")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Synthesis and Cytotoxic Property of Annonaceous Acetogenin Glycoconjugates
Authors Shi JF, Wu P, Cheng XL, Wei XY , Jiang ZH
Received 2 May 2020
Accepted for publication 1 September 2020
Published 17 November 2020 Volume 2020:14 Pages 4993—5004
DOI https://doi.org/10.2147/DDDT.S259547
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Jing-Fang Shi,1,2 Ping Wu,2 Xiao-Li Cheng,2 Xiao-Yi Wei,2 Zi-Hua Jiang3
1Guangdong Provincial Key Laboratory for Crop Germplasm Resources Preservation and Utilization, Agro-Biological Gene Research Center, Guangdong Academy of Agricultural Sciences, Guangzhou 510640, People’s Republic of China; 2Key Laboratory of Plant Resources Conservation and Sustainable Utilization, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou 510650, People’s Republic of China; 3Department of Chemistry, Lakehead University, Thunder Bay, Ontario P7B 5E1, Canada
Correspondence: Zi-Hua Jiang
Department of Chemistry, Lakehead University, 955 Oliver Road, Thunder Bay, Ontario P7B 5E1, Canada
Tel +1-807-766-7171
Email [email protected]
Xiao-Yi Wei
Key Laboratory of Plant Resources Conservation and Sustainable Utilization, South China Botanical Garden, Chinese Academy of Sciences, Xingke Road 723, Tianhe District, Guangzhou 510650, People’s Republic of China
Tel +86-20-37252538
Email [email protected]
Background: Annonaceous acetogenins (ACGs) are secondary metabolites produced by the Annonaceae family and display potent anticancer activity against various cancer cell lines. Squamocin and bullatacin are two examples of ACGs that show promising antitumor activity; however, preclinical data are not sufficient partly due to their being highly lipophilic and poorly soluble in water. These compounds also display high toxicity to normal cells. Due to these disadvantageous properties, the therapeutic potential of squamocin and bullatacin as antitumor agents has not been fully evaluated.
Methods: In order to enhance their water solubility and potentially improve their cancer targeting, squamocin and bullatacin were conjugated to a glucose or galactose to yield glycosylated derivatives by direct glycosylation or the Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC) reaction (the click reaction). The synthesized compounds were evaluated for their anticancer property against HeLa, A549 and HepG2 cancer cell lines using MTT assay.
Results: Nine glycosyl derivatives were synthesized and structurally characterized. Most of them show comparable in vitro cytotoxicity against HeLa, A549 and HepG2 cancer cell lines as their parent compounds squamocin and bullatacin. It appears that the type of sugar residue (glucose or galactose), the position at which the sugar residue is attached, and whether or not a linking spacer is present do not affect the potency of these derivatives much. The solubility of galactosylated squamocin 13 in phosphate buffer saline (PBS, pH = 7) is greatly improved (1.37 mg/mL) in comparison to squamocin (not detected in PBS).
Conclusion: The conjugation of a glucose or galactose to squamocin and bullatacin yields glycosyl derivatives with similar level of anticancer activity in tested cell lines. Further studies are needed to demonstrate whether or not these compounds show reduced toxicity to normal cells and their therapeutic potential as antitumor agents.
Keywords: annonaceous acetogenins, squamocin, bullatacin, glycosylated, cytotoxicity, anticancer, solubility
Introduction
Annonaceous acetogenins (ACGs) have been isolated exclusively from species of the annonaceae family, exhibiting potent growth inhibitory activity against various cancer cell lines.1–7 Bullatacin and squamocin (Figure 1) are two of the most cytotoxic acetogenins bearing adjacent tetrahydrofuran (THF) rings, which have been reported mainly in the seeds of these plants.8–11 The anticancer potency of bullatacin has been reported to be much higher than some of the anticancer drugs currently used in the clinic. For example, bullatacin was found to be 104–105 times more potent than doxorubicin against both A549 and MCF-7 cell lines,12 and 250 times more potent than doxorubicin against MCF-7/ADR, the human breast adenocarcinoma multidrug-resistant cell line.13 In addition, bullatacin was found to be 300 times more effective than taxol in treating L1210 murine leukemia in mouse model.14 Bullatacin and squamocin are promising anti-cancer agents worthy of further investigation. However, they display high toxicity toward normal cells as well10 and show poor solubility in water (less than 1 μg/mL).15 These properties prevent them from full evaluation of their therapeutic potential for cancer treatment. The preparation of derivatives for tumor-specific targeting by incorporation of different ligands could improve their activity and yield more suitable drugs.
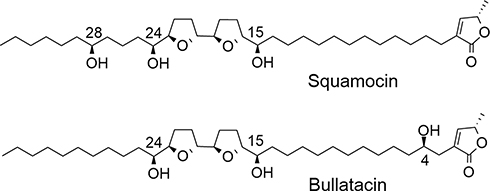 |
Figure 1 Structures of squamocin and bullatacin. |
Conjugation of carbohydrates with drugs has become increasingly important in bioorganic chemistry and chemical biology.16,17 Glycosylation of drug molecules has been employed to modify their physico-chemical properties, including polarity, solubility, and stability, without affecting their biological activities.18,19 For example, glycosylated porphyrins have been shown as promising photosensitizers in photodynamic therapy (PDT) for cancer owing to their good solubility and specific membrane interactions.20 Moreover, glycosylation has been widely explored as a targeting strategy to selectively deliver anticancer drugs to cancer cells. This strategy is based on the findings that cancer cells require more glucose due to their rapid growth and altered metabolism than normal cells do, which results in the expression of higher levels of glucose transporters on plasma membrane of cancer cells to facilitate the uptake of glucose. For example, glucose transporter-1 (GLUT1), which also transports other hexoses such as galactose, mannose, and glucosamine,21 is expressed at a level 100–300 times higher in malignant cells than in normal cells.22 Anticancer agents conjugated to a glucose (or another sugar that is also a glucose transporter substrate) may be taken up by cancer cells more rapidly via facilitation by glucose transporters and be more selective toward cancer cells than normal cells. A number of anticancer agents such as doxorubicin,23 daunorubicin,24 paclitaxel and docetaxel,25 8-hydroxyquinoline and derivatives,26 and benzodiazepins27 have been redesigned to improve their cancer targeting and selectivity.
Previously, we reported a series of biotin-conjugated squamocin/bullatacin derivatives that showed tumor cell growth inhibitory activity with higher selectivity toward biotin receptor (+) tumor cells than their parent squamocin/bullatacin.28 In this study, squamocin and bullatacin are conjugated to a glucose or galactose to improve their solubility in water and potentially their cancer targeting. The synthesis and preliminary studies of the anticancer activity of these new glycoconjugates are described.
Materials and Methods
Reagents and Instrumentation
Squamocin and bullatacin were obtained in our previous chemical investigation on the seeds of Annona squamosa.42 2,3,4,6-Tetra-O-acetyl-β-
Optical rotations were obtained on a Perkin-Elmer 341 polarimeter with MeOH as solvent. The 1H and 13C NMR spectra were collected on a Bruker Avance-600 instrument at 600 (1H) and 150 (13C) MHz. The 2D NMR (1H-1H COSY, HSQC, and HMBC) spectra were recorded on a Bruker Avance-600 instrument. Chemical shifts are reported as ppm (δ units) relative to tetramethylsilane (TMS) as an internal standard and the coupling constants as J in hertz. The splitting pattern abbreviations are as follows: s = singlet, d = doublet, dd = double doublet, t = triplet, m = multiplet. ESIMS data were obtained on an MDS SCIEX API 2000 LC/MS instrument. HRESIMS data were obtained on a Bruker maXis Q-TOF mass spectrometer. Preparative HPLC was run on a Shimadzu LC-6A pump and a Shimadzu RID-10A refractive index detector and all separations were carried out with an XTerra Prep MS C18 column (19 × 300 mm, 10 μm) at a flow rate of 5 mL/min except where otherwise stated.
