")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Synthesis and characterization of different immunogenic viral nanoconstructs from rotavirus VP6 inner capsid protein
Authors Bugli F, Caprettini V, Cacaci M, Martini C, Paroni Sterbini F, Torelli R, Della Longa S, Papi M, Palmieri V, Giardina B, Posteraro B, Sanguinetti M, Arcovito A
Received 3 January 2014
Accepted for publication 15 February 2014
Published 30 May 2014 Volume 2014:9(1) Pages 2727—2739
DOI https://doi.org/10.2147/IJN.S60014
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Francesca Bugli,1 Valeria Caprettini,2 Margherita Cacaci,1 Cecilia Martini,1 Francesco Paroni Sterbini,1 Riccardo Torelli,1 Stefano Della Longa,3 Massimiliano Papi,4 Valentina Palmieri,4 Bruno Giardina,5 Brunella Posteraro,1 Maurizio Sanguinetti,1 Alessandro Arcovito5
1Istituto di Microbiologia, Università Cattolica del Sacro Cuore, 2Dipartimento di Fisica, Sapienza Università di Roma, Rome, 3Dipartimento di Medicina Clinica, Sanità Pubblica, Scienze della Vita e dell’Ambiente, Università dell’Aquila, L’Aquila, 4Istituto di Fisica, 5Istituto di Biochimica e Biochimica Clinica, Università Cattolica del Sacro Cuore, Rome, Italy
Abstract: In order to deliver low-cost viral capsomeres from a large amount of soluble viral VP6 protein from human rotavirus, we developed and optimized a biotechnological platform in Escherichia coli. Specifically, three different expression protocols were compared, differing in their genetic constructs, ie, a simple native histidine-tagged VP6 sequence, VP6 fused to thioredoxin, and VP6 obtained with the newly described small ubiquitin-like modifier (SUMO) fusion system. Our results demonstrate that the histidine-tagged protein does not escape the accumulation in the inclusion bodies, and that SUMO is largely superior to the thioredoxin-fusion tag in enhancing the expression and solubility of VP6 protein. Moreover, the VP6 protein produced according to the SUMO fusion tag displays well-known assembly properties, as observed in both transmission electron microscopy and atomic force microscopy images, giving rise to either VP6 trimers, 60 nm spherical virus-like particles, or nanotubes a few micron long. This different quaternary organization of VP6 shows a higher level of immunogenicity for the elongated structures with respect to the spheres or the protein trimers. Therefore, the expression and purification strategy presented here – providing a large amount of the viral capsid protein in the native form with relatively simple, rapid, and economical procedures – opens a new route toward large-scale production of a more efficient antigenic compound to be used as a vaccination tool or as an adjuvant, as well as represents a top-quality biomaterial to be further modified for biotechnological purposes.
Keywords: Virus-like particles, protein-based nanotubes, SUMO fusion tag, human rotavirus vaccine
Introduction
Virus-like particles (VLPs) or nanotubes (NTs) are currently produced from a wide variety of viruses as self-assembled protein oligomers whose structure and physicochemical properties can be tuned by genetic engineering or chemical modifications, in order to satisfy a rapidly increasing number of application areas, including vaccine production, drug delivery by protein scaffolds, diagnostics, and gene therapy.1–3 In this context, there is great interest in the development of simpler and more efficient protocols for the expression and purification of self-assembling protein candidates.
Rotaviruses are the most common etiologic agents of severe diarrhea in infants and young children worldwide, and responsible for 400,000 deaths worldwide in children younger than 5 years.4 VP6 is a unique structural protein of the middle of three capsid layers of the rotavirus (see Figure 1). It contains 397 amino acids, and has a theoretical molecular mass of 45 kDa. VP6, which is the most conserved, immunogenic, and abundant rotavirus protein, is extremely stable, and spontaneously forms trimers that, depending on pH and ionic strength, can self-assemble to form nanostructures, such as spheres or NTs.5 Many rotavirus strains are characterized by conserved VP6 epitopes, and thus this antigen is considered the major diagnostic marker for acute rotavirus gastroenteritis.6
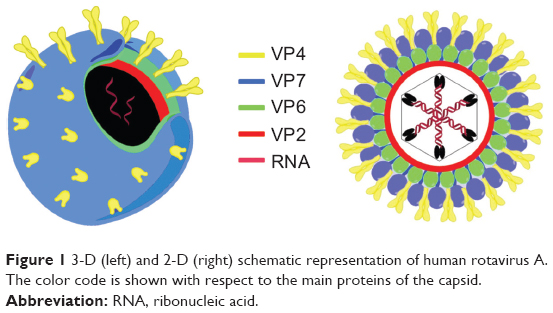 | Figure 1 3-D (left) and 2-D (right) schematic representation of human rotavirus A. The color code is shown with respect to the main proteins of the capsid. |
Moreover, despite the introduction of a live attenuated monovalent rotavirus vaccine in Mexico in 2006–2007,7 other authors have focused on the development of a subunit-based vaccine, and in this regard, VP6 has shown the optimal characteristics to be a potential candidate.8 Indeed, VLPs are a safe and efficacious class of vaccine, with approved members available for hepatitis B (based on the Australia antigen) and cervical cancer.9,10
Although the present demand for VLPs is primarily as products for vaccination, other applications, including diagnostics, biomedicine, material science, and nanotechnology, are increasingly attracting general scientific interest.11 Independently of their final purpose, VLPs are obtained when viral structural proteins are produced in heterologous expression systems in the absence of viral genetic material, and rotavirus-like particles have been frequently expressed in insect cell baculovirus expression.12 This system has proven to be fruitful and versatile. However, insect cell culture requires expensive media, and baculovirus manipulation requires highly skilled personnel.
To overcome this shortcoming, Zhao et al13 expressed VP6 from rotavirus A in Escherichia coli in the form of an inclusion body; however, as the inclusion bodies needed to be denatured and the purified protein refolded, the proposed strategy was complex, time-consuming, and weakly reproducible.
Several technological advances have widely improved recombinant protein expression in E. coli, including the development of strong promoters14 and the use of protein tags or removable protein-fusion domains.15 The fusion systems are characterized by their ability to enhance protein expression, due to a combination of different favorable effects, such as reducing proteolytic degradation of the recombinant protein and/or improving protein folding and solubility to facilitate purification and detection. A variety of partner structures have been used as fusion motifs, including maltose-binding protein, glutathione S-transferase, and thioredoxin (TRX).16–18 However, predicting which fusion tag will enhance the solubility of any protein in E. coli remains empirical.
