")
Back to Journals » International Journal of Nanomedicine » Volume 11
Systemic delivery and activation of the TRAIL gene in lungs, with magnetic nanoparticles of chitosan controlled by an external magnetic field
Authors Alvizo-Baez CA , Luna-Cruz IE , Vilches-Cisneros N, Rodríguez-Padilla C , Alcocer-González JM
Received 28 July 2016
Accepted for publication 22 September 2016
Published 2 December 2016 Volume 2016:11 Pages 6449—6458
DOI https://doi.org/10.2147/IJN.S118343
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Cynthia A Alvizo-Baez,1 Itza E Luna-Cruz,1 Natalia Vilches-Cisneros,2 Cristina Rodríguez-Padilla,1 Juan M Alcocer-González1
1Laboratory of Immunology and Virology, Biological Sciences Faculty, University Autonomous of Nuevo León, San Nicolás de los Garza, 2Pahologic Anatomy and Cytopathology Service of the University Hospital, University Autonomous of Nuevo León, Monterrey, Mexico
Abstract: Recently, functional therapies targeting a specific organ without affecting normal tissues have been designed. The use of magnetic force to reach this goal is studied in this work. Previously, we demonstrated that nanocarriers based on magnetic nanoparticles could be directed and retained in the lungs, with their gene expression under the control of a promoter activated by a magnetic field. Magnetic nanoparticles containing the TRAIL gene and chitosan were constructed using the ionic gelation method as a nanosystem for magnetofection and were characterized by microscopy, ζ-potential, and retention analysis. Magnetofection in the mouse melanoma cell line B16F10 in vitro induced TRAIL-protein expression and was associated with morphological changes indicative of apoptosis. Systemic administration of the nanosystem in the tail vein of mice with melanoma B16F10 at the lungs produced a very significant increase in apoptosis in tumoral cells that correlated with the number of melanoma tumor foci observed in the lungs. The high levels of apoptosis detected in the lungs were partially related to mouse survival. The data presented demonstrate that the magnetofection nanosystem described here efficiently induces apoptosis and growth inhibition of melanoma B16F10 in the lungs. This new approach for systemic delivery and activation of a gene based in a nanocomplex offers a potential application in magnetic gene delivery for cancer.
Keywords: magnetic nanoparticles, magnetofection, TRAIL, chitosan, apoptosis
Introduction
Successful gene delivery and expression remains a significant hurdle that must be overcome before genetic therapies gain clinical acceptance. As such, there is currently much emphasis on further developing nucleic acid-delivery systems, which can be classified into three categories: physical injection, viral vectors, and nonviral vectors.1 These systems refer to synthetic reagents and additives that protect the genes from degradation and allow them to overcome cellular barriers during the delivery process.2 The efficiency of nonviral gene delivery depends critically on the local concentration of plasmid DNA in the desired tissue and overcoming some barriers, including rapid degradation by nucleases.3 The study of gene therapies based on nonviral vectors is very relevant, because there is no known overall efficiency, particularly for such diseases as lung cancer, which has the highest mortality rate of all common cancers and a survival rate of less than 5 years. Out of the 8.2 million deaths caused by cancer in 2011 globally, mortality from lung cancers contributed the most, with 1.3 million deaths. Clearly, there is a lack of diagnostics and effective treatment regimens.4 Different approaches for the delivery of genes to the lungs, such as intravenous (IV) injection and nasal or intratracheal instillation, have been reported.5–7
The ideal anticancer therapies are those that can selectively kill cancer cells without affecting healthy cells. TRAIL is an example of a molecule that selectively kills malignant cells, but not normal cells. In 1995, Wiley et al demonstrated that recombinant, soluble TRAIL (or Apo2L) possessed the unique ability to induce apoptosis in a range of tumor cell lines, while having little cytotoxicity against normal cells and tissues.8 TRAIL is expressed constitutively in many normal tissues, including lymphocytes, spleen, thymus, prostate, ovary, and intestines (although not in the brain, liver, or testis). In fact, the presence of TRAIL in normal tissues suggests that normal cells contain mechanisms that protect them from apoptosis induction by TRAIL.9
Magnetofection is a nonviral transfection method that uses an external magnetic field to target cells with nucleic acids that are bound to magnetic nanoparticles (MNPs).10 MNP–nucleic acid complexes are added to cell-culture media and then onto the cell surface by applying a magnetic force.11 This technique has significant advantages over traditional transfection methods, including reduced process time: about 5–10 minutes. Moreover, magnetofection has the major safety advantage of exploiting natural uptake pathways (the endocytic mechanisms of cells during the transfection process, without disrupting the cell membrane), resulting in high cell viability posttransfection.12 The final conclusion in a large number of studies in this field is that exposure to magnetic fields with intensities under 0, 1 mT cannot produce a significant cellular change, as genotoxic or epigenetic activity. The last few years in particular have shown an increase in studies indicating evidence of genotoxic effects caused by exposure to magnetic fields alone ranging from 5 mT to 30 μT in vitro and from 5 mT to 100 μT in vivo. Exposure was 1–3 days. The discrepancies between the many studies so far conducted are probably due to differences in experimental parameters. These comprise physical features (such as frequency and flux intensity), duration, and mode of exposure, in addition to characteristics of the cells or animals exposed.13
In vivo magnetic fields are focused on the required site to promote transfection and also to deliver the therapeutic gene to a specific organ or site within the body.14 Many different tissues have been studied as potential magnetic drug-delivery targets. Delivering drug-filled magnetic particles to distinct areas in the liver has been the most successful, but it is also possible to increase particle concentrations magnetically in the lungs, vessel walls, brain, and tumors.15 Many researchers have described magnetofection methods where they modified the surface of iron oxide-based MNPs to increase transfection efficiency and reduce cytotoxicity. To achieve this, some investigators selected coating agents, such as chitosan.16 In 1995, Mumper et al17 were the first group to propose the use of chitosan as a DNA carrier. It showed significantly lower toxicity than poly-L-lysine and polyethylenimine. Additionally, it enhanced the transport of the drug across the cell membrane.18 The cationic properties of chitosan, together with its biocompatibility, make it attractive as an alternative biocompatible gene-delivery system.19
Chitosan NPs are formed according to an approximation of “bottom-up”, as a result of self-association processes under which the polymer chains are ordered nanoscopic structures, either by inter–intramolecular interactions or by covalent or noncovalent interactions. In these NPs or nanospheres, the drug can be entrapped or bonded to the solid polymer matrix. In the past decade, chitosan NPs have been extensively investigated, because they can control the drug-release rate, prolong the duration of therapeutic effectiveness, and deliver drugs to specific sites in the body.20
In this study, we demonstrated that IV injection of a nanosystem formed by MNPs covered with chitosan and the sequence of a pCEM-TRAIL gene containing a promoter activated by magnetic force had a therapeutic effect in B16F10 murine melanoma cells (Figure 1). Treatment also had a positive effect in a model of lung-tumor metastasis in C57BL/6 mice.