Preparation of Galacosylated Squamocin 4 and 5
2,3,4,6-Tetra-O-acetyl-
Squamocin (124 mg, 0.2 mmol), 3 (147 mg, 0.3 mmol) were dissolved in dried CH2Cl2 (5 mL) at 0°C. 4Ǻ molecular sieves (200 mg) and TMSOTf (0.02 mL, 0.1mmol) was added at nitrogen atmosphere and the mixture was stirred at 0°C for 30 min (Scheme 1). The reaction mixture was quenched with triethylamine and then concentrated under reduced pressure. The residue was dissolved in EtOAc and extracted with 1 N HCl, saturated aqueous NaHCO3 and saturated aqueous NaCl solution. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated to give crude product 245 mg.
The deprotection was performed by treatment with Et3N: MeOH: H2O (1:8:1) at 35°C for 30 h (Scheme 1). The reaction mixture was concentrated in vacuo and the crude product was separated by Sephadex LH-20 gel permeation chromatography using MeOH, followed by preparative HPLC using 88% MeOH to afford 4 (2 mg, 1%, tR = 43.6 min) and 5 (2 mg, 1%, tR = 65.1 min).
Characterization Data of Compound 4
A yellowish waxy solid; [α]20D +4.6 (c 0.06, MeOH); 1H NMR (600 MHz, CDCl3): 7.00 (1H, m, H-35), 5.00 (1H, m, H-36), 4.37 (1H, d, J = 7.6 Hz, H-1′), 4.03 (3H, m, H-15, 16, 4′), 3.86 (3H, m, H-20, 2′, 6′a), 3.80 (2H, m, H- 19, 6′b), 3.74 (1H, m, H-24), 3.62 (2H, m, H-23, 3′), 3.57 (1H, m, H-28), 3.53 (1H, m, H-5′), 2.26 (2H, t, J = 7.7 Hz, H-3), 1.95 (3H, m), 1.88 (1H, m), 1.82 (1H, m), 1.69 (1H, m), 1.45–1.58 (5H, m), 1.38–1.44 (4H, m), 1.41 (3H, d, J = 6 Hz, H-37), 1.20–1.37 (30H, m), 0.88 (3H, t, J = 6.9 Hz, H-34); 13C NMR (150 MHz, CDCl3): 174.2 (C-1), 149.2 (C-35), 134.5 (C-2), 102.3 (C-1′), 82.8 (2C, C-16, C-19), 82.2 (C-20), 81.3 (C-23), 81.2 (C-15), 74.8 (C-5′), 77.5 (C-36), 73.7 (C-3′), 71.8 (C-28), 71.7 (C-2′), 71.2 (C-24), 69.2 (C-4′), 61.5 (C-6′), 37.7 and 37.4 (C-27, C-29), 32.1 (C-25), 31.6 (C-32), 30.2 (C-14), 30.0, 29.9 (3C), 29.8 (2C), 29.7, 29.5, 29.4, 29.3, 29.0, 28.9, 27.6, 26.0, 25.4, 25.1, 24.9, 22.9 (C-33), 21.7 (C-26), 19.4 (C-37), 14.3 (C-34); HR-ESIMS m/z: 785.5413 [M + H]+ (calcd for C43H77O12, 785.5410).
Characterization Data of Compound 5
A yellowish waxy solid; [α]20D −1.2 (c 0.07, MeOH); 1H NMR (600 MHz, CDCl3): 7.00 (1H, m, H-35), 4.99 (1H, m, H-36), 4.30 (1H, d, J = 6.7 Hz, H-1′), 4.04 (1H, m, H-4′), 3.95 (2H, m, H-19, 20), 3.82 (5H, m, H-16, 23, 2′, 6′), 3.63 (3H, m, H-24, 28, 3′), 3.54 (1H, m, H-5′), 3.39 (1H, m, H-15), 2.28 (2H, t, J = 7.7 Hz, H-3), 1.93 (3H, m), 1.81 (1H, m), 1.72 (1H, m), 1.61 (1H, m), 1.45–1.56 (5H, m), 1.38–1.44 (4H, m), 1.40 (3H, d, J = 6 Hz, H-37), 1.20–1.35 (30H, m), 0.88 (3H, t, J = 6.9 Hz, H-34); 13C NMR (150 MHz, CDCl3): 174.2 (C-1), 149.2 (C-35), 134.5 (C-2), 103.5 (C-1′), 83.6 (C-16), 83.1 (C-19), 82.7 (C-20), 82.2 (C-23), 81.4 (C-28), 77.7 (C-36), 74.6 (C-15), 74.4 (C-5′), 73.6 (C-3′), 71.8 (C-24, C-2′), 69.9 (C-4′), 61.4 (C-6′), 35.6 and 34.1 (C-27, C-29), 33.2 (C-14), 32.6 (C-25), 32.1 (C-32), 30.0, 29.9 (4C), 29.8, 29.7, 29.5, 29.4, 29.1, 29.0, 28.9, 27.6, 25.8, 25.7, 25.4 (2C), 22.9 (C-33), 21.7 (C-26), 19.4 (C-37), 14.4 (C-34); HR-ESIMS m/z: 785.5415 [M + H]+ (calcd for C43H77O12, 785.5410).
Preparation of β-D-Galactopyranosyl Azide 8 and β-D-Glucopyranosyl Azide 9
Eighty microliters of sodium methoxide solution in methanol (5 M) was added to 2 mL dried MeOH, then 2,3,4,6-tetra-O-acetyl-β-
Eighty microliters of sodium methoxide was added to 2 mL dried MeOH, then 2,3,4,6-tetra-O-acetyl-β-
Preparation of Galactosyl Squamocin Derivatives 11–13
Squamocin (187 mg, 0.3 mmol), 5-hexynoic acid (40 mg, 0.36 mmol) and DMAP (3 mg, 0.03 mmol) were dissolved in 3 mL dried CH2Cl2. DCC (123 mg, 0.6 mmol) was added and the mixture was stirred under N2 atmosphere at room temperature for 16 h (Scheme 3). The resulting precipitate (DCU) was removed by filtration and the filtrate was concentrated under reduced pressure. The residue was dissolved in EtOAc and sequentially washed with 0.5% aqueous HCl solution, saturated aqueous NaHCO3 solution and saturated aqueous NaCl. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude products were separated by Sephadex LH-20 size exclusion chromatography using MeOH to give crude product 10A (204 mg).
The crude product 10A (204 mg) and β-
Characterization Data of Compound 11
A colorless waxy solid; [α]20D +19.6 (c 0.2, MeOH); 1H NMR (600 MHz, CDCl3): δ 7.92 (1H, s, H-6′), 7.00 (1H, m, H-35), 5.55 (1H, d, J = 8.0 Hz, H-7′), 5.00 (1H, m, H-36), 4.83 (1H, m, H-15), 3.94 (1H, m, H-16), 4.29 (1H, m, H-8′), 4.14 (1H, m, H-10′), 3.80 (7H, m, H-19, 20, 23, 9′, 11′, 12′), 3.59 (1H, m, H-24), 3.52 (1H, m, H-28), 2.75 (2H, m, H-4′), 2.36 (2H, m, H-2′), 2.26 (2H, m, H-3), 1.85–2.02 (4H, m), 1.74 (2H, m), 1.48–1.59 (6H, m), 1.38–1.47 (6H, m), 1.41 (3H, d, J = 6.0 Hz, H-37), 1.16–1.35 (30H, m), 0.87 (3H, t, J = 6.6 Hz, H-34); 13C NMR (150 MHz, CDCl3): δ 174.2 (C-1), 173.6 (C-1′), 149.2 (C-35), 147.2 (C-5′), 134.5 (C-2), 122.4 (C-6′), 88.3 (C-7′), 82.7 (C-20), 82.2 (C-23), 81.9 (C-19), 80.7 (C-16), 77.9 (C-11′), 77.7 (C-36), 75.7 (C-15), 74.0 (C-9′), 72.2 (C-24), 71.9 (C-28), 70.3 (C-8′), 69.0 (C-10′), 61.2 (C-12′), 37.8 and 37.3 (C-27, C-29), 33.8 (C-2′), 33.0 (C-25), 32.1 (C-32), 30.9 (C-14), 29.8 (3C), 29.7 (5C), 29.5, 29.4, 28.9, 28.6, 28.6, 27.6, 26.0, 25.5, 25.4 (2C), 24.7, 24.6 (C-4′), 22.9 (C-33), 22.4 (C-26), 19.4 (C-37), 14.3 (C-34); HR-ESIMS m/z: 922.6015 [M + H]+ (calcd for C49H84N3O13, 922.5999).