Recently, a novel expression system using a small ubiquitin related-modifier (SUMO) as a fusion partner that facilitates efficient expression of recombinant proteins in E. coli has been developed.19,20 The SUMO fusion tag has enhanced the expression and solubility of several proteins, including severe acute respiratory syndrome coronavirus 3CL protease, nucleocapsids, and membrane proteins. Moreover, a hexahistidine SUMO fusion construct has been shown to enhance expression and facilitate purification by means of Nickel-nitrilotriacetic acid (Ni+2-NTA) chromatography.21
Traditional gene-fusion systems require engineered cleavage sites, which are recognized by the proteases and are positioned between the fusion tag and the protein target. Cleavage by traditional proteases such as factor Xa or thrombin protease22 results in the generation of nonnative N-terminal sequences, due to the retention of several amino acids downstream from the cleavage site required for protease recognition.
Many structural and therapeutic proteins require specific N-terminal amino acids for biological activity, half-life, or structural stability. In this respect, releasing the target polypeptide from the fusion protein without N-terminal extension could be essential for viral structural proteins, such as VP6, that are able to self-assemble forming VLPs. The SUMO-associated protease 1 recognizes the tertiary structure of SUMO, and it is able to cleave a variety of fusion partners with remarkable fidelity, allowing the production of target proteins with a native N-terminal sequence.19,20,23
Taking into account the diverse favorable features of the SUMO-expression system, the aim of this study was to develop a platform for the production of VLPs from E. coli by the expression of the human rotavirus VP6 protein with a SUMO fusion system; this goal was preliminary to the following step, which was the purification of a large amount of the protein in its native form, and at the same time providing a comparison of SUMO with other fusion systems to determine whether it truly represents an advancement.
Materials and methods
Virus
Human rotavirus A ribonucleic acid (RNA) was extracted from a clinical stool specimen obtained from the Institute of Microbiology of the Catholic University of Rome from a 5-year-old patient suffering from acute diarrhea. The diarrheal stool sample was positive for the rotavirus antigen by rapid immunochromatographic assay for the detection of rotavirus and adenovirus antigens in stool specimens (Rapid Strip Rota/Adeno; Meridian Bioscience, Cincinnati, OH, USA).
Viral RNA extraction and VP6 cloning
Viral RNA was isolated using a commercial kit (QIAamp viral RNA minikit; Qiagen, Venlo, Netherlands) and the manufacturer’s recommended protocol adjustment for fecal samples. Briefly, this method involved centrifugation of samples at 4,000 g for 30 minutes before mixing supernatant with the lysis buffer. The extracted RNA was denatured at 97°C for 5 minutes. A reverse-transcription polymerase chain reaction (RT-PCR) was carried out using the Qiagen OneStep RT-PCR kit as previously described by Ushijima et al.24 A 1,194-base pair fragment of the VP6 gene was amplified with the forward primer VP6-for (5′-ATG GAG GTT CTG TAT TCA TTG TCA-3′; nucleotides 1–24) and the reverse primer VP6-rev (5′-TCA CTT AAT CAA CAT GCT TCT GAT-3′; nucleotides 1,170–1,194). The reaction was carried out with an initial RT step at 45°C for 30 minutes, followed by PCR activation at 95°C for 15 minutes, 35 cycles of amplification (60 seconds at 94°C, 60 seconds at 54°C, and 1 minute, 30 seconds at 72°C), and a final extension of 7 minutes at 72°C in a GeneAmp® PCR System 9700 thermal cycler (Thermo Fisher Scientific, Waltham, MA, USA). PCR products were run on agarose gel, stained with ethidium bromide, and visualized under ultraviolet light. After purification of the PCR product from the agarose gel, the VP6-coding region was ligated with the Zero Blunt® PCR kit (Invitrogen), and the complete nucleotide sequence was determined using the ABI Prism 3130 xl genetic analyzer (Thermo Fisher Scientific).
Escherichia coli cell strains used for cloning and expressing recombinant VP6
The E. coli strains Top-10 and BL21(DE3) (Thermo Fisher Scientific) were used as host cells in subcloning and VP6 expression, respectively. The recombinant plasmids were transformed into E. coli cells using standard methods.25 Following transformation, cells were grown on nutrient agar plates containing antibiotics (34 μg/mL chloramphenicol for BL21(DE3). Single antibiotic-resistant recombinant colonies were selected for growth-kinetic studies and protein expression.
Plasmids used in this work
The Zero Blunt PCR vector was obtained from Thermo Fisher Scientific, the pET15b vector was obtained from EMD Millipore (Billerica, MA, USA), the pHis-Trx vector was kindly provided by Professor Andrea Brancaccio from the Consiglio Nazionale delle Richerche, and pET-SUMO was obtained from Thermo Fisher Scientific. Other chemical reagents were of analytical grade.
Construction of pET15-b, pTRX, and pET-SUMO plasmids encoding recombinant VP6
The coding region of VP6 was amplified by PCR using as template the Zero Blunt VP6 construct previously obtained. Primers used for cloning the VP6 open-reading-frame sequence into pET15-b, pTRX, and pET-SUMO expression vectors are reported in Table 1. Restriction sites introduced at the initiation codon in the forward primers and just after the stop codon in the reverse primers are underlined. Primers were used for VP6 amplification with HiFi deoxyribonucleic acid (DNA) polymerase (Kapa Biosystems, Wilmington, MA, USA), following PCR conditions suggested by the manufacturer. The PCR product was digested with appropriate restriction enzymes (New England Biolabs, Ipswich, MA, USA), separated on 1% agarose gel and extracted from the gel using a Qiaex extraction kit (Qiagen). The pET15-b and pTRX vectors were digested with corresponding restriction enzymes and purified from a 1% agarose gel. The digested PCR fragment of VP6 was ligated into the linearized pET15-b and pTRX expression vectors using T4 DNA ligase (New England Biolabs). The resulting vectors, named pET15b-VP6 and pTRX-VP6, were confirmed by restriction analyses and introduced in E. coli BL21(DE3) cells by electroporation. The PCR product of VP6 was cloned into a pET-SUMO expression vector using the TA cloning technique (Invitrogen) following the manufacturer’s suggested protocols.
 | Table 1 Primers used in this study |
VP6 expression
E. coli BL21(DE3) transformants were inoculated in 5 mL of Luria–Bertani (LB) medium supplemented with appropriate antibiotics and cultivated overnight at 37°C. This preculture was used to inoculate (1:100) a 100 mL LB medium, which was cultivated at 37°C until reaching a 600 nm optical density (OD) of 0.6–0.8. Then, an aliquot of 10 mL was collected as an uninduced sample, and isopropyl β-D-1-thiogalactopyranoside (IPTG) was added at a final concentration of 0.5 mM.
