")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Synthesis of budesonide conjugates and their anti-inflammatory effects: a preliminary study
Authors Yan Y, Wang P, Li R, Sun Y, Zhang R, Huo C, Xing J, Dong Y
Received 26 October 2018
Accepted for publication 25 January 2019
Published 19 February 2019 Volume 2019:13 Pages 681—694
DOI https://doi.org/10.2147/DDDT.S192348
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Yan Yan,1,2 Pengchong Wang,2 Ruiying Li,3 Ying Sun,2 Rui Zhang,2 Chuanchuan Huo,2 Jianfeng Xing,2 Yalin Dong1
1Department of Pharmacy, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, China; 2Department of Pharmaceutics, School of Pharmacy, Xi’an Jiaotong University, Xi’an, Shaanxi, China; 3Department of Clinical Medicine, College of Medicine, Xi’an Jiaotong University, Xi’an, Shaanxi, China
Purpose: Budesonide (Bud) is a nonhalogenated glucocorticoid with high anti-inflammatory potency and low systemic side effects. However, the poor water solubility of Bud affects its dissolution and release behavior, thus influencing its anti-inflammatory effect. This study was aimed at synthesizing and evaluating novel conjugates of Bud, hoping to increase the anti-inflammatory activity of Bud by improving its water solubility.
Materials and methods: Seven novel Bud conjugates (3a–3g) were designed and synthesized in this study. Besides, the equilibrium solubility, cell viability, in vitro and in vivo anti-inflammatory activity, and the hydrolysis behavior of the conjugates in different pH solutions, rat and human plasma, and rat lung homogenate were studied in detail.
Results: As compared to Bud, the equilibrium solubility of 3a, 3c, and 3e was significantly increased; 3a, 3b, and 3c significantly inhibited the interleukin-6 production in lipopolysaccharide-induced A549 cells; 3a and 3e could significantly decrease the xylene-induced ear edema; and 3a and 3c were gradually and slowly hydrolyzed into Bud in the alveolar fluid and lung homogenate and broken down quickly in plasma.
Conclusion: The amino acid ester compounds budesonide-21-glycine ester (3a) and budesonide-21-alanine ester (3c) were selected as potential conjugates of Bud. This study would provide a theoretical and an experimental basis for the in vivo process of glucocorticoids and the treatment of inflammatory diseases.
Keywords: budesonide, glucocorticoid, anti-inflammatory effect, equilibrium solubility, hydrolysis behavior
Introduction
Glucocorticoid has received much attention and has been widely used for the clinical treatment of allergy, asthma, autoimmune disease, and sepsis due to its powerful anti-inflammatory and immunosuppressive effects.1,2 Budesonide (Bud) is a kind of nonhalogenated glucocorticoid and characterized by a strong first-pass effect, a short half-time, less absorption and metabolism, and a high affinity for glucocorticoid receptors.3 In clinic, Bud is mainly used for the treatment of asthma, allergic rhinitis, and ulcerative colitis by inhalation and enteroclysis. Bud, which is a continuously inhaled corticosteroid, is commonly administered for the treatment of human asthma.4,5 Compared with other glucocorticoids in this class, Bud has stronger local anti-inflammatory and pharmaceutical effects, a longer action time, and a less systemic action; therefore, it could significantly reduce the adverse reactions when used in local treatment.6,7 However, the poor water solubility of Bud affects its dissolution and release behavior, thus influencing the drug concentration at the inflammatory site and its local bioavailability.8,9
In order to discover new compounds with a higher activity and explore the relationship between the structure and activity of glucocorticoid receptor modulators, a recent study selected 77 prednisolone C-21 heteroaryl thioethers as target molecules10–12 and investigated the effect of molecular force field on their drug activity by means of molecular docking and establishing their three-dimensional quantitative structure–activity relationship (3D-QSAR) models. The result indicated that the introduction of hydrophilic groups into the C21 substituent position of glucocorticoid could enhance the drug activity.13 Therefore, we hypothesized that the anti-inflammatory activity of Bud would be increased by conjugating small hydrophilic groups to its 21-hydroxy group with an increase in water solubility.
In the present study, seven novel Bud conjugates were designed and synthesized by introducing small hydrophilic molecules into the 21-hydroxy group of Bud, that is, budesonide-21-glycine ester (3a), budesonide-21-phenylalanine ester (3b), budesonide-21-alanine ester (3c), budesonide-21-diethylamine acetate (3d), budesonide-21-dimethylamine acetate (3e), budesonide-21-N-methylpiperazine acetate (3f), and budesonide-21-morpholine acetate (3g). Furthermore, their equilibrium solubility, cell viability, in vitro and in vivo anti-inflammatory effect, and hydrolysis behavior in different pH solutions, rat and human plasma, and rat lung homogenate were studied in detail. Hopefully, the synthesized conjugates could improve the water solubility and thus increase the anti-inflammatory activity of Bud.
Materials and methods
Materials
Bud, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI), 4-dimethylaminopyridine (DMAP), Boc-glycine, Boc-phenylalanine, and Boc-alanine were purchased from Duodian Chemistry Co. Ltd. (Nanjing, China). Sodium iodide, diethylamine, dimethylamine hydrochloride, N-methylpiperazine, morpholine, lipopolysaccharide from E. coli O111:B4 (LPS-EB), and MTT were purchased from Aladdin Bio-Chem Technology Co. Ltd. (Shanghai, China). Human interleukin (IL)-6 ELISA kit was purchased from Dakewe Bioengineering Co. Ltd. (Shenzhen, China). The human alveolar adenocarcinoma cell line (A549) was kindly provided by Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). The blank plasma of healthy volunteers was given by the First Affiliated Hospital of Xi’an Jiaotong University (Xi’an, China).
Male Sprague Dawley rats (220–250 g) and male Kunming mice (20–25 g) were obtained from Laboratory Animal Center of Xi’an Jiaotong University, SCXK 2007-001, Shaanxi, China. The animals were housed under a 12-hour light–dark cycle at a constant ambient temperature (22°C–25°C), with normal chow and water ad libitum. They were allowed to acclimatize for 1 week before the experiments were started. All animal care and experimental protocols were in strict accordance with the Guidelines of the Laboratory Animal Center of Xi’an Jiaotong University and approved by the Institutional Animal Care and Use Committee of Xi’an Jiaotong University (No XJTULAC2016-004).
Synthesis of amino acid ester conjugates of Bud
Bud (1.5 g, 3.5 mmol), EDCI (1.35 g, 7.0 mmol), and DMAP (0.24 g, 2.0 mmol) were dissolved in dichloromethane (10 mL). Boc-glycine (1.02 g, 5.8 mmol) dissolved in dichloromethane was added dropwise into the above solution, and the mixture was continuously stirred at 30°C for 2 hours. The reaction was monitored by a thin-layer chromatography (TLC) method, with a developer of petroleum ether/ethyl acetate (2:1). The dichloromethane was removed by rotary evaporation, and the residue was dissolved in ethyl acetate and washed twice with hydrochloric acid (2 mol/L) and saturated sodium bicarbonate solution, respectively. The product was dried with anhydrous magnesium sulfate, and the evaporation of solvent in vacuum afforded a crystallized yellow solid. The solid reside was placed onto a silica gel column and eluted with petroleum ether/ethyl acetate (4:1); then, a crystallized white solid was obtained (2a, 1.8 g, 88.6%).
Compound 2a (0.3 g, 0.51 mmol) and trifluoroacetic acid (3.4 mL) were dissolved in dichloromethane (10 mL), and the mixture was cooled to 0°C and stirred for 4 hours. The reaction was monitored by a TLC method with a developer of petroleum ether/ethyl acetate (1:1). The mixture was adjusted to alkaline with a saturated sodium carbonate solution, washed twice with water, and dried with anhydrous magnesium sulfate. The product was then dried by rotary evaporation and washed twice with cold petroleum ether. Finally, a crystallized yellow solid was obtained (3a, budesonide-21-glycine ester; 0.28 g, 96.1%).
Budesonide-21-phenylalanine ester (3b) and budesonide-21-alanine ester (3c) were obtained similarly with the above methods. Boc-phenylalanine and Boc-alanine were used instead of Boc-glycine, respectively (Figure 1A).
