")
Back to Journals » International Journal of Nanomedicine » Volume 11
Synthesis, characterization, and evaluation of mPEG–SN38 and mPEG–PLA–SN38 micelles for cancer therapy
Authors Xie J, Zhang X, Teng M, Yu B, Yang S , Lee R, Teng L
Received 24 December 2015
Accepted for publication 8 March 2016
Published 26 April 2016 Volume 2016:11 Pages 1677—1686
DOI https://doi.org/10.2147/IJN.S103110
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Jing Xie,1 Xiaomin Zhang,2 Meiyu Teng,1 Bo Yu,2 Shuang Yang,1 Robert J Lee,1,3 Lesheng Teng1
1College of Life Sciences, Jilin University, Changchun, 2Hangzhou PushiKang Biotechnology Co., Ltd, Hangzhou, People’s Republic of China; 3Division of Pharmaceutics, College of Pharmacy, The Ohio State University, Columbus, OH, USA
Abstract: 7-Ethyl-10-hydroxy camptothecin (SN38) is a potent topoisomerase inhibitor and a metabolite of irinotecan. Its clinical development has been hampered by its poor solubility. To address this problem, methoxy poly(ethylene glycol)-2000 (mPEG2K)–SN38 and mPEG2K–poly(lactide) (PLA1.5K)–SN38 conjugates were prepared and then dispersed into an aqueous medium to form micelles. Physicochemical characteristics of SN38–polymer conjugate micelles, for example, micelle diameter, zeta potential, morphology, and drug content, were then evaluated. The results showed that the mean diameters of mPEG2K–SN38 and mPEG2K–PLA1.5K–SN38 micelles were ~130 and 20 nm, respectively. These two micelles had similar drug contents. mPEG2K–PLA1.5K–SN38 micelles were more homogeneous than mPEG2K–SN38 micelles. Moreover, in vitro drug release behavior of the micelles was studied by high performance liquid chromatography. SN38 release from mPEG2K–SN38 micelles was much faster than from mPEG2K–PLA1.5K–SN38 micelles. In vitro cytotoxicity, cellular uptake, and apoptosis assays of the SN38–polymer conjugate micelles were carried out on BEL-7402 human liver cancer cells. In vivo biodistribution and antitumor tumor efficacy studies were carried out in a nude mouse xenograft model derived from BEL-7402 cells. The results showed that mPEG2K–PLA1.5K–SN38 micelles were significantly more effective than mPEG2K–SN38 micelles in tumor inhibition, and the inhibitory effect of mPEG2K–PLA1.5K–SN38 micelles on tumor growth was significantly greater than that of mPEG2K–SN38 micelles (1,042 vs 1,837 mm) at 30 days. In conclusion, mPEG–PLA–SN38 is a promising anticancer agent that warrants further investigation.
Keywords: SN38, polymer conjugate, micelles, chemotherapy, liver cancer
Introduction
7-Ethyl-10-hydroxy camptothecin (SN38) is a potent topoisomerase I inhibitor.1–3 SN38 has shown activity against several malignancies, including colorectal, lung, gastric, cervical, lymphoma, and ovarian cancers.4,5 Irinotecan (camptothecin-11 [CPT-11]), a water-soluble prodrug of SN38, is currently approved for the treatment of metastatic colorectal cancer.6,7 However, only 3%–4% of administered dose of CPT-11 in a patient is converted to the active form of the drug SN38 and this conversion is affected by genetic polymorphism.8–10 CPT-11 as a prodrug is 100–1,000 times less potent than SN38 and causes gastrointestinal toxicity.11,12 Therefore, SN38 itself has been considered for use as an anticancer agent. However, SN38 has poor solubility.6 Another problem is that the active form of SN38 has a closed lactone ring in its structure. However, at physiological pH, the closed lactone ring can be converted into an inactive carboxylate form, resulting in drug inactivation.13
In recent years, much effort has been devoted to increasing the solubility of SN38.14,15 A promising approach is conjugation to a polymeric carrier.16,17 In 2008, a poly (ethylene glycol)–SN38 (PEG–SN38) was synthesized18 and shown to have significantly superior antitumor efficacy compared to irinotecan.19–21 Methoxy poly(ethylene glycol)-b-poly(lactide) (mPEG–PLA) is an amphiphilic block copolymer that can self-assemble into a micelle, which has a hydrophobic PLA core and a hydrophilic mPEG shell.22–24 Hydrophobic drugs can be incorporated into the PLA core. Meanwhile, the hydrophilic mPEG shell can reduce clearance of the micelles by the mononuclear phagocyte system.
In this study, mPEG–SN38 and mPEG–PLA–SN38 conjugates were synthesized, characterized, and evaluated as anticancer agents both in vitro and in vivo.
Materials and methods
Materials
N-Ethyl-N′-(3-dimethylaminopropyl) carbodiimide (EDC), 4-(dimethylamino) pyridine (DMAP), and 3-(4, 5-dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (Shanghai, People’s Republic of China). SN38 was obtained from Dalian Meilun Biotechnology Inc. (Dalian, People’s Republic of China) and its purity (>95%) was determined by 1H NMR and by high performance liquid chromatography (HPLC). Penicillin–streptomycin, RPMI1640, fetal bovine serum, and 0.25% (w/v) trypsin, 0.03% (w/v) EDTA solution were purchased from HyClone (Logan, UT, USA). Acetonitrile (HPLC grade) was obtained from Nanjing Xinhuayuan Chemical Agents Co. (Nanjing, People’s Republic of China). Succinic anhydride modified methoxy poly(ethylene glycol)-2000 (mPEG2K) (mPEG2K–SA) and block copolymer mPEG2K–PLA (mPEG2K–poly(lactide) [PLA1.5K]–SA) were purchased from Advanced Polymer Materials Inc. (Dorval, Quebec, Canada). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethyl indotricarbocyanine Iodide (DiR) was purchased from Caliper Life Sciences (Hopkinton, MA, USA).