After the addition of IPTG, each culture was incubated at 25°C overnight. Cells were pelleted by centrifugation at 5,000 g at 4°C for 15 minutes, the culture medium was discarded and cells resuspended in lysis buffer (300 mM NaCl, 20 mM K-phosphate buffer pH 7.8, 0.05% Triton X, 2 mM dithiothreitol, 5% glycerol and protease inhibitors). Lysozyme was added at a final concentration of 1 mg/mL, and samples were incubated in ice for 30 minutes. The homogenates were sonicated four times for 60 seconds, with 30 seconds’ interval between each sonication, and then centrifuged at 14,000 rpm to separate supernatants and pellets.
Total proteins in bacteria lysate were applied to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the expression yield was analyzed using Quantity One® quantitation software (Thermo Fisher Scientific) according to the relative band intensities of Coomassie blue stain.
Purification of recombinant SUMO-VP6 protein
BL21(DE3)-pET-SUMO/VP6 strains were cultured in 250 mL sterilized LB medium with vigorous shaking (220 rpm) at 37°C to an OD measured at 600 nm (A600) of 0.6. IPTG was then added to a final concentration of 0.25 mM to induce the expression of the recombinant protein at 25°C for 12 hours. Cultures were collected by centrifugation at 5,000 g at 4°C for 20 minutes, and the cell pellet frozen at -80°C. Whole-cell lysates were prepared according to the protocol previously described, except that 10 mM imidazole was added to lysis buffer to prevent nonspecific association of bacterial proteins with Ni2+-NTA resins (Qiagen). All buffers used in the purification were prepared using lipopolysaccharide (LPS)-free distilled water and included the use of Triton X-114 at a concentration of 0.05% during the middle washing steps. Surfactant Triton X-114 was successfully used to reduce endotoxins during histidine (his) fusion-protein purification.26 After centrifugation at 14,000 rpm for 30 minutes, the bacterial soluble lysate was filtered at 0.22 μm and applied to the column at a flow rate of 1 mL/minute to capture the His-SUMO-VP6 fusion proteins. The resin was then washed three times with 20 mL of wash buffer (lysis buffer plus 25 mM imidazole) and twice with 20 mL of the same buffer containing 50 mM imidazole. The fusion protein was eluted with a 10-column volume of elution buffer (lysis buffer plus 300 mM imidazole). The peak fractions with high OD ultraviolet values at 280 nm, which were detected by LP Data View (Bio-Rad Laboratories, Hercules, CA, USA) and contained the fusion protein, were analyzed on 10% SDS-PAGE and stained with Coomassie blue. A Pierce Limulus amebocyte lysate chromogenic endotoxin quantitation kit (Thermo Fisher Scientific) was used following the manufacturer’s instructions to determine the amount of LPS in purified VP6 preparation. Elution fractions with high SUMO-VP6 concentration were pooled and dialyzed overnight at 4°C against SUMO protease cleavage buffer: 20 mM Tris HCl, 150 mM NaCl, 1 mM dithiothreitol (pH 7.5).
Cleavage of the fusion protein and purification of released VP6
The SUMO-VP6 protein (200 μg) was reacted with 20 U of SUMO protease at 30°C for 6 hours. Since both SUMO and SUMO protease (Thermo Fisher Scientific) had 6x His tags, but VP6 did not, the cleaved SUMO-VP6 samples could be reapplied to the nickel column to obtain the purified VP6 by subtracting the 6x His-tagged proteins. Briefly, after the SUMO-VP6 was cleaved by the SUMO protease, the sample was loaded onto a nickel column with Ni-NTA resin. Most of the VP6 without 6x His tags was eluted in the flow-through (unbound) fractions, and the rest was recovered by washing the resin with lysis buffer. The purified protein was checked by SDS-PAGE, and the samples were stored at -80°C for further assay.
Production of polyclonal antiserum and immunoblot analysis
Female BALB/c mice (6–8 weeks old; 25–30 g) were housed in a filter-top cage with free access to food and water. Mice were maintained in our Unit for Laboratory Animal Medicine according to protocols that had been approved by an institutional animal-use committee.
Three female BALB/c mice were immunized in total, with recombinant purified VP6 dialyzed against phosphate-buffered saline (PBS). Animals were initially immunized subcutaneously with 25 μg of Ag in Freund’s complete adjuvant (final volume =200 μL [100:100 Freund’s complete adjuvant:VP6 in PBS]). Boosts with the same amount of Ag/adjuvant and the same procedure were given at 3-weekly intervals. Three weeks after the last immunization, mice were intraperitoneally injected with 10 μg of antigen in the presence of incomplete Freund’s adjuvant. Animals were bled 2 weeks after each boost, and sera from all bleedings were assayed for anti-VP6 antibody titer by enzyme-linked immunosorbent assay (ELISA) and immunoblot.
Western blot analysis of VP6 protein
Recombinant SUMO-VP6 fusion protein purified from the Ni2+-NTA column was electrophoresed on a 10% SDS-PAGE gel. The purity of the column-purified recombinant SUMO-VP6 and the specificity of the polyclonal antiserum were determined by immunoblot analysis of the protein.
The SDS-PAGE gel was electrotransferred to nitrocellulose membranes for 1 hour at 100 mV using a Bio-Rad Trans-Blot® apparatus. The nitrocellulose membranes were incubated with blocking solution (1× PBS, 5% fat-free milk powder, and 0.3% Tween 20) for 1 hour at room temperature and then incubated with either the anti-VP6 antiserum (1:2,500 dilution) or the mouse anti-His monoclonal antibody (Qiagen) in blocking solution for a further 2 hours. Preimmune serum collected from the same mouse was used as a control (data not shown). The membrane was washed three times with blocking solution and incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse immunoglobulin (Ig)-G antibody (Sigma-Aldrich, St Louis, MO, USA) for 1 hour. After the membrane had been washed three times with blocking solution and twice with Tris-buffered saline (50 mM Tris HCl, 150 mM NaCl, pH 7.5), antibodies were detected by adding to the membrane SuperSignal™ West Dura extended-duration substrate (Thermo Fisher Scientific). Results of all Western blot experiments were analyzed using a Bio-Rad ChemiDoc™ imaging system.