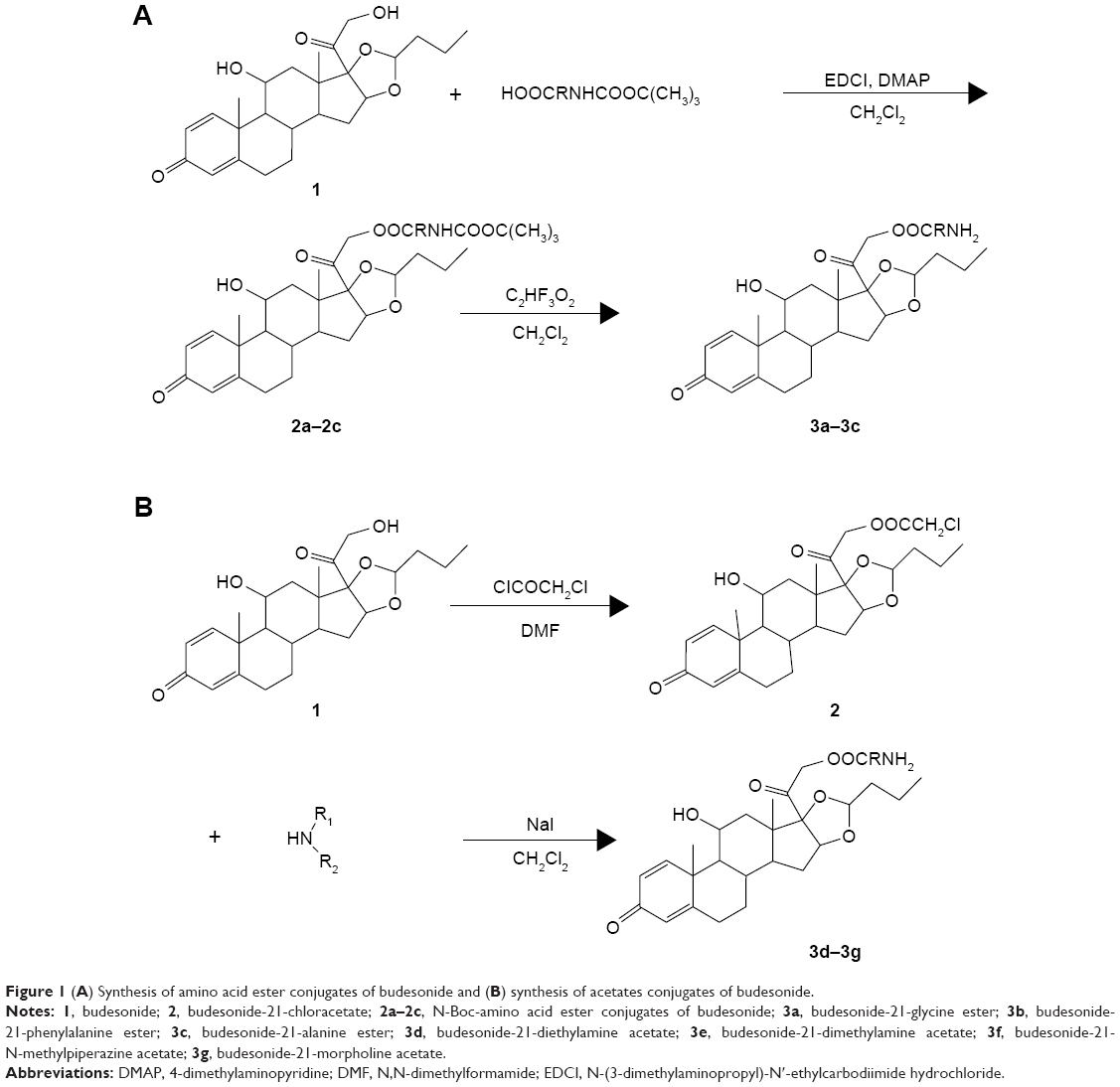 | Figure 1 (A) Synthesis of amino acid ester conjugates of budesonide and (B) synthesis of acetates conjugates of budesonide. |
Synthesis of acetates conjugates of Bud
Bud (2.0 g, 3.96 mmol) and chloroacetyl chloride (3.5 mL) were dissolved in N,N-dimethylformamide (10 mL), the mixture was stirred at room temperature for 30 minutes, and the reaction was monitored by a TLC method with a developer of petroleum ether/ethyl acetate (3:1). The mixture was dispersed in water (100 mL) and filtered, and the residue was washed twice with water. After drying, a pale yellow solid was obtained (4.5 g). The solid reside was placed onto a silica gel column and eluted with petroleum ether/ethyl acetate (6:1), and then, a crystallized white solid was obtained (2, budesonide-21-chloracetate; 2.84 g, 98.5%).
Compound 2 (0.3 g, 0.6 mmol), sodium iodide (0.09 g, 0.6 mmol), and diethylamine (0.72 mL) were dissolved in dichloromethane (10 mL), the mixture was refluxed for 30 minutes, and the reaction was monitored by a TLC method with a developer of petroleum ether/ethyl acetate (1:1). The dichloromethane was removed by rotary evaporation, and the residue was washed twice with water and saturated sodium chloride solution, respectively. The product was dried with anhydrous sodium sulfate, and the evaporation of solvent in vacuum afforded a crystallized yellow solid (3d, budesonide-21-diethylamine acetate; 0.16 g, 74.5%).
Budesonide-21-dimethylamine acetate (3e, 77.9%), budesonide-21-N-methylpiperazine acetate (3f, 62.4%), and budesonide-21-morpholine acetate (3g, 67.1%) were obtained similarly with the above methods. Dimethylamine hydrochloride, N-methylpiperazine, and morpholine were used instead of diethylamine, respectively (Figure 1B).
Characterization of the synthesized Bud conjugates
The structures of the compounds were confirmed by differential scanning calorimetry (DSC), proton nuclear magnetic resonance (1H NMR), and mass spectrometry (MS). The thermal behavior of each prodrug was analyzed by a differential scanning calorimeter (DSC822e; Mettler Toledo, Greifensee, Switzerland). Samples (5 mg) were placed in an aluminum crucible and heated at a scanning rate of 10°C/min from 25°C to 400°C under nitrogen atmosphere. The 1H NMR spectrum (in CDCl3) of each sample was recorded on a Varian 400 spectrometer (Varian, Palo Alto, CA, USA) at 400 MHz using tetramethylsilane as an internal standard. The MS analysis was performed by a TSQ Endura™ Triple Quadrupole Mass Spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) equipped with an EASY-Max NG ion source system.
Equilibrium solubility of the synthesized Bud conjugates
Each conjugate and Bud powder (0.5 g) were separately dispersed in distilled water (50 mL) and continuously stirred at 25°C for 72 hours. At 0, 12, 24, 36, 48, 60, and 72 hours, 1 mL of the supernatant was sampled and filtered with 0.45-μm microporous membrane.14 The drug concentration was measured by high-performance liquid chromatography (HPLC) method to determine the equilibrium solubility of a parent drug as well as the conjugates.
The contents of Bud and the synthesized conjugates in experimental samples were measured by HPLC (Shimadzu, Kyoto, Japan) using a Diamonsil® ODS-2 C18 column (Dikma Technologies Co. Ltd., Beijing, China).15 The detecting conditions were as follows: (1) Bud: the mobile phase was 70% of methanol solution containing 0.1% acetic acid; (2) 3a and 3b: the mobile phase was a mixture of methanol/phosphate buffer (pH 8.0, 70:30, v/v); (3) 3c: the mobile phase was a mixture of acetonitrile/phosphate buffer (pH 7.4, 70:30, v/v); and (4) 3d, 3e, 3f, and 3g: the mobile phase was a mixture of methanol/phosphate buffer (pH 8.0, 80:20, v/v). Samples were detected at 30°C, the wavelength was 242 nm, and the flow rate was 1.0 mL/min.
Cell viability of the synthesized Bud conjugates
The A549 cells were cultured in Roswell Park Memorial Institute-1640 medium, supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin and incubated at 37°C in a humidified atmosphere with 5% CO2.16 Throughout the experiment, the cells were used in the logarithmic phase of growth. They were plated into a 96-well plate at a density of 5×103 cells/well. The following day, the cells were treated with or without 200 μL of different concentration of drug solution for 72 hours. The medium was removed and replaced with a medium containing 5 mg/mL MTT and incubated for 4 hours. Then, the medium was removed, and the product was solubilized using dimethyl sulfoxide (DMSO). Finally, the absorbance was detected at 490 nm using a microplate reader (Bio-Tek Instruments Inc., Winooski, VT, USA) according to the manufacturer’s instructions.17 The inhibitory rate of cell growth was calculated using the following equation, and the 5% inhibiting concentration (IC5) was calculated from the dose–response curves.
|
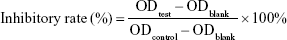
Ability of Bud conjugates to inhibit IL-6 production
The A549 cells were treated with test compounds (10−11–10−6 M) in 100 μL of medium and incubated in a 48-well plate for 1 hour. Then, LPS-EB solution (5 μL, 10 μg/mL) was added in each well and incubated for 24 hours. Finally, supernatants were collected and assayed for the IL-6 level by ELISA.18 The absorbance was detected at 450 nm by a microplate reader. The inhibitory rate of IL-6 production was calculated, and the half maximal inhibitory concentration of different compounds was calculated from the dose–response curves.
