")
Back to Journals » Clinical Epidemiology » Volume 12
Synthetic and External Controls in Clinical Trials – A Primer for Researchers
Authors Thorlund K , Dron L, Park JJH, Mills EJ
Received 12 December 2019
Accepted for publication 21 April 2020
Published 8 May 2020 Volume 2020:12 Pages 457—467
DOI https://doi.org/10.2147/CLEP.S242097
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Irene Petersen
Kristian Thorlund,1,2 Louis Dron,2 Jay JH Park,2,3 Edward J Mills1,2
1Department of Health Research Methods, Evidence & Impact (HEI), McMaster University, Hamilton, ON, Canada; 2MTEK Sciences, Vancouver, BC, Canada; 3Department of Medicine, University of British Columbia, Vancouver, BC, Canada
Correspondence: Kristian Thorlund
MTEK Sciences, 802-777 West Broadway, Vancouver, BC V5Z 1J5, Canada
Email [email protected]
Abstract: There has been a rapid expansion in the use of non-randomized evidence in the regulatory approval of treatments globally. An emerging set of methodologies have been utilized to provide greater insight into external control data used for these purposes, collectively known as synthetic control methods. Through this paper, we provide the reader with a set of key questions to help assess the quality of literature publications utilizing synthetic control methodologies. Common challenges and real-life examples of synthetic controls are provided throughout, alongside a critical appraisal framework with which to assess future publications.
Keywords: synthetic control, RCTs, real-world evidence
Current Challenges of Clinical Trial Investigations
Randomized clinical trials (RCTs) are the gold standard for the evaluation of experimental interventions. In RCTs, patients are usually randomized to either an experimental intervention arm or a control intervention arm that usually consists of placebo or standard-of-care (SOC). Patient recruitment and retention are two key factors for successful RCTs. The use of placebo, however, can impose recruitment and retention challenges that can halt the timelines of these placebo-controlled trials, as patients have been shown to be less willing to participate in placebo-controlled RCTs.1 The intent of clinical trials is research and not medical care, but still, patients often hope for some level of treatment.2
While the use of active control (ie SOC treatment) has been suggested to address ethical and logistics challenges of associated with placebos, it often presents with similar challenges. In a rapidly progressing field such as oncology, it is not unusual for the SOC to become updated during the course of the trial. An updated SOC can ultimately challenge the fundamental ethical basis of RCTs in “clinical equipoise”, a genuine uncertainty within the scientific and medical community as to which intervention is clinically superior, that justify randomizing patients to the control group.3 In rare diseases, it can be difficult to determine what should be used as an active control, given that there are often no established treatments in these areas. Many clinical trials on rare diseases are conducted with very few patients, translating to insufficient statistical power, or are performed as single-arm trials that make it difficult to compare against other therapeutic options without synthetic control methods.4,5
With the rise in precision medicine, these challenges have been amplified, as more diseases are being diagnosed and classified according to their genetic make-up, resulting in increased sub-stratified disease definitions. For instance, epidermal growth factor receptor (EGFR) is a key mutation for non-small cell lung cancer (NSCLC) patients, and there have been several trial programs that have been based on EGFR mutations for this disease. However, it is important to recognize that only a proportion of NSCLC patients will have an EGFR mutation, so conducting clinical trials that only recruit EGFR-positive patients versus NSCLC patients based on a broader disease classification only will be much more challenging. With these granularities in how diseases are now being classified, ‘rare diseases’ have become more paradoxically common in oncology and other disease areas. Investigators are experiencing increasing challenges of enrolling a sufficiently large number of patients within a reasonable window of time for their clinical trials. While it is difficult to dispute the value of properly conducted RCTs, and the routine, successful implementation of studies utilizing either a placebo or SOC arm, the availability of data sources and methodologies developed to utilize external data have evolved dramatically over recent years. We can optimize the use of external data set with synthetic control methods, but as this is a new concept to many researchers, improving the literacy in these methods is important. For this discussion, we define external data as any source of clinical data from potentially relevant sources, inclusive of clinical trial data, routine health record data, insurance claims data or patient registries. Synthetic controls are defined as cohorts of patients from external data and adjusted using any of a variety of statistical methodologies.
Introduction to Synthetic Controls for Clinical Evaluation
The synthetic control methods are statistical methods that can be used to evaluate the comparative effectiveness of an intervention using external control data. The US Food and Drug Administration (FDA) and European Medicines Agency (EMA) have recognized these issues and taken several initiatives to allow for these novel approaches to external control data.6,7 The FDA approved cerliponase alfa for a specific form of Batten disease, based on synthetic control study that compared the data of 22 patients studied in a single-arm trial versus independent external control group data with 42 untreated patients.8 Across 20 European countries, alectinib, a non-small cell lung cancer treatment, had an expansion of label based on synthetic control study based on an external data set of 67 patients.9 A kinase inhibitor, palbociclib, also had an expanded indication for men with HR+, HER2-advanced or metastatic breast cancer on the basis of external control data.10 The use of non-comparative data is not unique to rare diseases alone, as more common chronic diseases such as hepatitis C and previously treated rheumatoid arthritis have had treatments approved based on non-comparative data.11 Moreover, a recent review of 489 pharmaceutical technologies assessed by the National institute for Health and Care Excellence (NICE) identified 22 submissions that used external data and synthetic control methods to establish clinical efficacy.11 Of these, 13 (59%) utilized published RCT data for their external control, and six (27%) utilized observational data. Over half of the applications were made in the last two recent years alone, further confirming the increasing attention paid by both drug manufacturers and health technology assessment agencies on this topic.