Characterization Data of Compound 12
A colorless waxy solid; [α]20D +16.0 (c 0.2, MeOH); 1H NMR (600 MHz, CDCl3): δ 7.82 (1H, s, H-6′), 7.00 (1H, m, H-35), 5.58 (1H, br s, H-7′), 5.00 (2H, m, H-36, H-24), 4.32 (1H, m, H-8′), 4.18 (1H, m, H-10′), 4.05 (1H, m, H-23), 3.85 (2H, m, H- 9′, 11′), 3.83 (1H, m, H-16), 3.80 (4H, m, H-19, 20, 12′), 3.53 (1H, m, H-28), 3.39 (1H, m, H-15), 2.67 (2H, m, H-4′), 2.35 (2H, m, H-2′), 2.26 (2H, m, H-3), 1.85–2.02 (5H, m), 1.76 (2H, m), 1.48–1.62 (5H, m), 1.37–1.47 (6H, m), 1.41 (3H, d, J = 6.0 Hz, H-37), 1.16–1.36 (30H, m), 0.87 (3H, t, J = 7.0 Hz, H-34); 13C NMR (150 MHz, CDCl3): δ 174.3 (C-1), 174.2 (C-1′), 149.2 (C-35, C-5′), 134.5 (C-2), 88.3 (C-7′), 83.7 (C-16), 82.7 (C-20), 81.9 (C-19), 80.9 (C-23), 77.7 (C-11′, C-36), 75.1 (C-24), 74.7 (C-15), 73.9 (C-9′), 71.4 (C-28), 70.3 (C-8′), 69.3 (C-10′), 61.1 (C-12′), 37.8 and 37.1 (C-27, C-29), 33.9 (C-2′), 33.1 (C-14), 32.1 (C-32), 31.2 (C-25), 30.0, 29.9 (3C), 29.8 (2C), 29.7, 29.5, 29.4, 29.1, 28.8, 28.4, 27.6, 25.9, 25.7, 25.4 (2C), 24.9 (C-4′), 22.9, 22.9 (C-33), 22.0 (C-26), 19.4 (C-37), 14.4 (C-34); HR-ESIMS m/z: 922.6020 [M + H]+ (calcd for C49H84N3O13, 922.5999).
Characterization Data of Compound 13
A colorless waxy solid; [α]20D +15.6 (c 0.2, MeOH); 1H NMR (600 MHz, CDCl3): δ 7.85 (1H, s, H-6′), 7.00 (1H, m, H-35), 5.57 (1H, br s, H-7′), 5.00 (1H, m, H-36), 4.88 (1H, m, H-28), 4.28 (1H, m, H-8′), 4.16 (1H, m, H-10′), 3.91 (2H, m, H-19, 20), 3.83 (6H, m, H-16, 23, 9′, 11′, 12′), 3.75 (1H, m, H-24), 3.39 (1H, m, H-15), 2.68 (2H, m, H-4′), 2.33 (2H, m, H-2′), 2.26 (2H, m, H-3), 1.85–2.03 (6H, m), 1.79 (2H, m), 1.44–1.61 (10H, m), 1.41 (3H, d, J = 6.0 Hz, H-37), 1.20–1.37 (30H, m), 0.88 (3H, t, J = 7.0 Hz, H-34); 13C NMR (150 MHz, CDCl3): δ 174.2 (C-1), 173.5 (C-1′), 149.2 (C-35, C-5′), 134.5 (C-2), 88.3 (C-7′), 84.0 (C-16), 83.2 (C-19), 83.0 (C-20), 82.8 (C-23), 77.8 (C-11′), 77.7 (C-36), 74.6 (C-15), 74.3 (C-9′), 74.2 (C-28), 71.7 (C-24), 70.6 (C-8′), 69.2 (C-10′), 61.7 (C-12′), 34.6 and 34.5 (C-27, C-29), 34.0 (C-2′), 32.9 (C-14), 32.5 (C-25), 31.9 (C-32), 29.9 (4C), 29.8 (2C), 29.7, 29.5, 29.4 (2C), 29.2 (2C), 28.7, 27.6, 25.8, 25.6, 25.4 (2C), 24.8 (C-4′), 24.7, 22.8 (C-33), 22.4 (C-26), 19.4 (C-37), 14.3 (C-34); HR-ESIMS m/z: 922.6017 [M + H]+ (calcd for C49H84N3O13, 922.5999).
Preparation of Glucosylated Squamocin Derivatives 14 and 15
Squamocin (249 mg, 0.4 mmol), 5-hexynoic acid (67 mg, 0.6 mmol) and DMAP (7 mg, 0.06 mmol) were dissolved in 5 mL dried CH2Cl2. DCC (247 mg, 1.2 mmol) was added and the mixture was stirred under N2 atmosphere at room temperature for 16 h (Scheme 3). Following the same procedure as described for the preparation of 10A in the Experimental Section 3.4, intermediate product 10B (288 mg) was obtained.
The intermediate product 10B (288 mg) and β-
Characterization Data of Compound 14
A yellowish waxy solid; [α]20D +1.4 (c 1.1, MeOH); 1H NMR (600 MHz, CDCl3): δ 7.82 (1H, s, H-6′), 7.00 (1H, m, H-35), 5.59 (1H, d, J = 7.9 Hz, H-7′), 5.00 (1H, m, H-36), 4.96 (1H, m, H-24), 4.01 (2H, m, H-23, 8′), 3.82 (1H, m, H-20), 3.77 (6H, m, H-16, 19, 9′, 10′, 12′), 3.58 (1H, m, H-11′), 3.51 (1H, m, H-28), 3.35 (1H, m, H-15), 2.68 (2H, m, H-4′), 2.35 (2H, m, H-2′), 2.25 (2H, m, H-3), 1.94 (5H, m), 1.72 (2H, m), 1.52 (11H, m), 1.41 (3H, d, J = 6.0 Hz, H-37), 1.19–1.37 (30H, m), 0.87 (3H, t, J = 7.0 Hz, H-34); 13C NMR (150 MHz, CDCl3): δ 174.1 (C-1), 173.6 (C-2′), 149.2 (C-35), 134.4 (C-2), 129.0 (C-6′), 87.9 (C-7′), 83.6 (C-16), 82.6 (C-20), 81.8 (C-19), 80.8 (C-23), 79.2 (C-11′), 77.6 (C-36), 76.9 (C-9′), 75.1 (C-24), 74.4 (C-15), 72.7 (C-8′), 71.4 (C-28), 69.1 (C-10′), 61.1 (C-12′), 37.7 and 37.0 (C-27, C-29), 33.9 (C-2′), 33.1 (C-14), 32.0 (C-32), 31.3 (C-25), 29.9 (2C), 29.8 (3C), 29.7, 29.6, 29.5, 29.4, 29.0, 28.6, 28.5, 27.6 (2C), 25.9, 25.7, 25.3, 24.8 (2C), 24.5, 22.8 (C-33), 21.9 (C-26), 19.4 (C-37), 14.3 (C-34); HR-ESIMS m/z: 922.5996 [M + H]+ (calcd for C49H84N3O13, 922.5999).