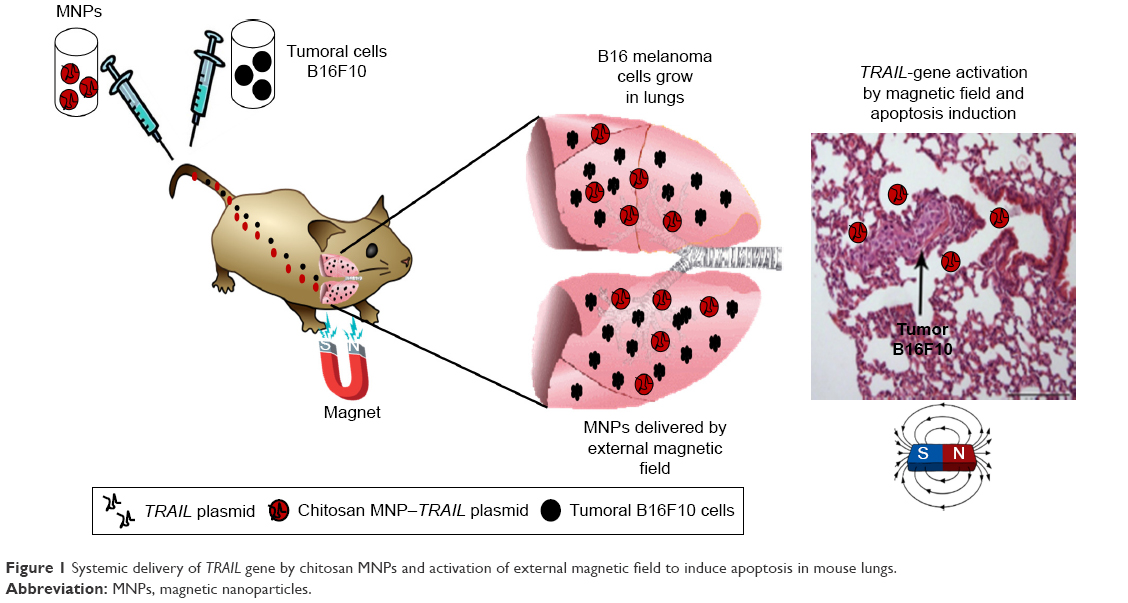 | Figure 1 Systemic delivery of TRAIL gene by chitosan MNPs and activation of external magnetic field to induce apoptosis in mouse lungs. |
Materials and methods
Chemicals
Chitosan (25 kDa) was purchased from Coyotefoods Biopolymer and Biotechnology Mexico. Cell-culture media, fetal bovine serum, and cell culture supplements were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Tripolyphosphate (TPP) was purchased from Sigma-Aldrich (St Louis, MO, USA). MNPs were obtained from OZ Biosciences (CombiMag; Marseille, France). In vivo DogtorMag™ transfection reagent designed for in vivo targeted transfection was also provided by OZ Biosciences. A plasmid purification kit was purchased from Thermo Fisher Scientific. A caspase 3 (active) red-staining kit was purchased from CTR Scientific (Monterrey, Mexico). A Wizard® Genomic DNA Purification kit, EcoR1, XbaI, and HindIII restriction enzymes, and DeadEnd fluorometric TUNEL (terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling) system were purchased from Promega Corporation (Fitchburg, WI, USA).
Plasmids
The plasmid PORF5-mTRAIL of 4,058 bp containing the TRAIL gene was purchased from InvivoGen (San Diego, CA, USA). The plasmid pCEM-Luc of 5,000 bp contains the firefly luciferase (Luc) gene under the control of the Hsp70 promoter, which has elements of response to an electromagnetic field (CEM), was obtained and characterized in our laboratory.21 The plasmids were purified with a DNA-purification kit (Promega).
Melanoma cell line
The mouse melanoma cell line B16F10 was provided by our laboratory cell bank. Cells were maintained at 37°C in 5% CO2. These tumors are synergic for C57BL/6 mice, and have historically been used as a model for human melanoma, based on the aggressive nature of the tumor, as well as its poor immunogenicity.
Animals
Male C57BL/6 mice (7–8 weeks old) from Harlan Laboratories (Mexico City, Mexico) were used in all experiments. This study was approved by the bioethics committee of Biological Sciences Faculty of the Autonomous University of Nuevo Leon (UANL). All animal-handling procedures were performed according to the Mexican Official Standard NOM-062-ZOO-1999, technical specifications for the production, care and use of laboratory animals.