In vivo anti-inflammatory study
To evaluate the anti-inflammatory effect of test compounds, the xylene-induced ear edema test was conducted.19 Mice in test groups received different amounts of conjugates at an equivalent dose of 200 μg/kg Bud, and mice in the control group received a solvent instead of a drug solution. Sixty mice were randomly divided into six groups (n=10): control group (2% DMSO), Bud group (200 μg/kg), 3a group (226 μg/kg), 3c group (233 μg/kg), 3d group (252 μg/kg), and 3e group (239 μg/kg). Mice in each group was administrated by intraperitoneal injection (0.1 mL/10 g of body weight). After 1 hour, each mouse received 20 μL of xylene on the right ear, and the left ear remained untreated. The mice were sacrificed 1 hour after xylene application, and 6-mm circular sections of the ears were taken. The sections of both sides of ears were weighed, and the ear edema was considered as the weight difference between the right ear and the left ear.20
Biological stability of the synthesized Bud conjugates
Analytical methods of Bud conjugates
The contents of 3a, 3c, 3d, and 3e in different samples were determined by HPLC using the methods described above. The linearity of concentrations for each compound was assessed by a calibration that obtained by plotting the peak area of each sample vs its concentration (0.2–500 μg/mL), respectively. The absolute recoveries of each compound from different samples were calculated by analyzing spiked samples at 5.0, 50.0, and 500.0 μg/mL, respectively, and each concentration level was tested three times. For precision evaluation, the samples (5.0, 50.0, and 500.0 μg/mL) were analyzed three times within 1 day (intraday) and within 5 days (interday) for calculating the relative standard deviation (RSD), respectively.
Stability of Bud conjugates in different pH solutions
Then, 3a, 3c, 3d, and 3e (1 mg) were incubated in different media including pH 1.2, 6.8, and 7.4 (containing 2% DMSO) with a final concentration of 200 μg/mL.21 The samples were constantly shaken at 37°C for 48 hours. At 0, 1, 2, 4, 8, 12, 24, and 48 hours, 200 μL of the mixture was collected, filtered with 0.45-μm microporous membrane, and analyzed by HPLC. The residual contents of each conjugate were calculated, and the hydrolysis curves were drawn.
Stability of Bud conjugates in rat plasma
Fresh rat plasma was diluted 5.7 times (v/v), and 3a, 3c, 3d, and 3e (1 mg) were added in the diluent (containing 2% DMSO) with a final concentration of 200 μg/mL and incubated at 37°C for 24 hours. At 0, 0.5, 1, 2, 4, 8, 12, and 24 hours, 200 μL of the mixture was collected, filtered with 0.22-μm microporous membrane, and analyzed by HPLC. The residual contents of each conjugate were calculated, and the hydrolysis curves were drawn.
Stability of Bud conjugates in human plasma
Blank plasma was collected from healthy volunteers. 3a, 3c, 3d, and 3e (1 mg) were added in human plasma (containing 2% DMSO) with a final concentration of 200 μg/mL. The stability study was conducted as above.
Stability of Bud conjugates in rat lung homogenate
Rat lung was collected, weighed, and then homogenized with three times of normal saline (m/v). Then, 3a, 3c, 3d, and 3e (1 mg) were added in rat lung homogenate (containing 2% DMSO) with a final concentration of 200 μg/mL. The stability study was conducted as described above.
Statistical analyses
Statistical analyses were performed with SPSS Version 19.0 (IBM Corporation, Armonk, NY, USA). The Shapiro–Wilk test was used to test distribution. The One-way analysis of variance followed by the Fisher’s least significant difference test was used to test normally distributed data. The results were presented as mean ± SD. P<0.05 was considered to be statistically significant.
Results
Preparation and characterization of Bud conjugates
Figure 1A shows the synthesis of amino acid ester conjugates of Bud. The esterification was reacted between the 21-hydroxyl group of Bud (1) and the carboxyl group of Boc-amino acids (Boc-glycine, Boc-alanine, or Boc-phenylalanine) under the catalysis of EDCI/DMAP to produce the N-Boc-amino acid ester conjugates of Bud (2a–2c). Finally, 2a–2c were deprotected with trifluoroacetic acid, and the final products (3a–3c) were obtained. Figure 1B shows the synthesis of acetates conjugates of Bud. The esterification was reacted between the 21-hydroxyl group of Bud (1) and chloroacetyl chloride to produce budesonide-21-chloracetate (2). Then, 2 was reacted with the amino group of diethylamine, dimethylamine, N-methylpiperazine, and morpholine, respectively, under the catalysis of sodium iodide to obtain the final products (3d–3g). The synthetic routes used in this study are simple with mild reaction conditions, less side reactions, and high yields. The products were characterized by 1H NMR, DSC, and MS. As shown in Figure 2, 3a–3g were successfully synthesized.
