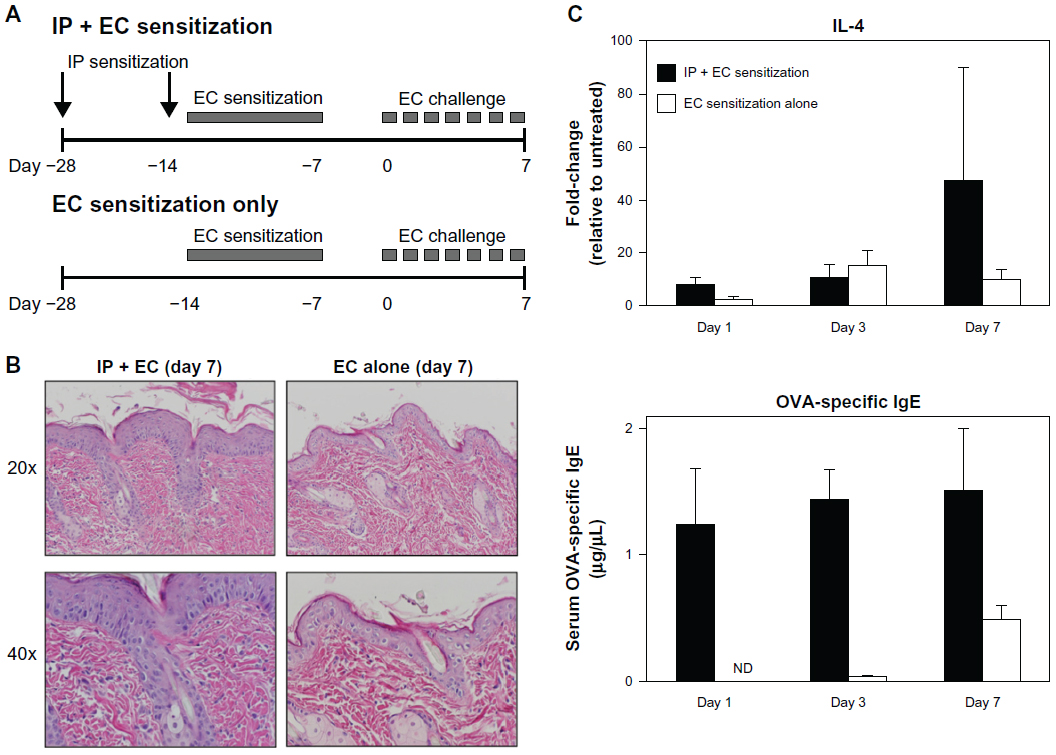 | Figure 7 Systemic sensitization augments local sensitization alone in elicitation of an AD phenotype. (A) Diagrams comparing OVA sensitization and challenge protocols with (above; previously described) and without (below) IP sensitization. (B) Mice sensitized by EC route alone (right) showed less pronounced histologic changes compared to mice sensitized by both IP and EC route (left). (C) Mice sensitized by EC route alone (white) showed less robust elevations in tissue IL-4 mRNA and circulating OVA-specific IgE compared to mice sensitized by both IP and EC route (black). |
At day seven of the EC challenge, mice that underwent a single EC sensitization alone showed less skin thickening, scale, and erythema than those that received IP sensitization in addition to EC sensitization. Histology revealed less prominent hyperkeratosis, acanthosis, dermal infiltration, and dermal thickening (Figure 7B). In general, the degree of morphologic changes present after 7 days of challenge following EC sensitization alone was comparable to that seen after only 24 hours of challenge following combined IP and EC sensitization (compare EC alone [day 7 images] in Figure 7B to day 1 images in Figure 2A). Correlating with these morphologic differences, levels of IL-4 expression and serum OVA-specific IgE were significantly lower in mice sensitized by EC route alone (Figure 7C). Thus, IP sensitization both reduced the length of exposures and yielded a more robust immune response.
Discussion
In this study, we present a mouse model of acute AD based on combined IP and EC sensitization, followed by EC challenge, with the protein allergen OVA. These mice demonstrated characteristic clinical and histologic features along with a Th2-dominant inflammatory profile, with significant inflammation as early as 24 hours of challenge and a robust response by 72 hours. Significantly, we have shown that our combined sensitization protocol is more effective than EC sensitization alone.