Construction and characterization of plasmid pCEM-TRAIL
The first objective of this investigation was to strip the TRAIL gene from the pORF5 vector, as well as the luciferase gene from pCEM-Luc, and finally to bind TRAIL and pCEM. To amplify the TRAIL fragment, we designed the following primer sequence: forward 5AAGCTT CATGGCTCCTTCCTCAGGGG and reverse 5TCTAGA TTA GTT AAT TAA AAA GGC TCC. The TRAIL sequence was amplified by polymerase chain reaction at temperatures of 60°C, 62°C, 64°C, 66°C, and 68°C, and run in an agarose gel (8%) to observe the 890 bp fragment. Then, the fragment was purified and cloned in the vector pGEM-T Easy. pCEM-Luc was digested with XbaI and HindIII to strip the luciferase sequence, and pCEM and TRAIL were ligated to form the pCEM-TRAIL vector.
Production and characterization of magnetic chitosan–DNA nanoparticles
Chitosan NPs were prepared as we previously described, based on the technique of ionic gelation, in which the positively charged amine groups interact with the negatively charged groups of TPP and plasmid.19 The spontaneous formation of NPs occurs because of the intermolecular bonds between the negative and positive charges. In brief, chitosan was dissolved in water to a concentration of 2 mg/mL and mixed with TPP (0.86 mg/mL), plasmid, MNP, and DogtorMag. The spontaneous formation of complexes occurred under agitation for 20 minutes at room temperature (37°C). The MNPs and plasmid were mixed at a ratio (v:w) of 1:1. The ratio of pCEM-TRAIL to chitosan was 1:30.
The DNA-binding ability of chitosan was evaluated by agarose-gel electrophoresis. Complexes containing 1 μg DNA and different proportions of chitosan were loaded into individual wells of a 1% agarose gel, electrophoresed at 80 V for 40 minutes, and stained with ethidium bromide.
In vitro gene transfection of B16F10 cells with magnetic chitosan–DNA nanoparticles
Cells (2×105) were seeded into a six-well plate in 1 mL of Dulbecco’s Modified Eagle’s Medium supplemented with fetal bovine serum and 1% antimycotic antibiotic and incubated at 37°C in 5% CO2 for 24 hours before transfection, so that the cells could reach 70% confluence. Complexes containing 1 μg of DNA formed as previously described were added to each well, and then a magnetic field was placed under the plate for magnetofection for 20 minutes and incubated for 24 hours posttransfection.
Detection of active caspase 3
Transfected cells were analyzed 24 and 48 hours after transfection with a red staining caspase 3 kit, which uses the caspase 3 inhibitor DEVD-FMK conjugated to sulforhodamine as the fluorescent in situ marker. The number of fluorescent cells was counted in the total image area using the ImageJ program.22
Western blot analysis
The TRAIL protein was detected in B16F10 cells. In brief, cells transfected with different treatments were lysed, and proteins were extracted and quantified in a nanodrop system. These proteins were run in a polyacrylamide gel (12%) at 100 V, 80 mA for 45 minutes. Then, the gel was transferred to a polyvinylidene difluoride membrane and run at 300 V, 100 mA for 3 hours. The membrane was then exposed to the primary and secondary antibodies. Finally, we developed the membrane by colorimetry.
Hematoxylin and eosin staining
After 48 hours of magnetofection with the complexes, the cells were stained with hematoxylin and eosin (H&E) to observe their morphological features by microscopy.
Survival measure and histological studies
For the pulmonary metastatic model, 200 μL B16F10 cell suspension (5×105) was injected IV into each mouse though the tail vein. We divided the mice into five groups of five mice per group, as shown in Table 1. Three days after tumor-cell implantation, the C57/BL6 mice received the treatments described (IV). While injecting the treatments, an Nd2Fe12B square magnet of 17×17×5 mm (length × length × height) specific for lung tissue was placed up the ribcage and removed at the end of administration according to the Oz Bioscience manual. The mice were monitored until death. After autopsy, lungs were collected and formalin-fixed and pulmonary nodules were counted. Following the counting of nodules, the lungs were dehydrated and embedded in paraffin using routine methods for immunohistopathological analysis. Paraffin sections (5 μm) were stained with H&E, and morphological characteristics were observed under microscopy. These experiments were repeated three times.
 | Table 1 Groups of mice for in vivo assays |
TUNEL assay
Cell apoptosis was analyzed using an in situ cell death-detection kit (Promega) based on the terminal TUNEL technique. Cells were cultured on glass culture slides coated with polylysine to avoid apoptotic cells losing adherence to the slides. After the cells were transfected with the NPs for the indicated time, the slides were fixed overnight in 100 g/L formaldehyde, treated with proteinase K and H2O2, and labeled with dUTP in a humidified chamber at 37° for 1 hour. The cells that did not receive TdT enzyme were used as the negative control. Apoptotic indices were calculated by counting the number of TUNEL-positive cells/total number of cells in a field ×100. A total of ten fields were counted in a random and blinded manner.