Animal immunization and sample collection
Native VP6 recombinant protein, VP6-VLPs, and VP6-NTs were separately suspended in PBS and scattered by water-bath sonication immediately prior to immunization. Doses were accurately adjusted to deliver 10 μg/mouse of each formulation. Female BALB/c mice (four mice/experimental group) were injected subcutaneously with complete Freund adjuvant on day 0 and boosted on days 21 and 42 with the same dose of antigen used during prime immunization. Blood samples were collected from the tail vein at study weeks 0 (preimmunization bleed), 3, and 6. Sera from mice receiving no antigen (naïve mice) were used as a negative control. Tail blood samples were incubated at 37°C for 20 minutes, centrifuged at 3500 g for 20 minutes, and the serum separated and stored at -20°C.
ELISA
Rotavirus VP6-specific antibodies were determined in sera by ELISA as described previously.27 Recombinant native VP6 in carbonate-bicarbonate buffer was used to coat flat-bottom, 96-well microtiter plates (Costar®; Sigma-Aldrich). After being blocked with 5% skim milk (Sigma-Aldrich) in PBS, the serum samples were diluted to 1:2,000 for IgG detection and 1:200 for IgA detection, with serial twofold dilutions up to 1:128,000 and 1:12,800, respectively, and were added to the plates and incubated for 2 hours at 37°C. All sera and secondary antibody dilutions were prepared in 1% skim milk +0.05% Tween 20 in PBS and added to the wells at a volume of 100 μL. During each step, plates were washed five times with 0.05% Tween 20 in PBS. For the detection of antigen-specific IgG and IgA response, HRP-labeled goat anti-mouse IgG or IgA (Sigma-Aldrich) was diluted 10,000-fold. The secondary antibody was incubated for 1 hour at 37°C, and o-phenylenediamine dihydrochloride (SigmaFast™ OPD; Sigma-Aldrich) was used as a substrate for HRP. After 30 minutes, the color reaction was stopped with 2 M sulfuric acid. Absorbance (OD) at a wavelength of 490 nm was measured in a microtiter plate reader (Bio-Rad Laboratories).
Transmission electron microscopy
Samples for transmission electron microscopy (TEM) were prepared by dialyzing the protein, with purification and concentration up to 250 mg/mL in 20 mM Tris HCl in the presence of 150 mM NaCl and 200 mM CaCl2 with a different buffer depending upon the desired pH and ionic strength. In the experiments reported in this work, we used 20 mM glycine HCl at pH 4.5 and 20 mM 2-(N-morpholino)ethanesulfonic acid at pH 6, always in the presence of 150 mM NaCl. All the samples were equilibrated for 24 hours at 4°C at their own pH. Subsequently, a small drop (10 μL) was dispersed on carbon-coated glow-discharged copper grids. The sample was allowed to adsorb on the carbon film for 5 minutes, and sample excess was removed with filter paper. After staining with a 2% uranyl acetate solution for 30 seconds in the dark at room temperature, staining excess was also removed with filter paper and the grid washed with ultrapure water. Grids were then air-dried for 1 hour before observation at room temperature. TEM micrographs were collected working with an acceleration voltage of 120 keV at a magnification of 30,000× (Libra 120; Carl Zeiss Meditec, Jena, Germany).
Atomic force microscopy
Atomic force microscopy (AFM) imaging was performed using a NanoWizard® II (JPK Instruments, Berlin, Germany) in the contact operation mode.28 The same samples that were characterized using TEM were also analyzed by means of AFM. No staining procedure was necessary in this case, so all samples were kept in an aqueous environment and at a controlled temperature (25°C) throughout the measurement procedure.29 This approach was aimed at minimizing perturbations and artifacts due to salt crystallization following dehydration that could hide or modify the real particle structure.30 The microscope probe consisted of an ultrasharp silicon nitride cantilever of nominal force constant k =0.1 N/m, with a tip radius of less than 10 nm (MikroMasch® NSC16; NanoWorld, Neuchâtel, Switzerland). Image analysis was performed using Gwyddion software (Czech Metrology Institute, Brno, Czech Republic). Samples were laid down on freshly cleaved mica disks (Ted Pella, Redding, CA, USA).
Dynamic light scattering
Dynamic light-scattering measurements on protein solutions were carried out with a Zetasizer Nano S (Malvern Instruments, Malvern, UK) equipped with a 4 mW He–Ne laser (633 nm). Measurements were performed at 25°C at an angle of 173°C from the incident beam. Peak-intensity analyses were used to determine hydrodynamic radius number distribution (Pn) in different buffer conditions.
Ethics statement
The mouse experiments were performed under a protocol approved by the Institutional Animal Use and Care Committee at the Univerità Cattolica del Sacro Cuore, Rome, Italy (permit AA 21, 13/12/2011) and authorized by the Italian Ministry of Health, according to Legislative Decree 116/92, which implemented European Directive 86/609/EEC on laboratory animal protection in Italy. Animal welfare was routinely checked by veterinarians of the Service for Animal Welfare.
The clinical stool sample used for viral RNA extraction had previously been used for diagnostic purposes and was disposed of in the following days. We submitted a brief information letter to the institutional review board (Ethics Committee, Università Cattolica del Sacro Cuore), which rapidly approved the use of this single stool sample not involved in a clinical trial but related to biotechnology purposes.
Results and discussion
Expression of VP6 fusion proteins
Many heterologous proteins expressed in E. coli fold improperly and aggregate, leading to the accumulation of inclusion bodies wherein the proteins remain essentially insoluble. Functional VP6 particles from human rotavirus A have been expressed in several eukaryotic vector systems, including animal, insect, and yeast cells.31,32 A recent study from Zhao et al reported the in vitro assembly into VLPs of the VP6 protein expressed in E. coli.13 According to their protocol, however, the VP6 protein accumulates in the bacterial cell as insoluble aggregates, and a complicated renaturation process is required to restore the native folding, which is competent to self-assemble in VLPs.
As demonstrated by the enormous progress in the optimization of the production of recombinant proteins, the expression level and solubility of proteins may be enhanced by the use of fusion partners that can be cleaved after production. In the present study, we compared the results of the expression in E. coli of VP6 fused to three different affinity tags: hexahistidine, TRX, and SUMO.
For all these tags, known to be successful for the expression of difficult targets, we constructed N-terminal fusions that could be separated from VP6 following expression using different proteases. VP6 fusion proteins were expressed in BL21(DE3) E. coli cells, and the expression levels using the different vectors were subjected to a pilot expression test. Based on the optimization experiments (data not shown), we concluded that the best expression conditions for the fusion proteins involved incubation to an A600 of 0.6, addition of IPTG to a final concentration of 0.25 mM, and then further incubation for 12 hours at 25°C. We used the optimized conditions for the expression of all three different VP6 fusion proteins.