Many mouse models of AD-like phenotypes have been reported.8 Nc/Nga mice are the best characterized of spontaneous strains. They are naturally occurring and hence arguably the purest model of intrinsic human disease, although the genetic defect responsible has not yet been identified. Among genetically engineered mice, AD-like features often occur secondary to other phenotypes; eg, abnormalities in epidermal integrity, cholesterol metabolism, immune regulation, or other structural or inflammatory defects in the skin. However, several engineered strains are based on skin-specific overexpression of pro-allergic Th2 cytokines (eg, IL-4, IL-13, and TSLP [thymic stromal lymphopoietin]) and are therefore uniquely suited for studying AD-specific adaptive immunity. Finally, AD-like phenotypes have been replicated by repeat EC application of protein allergens (OVA or house dust mite allergen), hapten (oxazolone or trinitrochlorobenzene), or Staphylococcus aureus superantigen (Staphylococcal Enterotoxin B; SEB).8
EC sensitization models are attractive in that they allow one to track inflammation following elicitation. The disadvantage, however, is that they are labor-intensive and time-consuming, requiring up to 50 days for a robust response. Here, we introduce a protein allergen-based model of AD in which EC sensitization is augmented by previous and concomitant IP exposure, providing a shorter and simpler protocol. As early as 29 days from the initiation of our protocol, these mice display characteristic histologic and immunologic features of human AD. Previous groups have demonstrated simplifications of the Spergel model that have likewise yielded robust inflammatory responses.27 However, we are the first to perform a direct comparison with and without IP sensitization, demonstrating that IP sensitization augments the EC response to allergens and yields significant inflammation after only 3 days of challenge.
Of greater significance, our model suggests that non-cutaneous modes of allergen sensitization may play a larger contributory role than traditionally believed. AD is classically believed to result from repeated sensitization to allergens via the skin, frequently in the setting of a genetic predisposition. Recent research on the role of barrier defects,28 particularly the discovery of filaggrin mutations as a major risk factor,29,30 has corroborated the notion that EC sensitization plays a primary pathogenetic role in AD. Furthermore, EC sensitization is thought to prime the respiratory epithelium for allergies and asthma later in life, a phenomenon that has been termed the atopic march.3 EC sensitization likely also plays a pathogenetic role in other associated allergies, most notably food allergy. In accordance with the atopic march, EC sensitization has been shown in mice to effectively prime for subsequent Th2-mediated allergic inflammation in both the airway and gastrointestinal tract in models of asthma, allergic rhinitis, eosinophilic esophagitis, and food allergy.4,31–34
Whether patients with AD can become sensitized by alternate routes of allergen exposure has rarely been addressed. Food allergies commonly appear early in life2 which suggests the gastrointestinal route as a possible mode of allergen sensitization for AD. Indeed, Li et al demonstrated that intragastric sensitization with cow’s milk or peanuts could effectively produce an AD-like phenotype in approximately one third of the mice.7 Although IP injection is not a physiologic means of allergen exposure, it can be viewed as sensitization at a distant site that then primes for allergic responses locally. IP sensitization has previously been shown to induce stronger respiratory and gastrointestinal responses to allergen compared to local sensitization alone, in animal models of asthma and food allergy, respectively.5,6 Here, we demonstrate for the first time that IP sensitization augments EC sensitization by directly comparing the inflammatory response with and without previous IP sensitization. More significantly, our results support the notion that the atopic march is not unidirectional. The early onset of AD may simply reflect its increased susceptibility to barrier disruption and allergen sensitization. This may underlie scenarios in which clinical AD appears later in life and/or after the appearance of respiratory or gut allergies or in which skin disease is exacerbated by cutaneous exposure to classically respiratory or food allergens.