Results and discussion
Preparation and characterization of chitosan magnetic nanoparticles
NPs analyzed by electronic and atomic force microscopy demonstrated a well-defined spherical shape, as reported previously (Figure 2).7 The chitosan–DNA NPs were prepared at an nitrogen:phosphate ratio of 1:60 with a ζ-potential of −25 mV, which resulted in optimum NPs of 200–250 nm (Figure 3), an appropriate size for transfection in vitro and in vivo. The spherical and compact morphology was a consequence of the chitosan-controlled gelation promoted by TPP.23
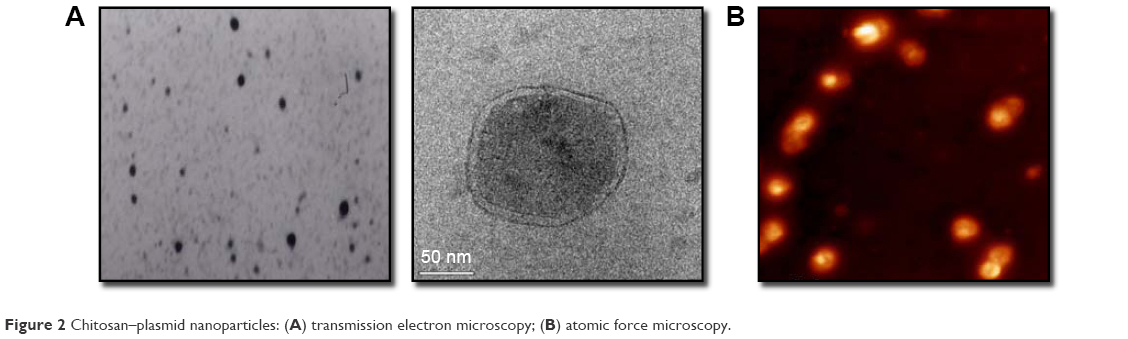 | Figure 2 Chitosan–plasmid nanoparticles: (A) transmission electron microscopy; (B) atomic force microscopy. |
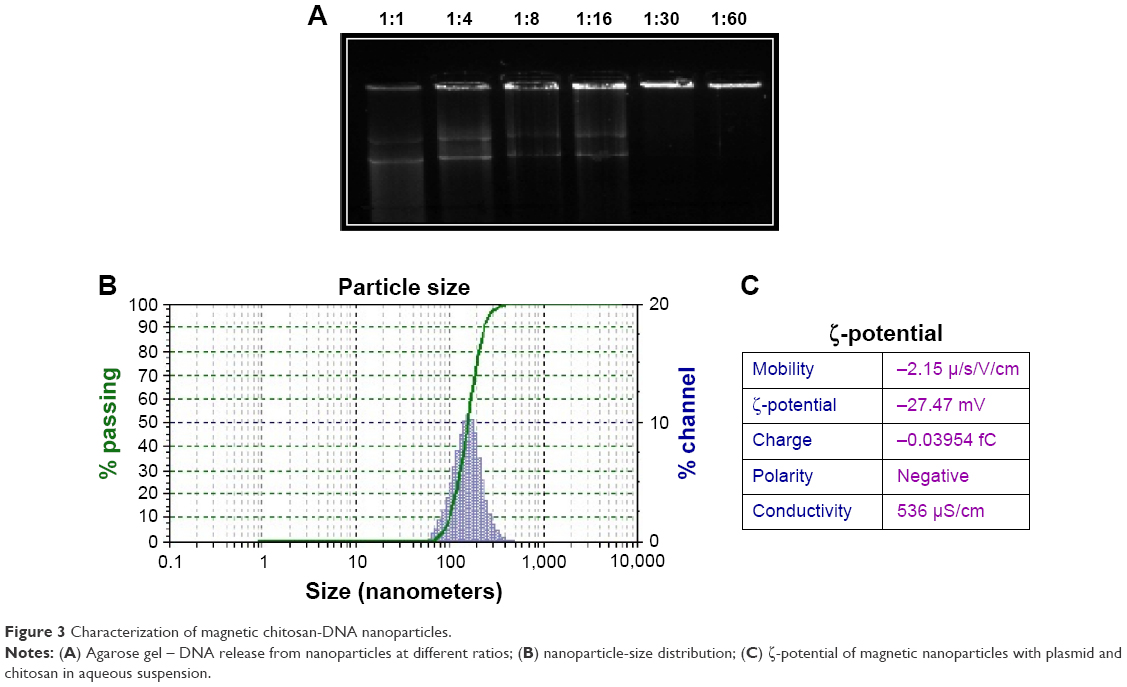 | Figure 3 Characterization of magnetic chitosan-DNA nanoparticles. |
In vitro gene expression of magnetic chitosan–DNA nanoparticles
Caspase 3 activation
Cell death triggered by TRAIL proceeds either directly from the death-inducing signaling complex formed upon TRAIL binding to its cognate agonistic receptors (TRAIL-R1 or TRAIL-R2), or indirectly via an amplification loop involving the mitochondria. Both pathways ultimately lead to the activation of caspase 3, the main caspase responsible for the execution of apoptosis.24 Cells were observed by fluorescence microscopy 24 and 48 hours after transfection. Cells treated with naked NPs showed a lower expression of active caspase 3 compared to chitosan–DNA and MNPs at 48 hours (Figure 4). This result suggests that the expression of caspase 3 is induced by the nanosystem with the magnetic field. Other authors have speculated that favorable postoperative survival is achieved by accelerated apoptotic cancer cell death when caspase 3 is expressed strongly, because caspase 3 plays an important role in the induction of apoptotic cell death in non-small-cell lung cancer.25 Apoptosis induction by TRAIL in lungs using an adenovirus as a vector has already been achieved.26 Others have already induced pulmonary apoptosis using different systems, with similar results.27