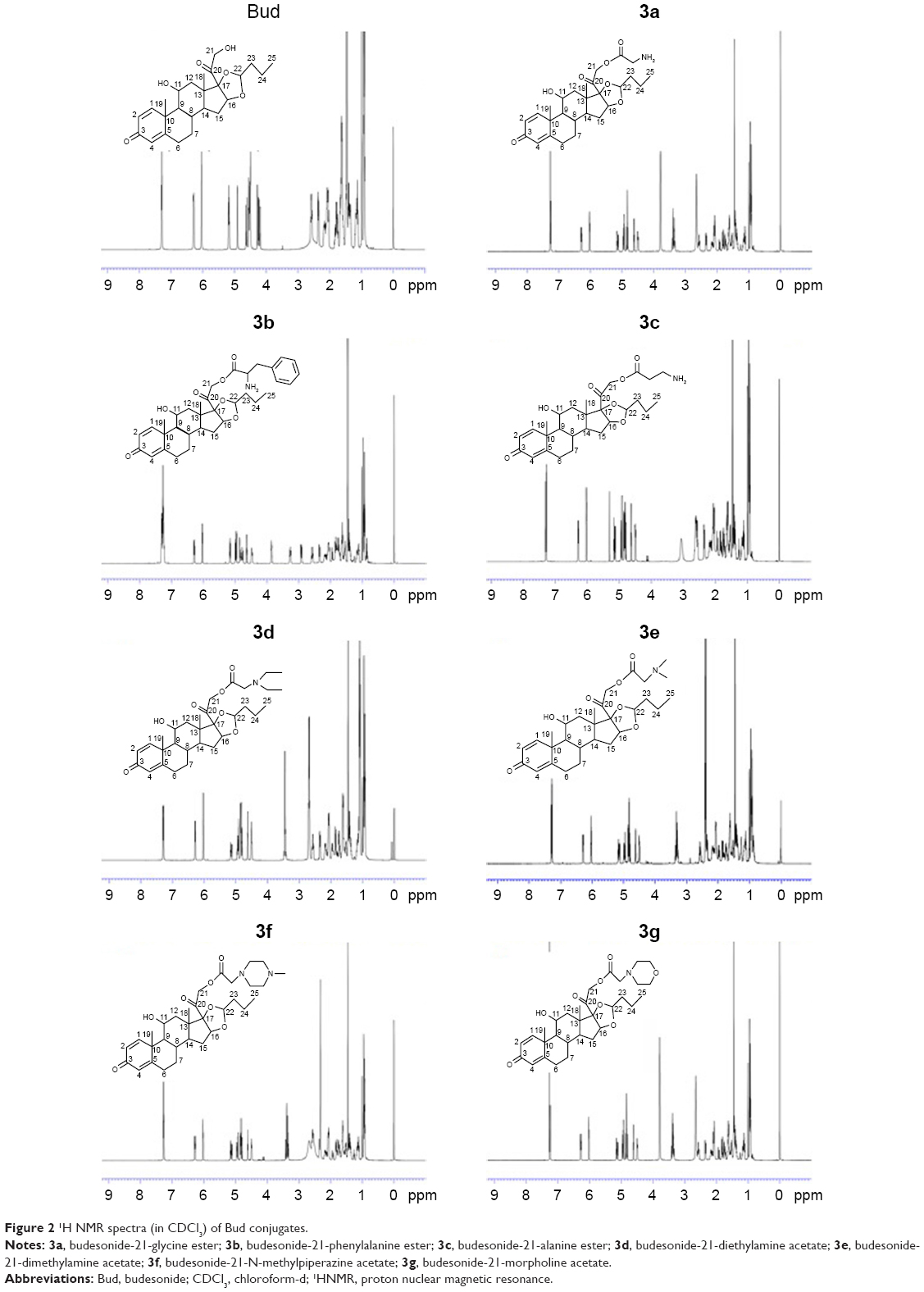 | Figure 2 1H NMR spectra (in CDCl3) of Bud conjugates. |
Bud: 1H NMR (CDCl3): δ 7.30 (dd, 1H, C1-H), 6.28 (dd, 1H, C2-H), 6.03 (s, 1H, C4-H), 4.58–4.45 (m, 3H, C21-H, C16-H), 4.27–4.15 (m, 2H, C24-H, C11-H), 2.36 (d, 2H, C23-H), 2.37–1.96 (m, 2H, C6-H), 1.95–1.42 (m, 3H, C8-H, C12-H, C7-H), 1.72 (m, 1H, C15-H), 1.54–1.42 (m, 5H, C12-H, C15-H, C14-H, C25-H), 1.54–1.18 (m, 3H, C19-H), 1.32 (m, 2H, C26-H), 0.95–0.81 (m, 5H, C7-H, C9-H, C18-H), 0.95 (m, 3H, C27-H); 3a: Yield: 0.24 g, 96.1%. mp: 106°C–107°C. 1H NMR (CDCl3): δ 7.29 (dd, 1H, C1-H), 6.25 (ddd, 1H, C2-H), 5.99 (d, 1H, C4-H), 4.72–4.95 (m, 3H, C21-H, C16-H), 4.46–4.59 (m, 2H, C24-H, C11-H), 3.54 (dt, 2H, C23-H), 2.55–2.31 (m, 2H, C6-H), 1.97–2.13 (m, 3H, C8-H, C12-H, C7-H), 1.79 (m, 1H, C15-H), 1.51–1.68 (m, 5H, C12-H, C15-H, C14-H, C25-H), 1.44 (s, 3H, C19-H), 1.37 (m, 2H, C26-H), 0.95–1.17 (m, 5H, C7-H, C9-H, C18-H), 0.90 (td, 3H, C27-H); 3b: Yield: 0.19 g, 68.7%. mp: 95°C–96°C. 1H NMR (CDCl3): δ 7.27 (m, 6H, C1-H, Ph-5H), 6.03–6.28 (ddd, 1H, C2-H, C4-H), 5.16 (dd, 1H, Ph-H), 4.79–4.97 (m, 3H, C16-H, C21-H), 4.63 (t, 1H, C25-H), 4.47 (dd, 1H, C11-H), 3.85 (dt, 1H, C23-H), 3.27 (ddd, 1H, C24-H), 2.92 (dd, 1H, C24-H), 2.58 (td, 1H, C6-H), 2.35 (dd, 1H, C6-H), 2.05–2.15 (m, 3H, C7-H, C8-H, C12-H), 1.55–1.84 (m, 6H, C12-H, C14-H, C15-H, C26-H), 1.46 (d, 3H, C19-H), 1.39 (m, 2H, C27-H), 1.11–1.17 (m, 2H, C7-H, C9-H), 0.98 (d, 3H, C18-H), 0.94 (td, 3H, C28-H). MS: m/z 578.27 [M+H]+; 3c: Yield: 0.23 g, 86.5%. mp: 84°C–85°C. 1H NMR (CDCl3): δ 7.29 (m, 1H, C1-H), 6.28 (ddd, 1H, C2-H), 6.03 (d, 1H, C4-H), 4.49–4.93 (m, 5H, C11-H, C16-H, C21-H, C25-H), 3.06 (m, 2H, C23-H), 2.59 (m, 3H, C6-H, C24-H), 2.34 (dd, 1H, C6-H), 2.04–2.16 (m, 3H, C7-H, C8-H, C12-H), 1.52–1.83 (m, 6H, C12-H, C14-H, C15-H, C26-H), 1.46 (s, 3H, C19-H), 1.41 (td, 2H, C27-H), 1.11–1.17 (m, 2H, C7-H, C9-H), 0.98 (d, 3H, C18-H), 0.93 (td, 3H, C28-H). MS: m/z 502.26 [M+H]+.
3d: Yield: 0.16 g, 74.5%. mp: 147°C–148°C. 1H NMR (CDCl3): δ 7.29 (m, 1H, C1-H), 6.28 (d, 1H, C2-H), 6.03 (s, 1H, C4-H), 4.82–4.93 (dd, 3H, C16-H, C21-H), 4.63 (t, 1H, C24-H), 4.50 (m, 1H, C11-H), 3.44 (m, 2H, C23-H), 2.57–2.69 (dd, 5H, C6-H, C1-H), 2.34 (d, 1H, C6-H), 2.08–2.16 (m, 3H, C7-H, C8-H, C12-H), 1.54–1.82 (m, 6H, C12-H, C14-H, C15-H, C25-H), 1.46 (s, 3H, C19-H), 1.40 (td, 2H, C26-H), 1.12–1.16 (m, 2H, C7-H, C9-H), 1.09 (t, 6H, C2-H), 0.98 (d, 3H, C18-H), 0.92 (m, 3H, C27-H). MS: m/z 544.39 [M+H]+; 3e: Yield: 0.17 g, 77.9%. mp: 135°C–136°C. 1H NMR (CDCl3): δ 7.28 (m, 1H, C1-H), 6.29 (ddd, 1H, C2-H), 6.03 (d, 1H, C4-H), 4.81–4.97 (m, 3H, C16-H, C21-H), 4.62 (t, 1H, C24-H), 4.51 (m, 1H, C11-H), 3.30 (m, 2H, C23-H), 2.57 (td, 1H, C6-H), 2.35–2.40 (m, 7H, C6-H, C1-H), 2.07–2.15 (m, 3H, C7-H, C8-H, C12-H), 1.62–1.83 (m, 4H, C12-H, C14-H, C15-H), 1.53 (m, 2H, C25-H), 1.46 (s, 3H, C19-H), 1.39 (td, 2H, C26-H), 1.15 (m, 2H, C7-H, C9-H), 0.99 (d, 3H, C18-H), 0.93 (m, 3H, C27-H). MS: m/z 516.27 [M+H]+; 3f: Yield: 0.13 g, 62.4%. mp: 174°C–175°C. 1H NMR (CDCl3): δ 7.27 (m, 1H, C1-H), 6.29 (ddd, 1H, C2-H), 6.03 (d, 1H, C4-H), 4.81–4.94 (m, 3H, C16-H, C21-H), 4.62 (t, 1H, C24-H), 4.50 (m, 1H, C11-H), 3.36 (m, 2H, C23-H), 2.64 (m, 4H, C1-H), 2.54 (m, 5H, C6-H, C2-H), 2.32–2.35 (m, 4H, C6-H, C3-H), 2.06–2.16 (m, 3H, C8-H, C7-H, C12-H), 1.60–1.81 (m, 4H, C12-H, C14-H, C15-H), 1.53 (m, 2H, C25-H), 1.46 (s, 3H, C19-H), 1.38 (m, 2H, C26-H), 1.11–1.16 (m, 2H, C7-H, C9-H), 0.98 (d, 3H, C18-H), 0.93 (m, 3H, C27-H). MS: m/z 571.26 [M+H]+; 3g: Yield: 0.14 g, 67.1%. mp: 161°C–162°C. 1H NMR (CDCl3): δ 7.26 (m, 1H, C1-H), 6.28 (ddd, 1H, C2-H), 6.03 (d, 1H, C4-H), 4.82–4.95 (m, 3H, C16-H, C21-H), 4.62 (t, 1H, C24-H), 4.50 (m, 1H, C11-H), 3.79 (s, 4H, C2-H), 3.36 (m, 2H, C23-H), 2.65 (s, 4H, C1-H), 2.57 (td, 1H, C6-H), 2.35 (dd, 1H, C6-H), 2.08–2.16 (m, 3H, C8-H, C7-H, C12-H), 1.61–1.82 (m, 4H, C12-H, C14-H, C15-H), 1.54 (m, 2H, C25-H), 1.45 (s, 3H, C19-H), 1.37 (m, 2H, C26-H), 1.11–1.16 (m, 2H, C7-H, C9-H), 0.98 (d, 3H, C18-H), 0.92 (m, 3H, C27-H). MS: m/z 558.26 [M+H]+.
Equilibrium solubility of Bud conjugates
Figure 3 presents the results of solubility study of conjugates. The data indicated an improvement in the solubility of the drug after binding to hydrophilic carriers. The equilibrium solubility values of 3a, 3b, 3c, 3d, 3e, 3f, and 3g in 72 hours were 0.4015, 0.0035, 0.3301, 0.0126, 0.0345, 0.0189, and 0.0104 g/100 g water, respectively. Particularly, the solubility values of 3a, 3c, and 3e were significantly increased 180-fold, 149-fold, and 16-fold (P<0.01) as compared to Bud (0.0022 g/100 g water).