From the conventional evidence-based medicine, the use of external data to create synthetic controls for clinical evaluations represents a radical paradigm shift. A healthy degree of scepticism on the use of synthetic controls is thus expected from the scientific community. Nevertheless, it is likely that there will be an increasing number of clinical trials that use external data as a synthetic control, so it is important for researchers to comprehend the validity and reliability of synthetic control studies. Here in this paper, we provide guidance on what questions researchers must ask when interpreting and critically evaluating the evidence from synthetic control-based clinical trials. For a critical evaluation of synthetic control clinical trials, it will be important for researchers to evaluate the external data that is used itself and the statistical methods used to create a synthetic control group. We have outlined eleven key questions in Table 1 that researchers can ask regarding the validity and quality of trials utilizing external data and synthetic control trials.
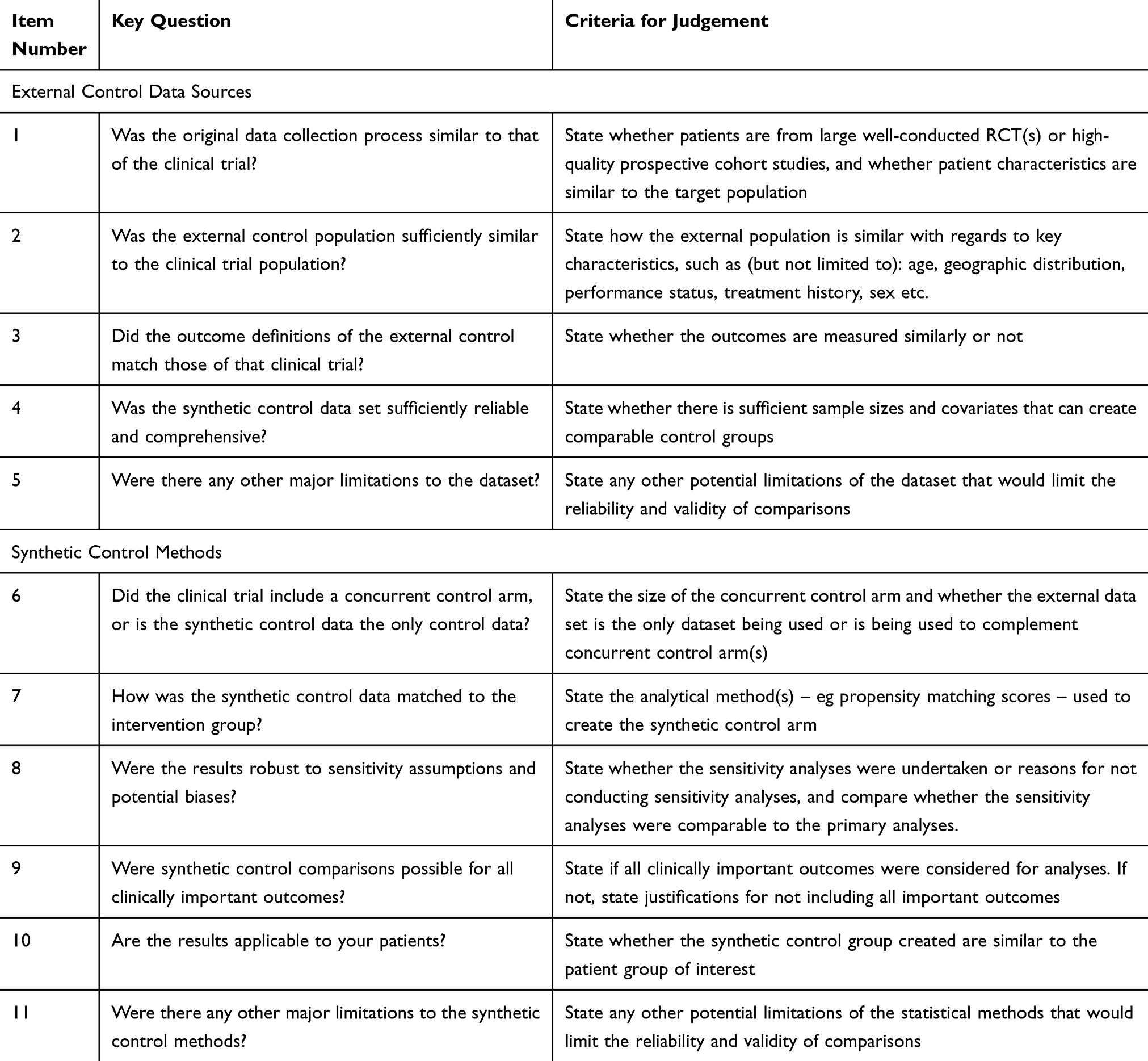 |
Table 1 Synthetic Control Quality Checklist |
“Synthetic” Control Data Set
It is important to consider the validity and reliability of the “synthetic” control data set that is used for clinical comparisons of different interventions. For this, it is important to consider the process of the original data collection, compare the populations of the datasets that are being compared, and the reliability and comprehensiveness of the datasets. We have outlined these important considerations in Table 1 and Figure 1.