Synthesis of mPEG–SN38 and mPEG–PLA–SN38 conjugates
The synthesis of the SN38–polymer conjugates has been described previously.25 Briefly, in a dried flask, 0.082 mmol SN38, 0.076 mmol mPEG2K–SA or 0.076 mmol mPEG2K–PLA1.5K–SA, 0.083 mmol EDC, and 0.082 mmol DMAP were dissolved in 3 mL of dry N,N-dimethylformamide (DMF). And then 20 μL dry pyridine was added. The reaction was continued under stirring for 24 hours at room temperature. The solvent was removed under vacuum and the crude product obtained was purified by column chromatography (dichloromethane:methanol =30:1). The obtained oily yellow liquid was dried in vacuo overnight to give the final product mPEG–SN38 (at 82.7% yield) or mPEG–PLA–SN38 (at 87.9% yield).
Preparation of SN38–polymer conjugate micelles
SN38–polymer conjugate micelles were prepared by a desolvation method. Two milligrams of mPEG–SN38 or mPEG–PLA–SN38 was dissolved in 5 mL acetone. Under magnetic stirring, the acetone solution was slowly added into 6 mL deionized water at a speed of 60 mL/h. The suspension was stirred for 15 minutes. Then, acetone was removed on a rotovap at 37°C, resulting in SN38–polymer conjugate micelles.
Characterization of SN38–polymer conjugate micelles
The particle diameter and zeta (ζ) potential of SN38–polymer conjugate micelles were measured on Malvern ZetaSizer (Malvern Instruments Ltd., Malvern, UK) at 25°C. The analysis was performed in triplicate and the results were expressed as mean ± standard deviation. The morphology of the micelles was observed by transmission electron microscopy (TEM). Samples were prepared by applying the SN38–polymer conjugate micelles suspension (2 mg/mL) onto a 100-mesh formvar-coated copper grid. Images were recorded using a JEOL JEM-200CX TEM at an acceleration voltage of 200 kV.
Measurement of drug content
The SN38 content in the two prodrug micelles was determined by HPLC. Briefly, 1.0 mL of 1.0 mg/mL micelle suspension was added to the same volume of acetonitrile. To trigger complete release of SN38 from the conjugate, 50 μL NaOH (0.3 mol/L) was then added to 100 μL of the sample solution, which was incubated for 15 minutes at 25°C. After the incubation, 50 μL of HCl (0.3 mol/L) was added to neutralize the solution pH.25,26 Finally, the preparation was filtered with 0.22 μm pore-size filter membranes and analyzed by HPLC. The mobile phase used was a linear gradient of water and acetonitrile at a flow rate of 1.0 mL/min over 30 minutes. The column effluent was detected at 370 nm. Drug concentration was calculated by the linear calibration curve of SN38.
In vitro release studies
In vitro release of SN38 from micelles was investigated in a phosphate-buffered saline (PBS) (pH 7.4) medium containing 0.2% Tween 80 by a dialysis method. The experiment was carried out as follows: 1.0 mL of SN38–polymer conjugate micelles (1 mg SN38 equivalent) was diluted with 9.0 mL PBS to a total volume of 10.0 mL. One mL of micelle suspension was used to determine the initial concentration and then the remaining 9.0 mL was introduced into a dialysis bag (molecular weight cut off =14 kDa). The end-sealed dialysis bag was immersed in 50 mL of PBS (pH 7.4) containing 0.2% Tween 80 at 37°C, which was shaken at a speed of 100 rpm for 32 hours. Samples of 1.0 mL each were withdrawn at preset time intervals (0, 1, 2, 4, 8, 12, 24, 36, and 48 hours) and replaced with an equal volume of fresh release medium. The concentrations of SN38 in samples were determined by the HPLC method described in “Measurement of drug content”.
Cellular uptake of fluorescein isothiocyanate-labeled SN38–polymer conjugate micelles
BEL-7402 cells were seeded in 35 mm glass-bottom microwell dishes (Nest Biotechnology Co., Ltd, Wuxi, Jiangsu, People’s Republic of China) at a seeding density of 6×105 cells per dish in 2.0 mL medium and incubated overnight. Then, the medium was replaced with suspension of fluorescein isothiocyanate (FITC)-labeled SN38–polymer conjugate micelles diluted in culture medium (20 μg/mL) and the cells were incubated for 4 hours. FITC-labeled SN38–polymer conjugate micelles were synthesized using the same method as that for SN38–polymer conjugate micelles with the exception of addition of 200 μg FITC into the acetone solution. After 4 hours incubation, cells were washed with PBS and fixed with 4% paraformaldehyde (v/v) at room temperature for 20 minutes, followed by staining of the cellular nuclei with 4′,6-diamidino-2-phenylindole (5 μg/mL). Finally, the cells were examined with a Carl Zeiss laser scanning microscope. Image analysis was performed using the LSM 5.10 software (Carl Zeiss, Oberkochen, Germany). The cells and experimental protocol was approved by the Institution Review Board of Jilin University.
In vitro cytotoxicity
Cytotoxicity of the SN38 conjugates was evaluated by the MTT method.27 BEL-7402 human liver cancer cells were cultured in RPMI1640 growth medium supplemented by 10% (v/v) fetal bovine serum and 1% (w/v) penicillin–streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Briefly, cells were seeded at a density of 5×103 cells per well in a 96-well plate and incubated overnight. Then, the medium was replaced with 100 μL of fresh medium containing serial dilutions of SN38 conjugate (at SN38 equivalent from 0.05 to 100 μg/mL). After 48 hours of incubation, cell survival was then measured as follows: 30 μL of MTT (5 mg/mL) solution was added to each well. The incubation was continued for another 4 hours. Then, the MTT derivative was dissolved with dimethylsulfoxide (100 μL) and the optical density of the solution was determined on a microplate reader at 492 nm.