Characterization Data of Compound 15
A yellowish waxy solid; [α]20D +0.8 (c 1.1, MeOH); 1H NMR (600 MHz, CDCl3): δ 7.71 (1H, s, H-6′), 7.00 (1H, m, H-35), 5.57 (1H, d, J = 8.0 Hz, H-7′), 4.99 (1H, m, H-36), 4.86 (1H, m, H-28), 4.03 (1H, m, H-8′), 3.91 (2H, m, H-19, 20), 3.82 (3H, m, H-16, 12′), 3.78 (4H, m, H-23, 24, 9′, 10′), 3.61 (1H, m, H-11′), 3.37 (1H, m, H-15), 2.68 (2H, m, H-4′), 2.32 (2H, m, H-2′), 2.25 (2H, m, H-3), 1.93 (4H, m), 1.86 (1H, m), 1.77 (1H, m), 1.52 (12H, m), 1.40 (3H, d, J = 6.8 Hz, H-37), 1.19–1.35 (30H, m), 0.88 (3H, t, J = 7.0 Hz, H-34); 13C NMR (150 MHz, CDCl3): δ 174.1 (C-1), 173.4 (C-2′), 149.1 (C-35), 146.9 (C-5′), 134.3 (C-2), 121.6 (C-6′), 87.8 (C-7′), 83.6 (C-16), 83.0 (C-19), 82.7 (C-20), 82.2 (C-23), 79.1 (C-11′), 77.6 (C-36), 76.9 (C-9′), 74.4 (C-15), 74.3 (C-28), 72.7 (C-8′), 71.5 (C-24), 69.0 (C-10′), 61.1 (C-12′), 34.4 and 34.3 (C-27, C-29), 33.9 (C-2′), 33.0 (C-14), 32.4 (C-25), 31.8 (C-32), 29.9, 29.8, 29.7 (2C), 29.6, 29.4, 29.3 (2C), 29.0, 29.0, 28.6, 27.5, 25.7, 25.4, 25.3, 24.9, 24.8, 24.6, 23.6, 22.7 (C-33), 22.2 (C-26), 19.3 (C-37), 14.2 (C-34); HR-ESIMS m/z: 922.5986 [M + H]+ (calcd for C49H84N3O13, 922.5999).
Preparation of Galactosylated Bullatacin 17
Galactosylated bullatacin derivative was synthesized using the same method as described for galactosylated squamocin derivatives 11–13. Briefly, bullatacin (93 mg, 0.15 mmol), 5-hexynoic acid (20 mg, 0.18 mmol) and DMAP (2 mg, 0.02 mmol) were dissolved in 3 mL dried CH2Cl2. DCC (62 mg, 0.3 mmol) was added and the mixture was stirred under N2 atmosphere at room temperature for 16 h (Scheme 4). The resulting precipitate (DCU) was removed by filtration and the filtrate was concentrated under reduced pressure. The residue was dissolved in EtOAc and sequentially washed with 0.5% aqueous HCl solution, saturated aqueous NaHCO3 solution and saturated aqueous NaCl. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude products were separated by Sephadex LH-20 gel permeation chromatography using MeOH to provide intermediate product 16A (93 mg).
The intermediate product 16A (93 mg) and β-
Characterization Data of Compound 17
A colorless waxy solid; [α]20D +20.3 (c 0.1, MeOH); 1H NMR (600 MHz, CDCl3): δ 7.83 (1H, s, H-6′), 7.20 (1H, m, H-35), 5.49 (1H, m H-7′), 5.03 (1H, m, H-36), 4.86 (1H, m, H-15), 4.28 (1H, m, H-8′), 4.10 (1H, m, H-10′), 3.99 (1H, m, H-16), 3.85 (3H, m, H-19, 20, 23), 3.81 (5H, m, H-4, 9′, 11′, 12′), 3.73 (1H, m, H-24), 2.70 (2H, m, H-4′), 2.48 (1H, d, H-3a), 2.40 (1H, d, H-3b), 2.36 (2H, m, H-2′), 1.88–1.99 (6H, m), 1.77 (2H, m), 1.58 (2H, m), 1.51 (2H, m), 1.43 (3H, m), 1.39 (3H, d, J = 6.7 Hz, H-37), 1.20–1.33 (33H, m), 0.86 (3H, t, J = 7.0 Hz, H-34); 13C NMR (150 MHz, CDCl3): δ 175.0 (C-1), 173.5 (C-2′), 152.3 (C-35), 147.1 (C-5′), 131.1 (C-2), 122.0 (C-6′), 88.4 (C-7′), 82.8 (C-19), 82.3 (C-20), 82.2 (C-23), 80.3 (C-16), 78.3 (C-11′), 77.9 (C-36), 74.1 (C-9′), 75.5 (C-15), 70.2 (C-8′), 70.0 (C-4), 71.7 (C-24), 69.0 (C-10′), 61.2 (C-12′), 37.5 (C-5), 33.8 (C-2′), 33.3 (C-3), 32.8 (C-25), 32.1 (C-32), 30.7 (C-14), 30.0, 29.8 (4C), 29.7 (2C), 29.6 (2C), 29.5 (2C), 28.8, 28.5, 28.2, 26.2, 25.7, 25.5, 24.9, 24.8, 24.5, 22.8 (C-33), 19.2 (C-37), 14.3 (C-34); HR-ESIMS m/z: 922.6013 [M + H]+ (calcd for C49H84N3O13, 922.5999).
Preparation of Glucosylated Bullatacin 18
Bullatacin (87 mg, 0.14 mmol), 5-hexynoic acid (22 mg, 0.21 mmol) and DMAP (2.4 mg, 0.02 mmol) were dissolved in 3 mL dried CH2Cl2. DCC (86 mg, 0.42 mmol) was added and the mixture was stirred under N2 atmosphere at room temperature for 16 h (Scheme 4). The crude product was purified in a similar way as described for the preparation of 16A to provide the intermediate product 16B (115 mg).
The intermediate product 16B (115 mg) and β-
Characterization Data of Compound 18
A yellowish waxy solid; [α]20D +4.5 (c 1.1, MeOH); 1H NMR (600 MHz, CDCl3): δ 7.75 (1H, s, H-6′), 7.15 (1H, m, H-35), 5.58 (1H, d, J = 7.9 Hz, H-7′), 5.06 (1H, m, H-36), 5.00 (1H, m, H-4), 4.01 (1H, m, H-8′), 3.94 (1H, m, H-19), 3.90 (1H, m, H-20), 3.83 (5H, m, H-16, 23, 24, 12′), 3.74 (2H, m, H-9′, 10′), 3.59 (1H, m, H-11′), 3.39 (1H, m, H-15), 2.63 (2H, m, H-4′), 2.55 (1H, m, H-3a), 2.47 (1H, m, H-3b), 2.31 (2H, m, H-2′), 1.95 (3H, m), 1.88 (2H, m), 1.78 (1H, m), 1.41–1.61 (6H, m), 1.43 (3H, m), 1.32 (3H, d, J = 6.7 Hz, H-37), 1.20–1.33 (33H, m), 0.88 (3H, t, J = 7.0 Hz, H-34); 13C NMR (150 MHz, CDCl3): δ 174.1 (C-1), 173.3 (C-2′), 152.2 (C-35), 147.0 (C-5′), 129.8 (C-2), 122.0 (C-6′), 87.8 (C-7′), 83.5 (C-16), 83.0 (C-20), 82.8 (C-19), 82.4 (C-23), 79.1 (C-11′), 78.1 (C-36), 76.9 (C-9′), 74.4 (C-15), 72.6 (C-8′), 72.3 (C-4), 71.6 (C-24), 69.1 (C-10′), 61.1 (C-12′), 34.1 (C-2′), 33.8 (C-14), 33.3 (C-5), 32.6 (C-25), 32.1 (C-32), 29.9, 29.8 (5C), 29.7 (2C), 29.6, 29.5 (3C), 29.1 (2C), 28.6, 26.3, 25.8, 25.4, 24.9, 24.6, 24.5, 22.9 (C-33), 19.1 (C-37), 14.3 (C-34); HR-ESIMS m/z: 922.6012 [M + H]+ (calcd for C49H84N3O13, 922.5999).
Aqueous Solubility of Squamocin and Galactosyl Derivative 13
HPLC method with refractive index detector was employed to measure the aqueous solubility of squamocin and its galactosyl derivative 13. Standard curve of compound 13 was found to be y=1.45*106x + 8.64*105 (R2 = 0.9970) by using six gradient concentrations (10, 5, 2.5, 1.25, 0.625, and 0.3125 mM) in methanol. Excess amount of squamocin/compound 13 was added to 1 mL of phosphate-buffered saline (PBS, pH = 7.0) and sonicated for 5 min to provide saturated solution. The mixture was centrifuged (12000 rpm) for 2 min and the supernatant was analyzed by HPLC for the content of squamocin/compound 13. The concentration of compound 13 was found to be 1.49 mM (1.37 mg/mL) in PBS (pH 7.0) at ambient temperature (around 28°C) while squamocin was not detected in PBS (pH 7.0).
Cell Culture
A549, HeLa and HepG2 cell lines were obtained from Kunming Cell Bank, Chinese Academy of Sciences. The cell lines were cultured in RPMI1640 (Invitrogen) cell culture medium supplemented with 10% FBS and 1% penicillin and streptomycin. All cell lines were maintained in a humidified atmosphere of 5% CO2 at 37°C.