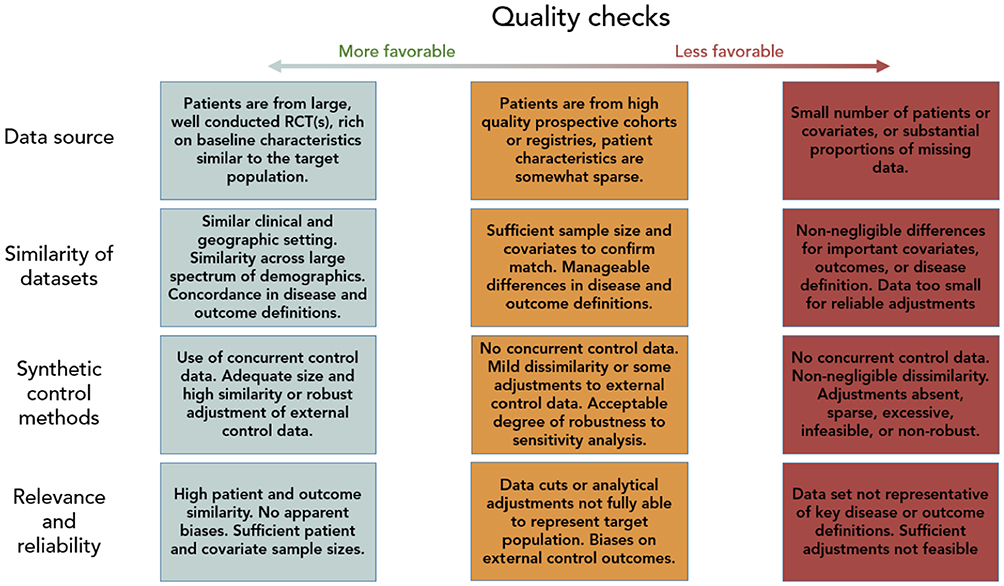 |
Figure 1 Quality check process. |
Was the Original Data Collection Process Similar to That of the Clinical Trial?
Ideally, synthetic controls should be informed by external control data from recent RCTs answering as similar a question as possible, and using as similar designs and implementation processes as possible. Examples of such RCTs would be those investigating a less efficacious intervention in the same population as well as those investigating a broader population where subgroup control data on the target population are available. The data collection in RCTs generally adheres to a high level of stringency. Particularly for RCTs conducted within the same disease areas and within the past 5–10 years, one can usually be reasonably confident that clinical outcome and covariate definitions, value ranges, biomarker kits and thresholds, and others were reasonably similar.
Control data from well-designed observational cohorts may also be adequate, particularly if they have some link to RCTs such as concurrent SOC surveillance, prospective evaluation of efficiency, or were designed to be hypothesis generation for future RCTs. Conversely, control data retrieved from electronic medical records reflect clinical practice and not the controlled environment in which RCTs typically establish efficacy. Pertinent to many future FDA submissions, such data will likely come from large commercial entities selling “real-world data”. Particularly, the data collection and curation processes from such sources may be highly heterogeneous, leaving uncertainty of unknown biases and systematic missing data patterns. The same limitations apply to large case series. Methods exist to minimize these sources of heterogeneity as discussed below, but the resulting dataset is still susceptible to sources of bias.
While publications reporting a clinical trial making use of a synthetic control will rarely provide exhaustive details on the data curation processes from external control sources, a brief description of the external data source(s) as well as a justification for its use may often be available. If confidence in the similarity of data recording processes cannot be asserted from this information, a comparison of published trial protocols may be necessary. If the external data come from non-RCT sources, some additional information is required to assert that the reported data variables are, in fact, sufficiently similar to combine. These recommendations are in line with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) group harmonized tripartite guideline E10.12
Is the External Control Population Sufficiently Similar to the Clinical Trial Population?
Evaluating the similarity of the external control population to the clinical trial population is a multi-faceted exercise. There are many factors that may differ between external control sources and the clinical trial, but not all may matter. Further, not all important factors may be reported or quantifiable (ie unknown confounders). The eligibility criteria for the considered external control sources should be similar, but this does not guarantee that key patient characteristics are similar. In the context of oncology, if two trials both recruited patients with stage III–IV cancer, but if one predominantly includes stage III patients and the other predominantly includes stage IV patients, these cannot be considered similar. Some account of similarities in the distributions of key baseline characteristics should, therefore, be provided by authors of clinical trials making use of synthetic controls. It is important to note that although patient characteristics may differ in the original external data set, this does not necessarily preclude their use for constructing control groups. Through appropriate statistical adjustments, subgroup analyses or sensitivity analyses, it may be possible to utilize the adjusted external data to create a synthetic control (see section: “Synthetic control methods”).
Other important factors may not be reported, either because of international shifts in clinical research practice or interventional guidelines. In these instances, it may not be possible to successfully generate synthetic controls if data are not available to bridge the gap between these shifts. For example, the World Health Organization previously recommended initiation of antiretroviral therapy (ART) for patients with HIV and a CD4+ cell count of <200 cells/µL in their 2006 guidelines, but in 2010 this was amended to <350 cells/µL, on the basis of updated randomized controlled evidence indicating a mortality benefit. As CD4+ cell count is an important prognostic indicator for HIV-related mortality,13 it would be challenging to incorporate and mix control population data from most trials published prior to 2010, particularly in the absence of assessable baseline CD4+ cell count. Similarly, for trials that incorporate a “baseline” standard of care, gradual changes in treatment options create a similar issue. For instance, NRTI backbone therapy in the treatment of HIV used to more frequently contain stavudine and didanosine, and has shifted toward emtricitabine and in particular, tenofovir on the basis of updated WHO guidance.14 These have associated efficacy and safety changes that influence the outcomes of trials and datasets for which they have been provided, corresponding to challenges in interpreting comparative data across distinct time periods.
Evolution of standard-of-care is not limited to the eligible population of interest. For example, in oncology, the combination of underlying therapies considered to be standard-of-care has historically changed rapidly over time.15,16 This issue is by no means isolated to oncology applications, as similar challenges are present in other disease topics, particularly in diseases that are considered chronic, where a sequence of treatments either alone in combination are common-place (eg rheumatoid arthritis, type-II diabetes).17,18
As such, researchers looking to utilize standard-of-care treatment data from external data sources should take appropriate care to ensure that this bears sufficient similarity to the research population of interest. Ensuring that data relating to features such as dosing, frequency, combination(s) and sequence of treatment administration are available from the external control source is therefore crucial.