To evaluate the ability of different fusion tags to enhance VP6 expression and solubility in E. coli, soluble and insoluble fractions of induced and uninduced bacteria were assessed by SDS-PAGE. As shown in Figure 2, there was a clear difference among the fusion tags with respect to their ability to enhance expression and solubility. The His6-VP6 construct was observed to have a high yield of expression, even if the total amount of the protein produced was insoluble.
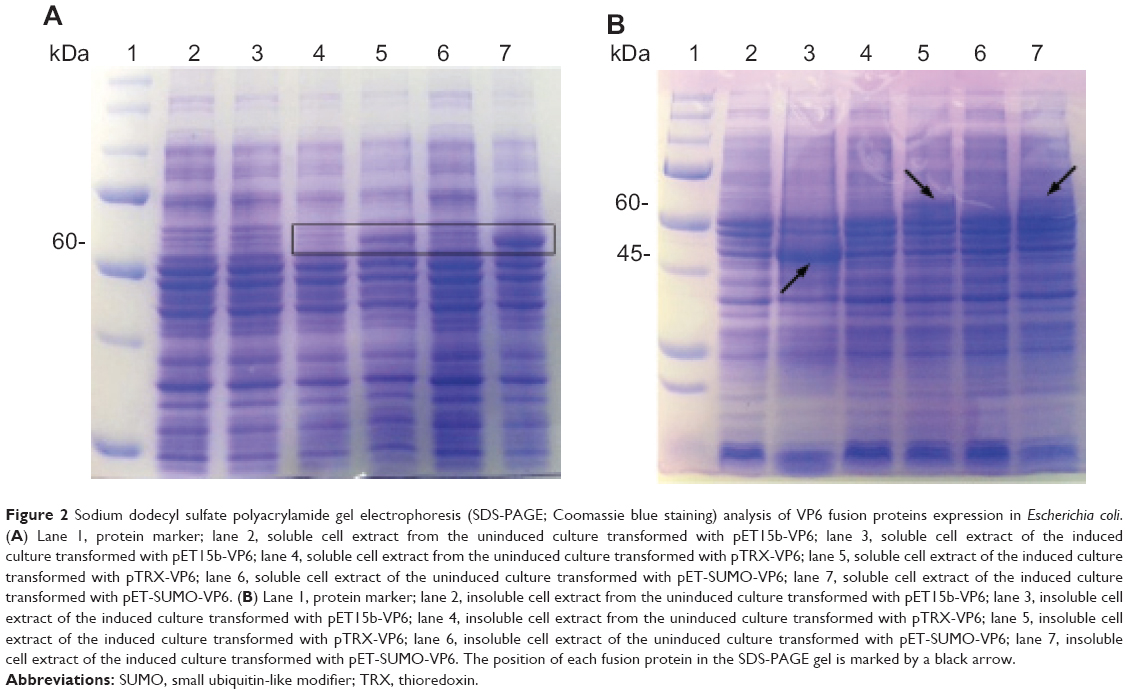 | Figure 2 Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; Coomassie blue staining) analysis of VP6 fusion proteins expression in Escherichia coli. (A) Lane 1, protein marker; lane 2, soluble cell extract from the uninduced culture transformed with pET15b-VP6; lane 3, soluble cell extract of the induced culture transformed with pET15b-VP6; lane 4, soluble cell extract from the uninduced culture transformed with pTRX-VP6; lane 5, soluble cell extract of the induced culture transformed with pTRX-VP6; lane 6, soluble cell extract of the uninduced culture transformed with pET-SUMO-VP6; lane 7, soluble cell extract of the induced culture transformed with pET-SUMO-VP6. (B) Lane 1, protein marker; lane 2, insoluble cell extract from the uninduced culture transformed with pET15b-VP6; lane 3, insoluble cell extract of the induced culture transformed with pET15b-VP6; lane 4, insoluble cell extract from the uninduced culture transformed with pTRX-VP6; lane 5, insoluble cell extract of the induced culture transformed with pTRX-VP6; lane 6, insoluble cell extract of the uninduced culture transformed with pET-SUMO-VP6; lane 7, insoluble cell extract of the induced culture transformed with pET-SUMO-VP6. The position of each fusion protein in the SDS-PAGE gel is marked by a black arrow. |
The TRX fusion tag was observed to have a lower enhancement of total expression, but a moderate quantity of recombinant protein (20%) was present in the soluble fraction. The SUMO-VP6 fusion yielded the best levels of expression, being the only tag able to preserve protein solubility, with about 80% of viral fusion protein expressed in a soluble form. Such different impacts of gene fusions on the expression and solubility of VP6 protein led us to proceed with large-scale expression and purification of just the SUMO-VP6 protein.
Large-scale purification of SUMO-VP6 fusion protein
We prepared soluble cell lysate from 250 mL of induced bacteria, and then we loaded it onto 1 mL of an Ni2+-NTA column. After being washed extensively, the SUMO-VP6 fusion protein was eluted in the presence of 300 mM imidazole. The elution profile of the first five fractions (over ten) is reported in Figure 3A as a 10% SDS-PAGE gel stained with Coomassie blue. All these fractions, obtained taking 1 μL out of 1.5 mL after purity and yield evaluations, were pooled, while the following fractions were discarded. Pooled elution fractions were analyzed by immunoblot using anti-His6 mouse monoclonal antibody (Figure 3B). Under reducing conditions, purified SUMO-VP6 runs as a single band of about 55 kDa, obtaining a purity greater than 99%. The final yield obtained was exceptionally high, estimated at a value equal to 50 mg/L. Moreover, we determined the amount of LPS in purified VP6 preparation was <3 EU/mg-1. Given that the next step would be the removal of the fusion partner, we decided to dialyze the sample against the specific SUMO protease cleavage-reaction buffer.