Cell Viability Assay
A549, HeLa and HepG2 cells (5 × 104/well) were seeded in flat-bottomed 96 well microplates. Then, squamocin/bullatacin and glycosylated squamocin/bullatacin derivatives dissolved in DMSO (10, 5, 2.5, 1.25, 0.625, 0.3125 μM as concentration gradient) were added to wells, respectively. The cells were incubated for 72h at 37°C in CO2 gas incubator and then treated with 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (20 μL/well, 5 mg/mL). After another 4 h incubation, the medium was removed and 150 μL of DMSO was added to each well. Absorbance in each well, including the blanks, was measured at 570 nm in a microtiter plate reader after the samples were swirled gently. Experiments were repeated at least three times. Growth inhibition rate was calculated as follows: inhibition rate (%) =x{1 - [∆OD (compound) - ∆OD (blank)]/[∆OD (control) - ∆OD (blank)]}× 100%.
Results and Discussion
Synthesis and Structure Characterization
Queiroz et al reported the synthesis of several glycosyl derivatives of squamocin in which the squamocin scaffold was modified through either acetylation of the hydroxyl group(s) during the glycosylation reaction or reduction of the α,β-unsaturated γ-lactone moiety during the deprotection of benzyl groups on the sugar residue.29 We aim to prepare glycosylated acetogenin derivatives wherein the acetogenin scaffold remains intact. The three hydroxyl groups in squamocin and bullatacin (Figure 1) play less important roles in their anticancer activity13,28 and sugar residue(s) may be attached to these hydroxyl groups without affecting their activities much.
We first tried the trichloroacetimidate as the glycosylation donor. Galactose trichloroacetimidate (3) was readily prepared from D-galactose in three steps (Scheme 1) according to literature procedure.30 Treatment of squamocin with imidate 3 in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) as the catalyst resulted in complicated product mixture comprising of mono- and di-glycosylated products (as indicated by mass spectrometry data). The glycosylation yield was also found to be very low partly due to the decomposition of the imidate. The glycosylated product was isolated and treated with Et3N-MeOH-H2O (1:8:1, v/v) to remove the acetyl groups on the sugar.31 Unfortunately, the γ-lactone ring of squamocin was not stable to allow for the complete removal of all acetyl groups. In the end, we managed to obtain two mono-glycosylated products 4 (glycosylated at C-15) and 5 (glycosylated at C-28) after purification by HPLC, albeit at only 1% yield. Due to the complications associated with glycosylation reaction and the removal of the acetyl protection groups on the sugar, alternative methods of attaching sugars to acetogenins are desirable.
The Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC) reaction, popularly known as the “click reaction”, is an efficient method to covalently link two molecular entities together and has been widely used to prepare various glycoconjugates32 and carbohydrate macrocycles alike.33 Here we use the CuAAC reaction to conjugate glucose or galactose with squamocin/bullatacin through an alkyne-functionalized linker. Thus, β-
Typically, reaction products were purified by silica gel chromatography and preparative HPLC. All products were structurally confirmed by the analysis of 1D (1H, 13C and DEPT) and 2D (COSY, HSQC and HMBC) NMR spectra, and HR-ESIMS data. The selected 1H and 13C chemical shifts of the glycosylated derivatives were listed in Tables 1 and 2, respectively, and compared with those of squamocin or bullatacin. The changes in 1H and 13C chemical shifts of these glycosylated derivatives with an ester linkage (11–15, 17 and 18) were similar with those observed for biotinylated acetogenins we reported earlier.28 When a secondary hydroxyl group was acylated, the chemical shift of the carbon (C-4, C-15, C-24, and C-28) bearing an oxygen atom moved downfield (Table 2). Up to 3.4–3.6 ppm downfield shift was observed for C-24 in compound 12 and compound 14 while the shift for C-15 was smaller (1.6 ppm) in compound 11. On the other hand, the chemical shifts of the neighboring carbons (C-5, C-14, C-16, C-23, C-25, C-27, and C-29) moved upfield (in a magnitude of 1.1–4.1 ppm) upon acylation of the hydroxyl group. When a hydroxyl group was glycosylated, the carbon bearing that hydroxyl group exhibited a significant downfield shift (7.1 ppm for C-15 in 4 and 9.8 ppm for C-28 in 5). Similarly, upfield shifts (1.9–3.2 ppm) for the neighboring carbons (C-14, C-16, C-27, and C-29) were observed as a result of direct glycosylation of that hydroxyl group (Table 2). In the case of 1H NMR data, a significant downfield shift (>1.5 ppm) was observed for the proton directly attached to the carbon bearing the hydroxyl group that was acylated while a smaller downfield shift (0.1–0.7 ppm) resulted from O-galactosylation of that hydroxyl group (Table 1). The 1H and 13C NMR spectra of all synthesized glycosyl derivatives of squamocin and bullatacin (4, 5, 11–15, 17 and 18) are provided in the Supplementary Material of this article.