Do the Outcome Definitions of the External Control Match Those of the Clinical Trial?
The definitions of clinical outcomes commonly change over time, and investigator preferences for definitions may differ across studies. For example, a popular composite outcome in cardiovascular trials, MACE (major adverse cardiovascular events), can vary significantly across studies in terms of the inclusion of individual outcomes (eg, in- or exclusion of revascularization) or the stringency of the definition of the individual outcome (eg unstable angina requiring vs not necessarily requiring hospitalization). Similarly, in oncology, the definition of disease progression may vary considerably. For example, the WHO response criteria19 were used prior to the widespread uptake of original RECIST (response evaluation criteria in solid tumors),20 with the two guidelines differing with regard to the number of measured lesions, the criteria for partial response and progressive disease. The RECIST guideline itself has been updated since to version 1.1,21 with differences from the prior version with regard to the number of measured lesions, requirements for absolute increases in lesion size increases for progressive disease and the integration of newer radiological measurement tools (FDG-PET). If the similarity of external control outcomes and the clinical trial outcomes is not justified in the report (publication) of the clinical trial, it may be necessary to double-check the outcome definition from publicly available sources such as clinicaltrials.gov in case the external control(s) come from recent RCTs. Other external sources may be difficult to access or verify and should be considered a bias risk to the validity of the synthetic control validity.
Is the Synthetic Control Data Set Sufficiently Reliable and Comprehensive?
Even if data collection processes, patient populations, clinical outcomes, and other pertinent factors are sufficiently similar to assert validity, the external control data must still be of a sufficient size to assert reliability, include sufficient variables to allow of statistical adjustments if necessary, and ideally come from at least two sources to assert some degree of replicability. Where no statistical adjustments are needed, a sufficient sample size is necessary for ensuring that the observed external control group effect is not a play of chance finding. Of course, with rare disease clinical trials where sample sizes are typically substantially smaller, it may not be feasible to apply such rigor. Where statistical adjustments are needed the associated sample size, and if applicable, the number of observed events must be sufficiently large to also support the adjustment for key variables. Whilst there is a commonly advocated minimum number of events per key variable discussed in several clinical guidelines,22,23 there is little evidence that any true rule of thumb can be used, and formal statistical assessment of sample size should be undertaken when adjustment is performed.24 The proportion of available covariates and the proportion of missing data is also highly important for the validity of statistical adjustments. For example, external data from prospective observational studies may only include data on covariates pertinent to the research question(s) studied. Commercial “real-world” databases may claim to house hundreds of covariates, but in reality, only have close to complete and analyzable data on a few of these.
When subgroups of external control data are used it is important to assess the reliability of the external control data based on the sample size rather of the subgroup than the sample size of the entire external control data source. In particular, commercial real-world databases may claim to house millions of patients, but when narrowing the eligibility criteria to a subset specifically matching that of the clinical trial of interest, data may only be available for a few hundred or even less patients, and these numbers may further decrease when restricting by the availability of clinical outcomes of interest. Having multiple congruent sources of external control data adds additional certainty, even if each source has a comparatively small sample size. For example, prior work utilizing Bayesian dynamic borrowing identified that when historic data with similar patient demographics were combined, uncertainty was reduced significantly per each additionally included trial external control data source.25 In contrast, when incongruent clinical trial data were combined, no additional improvement to uncertainty was noted regardless of the number of trials of external control data added.
Were There Any Other Major Limitations in the Dataset?
RCTs employing synthetic controls may have further limitations or biases, even if the employed external sources appear similar and unbiased and the methods used to match the data are appropriate. External RCTs, for example, may differ with respect to factors that either cannot be or are rarely recorded in a data set. Palliative care for rare diseases may comprise whichever is best practice and the given health centre setting, but these may differ substantially between centers and even between physicians.
Synthetic Control Methods
Did the Clinical Trial Include a Concurrent Control Arm or Is the Synthetic Control Data the Only Control Data?
Synthetic controls can broadly be used in two settings. First, external data can be used to augment the precision of a concurrent control group in a randomized clinical trial (eg, using 4:1 randomization between treatment and control). Second, they can be used to create a stand-alone control group solely from external data. The latter is more common for rare diseases where single-arm trials predominate and randomizing to control is unethical, infeasible or highly inefficient.
Synthetic controls used to augment the precision of a concurrent control group generally have higher validity since they can be validated with the control arm in the performed RCT. Particularly control data from similar RCTs are valuable in this setting. Even if pertinent RCTs are not highly identical, much strength can still be gained.25 Where it is either unethical or infeasible to enrol patients to a control intervention, external data should be selected from the best possible source of data and synthetic controls can be used as a substitute for an absent control arm. In this setting (often a rare disease setting), pertinent RCTs are typically not available. There are many examples where various types of historical data, but increasingly data from prospectively recruiting patient registries as well as subsets from commercial real-world databases are being used. The scientific validity of these depend on the accuracy of the match. N-of-1 data, where each enrolled patient has at least several months of historical data on SOC, are ideal, but often not available. Data from similar medical centers from similar geographical regions to the target trial of interest can also improve validity. Of course, the more recent the higher validity, although a few months’ buffer should be allowed for full data curation processes (including quality assurance) to be finalized. Aggregate estimates from published cohort studies or case series may also be considered, either to validate primary sources of external data or as a substitute in the absence of individual-level patient-level data. Aggregate estimates from published studies nonetheless face limitations as detailed reporting on patient demographics, and clinical settings can be relatively sparse, and as such, similarity can be difficult to validate.