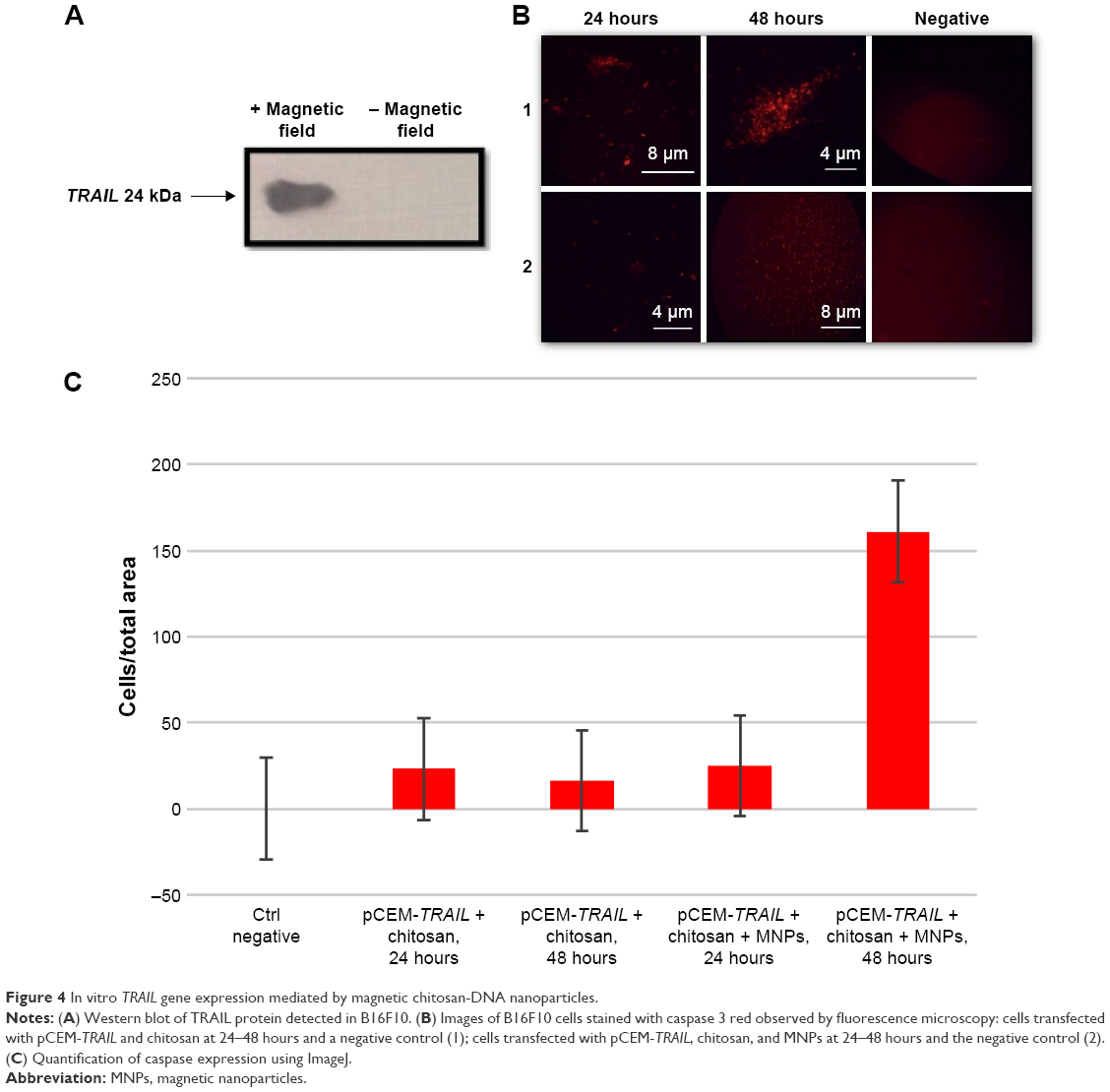 | Figure 4 In vitro TRAIL gene expression mediated by magnetic chitosan-DNA nanoparticles. |
H&E staining of cultured B16F10 melanoma cells
Cells were stained with H&E to observe by microscopy morphological features, such as nuclear condensation and cellular shrinkage 48 hours after transfection. The untransfected control cells showed morphologic hallmarks, such as stellate and elongation typical for normal B16F10 cells compared to cells treated with pCEM-TRAIL. When we observed cells transfected with chitosan–DNA complexes, a certain percentage of cells showed shrinkage. Finally, most of the cells treated with magnetic chitosan–DNA NPs were smaller and had lost their normal morphology (Figure 5). These results agree with the characteristics of apoptosis in B16F10 cells reported by others28,29 and with the morphological hallmarks of apoptosis.30
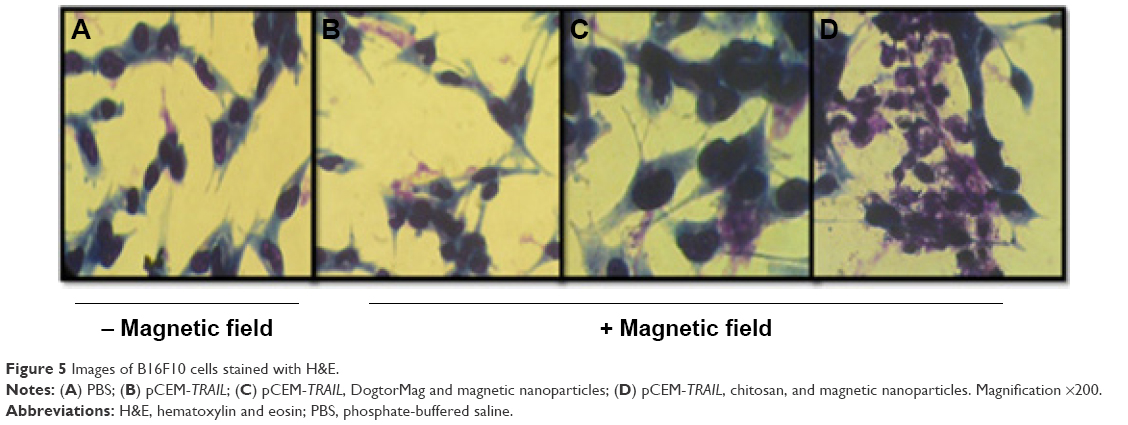 | Figure 5 Images of B16F10 cells stained with H&E. |
In vivo survival measure and histological studies
H&E staining of tissue and cell sections
H&E-stained tissue sections also showed few nodules in the lungs of mice treated with MNPs; in contrast, the untreated mice developed many melanomas (Figure 6). Control mice treated with phosphate-buffered saline had an extensive tumor burden in the lungs, whereas the mice treated with MNPs displayed fewer tumors. Recurrence of lung metastases often occurs, thereby indicating that conventional modalities are no longer effective against metastases. The development of passive or active drug-delivery systems for therapeutic applications represents an innovative trend in lung cancer therapy.31 In this study, we attempted to apply MNPs with a therapeutic gene activated by a magnetic field as a strategy to inhibit the development of melanoma in the lung.