Apoptosis analysis
BEL-7402 cells were seeded at a density of 1×106 cells/mL in each well and allowed to grow overnight. Then, the cells were incubated with CPT-11 (2 μM), mPEG–SN38 (2 μM SN38 equivalent), or mPEG–PLA–SN38 (2 μM SN38 equivalent) in cell culture medium for 12 hours at 37°C. Cells without drug treatment were used as a control. Cells were then washed with cold PBS three times. For apoptosis analysis, an Alexa Fluor® 488 Annexin V/propidium iodide apoptosis detection kit was used according to manufacturer’s protocol. Briefly, cells (1×105) were suspended in the binding buffer (200 μL). Then, 5 μL of Alexa Fluor® 488 Annexin V and 5 μL of propidium iodide (100 μg/mL) were added to each sample, which was incubated at room temperature for 15 minutes. Then, an additional binding buffer (300 μL) was added. The cells were then analyzed on a BD FACSCanto™ II flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
In vivo distribution study
DiR-labeled micelles were used to evaluate in vivo biodistribution of SN38–polymer conjugate micelles in BEL-7402 xenograft tumor-bearing mice. Micelles were prepared as described in “Preparation of SN38–polymer conjugate micelles”. Nude mice with xenograft tumors of ~500 mm3 were used in this study. First, 0.2 mL of DiR-labeled micelles were injected via tail vein (200 μg/kg DiR equivalent). The mice were anesthetized using isoflurane in oxygen and placed in the imaging chamber. Mice were purchased from the Laboratory Animal Center of Jilin University (Changchun, People’s Republic of China). The Institution Animal Ethics Committee (Jilin University) reviewed and approved the entire animal protocol prior to conducting the experiments. Fluorescent images were collected at 2, 6, and 24 hours using a Clairvivo OPT in vivo optical imaging system (SHIMADZU Corporation, Kyoto, Japan) with a 735 nm single laser. The imaging time was 5 seconds for each image. After completion of the image collection, the mice were sacrificed. Major organs including hearts, livers, spleens, lungs, kidneys, and tumors were excised and imaged under the same conditions. The experimental protocol was approved by the Laboratory Animal Center of Jilin University (license No. SCXK-(JI) 2011–0003).
In vivo antitumor efficacy
The antitumor efficacy of SN38–polymer conjugates was investigated in a BEL-7402 murine xenograft tumor model. Briefly, BALB/c nude mice (6 weeks, 18–23 g) were subcutaneously injected with 0.2 mL cell suspension containing 5×106 BEL-7402 cells in the right armpit. When tumor volumes reached ~100 mm3 (designated as day 0), mice were randomly divided into four groups (n=7) and treated as follows: PBS, CPT-11, mPEG–SN38, and mPEG–PLA–SN38 (at 10 mg/kg SN38 equivalent). The formulations were administered via tail vein using a 1.0 mL syringe every 3 days for four times. Throughout the study, tumor volume of each mouse was monitored every other day with a caliper, and calculated by the formula:
|
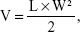
where L represents the longest diameter and W represents the shortest diameter perpendicular to length. The weight of the mice was measured regularly as an indicator of acute toxicity.
Statistical analysis
Data are presented as the mean ± standard deviation. The statistical significance of treatment outcomes was assessed using Student’s t-test (two-tailed). P-value of <0.05 was considered statistically significant in all analyses.
Results and discussion
Synthesis of mPEG2K–SN38 and mPEG2K–PLA1.5K–SN38 micelles
To synthesize PEG2K–SN38 and PEG2K–PLA1.5K–SN38, mPEG2K–SA was conjugated to SN38 through an esterification reaction between the carboxyl group on the polymer and the hydroxyl group on SN38, as shown in Figure 1A and 1B. The products were characterized by 1H NMR and fourier transform infrared spectroscopy. In the 1H NMR spectrum, the typical peak of phenolic hydroxyl (10.33 ppm) disappeared and the multiple peaks (7.38 ppm) of C-9 and C-11 divided appeared at 7.72 and 7.23 ppm. In fourier transform infrared spectroscopy spectrum, O–H vibration of phenolic hydroxyl of SN38 at 3,596 cm−1 and O–H vibration of carboxyl of PEG–SA and PEG–PLA–SA at 1,762 cm−1 disappeared and C=O stretching bands at 1,762 cm−1 was observed.25 These characteristic peaks confirmed the formation of ester bond. These data showed that mPEG2K–SN38 and mPEG2K–PLA1.5K–SN38 were successfully synthesized.
 | Figure 1 Schematic illustration of synthesis of SN38–polymer conjugate micelles. |
Preparation SN38–polymer conjugate micelles
The amphiphilic block copolymer PEG–PLA can be dissolved in organic solvents, such as chloroform, acetone, and DMF, and can self-assemble into micelles in an aqueous medium. The main driving forces of micelle formation are known to be the desolvation of the hydrophobic segment in aqueous medium, resulting in formation of a core. The hydrophilic segment stabilizes the core as tethered chains.28 Figure 1C and 1D illustrates the self-assembly of SN38–polymer conjugate micelles. As shown in Figure 1C, mPEG–SN38 self-assemble into micelles, the SN38 moiety coagulates and forms a core and mPEG segment forms a shell to stabilize the micelle. As shown in Figure 1D, mPEG–PLA–SN38 possesses two hydrophobic segments. During micelle assembly, the two hydrophobic segments form a double-layer core, SN38 inner layer and PLA outer layer. This provides greater protection of the SN38 and contributes to greater micelle stability.