How Was the Synthetic Control Data Matched to the Intervention Group?
There are many advanced statistical and computational techniques available to match external control data with a tangible degree of dissimilarity to the RCT being conducted or having finished. While there are too many individual methodologies to individually list, we have summarized many of the key methods and categories of methods in Table 2.
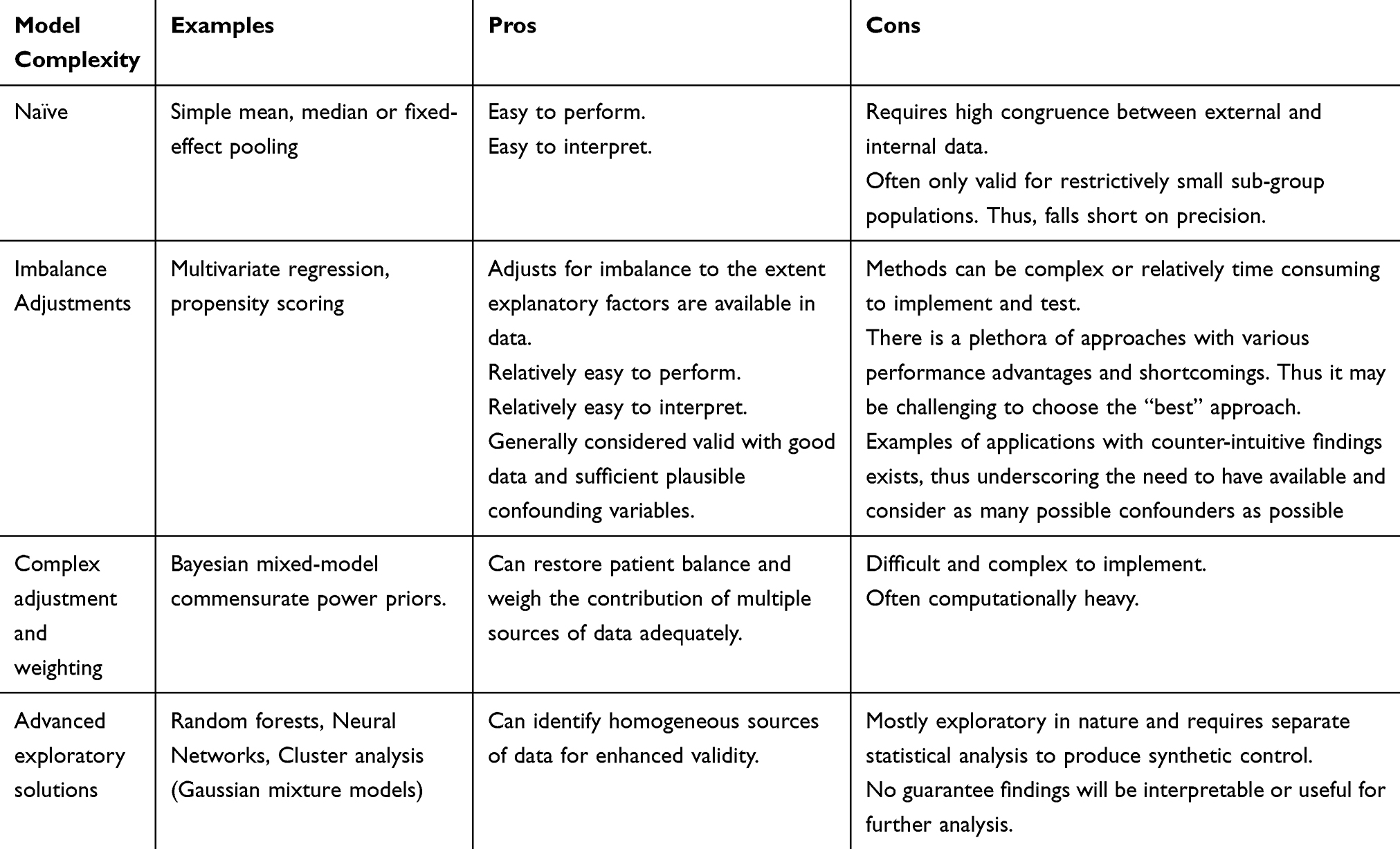 |
Table 2 A Summary of Commonly Used Models and Methods for Generating Synthetic Control Arms |
While many of these promise the world, the old garbage in garbage out principle always applies. As an illustrative example, imagine a synthetic control of 400 oncology patients, including 200 stage II patients, 195 stage III patients, but only 5 stage IV patients. In this instance, no reliable adjustment of stage IV patients exists due to this subgroup’s small sample size regardless of the analytical methods used, but a relationship between the response among stage II vs stage III patients can be quantified with reasonable reliability.
In settings where external control data are available from similar RCTs and where eligibility criteria and known patient characteristics are reasonably similar, one relatively straightforward method for use of synthetic control is via Bayesian analysis where the external data can be translated into a prior distribution and combined with the concurrent RCT data.25 This approach allows a flexible degree of weight to be put on the external data. Researchers reading an article utilizing a Bayesian approach will simply have to assert that the authors have justified the similarity of the external data and assigned an appropriate weight (precision) to the external data.
In settings where external control data differs notably with respect to eligibility criteria and patient characteristics, the simple and common approach is to restrict the external data to a subgroup of patients that match the concurrent RCT. The key with this approach is the ability to restrict to the population on all parameters that are known or likely to cause confounding. Once accurate subgroups have been obtained, external control data can be combined with concurrent control data using methods like the Bayesian approach. Restricting on several parameters can often substantially reduce the sample size of external data subgroups. Where this is true, some form of statistical adjustment may be preferred alongside a relaxation of the eligibility criteria applied to the external control data.