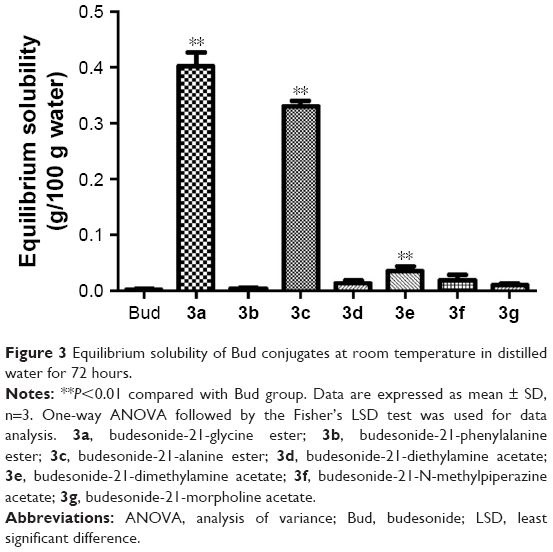 | Figure 3 Equilibrium solubility of Bud conjugates at room temperature in distilled water for 72 hours. |
Cytotoxicity of Bud conjugates
In order to select the experimental concentrations of Bud conjugates for this study, the cytotoxicity of conjugates to A549 cells was determined. The IC5 of Bud conjugates on A549 cell lines was regarded as the maximum safe concentration. As shown in Figure 4, Bud and the synthesized conjugates all showed certain cytotoxicity. However, the IC5 values of 3f and 3g (13.35±0.11 and 43.30±0.42 nM, respectively) were significantly lower than those of Bud and five other conjugates, which indicated its higher cytotoxicity. Combined with the result of solubility study, 3f and 3g were excluded from the following studies.
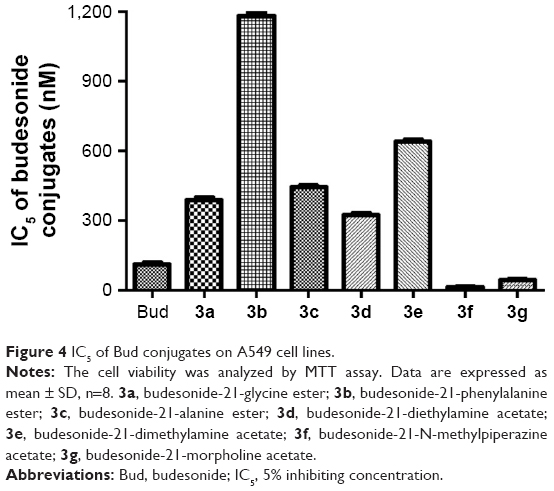 | Figure 4 IC5 of Bud conjugates on A549 cell lines. |
Inhibition of IL-6 in LPS-induced A549 cells
To determine the in vitro anti-inflammatory effect of Bud conjugates, the inhibition of IL-6 production in LPS-induced A549 cells was investigated. As shown in Figure 5, Bud and the five conjugates (1 nM) resulted in an effective anti-inflammatory effect, and their inhibitory rates on IL-6 were >50%. Compared with Bud, the conjugates significantly inhibited the IL-6 production in LPS-induced A549 cells (P<0.01); particularly, the inhibitory rates of 3a, 3b, and 3c were almost twice as much as that of Bud.
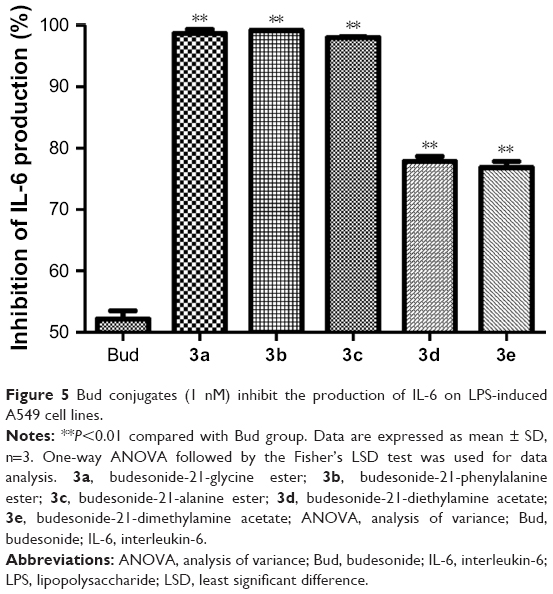 | Figure 5 Bud conjugates (1 nM) inhibit the production of IL-6 on LPS-induced A549 cell lines. |
In vivo anti-inflammatory study
To evaluate the in vivo anti-inflammatory activity of Bud conjugates, a xylene-induced mouse ear edema model was used; 3b was also excluded from this study because of its poor solubility. It was observed that there were significant differences in ear edema between the control group and test groups. Besides, as shown in Figure 6, Bud conjugates significantly decreased xylene-induced ear edema when compared with Bud. Especially, 3a and 3e inhibited ear edema by 30.67% and 46.67%, respectively, while Bud presented a relatively low inhibition of 17.19%.
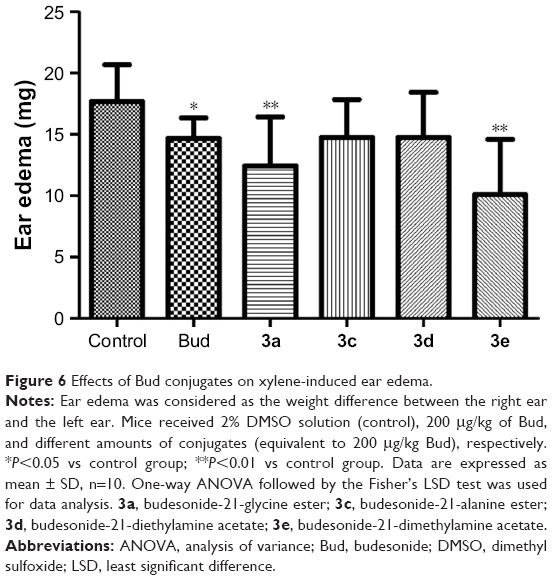 | Figure 6 Effects of Bud conjugates on xylene-induced ear edema. |
Biological stability of the synthesized Bud conjugates
A linear relationship was established between the concentration (0.2–500 μg/mL) and the peak area of each sample. Table 1 lists the regression equations of 3a, 3b, 3c, and 3e in different tissue samples for HPLC analysis. The absolute recoveries of the above four conjugates at each concentration (5.0, 50.0, and 500.0 μg/mL) were >95%. The recoveries had no significant difference between the three concentrations. For precision assay, both the intraday and interday RSDs in all tissue samples were <5%. These data validated that the HPLC method was acceptable and reproducible (Table 2).