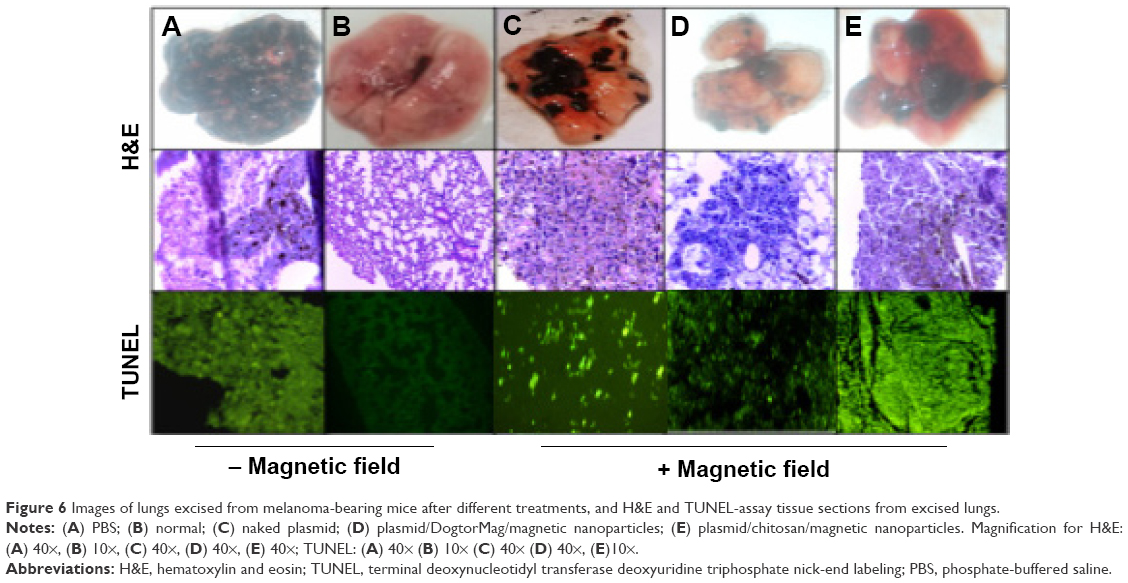 | Figure 6 Images of lungs excised from melanoma-bearing mice after different treatments, and H&E and TUNEL-assay tissue sections from excised lungs. |
TUNEL assay
TUNEL assays revealed a significantly higher proportion of TUNEL-positive cells in the MNP group. The results from these experiments support the use of MNPs for killing melanoma cells in vivo (Figure 7). In groups treated with naked plasmid and the magnetic field, we observed lower cell-death percentages of 10%–15%. In the group treated with MNPs, plasmid, DogtorMag, and the magnetic field, we observed up to 45% cell death. The group with the most cell death (80%) was that treated with NPs, chitosan, and magnet (Figure 6). No correlation was observed between the apoptosis levels detected by the TUNEL test with the survival rate, but there was a proportional relation between the apoptosis levels detected by TUNEL and the number of tumors in lungs observed macroscopically. The DNA degradation detected by TUNEL in pulmonary melanoma has been described by others using different treatments.26
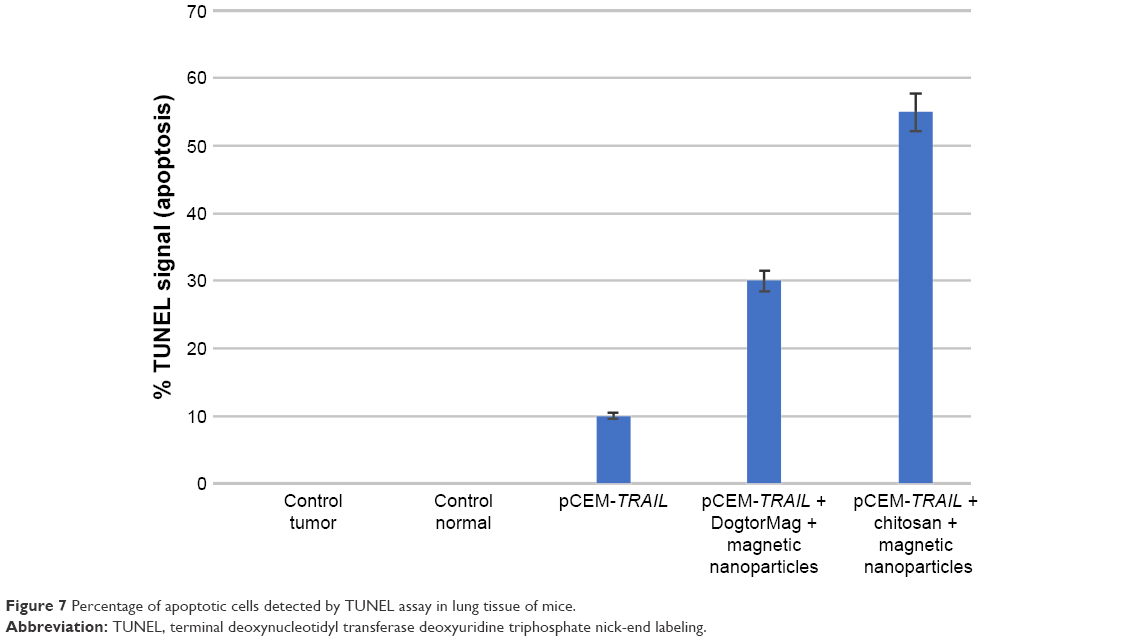 | Figure 7 Percentage of apoptotic cells detected by TUNEL assay in lung tissue of mice. |
Survival analysis of mice
In vivo studies in a melanoma pulmonary metastatic mice model were conducted to evaluate the survival rate of mice treated with our nanosystem. Figure 8 shows a Kaplan–Meier plot of the survival of B16F10 tumor-bearing mice after different treatments. NPs formed with the cationic lipid (DogtorMag) and naked plasmid showed better mouse survival, perhaps because a larger number of them were able to reach the lungs. It is necessary to find a balance between the grades of induced apoptosis to produce inhibition of tumors to correlate with better survival. Because a relatively small increase in the rate of epithelial cell apoptosis can result in considerable cell loss over time, a relatively minor upregulation of epithelial apoptosis, particularly of alveolar type II cells, could account for excessive epithelial loss and failure of reepithelialization.32
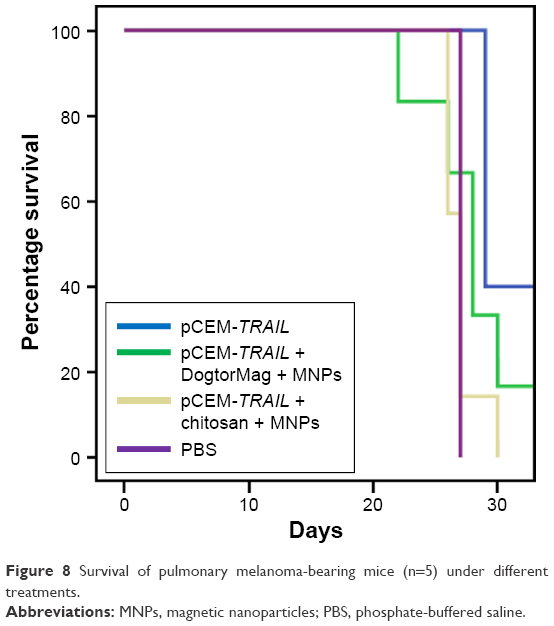 | Figure 8 Survival of pulmonary melanoma-bearing mice (n=5) under different treatments. |