Propensity score adjustment is a powerful set of statistical techniques designed for this type of setting. While propensity score methods require all confounders to be observed—as is the case with multivariate regression methods for addressing imbalance—there are some notable advantages. Firstly, it is easier to accommodate a larger number of linear and non-linear relationships compared to multivariate regression where limited sample sizes may present a greater problem (although care still needs to be taken when performing variable selection for propensity score methods26). Secondly, regression models where covariates are included in the outcome model will extrapolate regardless of the non-overlap of populations. Imagine a scenario where one trial for diabetes includes participants with a baseline HbA1c of >8mmol/mol, whereas the single-arm trial of interest only recruited patients with a baseline HbA1c of ≥6.5–7.9mmol/mol. A regression model would extrapolate between the two regardless of the fact that no true data exists to “bridge” these two populations, whereas propensity score adjustment creates a balanced pool of participants and responses.27 Lastly, propensity score matching has the advantage that it can be used to estimate average treatment effects on the treated or average treatment effects on the untreated.26 This may be mitigated by employing a doubly robust estimator, where covariates are included in both the treatment and outcome models.28 Of course, propensity score adjustments are no magic wand and applied examples with highly counter-intuitive findings have been observed in the literature.29 Thus, with the use of propensity score adjustments, it is always important to consider as many plausible for the model as possible. A schematic representation of propensity score adjustment in the context of adding data to a concurrent control arm is provided in Figure 2.
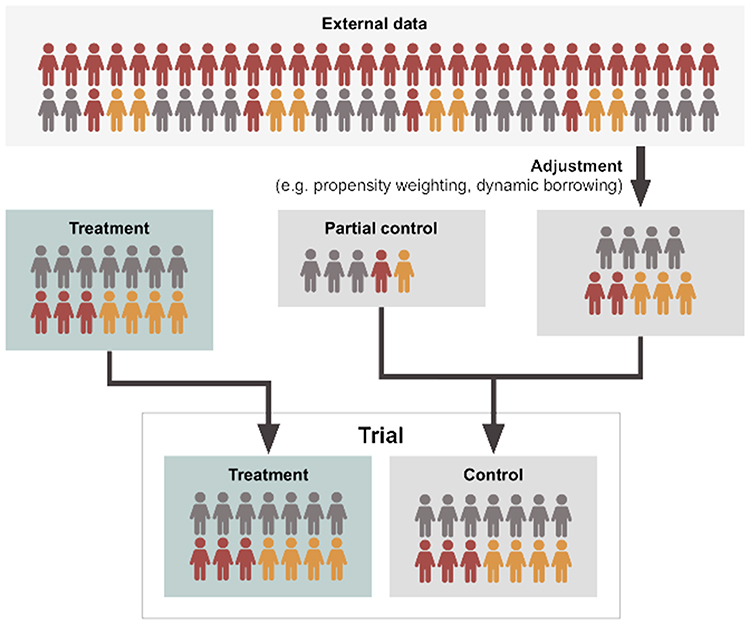 |
Figure 2 Schematic representation of dynamic borrowing or propensity-based methods. Adjustment methodologies refer to techniques such as propensity weighting or Bayesian dynamic borrowing as described in greater detail within Table 2. Here, an external dataset is adjusted utilizing statistical methodologies to make it more representative of a target population. This can either be centered on a target population existing as a partial control in a clinical trial, or a target treatment population. Depending on the method, a variable proportion of data from the external source is borrowed, adjusted for statistical (dis)similarity. |
Microsimulation represents an alternative methodology set to explore longer-term trends as a form of synthetic control. Microsimulations refer to Markov models wherein the unit of the simulation is an individual, rather than a population.30 This allows for high-resolution definitions of the key patient subgroup(s) over the full disease trajectory where no single external control data set covers such longevity. For example, the stages of non-alcoholic steatohepatitis are well studied in observational studies and some data are available in government and commercial patient registries. However, no study of sufficient size and longevity has covered the trajectory from early fibrosis to later endpoints like cirrhosis, liver cancer and liver decompensation. Microsimulations allow long-term outcomes to be simulated whilst accounting for well-established complex interactions between patient characteristics which are prognostic or predictive of the standard of care response. Microsimulations of synthetic controls can thereby be linked to controlled single-arm trials or uncontrolled prospective patient registries to aid in the estimation of a comparative effect, particularly in settings where a long-term clinical trial is infeasible. Other advantages of microsimulations include how the methods lend themselves to other health technologies such as diagnostic or screening tools. For example, the microsimulation of prostate-specific antigen screening strategies was previously pivotal in developing a nationwide screening strategy in the US.31 A schematic representation of microsimulation methods is provided in Figure 3.
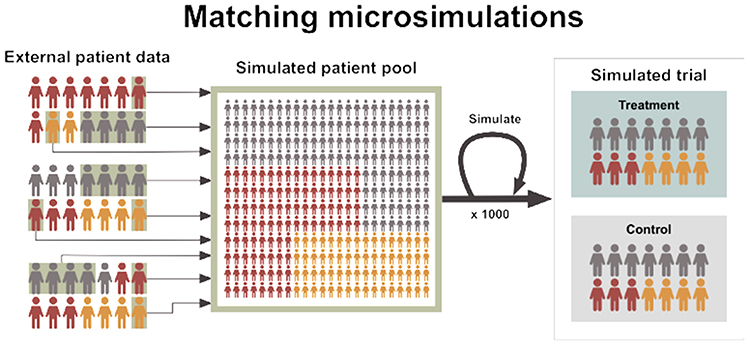 |
Figure 3 Matching microsimulations are shown as utilizing external individual-level patient data to construct simulated cohorts which can represent truly synthetic control groups at the individual-patient level. Here, external data informs patient trajectories for the outcome(s) of interest to the relevant trial population, which can then be analyzed and compared to data from an interventional treatment arm. |
Were the Results Robust to Sensitivity Assumptions and Potential Biases?