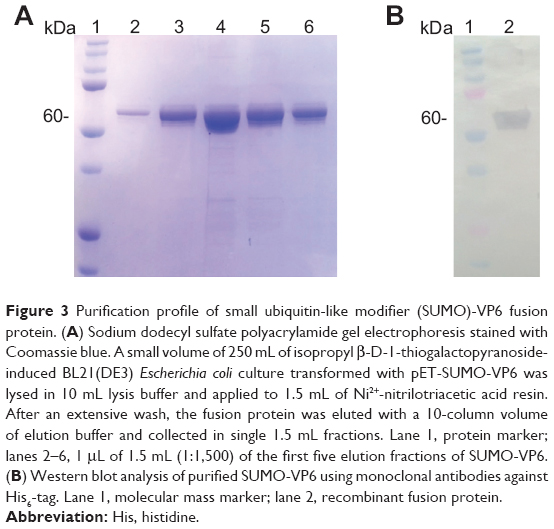 | Figure 3 Purification profile of small ubiquitin-like modifier (SUMO)-VP6 fusion protein. (A) Sodium dodecyl sulfate polyacrylamide gel electrophoresis stained with Coomassie blue. A small volume of 250 mL of isopropyl β-d-1-thiogalactopyranoside-induced BL21(DE3) Escherichia coli culture transformed with pET-SUMO-VP6 was lysed in 10 mL lysis buffer and applied to 1.5 mL of Ni2+-nitrilotriacetic acid resin. After an extensive wash, the fusion protein was eluted with a 10-column volume of elution buffer and collected in single 1.5 mL fractions. Lane 1, protein marker; lanes 2–6, 1 μL of 1.5 mL (1:1,500) of the first five elution fractions of SUMO-VP6. (B) Western blot analysis of purified SUMO-VP6 using monoclonal antibodies against His6-tag. Lane 1, molecular mass marker; lane 2, recombinant fusion protein. |
Enzymatic cleavage of SUMO-VP6 fusion protein and purification of recombinant native VP6
To remove the target VP6 protein from its SUMO partner, the fusion protein was incubated with SUMO protease at 30°C for 6 hours. The reaction mixture was analyzed by SDS-PAGE (Figure 4A). SUMO-VP6 was found to be cleaved almost completely, as demonstrated by the disappearance of the 55 kDa fusion protein and the appearance of two new bands corresponding to the expected molecular weights of SUMO (~11 kDa) and VP6 (~45 kDa).
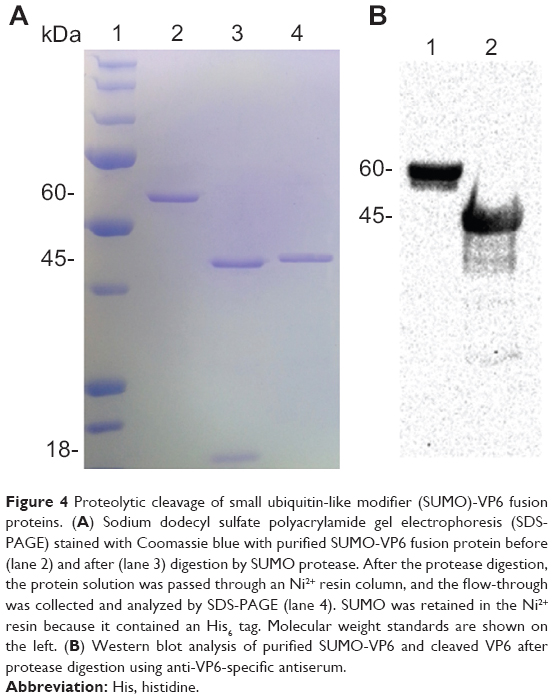 | Figure 4 Proteolytic cleavage of small ubiquitin-like modifier (SUMO)-VP6 fusion proteins. (A) Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) stained with Coomassie blue with purified SUMO-VP6 fusion protein before (lane 2) and after (lane 3) digestion by SUMO protease. After the protease digestion, the protein solution was passed through an Ni2+ resin column, and the flow-through was collected and analyzed by SDS-PAGE (lane 4). SUMO was retained in the Ni2+ resin because it contained an His6 tag. Molecular weight standards are shown on the left. (B) Western blot analysis of purified SUMO-VP6 and cleaved VP6 after protease digestion using anti-VP6-specific antiserum. |
In contrast to the fusion protein and the SUMO moiety, which contained N-terminal hexahistidine tags, VP6 released from the SUMO partner was unable to bind to Ni2+-NTA resin. Therefore, the cleavage-reaction mixture was reapplied to the affinity column, which detained most of the SUMO moiety and a small percentage of uncleaved fusion protein. VP6 was purified in the pass-through fraction with 99% homogeneity (Figure 4, lane 4). Given that VP6 recovered from flow-through (after passage through Ni2+-NTA resin) is untagged, recognition of cleaved VP6 protein by immunoblot was carried out using the specific anti-VP6 antiserum produced in mice, as described in the Materials and methods section. Figure 4B shows the result of the immunoblot of SUMO-VP6 and cleaved native VP6 with specific antiserum used at a dilution of 1:2,000. Purified native VP6 protein was stored overnight at 4°C to check for possible formations of protein precipitates, but the day after, the sample was completely clear, and no precipitates were visible after high-speed centrifugation.
Since VP6 remains soluble after the cleavage and removal of SUMO, it suggests an important benefit of the SUMO-expression system. SUMO is not only promoting the solubility through its attachment to the N-terminal of these proteins, but it actually may assist the folding of VP6 in a particular conformation that enhances solvent interactions.
VLP and NT production
In order to verify the self-assembling ability of the recombinant VP6 produced using the SUMO fusion tag, we prepared a series of samples to be characterized by means of TEM and AFM. The protein was first concentrated up to 250 μg/mL in the SUMO protease cleavage buffer at pH 7.5, and then was dialyzed to a different buffer at pH 4.5 and 6 (see the Materials and methods section for details) to identify different kinds of superstructures formed by VP6, according to what was reported by Lepault et al.5 The result of this approach is shown in Figures 5 and 6.