DCs survey antigens at the skin surface and are thus a critical bridge between the altered epithelial barrier and Th2-mediated inflammatory response in AD. Several DC subsets have been described in the skin. In the steady state, these include epidermal LC (langerin+MHCII+CD1a+), dermal myeloid DCs (divided into a majority langerin−CD103− and a minority langerin+CD103+ subpopulation),35,36 and plasmacytoid DCs (IFN-α producing APCs implicated in anti-viral immunity).37 During states of inflammation, additional inflammatory DCs appear whose phenotype varies depending on the particular immune milieu of the skin.25 In human AD, IDECs have been characterized by a CD1a+, CD11c+, CD11bhi, HLA-DR+, FcεRIhi phenotype, and positivity for CD206/macrophage mannose receptor, CD36, and the costimulatory molecules CD80 and CD86.19–23 While LCs are considered important and required for the initial response to allergen,17,18 IDECs are thought to be more important for maintenance of inflammation.24,25
Phenotypic and functional characterization of these important immune sentinels has been limited, particularly in mouse models. Here, we have shown a dramatic influx and/or proliferation of langerin−CD103− DCs in acute AD and have defined a panel of markers (CD11b, CD206, CD86, and CD36) that correspond to the IDECs described in human AD and can hopefully be used for reliable identification and/or isolation of these cells. Of note, none of these markers are specific for these cells. We feel that the full panel of these four markers, in addition to CD11c, is necessary as well as sufficient to identify the inflammatory DCs seen in murine AD. Importantly, dermal DCs in unchallenged control skin show little to no expression of these markers (data not shown), suggesting that the above population is indeed a novel population recruited from peripheral precursors, although upregulation of these markers by steady-state dermal DCs during inflammation cannot be excluded.
The role of the αEβ7 integrin on T lymphocytes is unknown. It is preferentially expressed on CD8+ and CD4+ Treg subsets but is also present on effector CD4+ cells.9,38,39 It has been shown to be upregulated by proinflammatory stimuli and antigen presentation, and αEβ7+ cells are increased in various inflammatory conditions including human AD. αEβ7 binds selectively to E-cadherin, an adhesion molecule highly expressed on epithelia;10 and a role in tissue homing was first suggested by its recognition on T cells almost exclusively within epithelial tissues.9 Within the skin, impaired recruitment and/or retention of T cell subsets has indeed been demonstrated during both steady state and inflammatory conditions using CD103 blockade or knockout mice.14,40 In contrast, intact entry and retention of CD8+ T cells has been demonstrated within intestinal epithelium in knockout mice, suggesting at most a redundant role for αEβ7 in cellular migration.41 Cross-linking studies using anti-CD103 monoclonal antibodies have revealed co-stimulation of cytotoxic and proliferative responses among intraepithelial lymphocytes, suggesting a primary role in cellular activation.42–44 Our findings support a dispensable role for αEβ7 in tissue homing in AD. We observed an upregulation of CD103 on CD4+ effector and regulatory subsets within SDLNs in our AD mice. However, expression was seen on only approximately 50% of the infiltrating CD4+ cells within inflamed skin. More significantly, we have observed intact T cell infiltration in Cd103−/− mice subject to AD inflammation (unpublished observations), suggesting an alternate or additional mechanism for tissue recruitment of these T cell subsets to the skin.
In summary, we present a model of AD based on systemic and local sensitization with the protein allergen OVA. Our model employs a simpler and more effective protocol than previously described models and underscores the potential importance of non-cutaneous modes of allergen exposure in human disease. Moreover, we have identified a unique IDEC-like inflammatory DC population previously uncharacterized in mouse models and have characterized expression of the αEβ7 integrin on lesional T cells. We hope that these findings facilitate further investigation of DC and T cell biology in AD.
Acknowledgments
We thank Dr Michelle Lin and Cliff Rims for technical assistance. This work was supported by a Dermatology Foundation Career Development Award (JY) and NIH grants AR051683 (JY), HL082658 (WCP), HL089455 (WCP).
Disclosure
The authors report no conflicts of interest in this work.