Clinical trial investigators employing synthetic controls face tough analytical decisions. With synthetic controls, there is no absolute best approach. There will be several ways to improve the similarity of external data by means of restriction to subgroups and statistical adjustments for several different parameters. The weighting of external control data is also not a given. Researchers reading a paper reporting use of synthetic controls should, therefore, assert that efforts have been made to investigate the robustness of sensitivities of the underlying methods for employing synthetic controls. For example, have the authors run multiple comparisons using different methods, and do these methods show concordant results? For example, in the analysis of blinatumomab for ALL, the authors demonstrated four differing sensitivity analyses, all of which demonstrated high levels of concordance, improving confidence in the validity of the results.32 If no sensitivity analyses have been conducted, the validity may be unclear. If sensitivity analyses have been conducted, these can be used to assert whether the approach is valid or not. Of course, in many settings and particularly for rare diseases, external control data may be sparse and confidence (or credible) intervals may remain large, yet having a synthetic control may be better than nothing. As such, the sensitivities of the employed synthetic control approaches need to be interpreted with context to the clinical and statistical limitations which may be fixed.
How Can I Apply the Results to Patient Care?
Were Synthetic Control Comparisons Possible for All Clinically Important Outcomes?
External data sources used to create synthetic controls may not always provide evidence on the same outcomes as the concurrent RCT. External data may either not contain some outcome(s) of interest at all, or the outcome definitions may differ substantially. When outcomes are not available in the external data, it is important to interpret the strength and quality of the evidence by each individual outcome, using the same standard practice for individual RCTs.33 For example, external data from oncology RCTs may contain progression-free survival outcome data multiple data sources, but overall survival outcome data may be limited to one smaller external RCT. Where outcome definitions differ, sufficient data may still have been available to maintain adequate similarity. For example, if an external data set used eGFR<45 mL/min/1.73m2 to define kidney failure, but the concurrent RCT used a cut-off of eGFR<30 mL/min/1.73m2, the use of external data may be challenged. However, if individual patient-level external data are available contains the eGFR values for each patient, then the proportion of patients with an eGFR<30mL/min/1.73m2 can be constructed directly. Readers of papers reporting on synthetic controls should assess the extent to which such measures were taken before asserting the strength and quality of the evidence for each outcome.
Are the Results Applicable to Your Patients?
As with conventional RCTs, those employing synthetic controls should provide inferences for population groups and clinical settings that are generalizable to clinical practice. If the synthetic control, whether stand-alone or used with a concurrent control, does not appear to be ideally matched or adjusted, the generalizability may suffer. Likewise, if adjustments were made to the RCT data set as well as the external control to let the two sources “meet in the middle”, the population and clinical setting the produced inferences are representing may either be unclear or non-generalizable to the clinical setting of interest in patient care. Researchers reviewing a trial employing synthetic controls should, therefore, always ask the question whether they employed matching or adjustment techniques have distorted inferences substantially away from the clinical setting of interest. Were there any other limitations in the synthetic control methods?
RCTs employing synthetic controls may have further limitations or biases, even if the employed external sources appear similar and unbiased and the methods used to match the data are appropriate. External RCTs, for example, may differ with respect to factors that either cannot be or are rarely recorded in a data set. Palliative care for rare diseases may comprise whichever is best practice and the given health centre setting, but these may differ substantially between centers and even between physicians.
Conclusion
While synthetic control methods hold great promise, particularly in the context of patient populations traditionally challenging to recruit or assess in randomized clinical trials, these methods do not constitute a “cure-all”. They are likely to be increasingly referenced and utilized within the regulatory and peer-reviewed literature space owing to co-existing improvements in medical record collection34 and statistical methodologies. As such, establishing criteria with which they should be assessed is an important step towards collective improvements in this space, and in ensuring that appropriate conclusions are drawn from this type of work. Through our checklist, we aim to provide researchers, clinicians and policy-makers with a quality assessment methodology for both readers and groups involved in relation to synthetic control research to improve the clarity of reporting. We recognise that each synthetic control project contains its own unique challenges with regards to generalisability of results, interpretation and associated statistical methodology, but believe our key questions (Table 1) cover most of the key recurring themes discussed elsewhere in the literature.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Groth SW. Honorarium or coercion: use of incentives for participants in clinical research. J N Y State Nurses Assoc. 2010;41(1):11.
2. Chiodo GT, Tolle SW, Bevan L. Placebo-controlled trials: good science or medical neglect? West J Med. 2000;172(4):271–273. doi:10.1136/ewjm.172.4.271
3. Hey SP, London AJ, Weijer C, Rid A, Miller F. Is the concept of clinical equipoise still relevant to research? BMJ. 2017;359:j5787.
4. Joppi R, Bertele V, Garattini S. Orphan drugs, orphan diseases. The first decade of orphan drug legislation in the EU. Eur J Clin Pharmacol. 2013;69(4):1009–1024. doi:10.1007/s00228-012-1423-2
5. Sasinowski FJ, Panico EB, Valentine JE. Quantum of effectiveness evidence in FDA’s approval of orphan drugs: update, July 2010 to June 2014. Ther Innov Regul Sci. 2015;49(5):680–697. doi:10.1177/2168479015580383
6. FDA. Submitting Documents Using Real-World Data and Real-World Evidence to FDA for Drugs and Biologics Guidance for Industry; 2019.