Characterization of SN38–polymer conjugate micelles
Physicochemical characterization
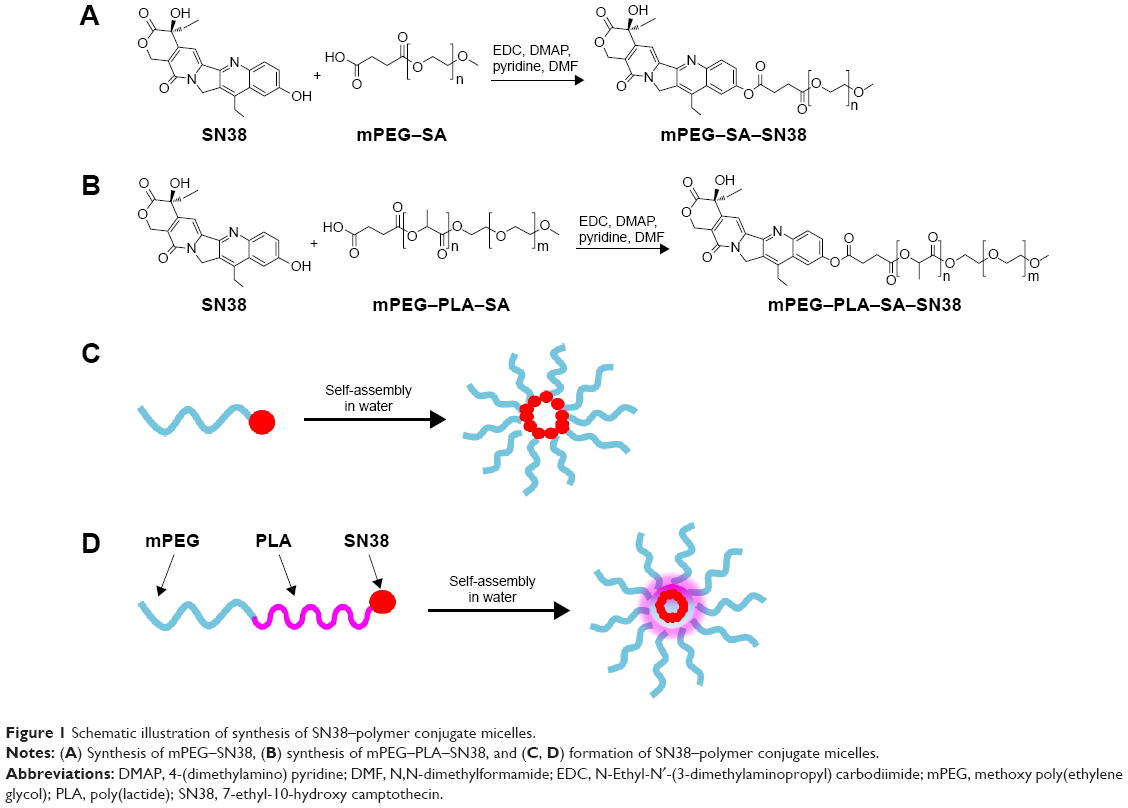
The physicochemical characteristics of SN38–polymer conjugate micelles, including particle diameter, zeta potential, and drug content, are summarized in Table 1. The mean diameter of mPEG2K–SN38 micelles was ~130 nm. However, the mean diameter of mPEG2K–PLA1.5K–SN38 micelles was much smaller at 20 nm (Figure 2A and B). This difference might be due to differences in the colloidal stability of the micelle preparations. The lipophilicity of SN38 is weaker than that of PLA1.5K–SN38, resulting in hydrophobic cores that are less stable. Smaller particle diameters can reduce reticuloendothelial system uptake, prolong circulation time of micelles in the blood, and facilitate extravasation from leaky capillaries at the tumor site.29 Hence, the more stable mPEG2K–PLA1.5K–SN38 micelles are likely to exhibit greater enhanced permeability and retention effect. The SN38–polymer conjugate micelles exhibited a negative surface charge. The zeta potential of mPEG2K–PLA1.5K–SN38 micelles was greater than that of mPEG2K–SN38 micelles. As shown in Table 1, the drug load of mPEG2K–SN38 micelles was higher than that of mPEG2K–PLA1.5K–SN38 micelles. TEM images of the SN38–polymer conjugate micelles, shown in Figure 2C and D, reveal that the SN38–polymer conjugate micelles are spherical in shape and are rather homogeneous in aqueous solution. The images confirmed that the particle diameters of mPEG2K–PLA1.5K–SN38 micelles was much smaller than those of mPEG2K–SN38 micelles. The particle diameter observed under TEM correlated well with the results from dynamic light scattering (Figure 2A and B), indicating SN38–polymer conjugate micelles are completely dispersed in aqueous solution.