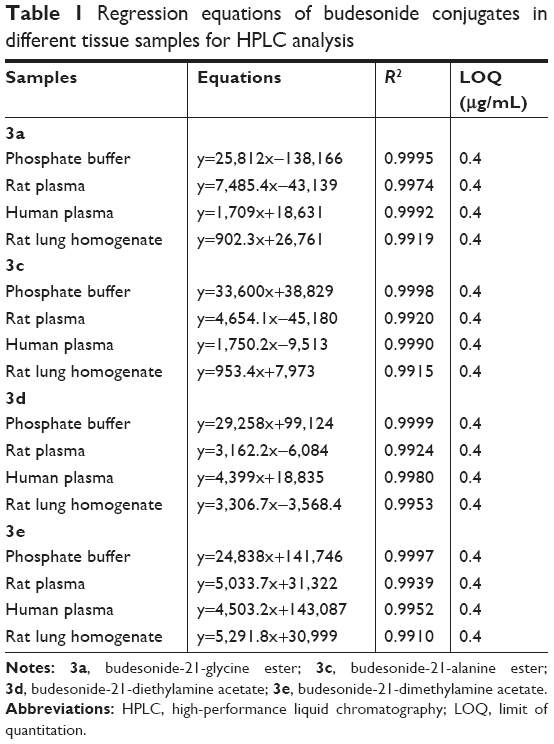 | Table 1 Regression equations of budesonide conjugates in different tissue samples for HPLC analysis |
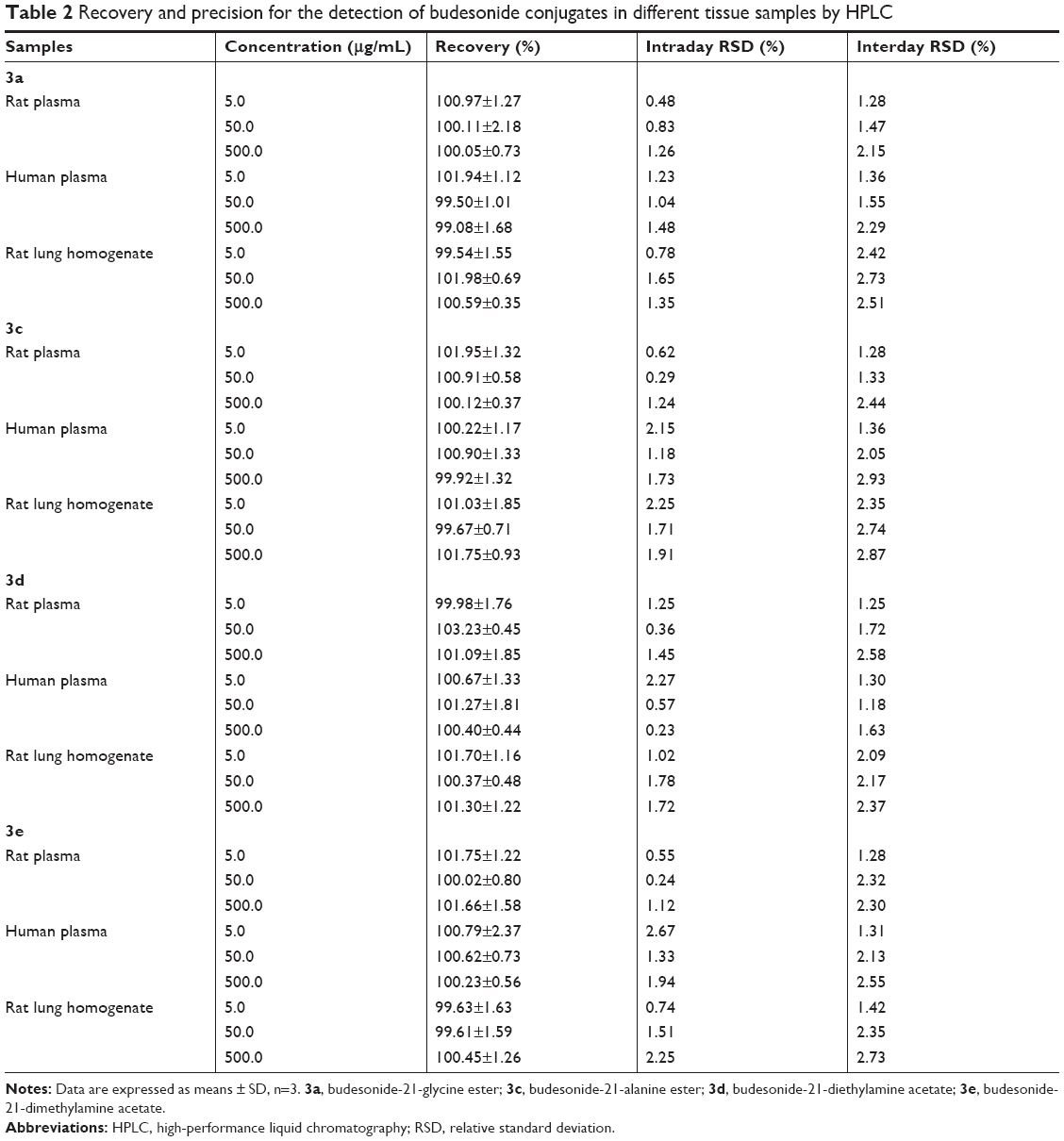 | Table 2 Recovery and precision for the detection of budesonide conjugates in different tissue samples by HPLC |
Bud conjugates have an ester bond that is susceptible to acidic and basic conditions. Figure 7 shows the drug content that was determined following the hydrolysis of the conjugates in different pH solutions. The amount of Bud released in hydrochloric acid solution (pH 1.2) and phosphate buffer solutions (pH 6.8 and 7.4) were calculated to evaluate the hydrolysis behavior of conjugates over time; 3a, 3d, and 3e were barely hydrolyzed in pH 1.2 solution with the remaining 76.15%, 93.35%, and 88.53% of conjugates in 48 hours, respectively. However, conjugates hydrolyzed more quickly under neutral and basic conditions, especially in pH 7.4; 3a, 3d, and 3e were almost completely hydrolyzed in 24 hours.
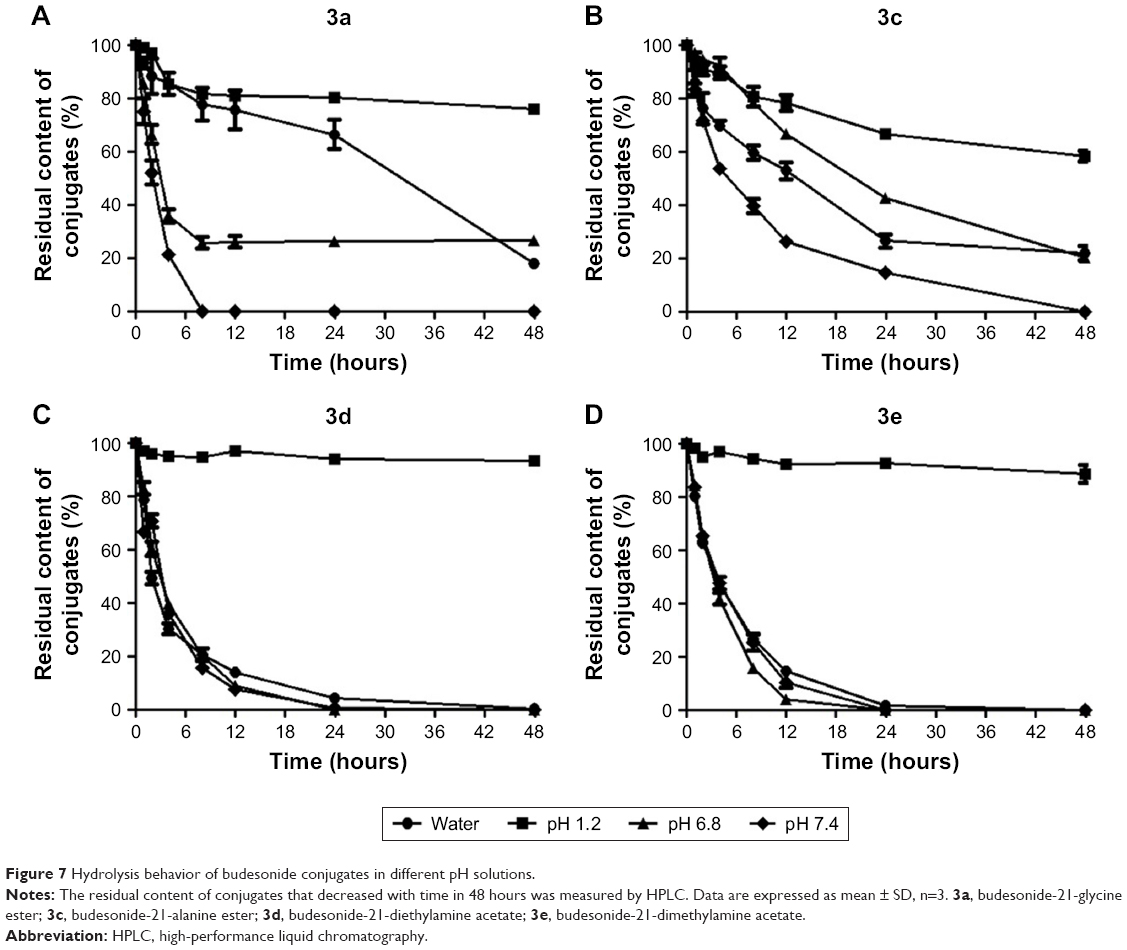 | Figure 7 Hydrolysis behavior of budesonide conjugates in different pH solutions. |
Figure 8 shows the residual content of conjugates after incubation with rat plasma, human plasma, and rat lung homogenate for 24 hours. As shown in Figure 8A, the four conjugates were gradually hydrolyzed in rat plasma by the action of carboxylesterase; 3a and 3e were rapidly hydrolyzed in 6 hours, <6% of 3c was left in 12 hours, and 3d was completely hydrolyzed in 24 hours. Figure 8B shows the hydrolysis behavior of conjugates in human plasma. Similar to the above result, 3a was rapidly hydrolyzed in 6 hours, <2% of 3e was left in 8 hours, and 3c and 3d were completely hydrolyzed in 8 and 24 hours, respectively.