The assays described are the results of several repeated experiments, the groups of mice behaving in the same way. The administration of 50 μg of naked plasmid produce an adequate expression of transgene in the lungs of mice.7 It is possible that the level of apoptosis occurring in these appropriate conditions improved mouse survival. On the other hand, when the same amount of plasmid (50 μg) was administered in the chitosan MNPs, it produced a high level of apoptosis in the lung, as demonstrated in the TUNEL assay. The excessive cellular death induced by the expression of TRAIL in the lung can potentially cause lung dysfunction by a strong inflammatory process resulting from the removal of apoptotic cells. The alteration in lung function and the presence of tumor cells remaining may be the cause of the lack of relationship with the survival obtained in mice.33–36 It may be important to develop cell-specific antiapoptotic strategies, rather than strategies aimed at blocking apoptosis of types of cells within the lungs. Other studies have reported similar survival profiles for pulmonary melanoma by IV administration of BSA-gel capsules and polyethylenimine–p53 complexes by aerosol delivery.6,35 There have been similar studies for targeted gene delivery to the lungs using magnetized aerosols comprising superparamagnetic oxide NPs.37
Therefore, in this study the magnetic field had two roles: the first to attract the MNPs when they reached the perimeter of the magnetic field and exert a potentiating effect of transfection, and second to induce activation of the promoter present in the plasmid that controls the expression of TRAIL. We have presented in vitro data that prove these effects. In in vivo assays, we only demonstrated that there was a significant difference in the activation of TRAIL-induced apoptosis in the lungs of mice, indicating the influence of the magnetic field in the attraction of the NPs and magnetofection under these conditions. However, in survival assays, this could not be demonstrated, though our previous studies and those of other groups support the in vivo effect of magnetofection.
Conclusion
According to our results, the nanosystem formed by MNPs, plasmid, and chitosan is stable and can efficiently magnetotransfect cells. We used this nanosystem to cause cell death in the B16F10 cell line by apoptosis and in the lungs of mice. In vitro, we observed the expression of active caspase 3, morphological changes in cells, and TRAIL protein expression. In vivo, we observed a significant reduction in tumor numbers. In addition, TUNEL assay in lung tissue revealed up to 80% of apoptotic cells. These results support the applicability of the extensively studied NPs, specifically in the fields of drug delivery and cancer treatment. However, many challenges must be overcome to expedite the translation of NP-based therapies.
Acknowledgment
We acknowledge Conacyt for providing the funds to do this investigation.
Disclosure
The authors report no conflicts of interest in this work.