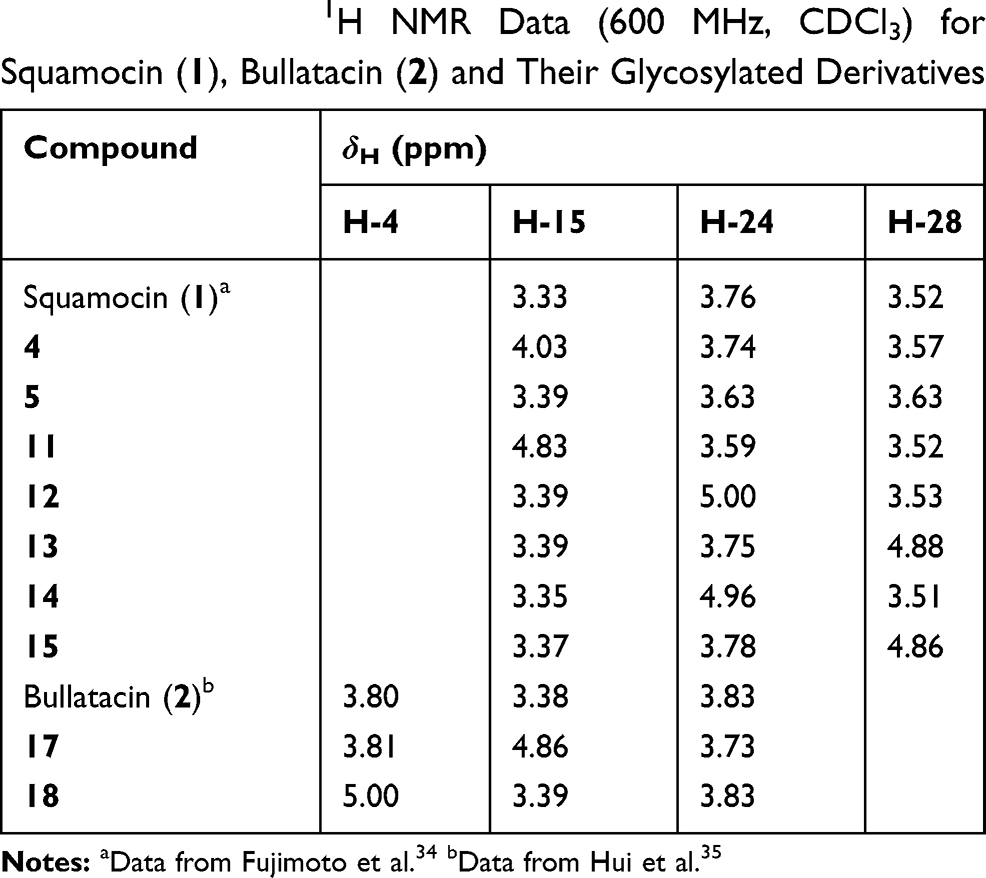 |
Table 1 Selected 1H NMR Data (600 MHz, CDCl3) for Squamocin (1), Bullatacin (2) and Their Glycosylated Derivatives |
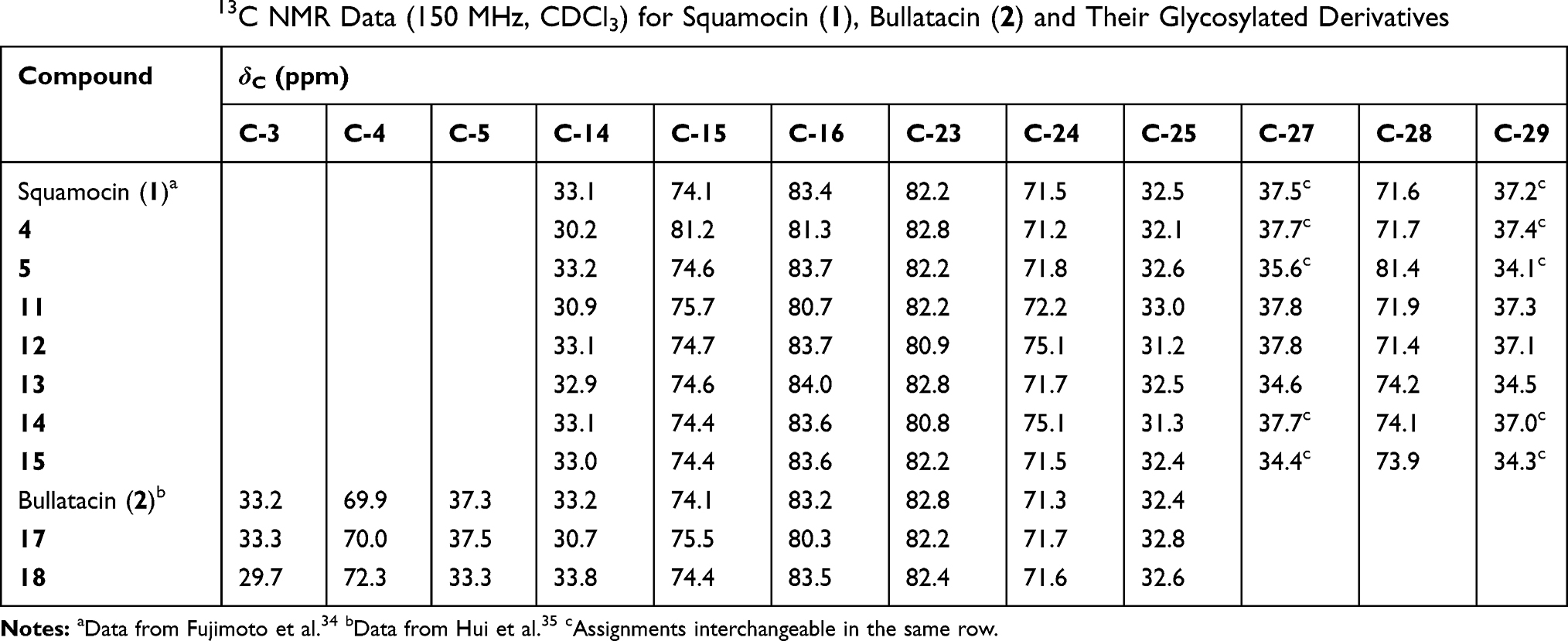 |
Table 2 Selected 13C NMR Data (150 MHz, CDCl3) for Squamocin (1), Bullatacin (2) and Their Glycosylated Derivatives |
Solubility
Squamocin and bulattacin are poorly soluble in water, which is a major drawback in the further exploitation of their potential uses. The attachment of a sugar residue is expected to increase their water solubility. Squamocin derivative 13 bearing a galactose residue was suspended in the phosphate buffer saline (PBS, pH = 7.0) and the mixture sonicated for 5 min. The supernatant was analyzed by HPLC for the content of 13. The solubility of 13 in PBS was found to be 1.37 mg/mL (1.49 mM) at ambient temperature (around 28°C). On the other hand, the solubility of squamocin in PBS was close to zero because no squamocin was detected in the supernatant of the PBS saturated with squamocin (Table 3). The data clearly showed that the attachment of one sugar residue to squamocin significantly improved its solubility in water.
 |
Table 3 Solubility of Squamocin and Its Derivative 13 |
Biological Evaluation
The cytotoxicity of the synthesized glycoconjugate derivatives against three tumor cell lines (Table 4) were evaluated using MTT assay. The IC50 values of these compounds were calculated based on three parallel experiments and presented in Table 4. Overall, when compared to squamocin (1) and bullatacin (2), the majority of these derivatives show similar level of anticancer activity against all three tumor cell lines with their IC50 values in µM range. Compound 15 bearing a glucose residue is slightly more active than squamocin. A few derivatives (eg, 11, 17 and 18) show up to 8–10 times higher IC50 than squamocin or bullatacin against certain cell lines (HeLa or A549), suggesting that these compounds exhibit some selectivity toward certain type of cancer cells. The data also suggest that all glycosyl derivatives either via glycosidic linkage (4 and 5) or triazolyl-ester linkage retain the anticancer activity. Furthermore, the position at which the glycosyl residue is attached does not affect their potency much, eg, compounds 4 and 5 having similar level of potency. The glucosyl derivatives (14, 15 and 18) are similarly active as the galactosyl derivatives (12, 13 and 17).
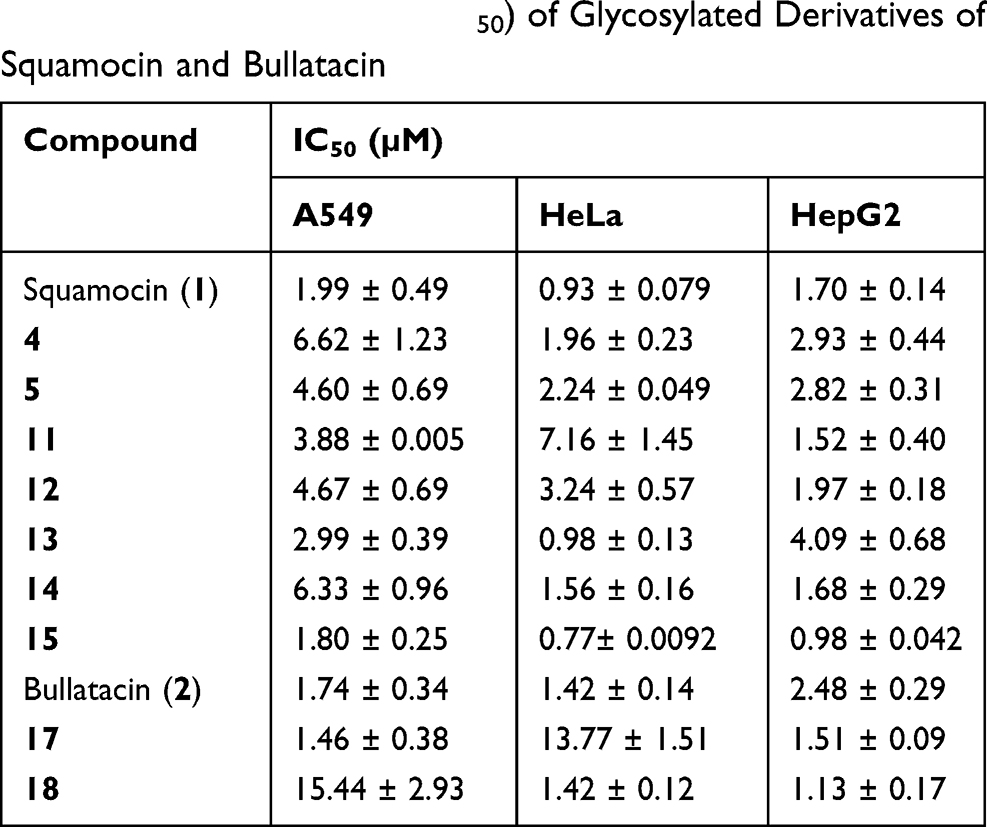 |
Table 4 Cytotoxic Activity (IC50) of Glycosylated Derivatives of Squamocin and Bullatacin |
Numerous studies have been conducted to elucidate the mechanism of action of Annonaceous acetogenins (ACGs) for their potent cytotoxic activity. ACGs are potent inhibitors of mitochondrial complex I in the electron transport system, with squamocin having the lowest IC50 value reported for this class of compounds.36 They are also powerful inhibitors of the NADH oxidases peculiar to the plasma membranes of cancer cells.37 Acetogenins cause cell cycle arrest at different phases in different cancer cells and are strong apoptosis inducers. Squamocin arrested T24 bladder cancer cells at the G1 phase, enhanced caspase-3 activity, cleaved the functional protein of PARP and caused cell apoptosis.38 Squamocin also inhibited the proliferation of chronic myeloid leukemia (K562) cells via G2/M arrest in association with the induction of p21, p27 and the reduction of Cdk1 and Cdc25C kinase activities.39 Three other THF-containing acetogenins (squamostatin A, squamocin M and corossolone) also showed potent antiproliferative activity against human nasopharyngeal carcinoma (NPC) cell lines and induced G2/M phase arrest, mitochondrial damage and apoptosis, and increased cytosolic and mitochondrial Ca2+ in NPCs.40 Furthermore, Dzhemilev et al recently showed that muricadienin, a linear acetogenin containing a 1Z,5Z-diene unit, induced apoptosis in the HEK293 kidney cancer cells and exhibited a moderate inhibitory activity against topoisomerases I and IIα.41 ACGs are versatile anticancer agents causing tumor cell death by different mechanisms. For these glycosylated acetogenins described here, more studies are required to demonstrate their cancer selectivity and mechanism(s) of action.