 | Table 1 The physicochemical characteristics of SN38–polymer conjugate micelles (n=3) |
 | Figure 2 Characteristics of SN38–polymer conjugate micelles. |
In vitro release of SN38 from the polymer conjugate micelles
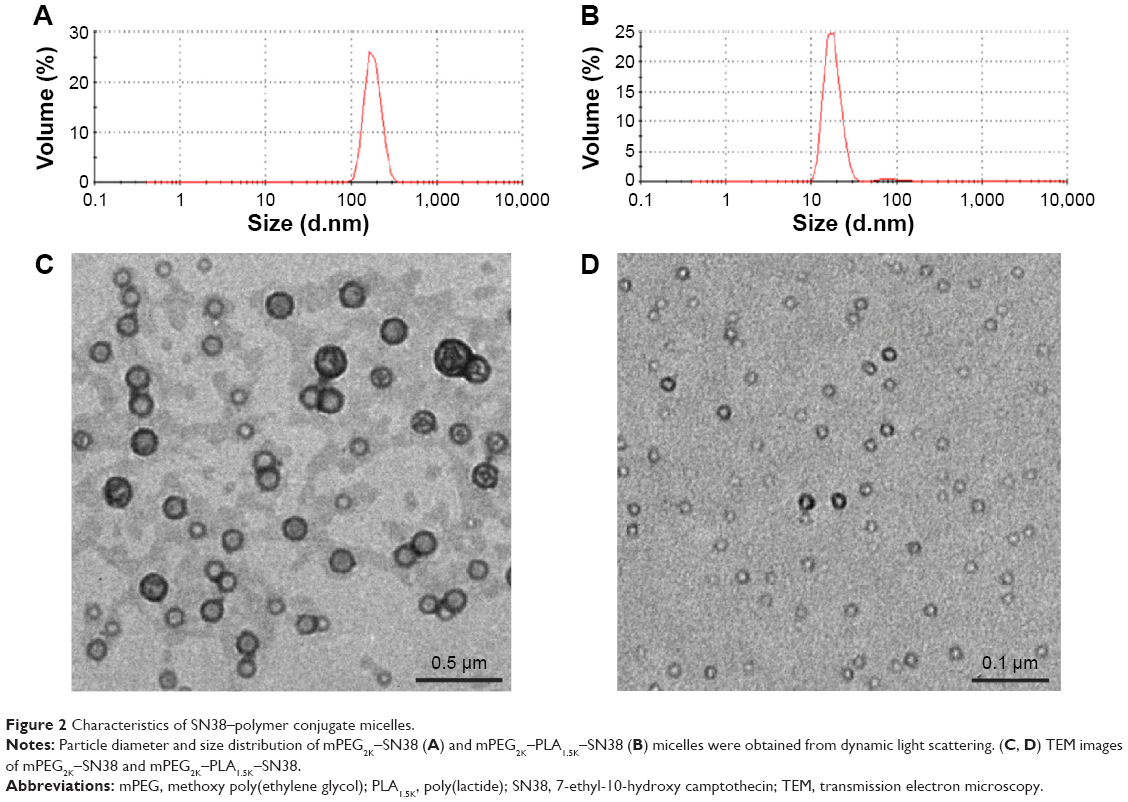
PBS (pH 7.4) was chosen as an in vitro release medium to simulate blood pH condition. The polymer conjugate micelles were incubated at 37°C in PBS (pH 7.4) containing 0.2% Tween 80 and examined for the release of SN38. As shown in Figure 3, SN38 was released from mPEG2K–SN38 micelles at a much faster rate than from mPEG2K–PLA1.5K–SN38 micelles. Approximately 50% of SN38 has been released from mPEG2K–SN38 micelles at 20 hours. Meanwhile, only ~20% was released from mPEG2K–PLA1.5K–SN38 micelles at the same time point. The reason for this difference is that mPEG2K–PLA1.5K–SN38 micelles contain a double-layer core, which is much more stable than a single-layer core in mPEG–SN38 micelles. As reported previously, rapid release of poorly water-soluble drugs from drug carriers may induce subsequent drug precipitation or their transfer to plasma proteins in vivo as a result of instant dilution in the plasma, resulting in drug inactivation.30 Therefore, the slower release of SN38 from the mPEG2K–PLA1.5K–SN38 micelles might be an advantage for its in vivo efficacy.
 | Figure 3 Cumulative release of SN38 (%) from the micelles in PBS (pH 7.4) containing 0.2% Tween 80 medium at 37°C. |
Cellular uptake
FITC was used as a probe for evaluation of cellular uptake of SN38–polymer conjugate micelles. The intracellular accumulation and distribution of FITC in BEL-7402 cells was studied using confocal microscopy (Figure 4A). After incubation of the BEL-7402 cells with SN38–polymer conjugate micelles for 4 hours, the mPEG2K–PLA1.5K–SN38 group displayed a higher fluorescence intensity in the cytoplasm than the mPEG2K–SN38 group. This could be due to the smaller diameter and greater stability of mPEG2K–PLA1.5K–SN38 micelles.
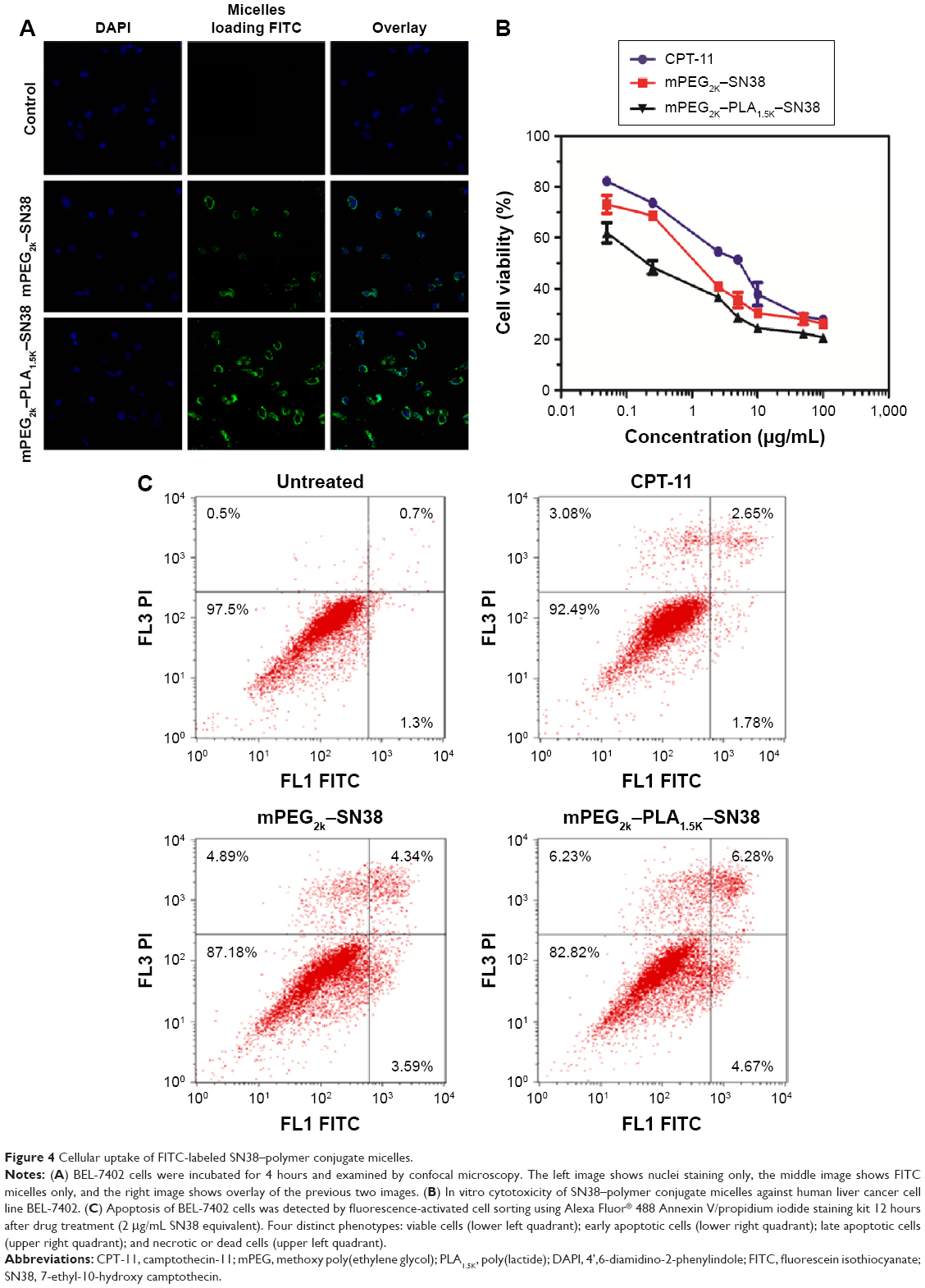 | Figure 4 Cellular uptake of FITC-labeled SN38–polymer conjugate micelles. |
In vitro cytotoxicity