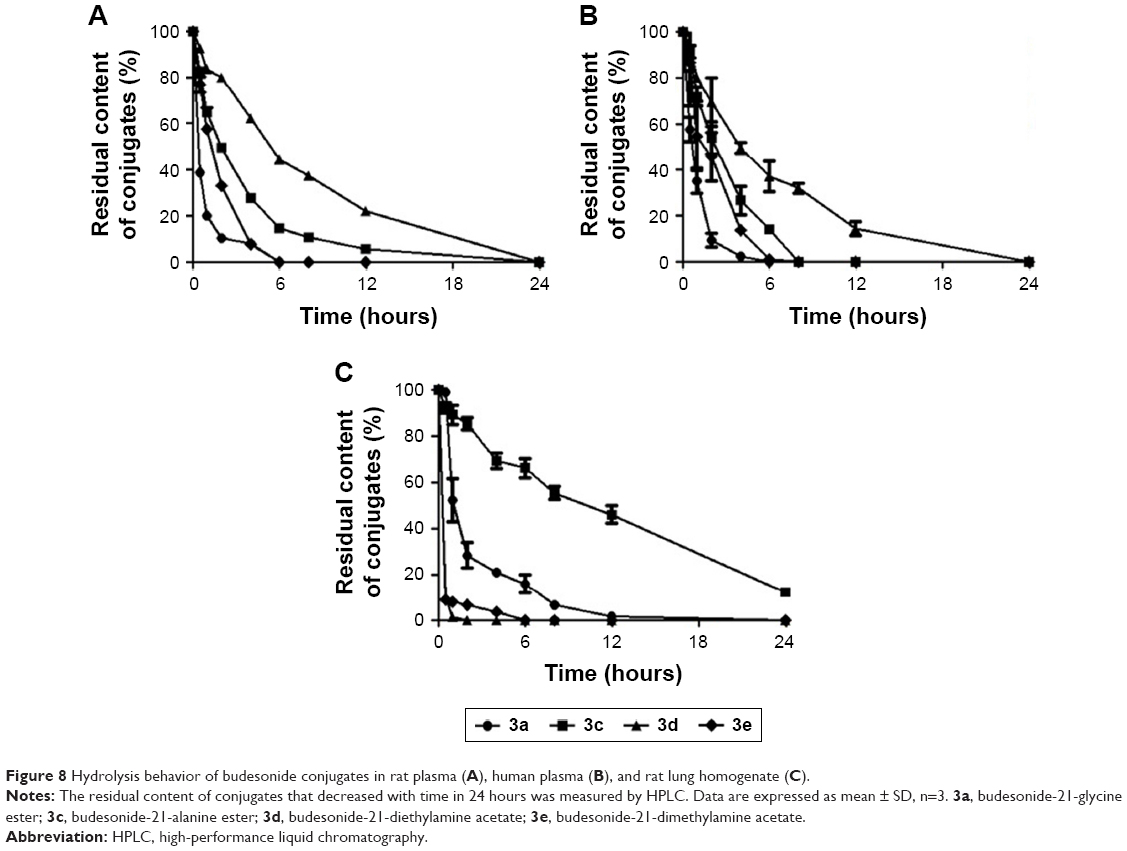 | Figure 8 Hydrolysis behavior of budesonide conjugates in rat plasma (A), human plasma (B), and rat lung homogenate (C). |
When conjugates were incubated in the rat lung homogenate, a rapid increase was observed in the hydrolysis profile. As shown in Figure 8C, 3d and 3e were rapidly and completely hydrolyzed in 2 and 6 hours, respectively; 84.04% of 3a was hydrolyzed in 6 hours and then almost completely hydrolyzed in 12 hours; 53.86% and 87.75% of 3c were hydrolyzed after 12 and 24 hours of incubation, respectively. Hydrolysis behaviors of conjugates were different in three conditions and were more significant in rat lung homogenate.
Discussion
Bud is a kind of nonhalogenated glucocorticoid developed in recent years, with the local anti-inflammatory activity superior to the systemic effect. Clinically, it is mainly used in the treatment of asthma, allergic rhinitis, and ulcerative colitis by inhalation and enteroclysis. Based on the study of quantitative structure–activity relationship of glucocorticoid receptor modulators, the hydrophilic groups at the C21 substituent position of glucocorticoids could enhance the drug activity. Besides, the poor water solubility of drugs affects its dissolution and release behavior and thus influences the drug concentration at the inflammatory site and its local bioavailability.
In this study, seven novel Bud conjugates were synthesized (3a–3g); moreover, the equilibrium solubility, cell viability, in vitro and in vivo anti-inflammatory effect, and the hydrolysis behavior of the synthesized conjugates were investigated in detail. The analytical methods of Bud conjugates by HPLC in different pH solutions and biological samples were established for detecting the content of compounds in different medium. As shown in Figure 3, the equilibrium solubility study suggested that the solubility of 3a, 3c, and 3e was significantly increased than that of Bud (180-fold, 149-fold, and 16-fold, respectively; P<0.01), which has achieved the goal of enhancing the water solubility of Bud. Besides, the results also showed that the solubilization effect of amino acid esters was more effective than that of acetates.
Bud is a first-line drug for the treatment of corticosteroid dependent and independent asthma, which exerts its potent anti-inflammatory activity primarily by reducing the number of inflammatory cells and inhibiting the expression of inflammatory factors, such as IL-6, IL-8, and tumor necrosis factor-α.22 A549 cell is one of the main cell models used in the study of airway epithelial cell, which is first suffered damage when the respiratory tract is exposed to hazardous substance.23 In this study, the in vitro inflammation model was established by using LPS-treated A549 cell lines. LPS was used as a stimulating factor, which is existed in the outer membrane of Gram-negative bacteria and elicits strong immune responses in animals. The inhibition of IL-6 by Bud and its conjugates at different concentrations in LPS-induced A549 cells was evaluated to reflect their anti-inflammatory effects. The data showed that the inhibitory rates of 3a, 3b, 3c, 3d, 3e, and Bud at 1 nM were all above 50%. Besides, the inhibitory effect of the conjugates was significantly higher than that of Bud (P<0.01). Therefore, the synthesized conjugates possessed a desirable in vitro anti-inflammatory activity at a relatively low concentration. This result is in accordance with those reported in the previous papers. Wang et al established 3D-QSAR models for a series of prednisolone conjugates by using the comparative molecular field analysis and comparative molecular similar indices analysis.13 The 3D-QSAR contour maps suggested that hydrophilic substituents at C21 could enhance the anti-inflammatory activity of prednisolone.
Based on the results of the equilibrium solubility and the in vitro inflammation study, 3a, 3c, 3d, and 3e were screened for further studies. The in vivo study on the anti-inflammatory activity of Bud conjugates was evaluated by the xylene-induced ear edema test. Xylene-induced ear edema is an acute and highly reproducible inflammatory response and characterized by releasing inflammatory mediators, which stimulate ear edema by increasing vascular permeability and promoting vasodilation. Therefore, the xylene-induced ear edema test presented fluid accumulation at the treatment site, and the inhibition of this fluid accumulation was regarded as an anti-inflammatory effect.24 Figure 6 shows the significant inhibition of ear edema by conjugates. Similar to the in vitro study’s findings, Bud and its conjugates significantly decreased ear edema when compared with the control (P<0.01); particularly, 3a and 3e were significantly more effective than Bud. Hereby, 3a, 3c, 3d, and 3e have been demonstrated with a desirable anti-inflammatory effect by in vitro and in vivo models, which also provide further evidence that the hydrophilic groups at the C21 substituent position enhance the drug activity, and the hydrophilic conjugates of Bud possess a high research value.
Bud conjugates are susceptible and instable in aqueous solutions because the existing ester bond is to be easily hydrolyzed. In the present study, the hydrolysis behavior of the synthesized conjugates was evaluated in different pH solutions, rat and human plasma, and rat lung homogenate, respectively. The pH value of the exhaled breath condensate in patients with bronchial asthma is about 6.5–7.0.25 Therefore, 3a, 3c, 3d, and 3e were incubated in hydrochloric acid solution (pH 1.2) and phosphate buffer solutions (pH 6.8 and 7.4), respectively. At time intervals, drug solutions were sampled and analyzed by HPLC to calculate the residual content of conjugates. As a result, Bud conjugates were easier to be hydrolyzed under a weakly alkaline condition, they presented a pH-dependent hydrolysis manner, and their hydrolysis rate increased with the pH value of the medium. Besides, the acetates conjugates of Bud showed a higher rate than that of the amino acid ester conjugates. Previously, Varshosaz et al prepared three dextran–Bud conjugates with different molecular weights of dextran (molecular weight =10,000, 70,000, and 500,000) and different degrees of substitution by using budesonide-21-hemisuccinate as a starting material.26,27 Their chemical stability was studied in 0.1 N HCl and phosphate buffer solutions of pH 6.8 and 7.4. The data showed that the conjugates were easier to be hydrolyzed in alkaline media, which is similar to our findings. This indicated that the hydrolysis of conjugates was mediated by the cleavage of ester bond, and the main affecting factor on their hydrolysis behavior is the pH value of release media.
Second, Bud conjugates were incubated with rat and human plasma. The activity of carboxylesterase in rat plasma is about 5.7 times that in human plasma; therefore, fresh rat plasma was diluted to 5.7 times with normal saline and is then used in the biological stability study.28,29 As a result, the hydrolysis rate of the conjugates in plasma is 3a>3e>3c>3d; particularly, about 90.0% of 3a was rapidly hydrolyzed in 2 hours, and 85.7% of 3d was hydrolyzed in 12 hours in human plasma. This result suggested that most conjugates would be broken down quickly into Bud in the blood and then be metabolized in a few hours; therefore, they would not significantly affect the pharmacokinetic process of Bud and ensure the safety of the conjugates.