In conclusion, we have prepared nine glycosyl derivatives of squamocin and bullatacin and evaluated their growth inhibitory activity with three cancer cell lines, including A549, HeLa and HepG2. Most squamocin/bullatacin glycoconjugates show similar in vitro cytotoxicity against the tested cell lines as squamocin and bullatacin, respectively. Among the synthesized compounds, compound 15 displays slightly higher activity than squamocin while some other derivatives show up to 8–10 times lower activity in A549 or HeLa cell line than squamocin and bullatacin, respectively. The type of sugar residue (glucose or galactose), the position of attachment, and whether or not a linking spacer is present do not seem to affect the potency much. The solubility of galactosylated squamocin 13 in phosphate buffer saline (PBS, pH = 7) at ambient temperature is greatly improved (1.37 mg/mL) in comparison to squamocin (not detectable). Further studies are needed to show whether or not these compounds exhibit reduced toxicity to normal cells, the mechanism of action, and their therapeutic potential for cancer.
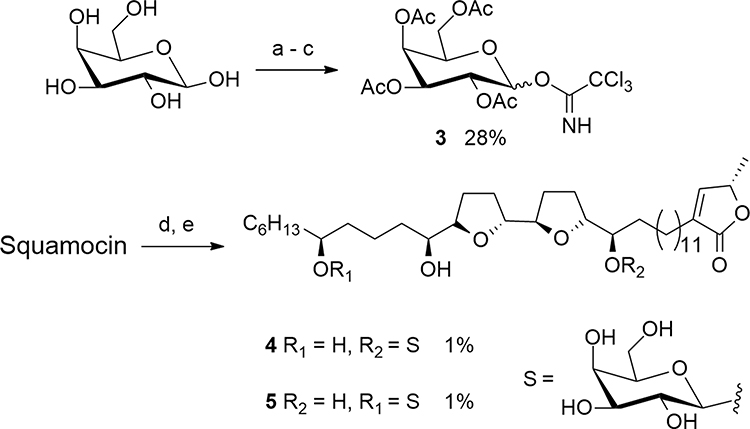 |
Scheme 1 Synthesis of galactosyl squamocin 4 and 5. Reagents and conditions: (a) Ac2O, pyridine, rt, N2, 16 h, 100%. (b) hydrazine acetate, DMF, 40 °C, 4 h, 91%. (c) trichloroacetonitrile, DBU, 0°C, 7 h, 31%. (d) 3, TMSOTf, CH2Cl2, rt, 16 h. (e) Et3N:MeOH:H2O (1:8:1, v/v), 35°C, 30 h. |
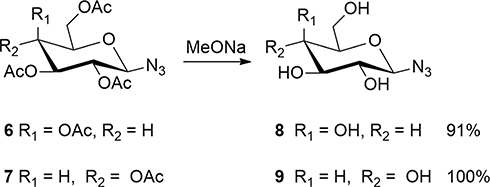 |
Scheme 2 Synthesis of glycosyl azides 8 and 9. |
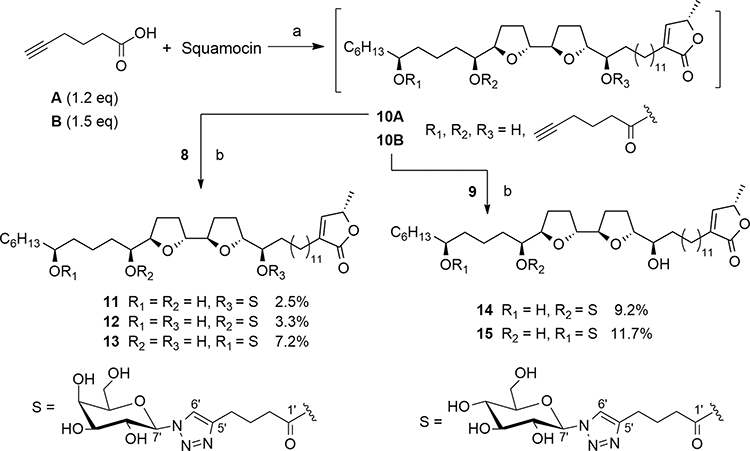 |
Scheme 3 Synthesis of 11–15 by click chemistry. Reagents and conditions: (a) DCC, DMAP, CH2Cl2, rt, N2, 16 h. (b) CuSO4·5H2O, sodium ascorbate, MeOH/H2O, 50 °C, 16 h. |
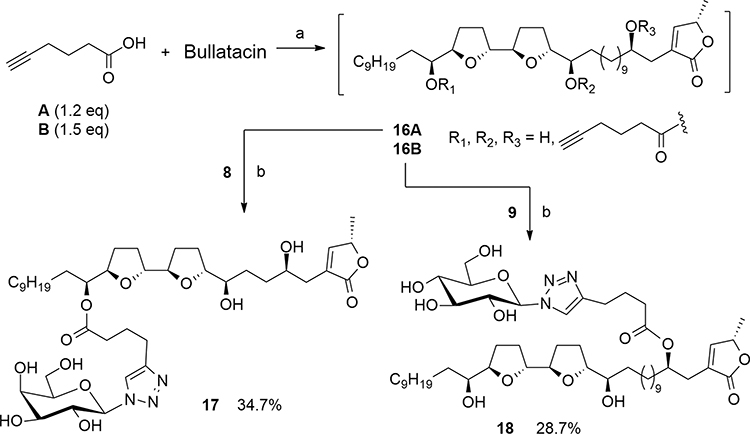 |
Scheme 4 Synthesis of 17 and 18 by click chemistry. Reagents and conditions: (a) DCC, DMAP, CH2Cl2, rt, N2, 16 h. (b) CuSO4·5H2O, sodium ascorbate, MeOH/H2O, 50 °C, 16 h. |
Acknowledgments
We thank Mr. Yunfei Yuan, South China Botanical Garden, Chinese Academy of Sciences, for NMR spectroscopic measurements, and Ms. Aijun Sun, South China Sea Institute of Oceanology, Chinese Academy of Sciences, for HRESIMS measurements. This work was supported by an NSFC grant (grant no. 31470423).
Disclosure
The authors declare no conflicts of interest.
References
1. Chang FR, Wu YC. Novel cytotoxic annonaceous acetogenins from Annona muricata. J Nat Prod. 2001;64:925–931.
2. Kim GS, Zeng L, Alali F, et al. Two new mono-tetrahydrofuran ring acetogenins, annomuricin E and muricapentocin, from the leaves of Annona muricata. J Nat Prod. 1998;61:432–436.
3. Ko YM, Wu TY, Wu YC, Chang FR, Guh JY, Chuang LY. Annonacin induces cell cycle-dependent growth arrest and apoptosis in estrogen receptor- α-related pathways in MCF-7 cells. J Ethnopharmacol. 2011;137:1283–1290.
4. Liaw CC, Chang FR, Lin CY, et al. New cytotoxic monotetrahydrofuran annonaceous acetogenins from Annona muricata. J Nat Prod. 2002;65:470–475.
5. McLaughlin JL. Paw paw and cancer: annonaceous acetogenins from discovery to commercial products. J Nat Prod. 2008;71:1311–1321.