References
Bieber T. Atopic dermatitis. Ann Dermatol. 2010;22(2):125–137. | |
Schäfer T. The impact of allergy on atopic eczema from data from epidemiological studies. Curr Opin Allergy Clin Immunol. 2008;8(5):418–422. | |
Ker J, Hartert TV. The atopic march: what’s the evidence? Ann Allergy Asthma Immunol. 2009;103(4):282–289. | |
Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J Clin Invest. 1998;101(8):1614–1622. | |
Zhang Y, Lamm WJ, Albert RK, Chi EY, Henderson WR, Lewis DB. Influence of the route of allergen administration and genetic background on the murine allergic pulmonary response. Am J Respir Crit Care Med. 1997;155(2):661–669. | |
Knippels LM, Penninks AH, Smit JJ, Houben GF. Immune-mediated effects upon oral challenge of ovalbumin-sensitized Brown Norway rats: further characterization of a rat food allergy model. Toxicol Appl Pharmacol. 1999;156(3):161–169. | |
Li XM, Kleiner G, Huang CK, et al. Murine model of atopic dermatitis associated with food hypersensitivity. J Allergy Clin Immunol. 2001;107(4):693–702. | |
Oyoshi MK, He R, Kumar L, Yoon J, Geha RS. Cellular and molecular mechanisms in atopic dermatitis. Adv Immunol. 2009;102:135–226. | |
Agace WW, Higgins JM, Sadasivan B, Brenner MB, Parker CM. T-lymphocyte-epithelial-cell interactions: integrin alpha(E)(CD103)beta(7), LEEP-CAM and chemokines. Curr Opin Cell Biol. 2000;12(5):563–568. | |
Higgins JM, Mandlebrot DA, Shaw SK, et al. Direct and regulated interaction of integrin alphaEbeta7 with E-cadherin. J Cell Biol. 1998;140(1):197–210. | |
Ericsson A, Svensson M, Arya A, Agace WW. CCL25/CCR9 promotes the induction and function of CD103 on intestinal intraepithelial lymphocytes. Eur J Immunol. 2004;34(10):2720–2729. | |
Koch SD, Uss E, van Lier RA, ten Berge IJ. Alloantigen-induced regulatory CD8+CD103+ T cells. Hum Immunol. 2008;69(11):737–744. | |
Robinson PW, Green SJ, Carter C, Coadwell J, Kilshaw PJ. Studies on transcriptional regulation of the mucosal T-cell integrin alphaEbeta7 (CD103). Immunology. 2001;103(2):146–154. | |
Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J Immunol. 2005;174(9):5444–5455. | |
Teraki Y, Shiohara T. Preferential expression of alphaEbeta7 integrin (CD103) on CD8+ T cells in the psoriatic epidermis: regulation by interleukins 4 and 12 and transforming growth factor-beta. Br J Dermatol. 2002;147(6):1118–1126. | |
de Vries IJ, Langeveld-Wildschut EG, van Reijsen FC, Bihari IC, Bruijnzeel-Koomen CA, Thepen T. Nonspecific T-cell homing during inflammation in atopic dermatitis: expression of cutaneous lymphocyte-associated antigen and integrin alphaE beta7 on skin-infiltrating T cells. J Allergy Clin Immunol. 1997;100(5):694–701. | |
Elentner A, Finke D, Schmuth M, et al. Langerhans cells are critical in the development of atopic dermatitis-like inflammation and symptoms in mice. J Cell Mol Med. 2009;13(8B):2658–2672. | |
Dubrac S, Schmuth M, Ebner S. Atopic dermatitis: the role of Langerhans cells in disease pathogenesis. Immunol Cell Biol. 2010;88(4):400–409. | |
Wollenberg A, Kraft S, Hanau D, Bieber T. Immunomorphological and ultrastructural characterization of Langerhans cells and a novel, inflammatory dendritic epidermal cell (IDEC) population in lesional skin of atopic eczema. J Invest Dermatol. 1996;106(3):446–453. | |
Dijkstra D, Stark H, Chazot PL, et al. Human inflammatory dendritic epidermal cells express a functional histamine H4 receptor. J Invest Dermatol. 2008;128(7):1696–1703. | |
Schuller E, Teichmann B, Haberstok J, Moderer M, Bieber T, Wollenberg A. In situ expression of the costimulatory molecules CD80 and CD86 on langerhans cells and inflammatory dendritic epidermal cells (IDEC) in atopic dermatitis. Arch Dermatol Res. 2001;293(9):448–454. | |
Wollenberg A, Wen S, Bieber T. Phenotyping of epidermal dendritic cells: clinical applications of a flow cytometric micromethod. Cytometry. 1999;37(2):147–155. | |