References
Bordelon H, Biris AS, Sabliov CM, Monroe WT. Characterization of plasmid DNA location within chitosan/PLGA/pDNA nanoparticle complexes designed for gene delivery. J Nanomater. 2011;2011:952060. | ||
Shi Y, Zhou L, Wang R, et al. In situ preparation of magnetic non-viral gene vectors and magnetofection in vitro. Nanotechnology. 2010;21:115103. | ||
Chorny M, Polyak B, Alferiev IS, Walsh K, Friedman G, Levy RJ. Magnetically driven plasmid DNA delivery with biodegradable polymeric nanoparticles. FASEB J. 2007;21:2510–2519. | ||
Wing-Hin L, Ching-Yee L, Traini D, Young PM. Inhalation of nanoparticles-based drug for lung cancer treatment: advantages and challenges. Asian J Pharm Sci. 2015;10:481–489. | ||
Densmore CL, Orson FM, Xu B, et al. Aerosol delivery of robust polyethyleneimine-DNA complexes for gene therapy and genetic immunization. Mol Ther. 2000;1:180–188. | ||
Gautam A, Densmore CL, Xu B, Waldrep JC. Enhanced gene expression in mouse lung after PEI-DNA aerosol delivery. Mol Ther. 2000;2:63–70. | ||
Alvizo-Báez CA, Luna-Cruz IE, Rodríguez-Padilla MC, Alcocer-González JM. Magnetic nanoparticles of chitosan for targeted delivery system of plasmids to the lungs. J Nanotechnol. 2014;2014:313415. | ||
Wiley SR, Schooley K, Smolak PJ, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. | ||
Kagawa S, He C, Gu J, et al. Antitumor activity and bystander effects of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene. Cancer Res. 2001;61:3330–3338. | ||
Dobson J. Gene therapy progress and prospects: magnetic nanoparticle-based gene delivery. J Gene Ther. 2006;13:283–287. | ||
Kami D, Takeda S, Itakura Y, Gojo S, Watanabe M, Toyoda M. Application of magnetic nanoparticles to gene delivery. Int J Mol Sci. 2011;12:3705–3722. | ||
Estelrich J, Escribano E, Queralt J, Busquets MA. Iron oxide nanoparticles for magnetically-guided and magnetically-responsive drug delivery. Int J Mol Sci. 2015;16:8070–8101. | ||
Mihai C, Rotinberg P, Brinza F, Vochita G. Extremely low-frequency electromagnetic fields cause DNA strand breaks in normal cells. J Environ Health Sci Eng. 2014;12:15. | ||
Kavaz D, Cirak T, Ozturk E, Bayram C, Denkbas EB. Preparation of magnetic chitosan nanoparticles for diverse biomedical applications. In: Vaseashta AK, Mihailescu IN, editors. Functionalized Nanoscale Materials, Devices and Systems. Heidelberg: Springer; 2008:313–320. | ||
Häfeli UO, Riffle JS, Harris-Shekhawat L, et al. Cell uptake and in vitro toxicity of magnetic nanoparticles suitable for drug delivery. Mol Pharm. 2009;6:1417–1428. | ||
Hashimoto M, Hisano Y. Directional gene-transfer into the brain by an adenoviral vector tagged with magnetic nanoparticles. J Neurosci Methods. 2011;194:316–320. | ||
Mumper RJ, Wang JJ, Claspell JM, et al. Novel polymeric condensing carriers for gene delivery. Proc Int Symp Controlled Release Bioact Mater. 1995;22:178–179. | ||
Sailaja AK, Amareshwar P, Chakravarty P. Chitosan nanoparticles as a drug delivery system. Res J Pharm Biol Chem Sci. 2010;1:474–484. | ||
Roy K, Mao HQ, Huang SK, Leong KW. Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat Med. 1999;4:387–391. | ||
Kafshgari MH, Khorram M, Khodadoost M, Khavari S. Reinforcement of chitosan nanoparticles obtained by an ionic cross-linking process. Iran Polym J. 2011;20:445–456. | ||
Rodriguez-de-la-Fuente AO, Alcocer-Gonzalez JM, et al. Effect of 60 Hz electromagnetic fields on the activity of Hsp70 promoter: an in vitro study. Cell Biol Int. 2009;33:419–433. | ||
US National Institutes of Health. ImageJ. Available from: http://imagej.nih.gov/ij. Accessed October 8, 2016. | ||
Gaspar VM, Sousa F, Queiroz JA, Correia IJ. Formulation of chitosan-TPP-pDNA nanocapsules for gene therapy applications. Nanotechnology. 2011;22:015101. | ||
Micheau O, Mérino D. Controlling TRAIL-mediated caspase-3 activation. Leuk Res. 2004;18:1578–1580. | ||
Koomägi R, Volm M. Relationship between the expression of caspase-3 and the clinical outcome of patients with non-small cell lung cancer. Anticancer Res. 2000;20:493–496. | ||
Shi J, Zheng D, Liu Y, et al. Overexpression of soluble TRAIL induces apoptosis in human lung adenocarcinoma and inhibits growth of tumor xenografts in nude mice. Cancer Res. 2005;65:1687–1692. | ||
Zamora-Avila D, Zapata-Benavides P, Franco-Molina M, et al. WT1 gene silencing by aerosol delivery of PEI-RNAi complexes inhibits B16-F10 lung metastases growth. Cancer Gene Ther. 2009;16:892–899. | ||
Pereyra F, Arruda D, Figueiredo C, et al. FTY720 induces apoptosis in B16F10-NEX2 murine melanoma cells, limits metastatic development in vivo, and modulates the immune system. Clinics (Sao Paulo). 2013;68:1018–1027. | ||
Strange R, Li F, Saurer S, Burkhardt A, Friis R. Apoptotic cell death and tissue remodeling during mouse mammary gland involution. Development. 1992;115:49–58. | ||
Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Int J Cardiovasc Res. 2000;45:528–537. | ||
Martin TR, Hagimoto N, Nakamura M, Matute-Bello G. Apoptosis and epithelial injury in the lungs. Proc Am Thorac Soc. 2005;2:214–220. | ||
Taube C, Schuler M. Apoptosis in the physiology and diseases of the respiratory tract. In: Reed JC, Green DR, editors. Apoptosis: Physiology and Pathology. New York: Cambridge University Press; 2011:221–230. | ||
Henson PM, Tuder RM. Apoptosis in the lung: induction, clearance and detection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L601–L611. | ||
Drakopanagiotakis F, Xifteri A, Polychronopoulos V, Bouros D. Apoptosis in lung injury and fibrosis. Eur Respir J. 2008;32:1631–1638. | ||
Shen H, Hui S, Meng X, et al. Biodegradable chitosan/alginate BSA-gel-capsules for pH-controlled loading and release of doxorubicin and treatment of pulmonary melanoma. J Mater Chem B Mater Biol Med. 2013;1:3906–3917. | ||
Kuwano K. Epithelial cell apoptosis and lung remodeling. Cell Mol Immunol. 2007;4:419–428. | ||
Heidsieck A, Gleich B, Bergemann C, Aneja M, Rudolph C. Magnetized aerosols comprising superparamagnetic iron oxide nanoparticles improve targeted drug and gene delivery to the lung. Pharm Res. 2012;29:1308–1318. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.