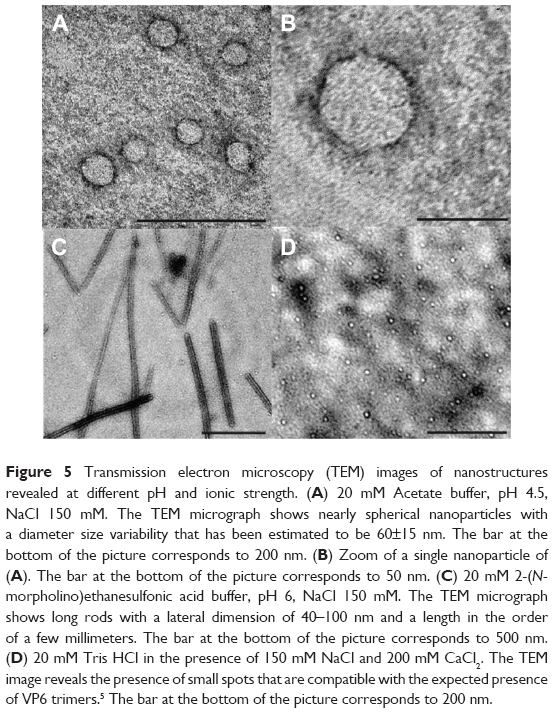 | Figure 5 Transmission electron microscopy (TEM) images of nanostructures revealed at different pH and ionic strength. (A) 20 mM Acetate buffer, pH 4.5, NaCl 150 mM. The TEM micrograph shows nearly spherical nanoparticles with a diameter size variability that has been estimated to be 60±15 nm. The bar at the bottom of the picture corresponds to 200 nm. (B) Zoom of a single nanoparticle of (A). The bar at the bottom of the picture corresponds to 50 nm. (C) 20 mM 2-(N-morpholino)ethanesulfonic acid buffer, pH 6, NaCl 150 mM. The TEM micrograph shows long rods with a lateral dimension of 40–100 nm and a length in the order of a few millimeters. The bar at the bottom of the picture corresponds to 500 nm. (D) 20 mM Tris HCl in the presence of 150 mM NaCl and 200 mM CaCl2. The TEM image reveals the presence of small spots that are compatible with the expected presence of VP6 trimers.5 The bar at the bottom of the picture corresponds to 200 nm. |
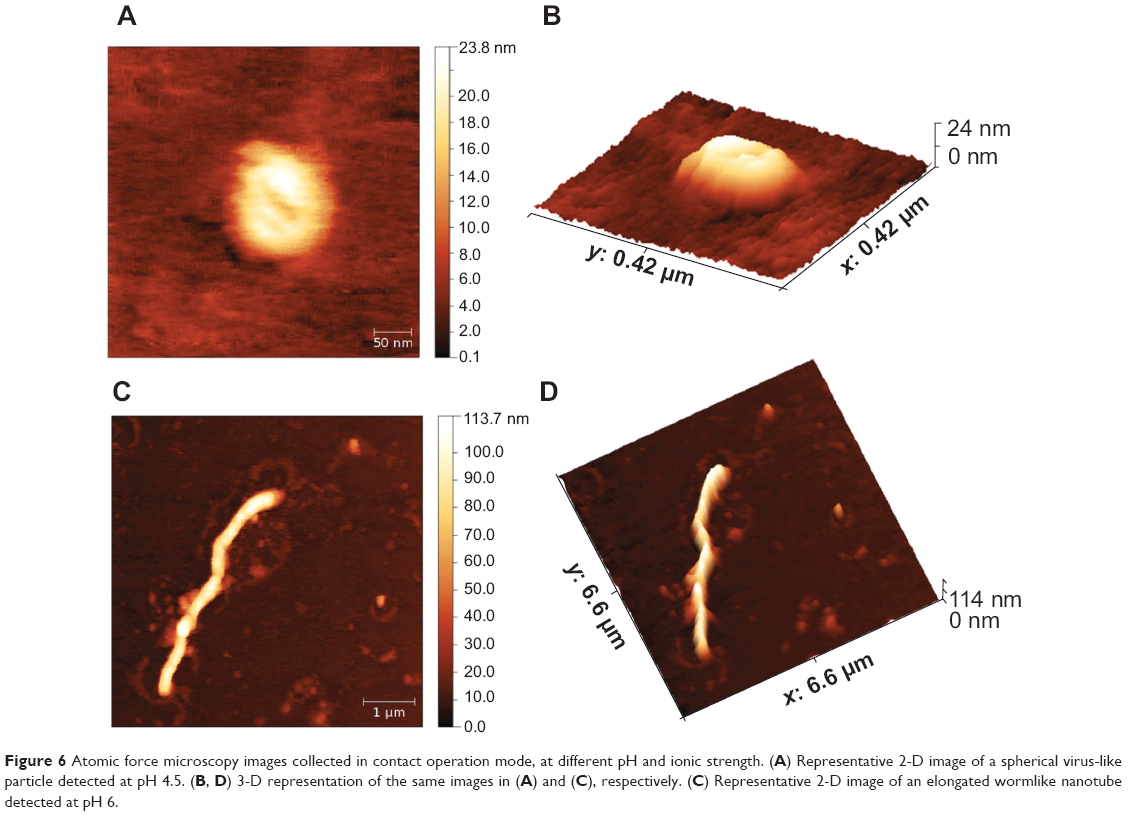 | Figure 6 Atomic force microscopy images collected in contact operation mode, at different pH and ionic strength. (A) Representative 2-D image of a spherical virus-like particle detected at pH 4.5. (B, D) 3-D representation of the same images in (A) and (C), respectively. (C) Representative 2-D image of an elongated wormlike nanotube detected at pH 6. |
In Figure 5A, the VLPs formed at pH 4.5 can be clearly identified using TEM. As expected, they appear as single roughly spherical particles with a size distribution of the diameter that has been calculated in the order of 60±15 nm. This result is in agreement with the determination of 75 nm that was reported by Lepault et al using a higher-resolution setup by means of cryo-electron microscopy.5 Moreover, the homogeneity seems quite high, with few particles forming small aggregates being clearly visible, which in any case seem to be characterized by single particles in close contact with more than an amorphous amount of protein material. In Figure 5B, a zoom of a single particle is shown to appreciate the morphology and the geometry of this VLP. In Figure 5C, the results obtained at pH 6 are shown. In this case, elongated structures that are a few micron in length and with a lateral dimension in the order of 40–100 nm may be indicative of the formation of NTs, as reported by Lepault et al at this pH value.5 Finally, in Figure 5D, the TEM image of the sample at pH 7.5 and in the presence of 200 mM CaCl2 shows a condition disfavoring the formation of ordered structures, both VLPs and NTs. As expected, no ordered, tubular, or spherical structures appeared, while, even though a higher resolution is necessary, the presence of small spots is evident and probably due to the presence of trimers of VP6.
To characterize in a liquid environment the ordered superstructures found at pH 4.5 and 6, we also performed AFM measurements on the same samples that were inspected by TEM. Our results are reported in Figure 6. At pH 4.5, using AFM and averaging a large number of images on different sample spots, we obtained two different distributions relative to the particle shape that appears as an oblate spheroid. We measured 90±20 nm and 23±5 nm for the circular diameter and the height, respectively.
As commonly observed on AFM micrographs, the shape of soft or flexible hollow particles, due to the pressure exerted by the AFM tip, appears ellipsoidal. Also, taking into account the well-known geometrical relations between a sphere and a circular spheroid, the real radius of the uncompressed particle can be easily recovered and a diameter of about 76 nm has been then recalculated.33 In Figure 6A, we have highlighted the typical ellipsoidal shape detected, showing a representative single particle at pH 4.5, and Figure 6C shows a 3-D representation of the same image.
Finally, a typical NT, revealed at pH 6, is shown in 2-D and 3-D representations (Figure 6, B and D). In this condition, we observed long rods with a contour length in the range of about 5–10 mm and a thickness around 100 nm. According to the results obtained by TEM and AFM, we demonstrated that the ability of VP6 protein to self-assemble into both VLPs and NTs was unaltered using this novel expression and purification assay.