Liver cancer is one of deadliest malignant tumors in humans.31,32 The cytotoxicity of SN38–polymer conjugate micelles was measured in human liver cancer BEL-7402 cells. As shown in Figure 4B, mPEG2K–PLA1.5K–SN38 micelles were highly cytotoxic with a half maximal inhibitory concentration of ~0.25 μg/mL. This suggests that mPEG2K–PLA1.5K–SN38 micelles effectively protect the active structure of SN38 and deliver greater concentration of the drug into the cells. mPEG2K–PLA1.5K–SN38 micelles were more potent than CPT-11 and mPEG2K–SN38 micelles. This result is consistent with the observed higher cellular uptake of mPEG2K–PLA1.5K–SN38 micelles, shown in Figure 4A.
Apoptosis assay
In order to understand the mechanism of antitumor activity, a fluorescence-activated cell sorting-based apoptosis analysis was performed using an Alexa Fluor 488 Annexin V/propidium iodide double-staining assay kit.33 The results are shown in Figure 4C. Greater apoptosis in BEL-7402 cells was obtained upon treatment with SN38–polymer conjugate micelles compared with the control group and CPT-11-treated group. mPEG2K–PLA1.5K–SN38-treated group showed greater apoptosis than PEG2K–SN38-treated group. This result is consistent with the results of cellular uptake and cytotoxicity.
In vivo biodistribution study using vital imaging
DiR is an indocarbocyanine dye with poor water solubility.34 It is often used for in vivo biodistribution studies because its emission wavelength is above 700 nm, which can effectively eliminate emission interference.35 In order to determine the in vivo biodistribution of SN38–polymer conjugate micelles, DiR-labeled micelles were injected intravenously into BEL-7402 liver cancer tumor-bearing mice through tail vein, and time-dependent distribution was observed using near infrared ray imaging in live animals. If the micelles were stable, DiR could be efficiently transported to the tumor site, otherwise the released DiR would be quickly eliminated. The fluorescent images of tumor-bearing mice are shown in Figure 5. As seen in Figure 5A, fluorescence signal was observed in the tumor as early as 2 hours after injection of DiR-labeled mPEG2K–PLA1.5K–SN38 micelles, and the fluorescence signal was increased at 24 hours. Fluorescent images of major organs harvested at the same point can be seen in Figure 5B. During the first 2 hours, every organ took up DiR. However, after 24 hours, DiR mostly accumulated in the tumor. Conversely, DiR-labeled mPEG2K–SN38 micelles had little distribution in the tumor and extensively accumulated in liver. At 24 hours, the distribution was also nearly the same as before (Figure 5C and D). These results indicated that the mPEG2K–PLA1.5K–SN38 micelles are selectively taken up by tumors by passive targeting.29,36