7. EMEA CfMPfHUJL. Guideline on Clinical Trials in Small Populations; 2006.
8. FDA approves first treatment for a form of batten disease [press release]. Online, April 2017.
9. Petrone J. Roche pays $1.9 billion for Flatiron’s army of electronic health record curators. Nat Biotechnol. 2018;36(4):289–290. doi:10.1038/nbt0418-289
10. Stalder RZ, Wrobel BJ, Boehncke L, Brembilla W-H, Costantino N. The janus kinase inhibitor tofacitinib impacts human dendritic cell differentiation and favours M1 macrophage development. Exp Dermatol. 2019;12:12.
11. Anderson M, Naci H, Morrison D, Osipenko L, Mossialos E. A review of NICE appraisals of pharmaceuticals 2000–2016 found variation in establishing comparative clinical effectiveness. J Clin Epidemiol. 2018;105:50–59.
12. Group IEW. ICH Harmonised Tripartite Guideline: Choice of Control Group and Related Issues in Clinical Trials E10; 2000.
13. Mills EJ, Bakanda C, Birungi J, Yaya S, Ford N. The prognostic value of baseline CD4(+) cell count beyond 6 months of antiretroviral therapy in HIV-positive patients in a resource-limited setting. AIDS. 2012;26(11):1425–1429. doi:10.1097/QAD.0b013e328354bf43
14. Organization WH. Antiretroviral Therapy for HIV Infection in Adults and Adolescents: Recommendations for a Public Health Approach-2010 Revision; 2010.
15. Moffett P, Moore G. The standard of care: legal history and definitions: the bad and good news. West J Emerg Med. 2011;12(1):109–112.
16. Markman M. Standard of care versus standards of care in oncology: a not so subtle distinction. J Oncol Pract. 2007;3(6):291. doi:10.1200/JOP.0761502
17. Mian AN, Ibrahim F, Scott IC, et al. Changing clinical patterns in rheumatoid arthritis management over two decades: sequential observational studies. BMC Musculoskelet Disord. 2016;17(1):44. doi:10.1186/s12891-016-0897-y
18. Mohan V, Cooper ME, Matthews DR, Khunti K. The standard of care in type 2 diabetes: re-evaluating the treatment paradigm. Diabetes Ther. 2019;10(1):1–13. doi:10.1007/s13300-019-0573-y
19. Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer. 1981;47(1):207–214. doi:10.1002/1097-0142(19810101)47:1<207::AID-CNCR2820470134>3.0.CO;2-6
20. Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. JNCI. 2000;92(3):205–216. doi:10.1093/jnci/92.3.205
21. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. doi:10.1016/j.ejca.2008.10.026
22. Moons KG, Altman DG, Reitsma JB, et al. Transparent reporting of a multivariable prediction model for Individual Prognosis or Diagnosis (TRIPOD): explanation and elaboration. Ann Intern Med. 2015;162(1):W1–73. doi:10.7326/M14-0698
23. Moons KG, de Groot JA, Bouwmeester W, et al. Critical appraisal and data extraction for systematic reviews of prediction modelling studies: the CHARMS checklist. PLoS Med. 2014;11(10):e1001744. doi:10.1371/journal.pmed.1001744
24. van Smeden M, de Groot JAH, Moons KGM, et al. No rationale for 1 variable per 10 events criterion for binary logistic regression analysis. BMC Med Res Methodol. 2016;16(1):163.
25. Dron L, Golchi S, Hsu G, Thorlund K.Minimizing control group allocation in randomized trials using dynamic borrowing of external control data – an application to second line therapy for non-small cell lung cancer. Contemp Clin Trials Comm.2019;16:100446. doi:10.1016/j.conctc.2019.100446
26. Williamson E, Morley R, Lucas A, Carpenter J. Propensity scores: from naive enthusiasm to intuitive understanding. Stat Methods Med Res. 2012;21(3):273–293. doi:10.1177/0962280210394483
27. Morgan SL, Winship C. Counterfactuals and Causal Inference: Methods and Principles for Social Research.
28. Lunceford JK, Davidian M. Stratification and weighting via the propensity score in estimation of causal treatment effects: a comparative study. Stat Med. 2004;23(19):2937–2960. doi:10.1002/sim.1903
29. Freemantle N, Marston L, Walters K, Wood J, Reynolds MR, Petersen I. Making inferences on treatment effects from real world data: propensity scores, confounding by indication, and other perils for the unwary in observational research. BMJ. 2013;347(nov11 3):f6409. doi:10.1136/bmj.f6409
30. Siebert U, Alagoz O, Bayoumi AM, et al. State-transition modeling: a report of the ISPOR-SMDM modeling good research practices task force-3. Value Health. 2012;15(6):812–820. doi:10.1016/j.jval.2012.06.014
31. Etzioni R, Penson DF, Legler JM, et al. Overdiagnosis due to prostate-specific antigen screening: lessons from U.S. prostate cancer incidence trends. JNCI J Natl Cancer Inst. 2002;94(13):981–990. doi:10.1093/jnci/94.13.981
32. Gokbuget N, Kelsh M, Chia V, et al. Blinatumomab vs historical standard therapy of adult relapsed/refractory acute lymphoblastic leukemia. Blood Cancer J. 2016;6(9):e473. doi:10.1038/bcj.2016.84
33. Guyatt G, Oxman AD, Akl EA, et al. GRADE guidelines: 1. Introduction—GRADE evidence profiles and summary of findings tables. J Clin Epidemiol. 2011;64(4):383–394.
34. OECD. Readiness of Electronic Health Record Systems to Contribute to National Health Information and Research; 2017.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.