6. Zeng L, Ye Q, Oberlies NH, et al. Recent advances in annonaceous acetogenins. Nat Prod Rep. 1996;13:275–306.
7. Sun S, Liu J, Zhou N, Zhu W, Dou QP, Zhou K. Isolation of three new annonaceous acetogenins from Graviola fruit (Annona muricata) and their anti-proliferation on human prostate cancer cell PC-3. Bioorg Med Chem Lett. 2016;26:4382–4385.
8. Nakanishi Y, Chang FR, Liaw CC, Wu YC, Bastow KF, Lee KH. Acetogenins as selective inhibitors of the human ovarian 1A9 tumor cell line. J Med Chem. 2003;46:3185–3188.
9. Castillo-Sánchez LHC, Jiménez-Osornio JJ, Delgado-Herrera MA. Secondary metabolites of the annonaceae, solanaceae and meliaceae families used as biological control of insects, Trop. Subtrop Agroecosyst. 2010;12:445–462.
10. Coria-Téllez AV, Montalvo-Gónzalez E, Yahia EM, Obledo-Vázquez EN. Annona muricata: a comprehensive review on its traditional medicinal uses, phytochemicals, pharmacological activities, mechanisms of action and toxicity. Arabian J Chem. 2018;11:662–691.
11. Landolt JL, Ahammadsahib KI, Hollingworth RM, et al. Determination of structure-activity relationships of annonaceous acetogenins by inhibition of oxygen uptake in rat liver mitochondria. Chem Bio Interact. 1995;98:1–13.
12. Hopp DC, Alali FQ, Gu ZM, McLaughlin JL. Three new bioactive bis-adjacent THF-ring acetogenins from the bark of Annona squamosa. Bioorg Med Chem. 1998;6:569–575.
13. Oberlies NH, Chang CJ, McLaughlin JL. Structure-activity relationships of diverse annonaceous acetogenins against multidrug resistant human mammary adenocarcinoma (MCF-7/Adr) cells. J Med Chem. 1997;40:2102–2106.
14. Ahammadsahib KI, Hollingworth RM, McGovren JP, Hui YH, McLaughlin JL. Mode of action of bullatacin: a potent antitumor and pesticidal annonaceous acetogenin. Life Sci. 1993;53:1113–1120.
15. Hong J, Li Y, Xiao Y, et al. Annonaceous acetogenins (ACGs) nanosuspensions based on a self-assembly stabilizer and the significantly improved anti-tumor efficacy. Colloids Surf B. 2016;145:319–327.
16. Moradi SV, Hussein WM, Varamini P, Simerska P, Toth I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides, Chem. Sci. 2016;7:2492–2500.
17. Hwu JR, Hsu CI, Hsu MH, Liang YC, Huang RCC, Lee YC. Glycosylated nordihydroguaiaretic acids as anti-cancer agents. Bioorg Med Chem Lett. 2011;21:380–382.
18. Mandai T, Okumoto H, Oshitari T, et al. Synthesis and biological evaluation of water soluble taxoids bearing sugar moieties. Heterocycles. 2001;54:561–566.
19. Calvaresi EC, Hergenrother PJ. Glucose conjugation for the specific targeting and treatment of cancer. Chem Sci. 2013;4:2319–2333.
20. Li HP. Study on synthesis and biological activity of a galactosylated piperazinyl porphyrin. Bioorg Med, Chem Lett. 2006;16:6298–6301.
21. Augustin R. The protein family of glucose transport facilitators: it’s not only about glucose after all. IUBMB Life. 2011;62:315–333.
22. Carvalho KC, Cunha IW, Rocha RM, et al. GLUT1 expression in malignant tumors and its use as an immunodiagnostic marker. Clinics. 2011;66:965–972.
23. Thomas M, Clarhaut J, Strale PO, Tranoy-Opalinski I, Roche J, Papot S. A galactosidase-responsive “trojan horse” for the selective targeting of folate receptor-positive tumor cells. ChemMedChem. 2011;6:1006–1010.
24. Michelle DG, Pinedo HM, Razi Q, Haisma HJ, Epie B. Cytosolic beta-glycosidases for activation of glycoside prodrugs of daunorubicin, Biochem. Pharmacol. 2003;65:1875–1881.
25. Katsuhiko M, Katsuyoshi N, Koji H, et al. In vivo antitumor activity of novel water-soluble taxoids. Biol Pharm Bull. 2008;31:1155–1158.
26. Oliveri V, Giuffrida ML, Vecchio G, Aiello C, Viale M. Gluconjugates of 8-hydroxyquinolines as potential anti-cancer prodrugs. Dalton Trans. 2012;41:4530.
27. Kamal A, Tekumalla V, Krishnan A, Palbhadra M, Bhadra U. Development of pyrrolo[2,1-c][1,4]benzodiazepine β-galactoside prodrugs for selective therapy of cancer by ADEPT and PMT. ChemMedChem. 2010;3:794–802.
28. Shi JF, Wu P, Jiang ZH, Wei XY. Synthesis and tumor cell growth inhibitory activity of biotinylated annonaceous acetogenins. Eur J Med Chem. 2014;71:219–228.
29. Queiroz EF, Roblot F, Duret P, et al. Synthesis, spectroscopy and cytotoxicity of glycosylated acetogenin derivatives as promising molecules for cancer therapy, J. Med Chem. 2000;8:1604–1610.
30. Roussel F, Knerr L, Grathwohl M, Schmidt RR. O-Glycosyl trichloroacetimidates bearing Fmoc as temporary hydroxy protecting group: a new access to solid-phase oligosaccharide synthesis. Org Lett. 2000;2:3043–3046.
31. Duret P, Figadère B, Hocquemiller R, Cavé A. Epimerization of annonaceous acetogenins under basic conditions. Tetrahedron Lett. 1997;38:8849–8852.
32. Dwivedi P, Mishra KB, Mishra BB, Tiwari VK. Click inspired synthesis of triazole-linked vanillin glycoconjugates. Glycoconjugate J. 2017;34:61–70.
33. Lewandowski B, Jarosz S. Application of 1′,2,3,3′,4,4′-hexa-O-benzylsucrose in the preparation of sucrose macrocycles via a click chemistry route: regioselectivity study. Synth Commun. 2015;43:2161–2168.
34. Fujimoto Y, Eguchi T, Kakinuma K, Ikekawa N, Sahai M, Gupta YK. Squamocin, a new cytotoxic bis-tetrahydrofuran containing acetogenin from Annona squamosa. Chem Pharm Bull (Tokyo). 1988;36:4802–4806.
35. Hui YH, Rupprecht JK, Liu YM, et al. Bullatacin and bullatacinone: two highly potent bioactive acetogenins from Annona bullata. J Nat Prod. 1989;52:463–477.
36. Qayed WS, Aboraia AS, Abdel-Rahman HM, Youssef AF. Annonaceous acetogenins as a new anticancer agent. Pharma Chem. 2015;7:24–35.
37. Alali FQ, Liu XX, McLaughlin JL. Annonaceous acetogenins: recent progress. J Nat Prod. 1999;62:504–540.
38. Yuan SSF, Chang HL, Chen HW, et al. Selective cytotoxicity of squamocin on T24 bladder cancer cells at the S-phase via a Bax-, Bad-, and caspase-3-related pathways. Life Sci. 2006;78:869–874.
39. Lu MC, Yang SH, Hwang SL, et al. Induction of G2/M phase arrest by squamocin in chronic myeloid leukemia (K562) cells. Life Sci. 2006;78:2378–2383.
40. Juang SH, Chiang CY, Liang FP, et al. Mechanistic study of tetrahydrofuran-acetogenins in triggering endoplasmic reticulum stress response-apotoposis in human nasopharyngeal carcinoma. Sci Rep. 2016;6:39251. doi:10.1038/srep39251
41. Dzhemilev UM, D’Yakonov VA, Tuktarova RA, et al. Short route to the total synthesis of natural muricadienin and investigation of its cytotoxic properties. J Nat Prod. 2016;79:2039–2044.
42. Xie HH, Wei XY, Wang JD, Liu MF, Yang RZ. A new cytotoxic acetogenin from the seeds of Annona bullata. Chin Chem Lett. 2003;14:588–590.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.