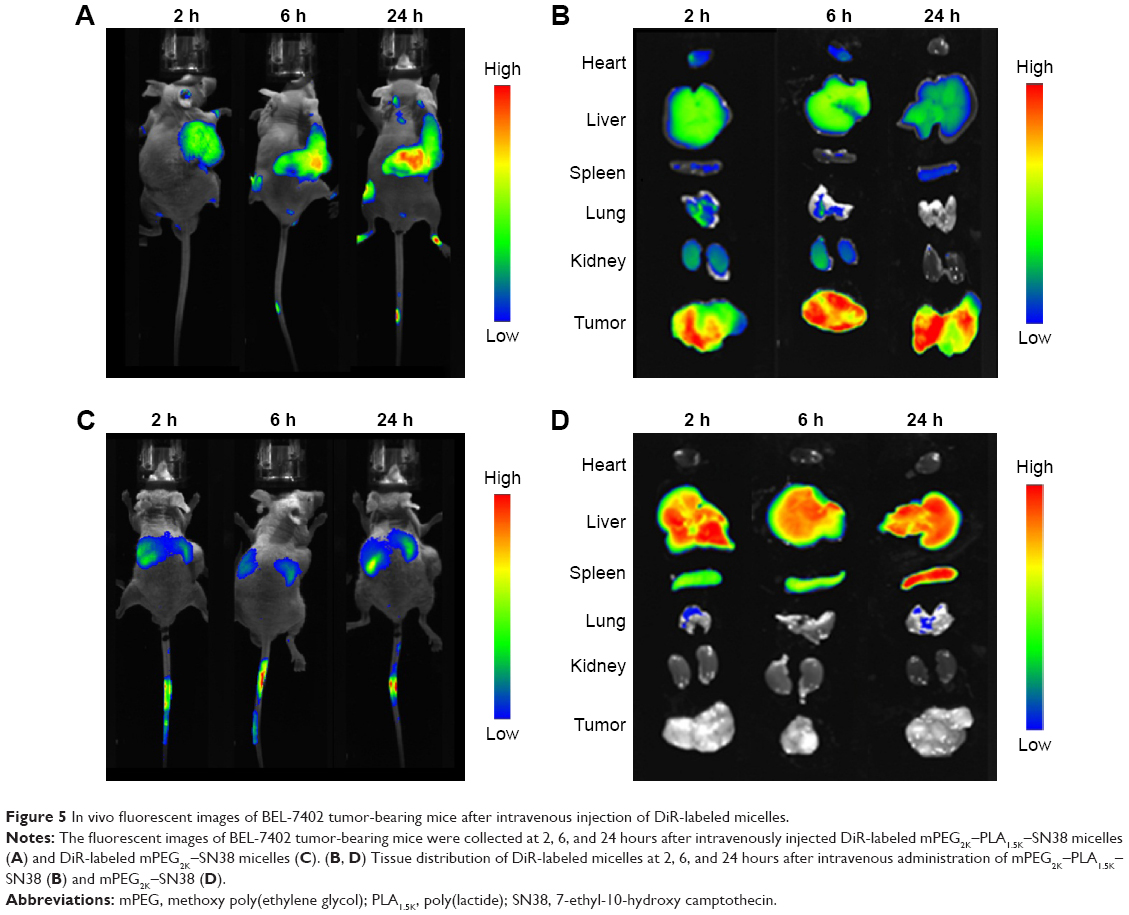 | Figure 5 In vivo fluorescent images of BEL-7402 tumor-bearing mice after intravenous injection of DiR-labeled micelles. |
In vivo antitumor efficacy
Antitumor efficacy of SN38–polymer conjugate micelles was evaluated using a nude mouse xenograft model from human liver cancer BEL-7402 cells. As shown in Figure 6A, the inhibitory effect of mPEG2K–PLA1.5K–SN38 micelles on tumor growth was significantly greater than that of mPEG2K–SN38 micelles at the same dosage (P<0.01). This was in accord with the observations in cellular uptake and cytotoxicity studies and the in vivo biodistribution results (Figure 5), which showed higher tumor uptake of mPEG2K–PLA1.5K–SN38 micelles. On the other hand, the body weight change (Figure 6B) observed in this experiment was not significant and suggested the lack of a significant difference in the toxicity among the treatment groups.
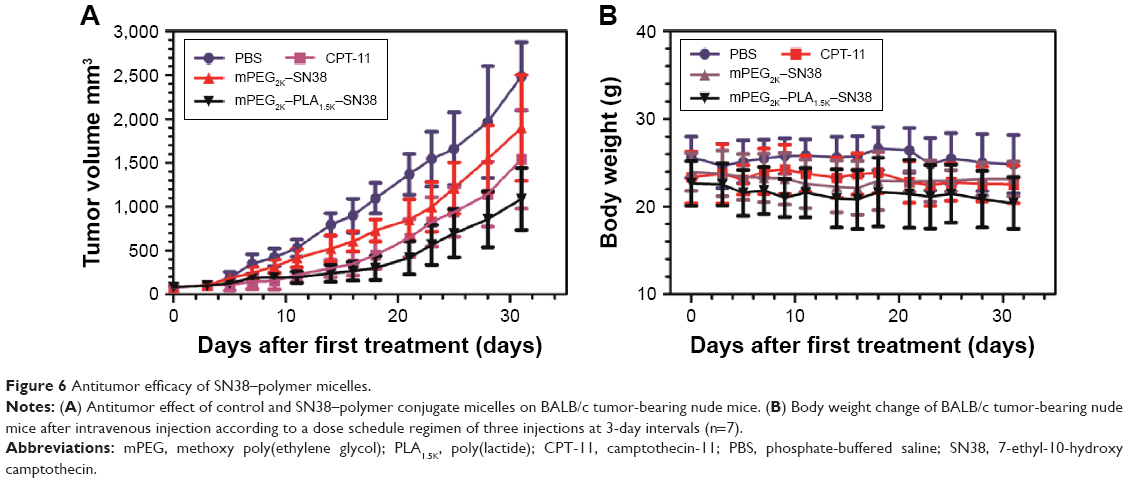 | Figure 6 Antitumor efficacy of SN38–polymer micelles. |
Conclusion
mPEG–PLA–SN38 and mPEG–SN38 are amphiphilic polymer conjugates that self-assemble into micelles in an aqueous media. In vitro and in vivo experiments were carried out to compare the properties and efficacy of these two prodrugs. Dynamic light scattering and TEM analysis of the micelles revealed that the size of mPEG–PLA–SN38 micelles was much smaller than PEG–SN38 micelles and the morphologies of the former were also more homogeneous. In vitro release of SN38 from mPEG–PLA–SN38 micelles was slower. In vitro cytotoxicity, cellular uptake, and apoptosis assay all showed that mPEG–PLA–SN38 micelles were more efficient than mPEG–SN38 micelles. In vivo biodistribution study using DiR-labeled mPEG–PLA–SN38 micelles showed much greater delivery to the tumor than mPEG–SN38 micelles. Finally, in vivo antitumor efficacy study showed mPEG–PLA–SN38 micelles possessed greater antitumor activity than mPEG–SN38 micelles as well. Collectively, mPEG–PLA–SN38 has been shown to be a promising anticancer agent that deserves further investigation.
Disclosure
The authors report no conflicts of interest in this work.