Finally, the hydrolysis character of conjugates was investigated in rat lung homogenate, and the hydrolysis rate of the conjugates presented a trend as 3d>3e>3a>3c; particularly, 3d was rapidly and completely hydrolyzed in 2 hours, and 53.8% of 3c was hydrolyzed in 12 hours. Taken together, 3a and 3c would be gradually and slowly hydrolyzed into Bud in the pH environment of alveolar fluid and the lung homogenate; thus, they could have a therapeutic effect through both the form of Bud conjugates and the produced parent drug, as well as prolong the action time of Bud. While absorbed into the blood, 3a and 3c could be quickly broken down into Bud and thereby reduce the possible systemic adverse reactions of 3a and 3c.
Above all, the amino acid ester compounds 3a and 3c were screened as potential conjugates of Bud. Their in vitro and in vivo characteristics would provide a theoretical and an experimental basis for the intracorporal process of glucocorticoids and the treatment of inflammatory diseases. However, further study is required to confirm their pharmacokinetic and pharmacodynamic properties in detail. Currently, studies are made to evaluate the therapeutic effect of 3a and 3c on asthma guinea pig model induced by ovalbumin.
Conclusion
In this study, seven novel conjugates of Bud (3a–3g) were designed and synthesized. Based on their equilibrium solubility, cell viability, in vitro and in vivo anti-inflammatory effect, and their hydrolysis behavior in different media, two amino acid ester compounds (3a and 3c) were screened as potential conjugates of Bud. As a result, the equilibrium solubility of 3a and 3c was significantly increased when compared to Bud, and they showed a significant anti-inflammatory effect in both in vitro and in vivo experiments. On this basis, further studies are made to evaluate their in vivo pharmacokinetics and anti-asthmatic effects in order to provide an experimental basis for further application.
Acknowledgment
This study was funded by National Natural Science Foundation of China (No. 81773663).
Disclosure
The authors report no conflicts of interest in this work.
References
Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. 2013;154(3):993–1007. | ||
Varga G, Ehrchen J, Brockhausen A, et al. Immune suppression via glucocorticoid-stimulated monocytes: a novel mechanism to cope with inflammation. J Immunol. 2014;193(3):1090–1099. | ||
Ali H, Weigmann B, Neurath MF, et al. Budesonide loaded nanoparticles with pH-sensitive coating for improved mucosal targeting in mouse models of inflammatory bowel diseases. J Control Release. 2014;183:167–177. | ||
Pauwels RA, Löfdahl CG, Postma DS, et al. Effect of inhaled formoterol and budesonide on exacerbations of asthma. Formoterol and Corticosteroids Establishing Therapy (facet) International Study Group. N Engl J Med. 1997;337(20):1405–1411. | ||
Maeda K, Yamaguchi M, Nagase H, et al. Utility and effectiveness of Symbicort® Turbuhaler® (oral inhalation containing budesonide and formoterol) in a patient with severe asthma after permanent tracheostomy. J Pharm Health Care Sci. 2018;4(1):24–28. | ||
Esmailpour N, Högger P, Rohdewald P. Binding kinetics of budesonide to the human glucocorticoid receptor. Eur J Pharm Sci. 1998;6(3):219–223. | ||
Löfberg R, Rutgeerts P, Malchow H, et al. Budesonide prolongs time to relapse in ileal and ileocaecal Crohn’s disease. A placebo controlled one year study. Gut. 1996;39(1):82–86. | ||
Liu Y, Bos IST, Oenema TA, et al. Delivery system for budesonide based on lipid-DNA. Eur J Pharm Biopharm. 2018;130:123–127. | ||
Bhatt H, Naik B, Dharamsi A. Solubility enhancement of budesonide and statistical optimization of coating variables for targeted drug delivery. J Pharm. 2014;2014(2):1–13. | ||
Biju P, McCormick K, Aslanian R, et al. Steroidal C-21 mercapto derivatives as dissociated steroids: discovery of an inhaled dissociated steroid. Bioorg Med Chem Lett. 2011;21(21):6343–6347. | ||
Biju P, McCormick K, Aslanian R, et al. Steroidal C-21 heteroaryl thioethers (Part 2): discovery of orally bioavailable selective glucocorticoid receptor modulators (dissociated steroids). Bioorg Med Chem Lett. 2012;22(2):1086–1090. | ||
Biju P, Wang H, Anthes J, et al. Steroidal C-21 heteroaryl thioethers. Part 3: pregn-4-eno-[3,2-c]pyrazole fused A ring modified steroids as selective glucocorticoid receptor modulators (dissociated steroids). Bioorg Med Chem Lett. 2012;22(9):3291–3295. | ||
Wang JH, Wang B, Li Y, et al. Study on quantitative structure-activity relationship of prednisolone derivatives as glucocorticoid receptor modulators. J Mol Sci. 2013;29(3):205–211. | ||
Mehvar R. Simultaneous analysis of dextran-methylprednisolone succinate, methylprednisolone succinate, and methylprednisolone by size-exclusion chromatography. J Pharm Biomed Anal. 1999;19(5):785–792. | ||
Li N, Tattam B, Brow KF, Seale JP. Quantification of epimeric budesonide and fluticasone propionate in human plasma by liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry. J Chromatogr B Biomed Sci Appl. 2001;761(2):177–185. | ||
Chu B, Wang J, Wang Y, Yang G. Knockdown of PKM2 induces apoptosis and autophagy in human A549 alveolar adenocarcinoma cells. Mol Med Rep. 2015;12(3):4358–4363. | ||
Lei Y, Li HX, Jin WS, et al. The radiosensitizing effect of paeonol on lung adenocarcinoma by augmentation of radiation-induced apoptosis and inhibition of the PI3K/Akt pathway. Int J Radiat Biol. 2013;89(12):1079–1086. | ||
Yuan F, Nelson RK, Tabor DE, et al. Dexamethasone prodrug treatment prevents nephritis in lupus-prone (NZB × NZW)F1 mice without causing systemic side effects. Arthritis Rheum. 2012;64(12):4029–4039. | ||
Wang Y, Chen P, Tang C, et al. Antinociceptive and anti-inflammatory activities of extract and two isolated flavonoids of Carthamus tinctorius L. J Ethnopharmacol. 2014;151(2):944–950. | ||
Yan Y, Zhang H, Sun J, et al. Enhanced transdermal delivery of sinomenine hydrochloride by ethosomes for anti-inflammatory treatment. J Drug Deliv Sci Technol. 2016;36:201–207. | ||
Varshosaz J, Emami J, Tavakoli N, et al. Synthesis and evaluation of dextran-budesonide conjugates as colon specific prodrugs for treatment of ulcerative colitis. Int J Pharm. 2009;365(1–2):69–76. | ||
von Asmuth EJ, Dentener MA, Ceska M, Buurman WA. IL-6, IL-8 and TNF production by cytokine and lipopolysaccharide-stimulated human renal cortical epithelial cells in vitro. Eur Cytokine Netw. 1994;5(3):301–310. | ||
Liu YY, Han JY, Lin SC, Liu ZY, Jiang WT. Effect of CDH1 gene methylation on transforming growth factor (TGF-β)-induced epithelial-mesenchymal transition in alveolar epithelial cell line A549. Genet Mol Res. 2014;13(4):8568–8576. | ||
Oh YC, Jeong YH, Cho WK, et al. Anti-inflammatory and analgesic effects of Pyeongwisan on LPS-stimulated murine macrophages and mouse models of acetic acid-induced writhing response and xylene-induced ear edema. Int J Mol Sci. 2015;16(1):1232–1251. | ||
Walsh BK, Mackey DJ, Pajewski T, et al. Exhaled-breath condensate pH can be safely and continuously monitored in mechanically ventilated patients. Respir Care. 2006;51(10):1125–1131. | ||
Varshosaz J, Emami J, Tavakoli N, et al. Synthesis and evaluation of dextran-budesonide conjugates as colon specific prodrugs for treatment of ulcerative colitis. Int J Pharm. 2009;365(1–2):69–76. | ||
Varshosaz J, Emami J, Ahmadi F, et al. Preparation of budesonide-dextran conjugates using glutarate spacer as a colon-targeted drug delivery system: in vitro/in vivo evaluation in induced ulcerative colitis. J Drug Target. 2011;19(2):140–153. | ||
Sterri SH, Fonnum F. Carboxylesterases in guinea-pig plasma and liver. tissue specific reactivation by diacetylmonoxime after soman inhibition in vitro. Biochem Pharmacol. 1987;36(22):3937–3942. | ||
Ozaki H, Sugihara K, Watanabe Y, et al. Comparative study of the hydrolytic metabolism of methyl-, ethyl-, propyl-, butyl-, heptyl- and dodecylparaben by microsomes of various rat and human tissues. Xenobiotica. 2013;43(12):1064–1072. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.