Wollenberg A, Mommaas M, Oppel T, Schottdorf EM, Günther S, Moderer M. Expression and function of the mannose receptor CD206 on epidermal dendritic cells in inflammatory skin diseases. J Invest Dermatol. 2002;118(2):327–334. | |
Novak N, Valenta R, Bohle B, et al. FcepsilonRI engagement of Langerhans cell-like dendritic cells and inflammatory dendritic epidermal cell-like dendritic cells induces chemotactic signals and different T-cell phenotypes in vitro. J Allergy Clin Immunol. 2004;113(5):949–957. | |
Johnson-Huang LM, McNutt NS, Krueger JG, Lowes MA. Cytokine-producing dendritic cells in the pathogenesis of inflammatory skin diseases. J Clin Immunol. 2009;29(3):247–256. | |
Henderson WR, Lewis DB, Albert RK, et al. The importance of leukotrienes in airway inflammation in a mouse model of asthma. J Exp Med. 1996;184(4):1483–1494. | |
Yatsuzuka R, Inoue T, Jiang S, Nakano Y, Kamei C. Development of new atopic dermatitis models characterized by not only itching but also inflammatory skin in mice. Eur J Pharmacol. 2007;565(1–3):225–231. | |
Elias PM, Schmuth M. Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Allergy Asthma Rep. 2009;9(4):265–272. | |
O’Regan GM, Sandilands A, McLean WH, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. 2009;124(3 Suppl 2):R2–R6. | |
van den Oord RA, Sheikh A. Filaggrin gene defects and risk of developing allergic sensitisation and allergic disorders: systematic review and meta-analysis. BMJ. 2009;339:b2433. | |
Akei HS, Mishra A, Blanchard C, Rothenberg ME. Epicutaneous antigen exposure primes for experimental eosinophilic esophagitis in mice. Gastroenterology. 2005;129(3):985–994. | |
Akei HS, Brandt EB, Mishra A, et al. Epicutaneous aeroallergen exposure induces systemic TH2 immunity that predisposes to allergic nasal responses. J Allergy Clin Immunol. 2006;118(1):62–69. | |
He R, Oyoshi MK, Jin H, Geha RS. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci U S A. 2007;104(40):15817–15822. | |
Vaali K, Puumalainen TJ, Lehto M, et al. Murine model of food allergy after epicutaneous sensitization: role of mucosal mast cell protease-1. Scand J Gastroenterol. 2006;41(12):1405–1413. | |
Henri S, Poulin LF, Tamoutounour S, et al. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med. 2010;207(1):189–206. | |
Poulin LF, Henri S, de Bovis B, Devilard E, Kissenpfennig A, Malissen B. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med. 2007;204(13):3119–3131. | |
Merad M, Manz MG. Dendritic cell homeostasis. Blood. 2009;113(15):3418–3427. | |
Lehmann J, Huehn J, de la Rosa M, et al. Expression of the integrin alpha Ebeta 7 identifies unique subsets of CD25+ as well as CD25- regulatory T cells. Proc Natl Acad Sci U S A. 2002;99(20):13031–13036. | |
Sather BD, Treuting P, Perdue N, et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med. 2007;204(6):1335–1347. | |
Schön MP, Schön M, Parker CM, Williams IR. Dendritic epidermal T cells (DETC) are diminished in integrin alphaE(CD103)-deficient mice. J Invest Dermatol. 2002;119(1):190–193. | |
Lefrançois L, Parker CM, Olson S, et al. The role of beta7 integrins in CD8 T cell trafficking during an antiviral immune response. J Exp Med. 1999;189(10):1631–1638. | |
Roberts K, Kilshaw PJ. The mucosal T cell integrin alpha M290 beta 7 recognizes a ligand on mucosal epithelial cell lines. Eur J Immunol. 1993;23(7):1630–1635. | |
Russell GJ, Parker CM, Cepek KL, et al. Distinct structural and functional epitopes of the alpha E beta 7 integrin. Eur J Immunol. 1994;24(11):2832–2841. | |
Sarnacki S, Bègue B, Buc H, Le Deist F, Cerf-Bensussan N. Enhancement of CD3-induced activation of human intestinal intraepithelial lymphocytes by stimulation of the beta 7-containing integrin defined by HML-1 monoclonal antibody. Eur J Immunol. 1992;22(11):2887–2892. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.