References
Pommier Y. DNA Topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109(7):2894–2902. | ||
Ulukan H, Swaan PW. Camptothecins: a review of their chemotherapeutic potential. Drugs. 2002;62(14):2039–2057. | ||
Zeghari-Squalli N, Raymond E, Cvitkovic E, Goldwasser Fo. Cellular pharmacology of the combination of the DNA topoisomerase I inhibitor SN-38 and the diaminocyclohexane platinum derivative oxaliplatin. Clin Cancer Res. 1999;5(5):1189–1196. | ||
Wu MH, Yan B, Humerickhouse R, Dolan ME. Irinotecan activation by human carboxyl esterases in colorectal adenocarcinoma cells. Clin Cancer Res. 2002;8(8):2696–2700. | ||
Zhang JA, Xuan T, Parmar M, et al. Development and characterization of a novel liposome-based formulation of SN-38. Int J Pharm. 2004;270:93–107. | ||
Atyabi F, Farkhondehfai A, Esmaeili F, Dinarvand R. Preparation of pegylated nano-liposomal formulation containing SN-38: In vitro characterization and in vivo biodistribution in mice. Acta Pharm. 2009;59(2):133–144. | ||
Mert O, Esendagl G, Dogan AL, Demir AS. Injectable biodegradable polymeric system for preserving the active form and delayed-release of camptothecin anticancer drugs. RSC Advances. 2012;2(1):176–185. | ||
Ledermann JA, Leonard P, Seymour M. Recommendation for caution with irinotecan, fluorouracil, and leucovorin for colorectal cancer. N Engl J Med. 2001;345(2):145–146. | ||
Chabot G. Clinical pharmacokinetics of irinotecan. Clin Pharmacokinet. 1997;33(4):245–259. | ||
Danks M. Topoisomerase enzymes as drug targets. In: Schwab M, editor. Encyclopedic Reference of Cancer. Berlin/Heidelberg: Springer; 2001:900–903. | ||
Slatter JG, Schaaf LJ, Sams JP, et al. Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) Following I.V. infusion of [14C]CPT-11 in cancer patients. Drug Metab Dispos. 2000;28(4):423–433. | ||
Shimada Y, Rothenberg M, Hilsenbeck SG, Burris HAI, Degen D, Von Hoff DD. Activity of CPT-11 (irinotecan hydrochloride), a topoisomerase I inhibitor, against human tumor colony-forming units. Anti-Cancer Drugs. 1994;5(2):202–206. | ||
Ulukan H, Muller MT, Swaan PW. Downregulation of topoisomerase I in differentiating human intestinal epithelial cells. Int J Cancer. 2001;94(2):200–207. | ||
Ebrahimnejad P, Dinarvand R, Sajadi A, et al. Preparation and in vitro evaluation of actively targetable nanoparticles for SN-38 delivery against HT-29 cell lines. Nanomed Nanotech Biol Med. 2010;6(3):478–485. | ||
Lei S, Chien P-Y, Sheikh S, Zhang A, Ali S, Ahmad I. Enhanced therapeutic efficacy of a novel liposome-based formulation of SN-38 against human tumor models in SCID mice. Anti Cancer Drugs. 2004;15(8):773–778. | ||
Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6(9):688–701. | ||
Li C, Wallace S. Polymer-drug conjugates: Recent development in clinical oncology. Adv Drug Deliv Rev. 2008;60(8):886–898. | ||
Zhao H, Rubio B, Sapra P, et al. Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjug Chem. 2008;19(4):849–859. | ||
Çirpanli Y, Bilensoy E, Lale Doğan A, Çaliş S. Comparative evaluation of polymeric and amphiphilic cyclodextrin nanoparticles for effective camptothecin delivery. Eur J Pharm Biopharm. 2009;73(1):82–89. | ||
Sapra P, Kraft P, Mehlig M, et al. Marked therapeutic efficacy of a novel polyethylene glycol-SN38 conjugate, EZN-2208, in xenograft models of B-cell non-Hodgkin’s lymphoma. Haematologica. 2009;94(10):1456–1459. | ||
Sapra P, Zhao H, Mehlig M, et al. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin Cancer Res. 2008;14(6):1888–1896. | ||
Dong Y, Feng S-S. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25(14):2843–2849. | ||
Kim J-H, Emoto K, Iijima M, et al. Core-stabilized polymeric micelle as potential drug carrier: increased solubilization of taxol. Polym Adv Technol. 1999;10(11):647–654. | ||
Kim SC, Kim DW, Shim YH, et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72:191–202. | ||
Lu L, Zheng Y, Weng S, et al. Complete 1 regression of xenograft tumors using biodegradable mPEG-PLA-SN38 block copolymer micelles. Colloids Surf B Biointerfaces. 2016;142:417–423. | ||
Koizumi F, Kitagawa M, Negishi T, et al. Novel SN-38a-incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor – secreting bulky tumors. Cancer Res. 2006;66(20):10048–10056. | ||
Kim SY, Lee YM, Baik DJ, Kang JS. Toxic characteristics of methoxy poly(ethylene glycol)/poly(ε-caprolactone) nanospheres; in vitro and in vivo studies in the normal mice. Biomaterials. 2003;24(1):55–63. | ||
Liggins RT, Hunter WL, Burt HM. Solid-state characterization of paclitaxel. J pharm sci. 1997;86(12):1458–1463. | ||
Maeda H. Macromolecular therapeutics in cancer treatment: The EPR effect and beyond. J Control Release. 2012;164(2):138–144. | ||
Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. J Control Release. 2001;73(2–3):137–172. | ||
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007; 132(7):2557–2576. | ||
Curley SA, Izzo F. Radiofrequency ablation of primary and metastatic hepatic malignancies. Annals of Surgery. 1999;230(1):1–8. | ||
Logue SE, Elgendy M, Martin SJ. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat Protoco. 2009;4(9):1383–1395. | ||
Xia H, Gao X, Gu G, et al. Penetratin-functionalized PEG -PLA nanoparticles for brain drug delivery. International Journal of Pharmaceutics. 2012;436(1–2):840–850. | ||
Zheng N, Dai W, Du W, et al. A novel lanreotide-encoded micelle system targets paclitaxel to the tumors with overexpression of somatostatin receptors. Mol Pharm. 2012;9(5):1175–1188. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63(3):170–183. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.