It is important to note that the images obtained using TEM and AFM techniques were collected directly from the samples after dialysis to the desired pH, to evaluate the homogeneity of the samples obtained by self-assembly of the whole batch of protein purified. Perhaps a higher degree of homogeneity, especially for the spherical particles, may be obtained using a subset of protein fractions and/or introducing a gel-filtration step after dialysis. These possibilities are future intriguing perspectives that are beyond the scope of this work. Finally, once formed, these structures are quite stable if the pH is changed, as demonstrated by means of dynamic light scattering, comparing the size distribution obtained in the same starting buffers with what is found diluting the different preparations 1:10 in PBS at pH 7.4 (Figure S1).
Immunogenic properties of VP6 assembly
A first application to test the potential use of the different VP6 self-assembly products was accomplished by administering to different groups of animals native protein, the VLP-VP6, and NT-VP6, according to the procedure described in the Materials and methods section. Inoculated mice were examined for total serum rotavirus VP6-specific IgG and IgA antibodies at 3 and 6 weeks after the initial immunization. Serum antibody response was determined by indirect ELISA. Antibody titers obtained by VP6 antigen formulations are given in Figure 7. All the mice that received subcutaneous administration of these different VP6 formulations displayed significantly enhanced levels of systemic IgG response in contrast to the naïve group. Nevertheless, NT-VP6 particle immunization resulted in a relatively higher level of IgG response. VP6-specific serum IgA antibodies were slightly stimulated by the three VP6 formulations, as expected from the poorly suitable route of administration of various antigens. Even in this case, although with minor differences, NTs were the most immunogenic among the different nanoconstructs. These results suggest that the tubular structural conformation is the most immunogenic one.
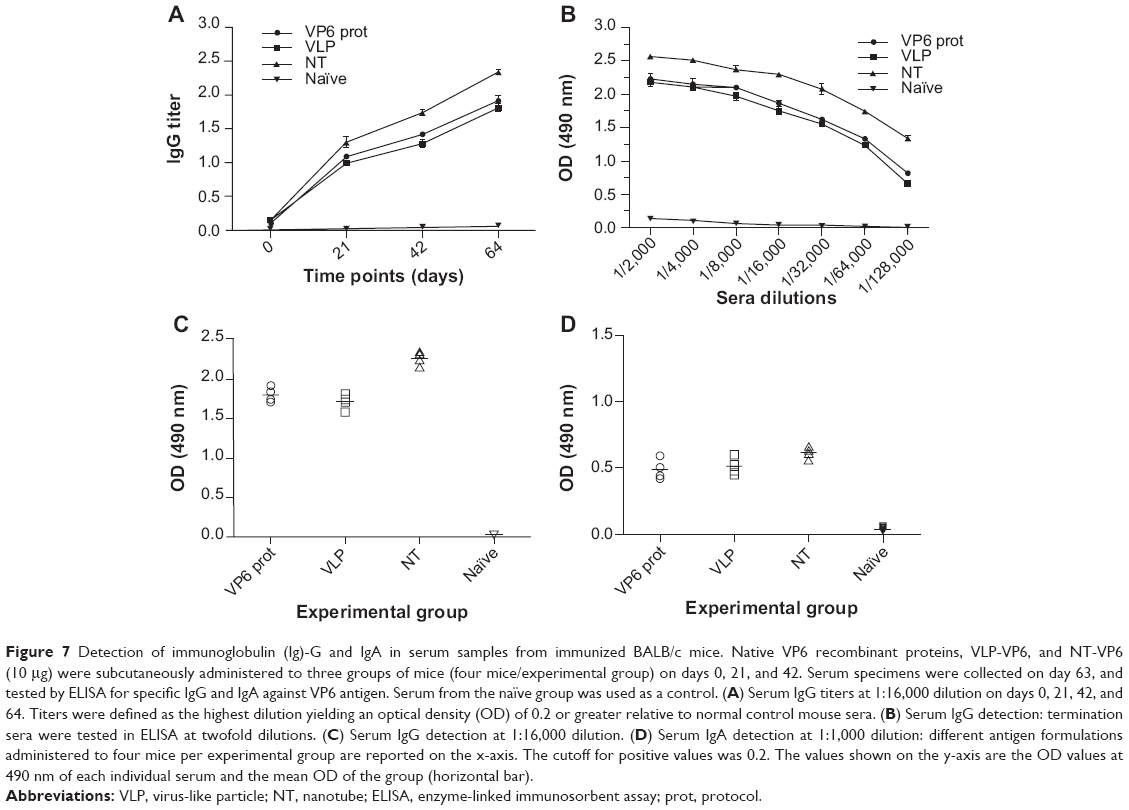 | Figure 7 Detection of immunoglobulin (Ig)-G and IgA in serum samples from immunized BALB/c mice. Native VP6 recombinant proteins, VLP-VP6, and NT-VP6 (10 μg) were subcutaneously administered to three groups of mice (four mice/experimental group) on days 0, 21, and 42. Serum specimens were collected on day 63, and tested by ELISA for specific IgG and IgA against VP6 antigen. Serum from the naïve group was used as a control. (A) Serum IgG titers at 1:16,000 dilution on days 0, 21, 42, and 64. Titers were defined as the highest dilution yielding an optical density (OD) of 0.2 or greater relative to normal control mouse sera. (B) Serum IgG detection: termination sera were tested in ELISA at twofold dilutions. (C) Serum IgG detection at 1:16,000 dilution. (D) Serum IgA detection at 1:1,000 dilution: different antigen formulations administered to four mice per experimental group are reported on the x-axis. The cutoff for positive values was 0.2. The values shown on the y-axis are the OD values at 490 nm of each individual serum and the mean OD of the group (horizontal bar). |
Conclusion
In modern nanotechnology, two different approaches have been identified in the creation of nanoparticles: the bottom-up approach, where materials and devices are built from molecular components that chemically self-assemble, and the top-down approach, where nano-objects are made from larger entities without atomic-level control. The creation of VLPs using the molecular biology framework clearly belongs to the first type. A prerequisite for the success of this method is the possibility of obtaining the viral protein in large amounts and with a high degree of purity. The main result of the present work was the definition, step by step, of a fast and highly reproducible protocol to obtain a large quantity of VP6. This protein is able to self-assemble at two different pH values, 6 and 4.5, forming NTs or VLPs, as previously demonstrated.5 Comparing the particle shape obtained by means of TEM and AFM measurements, we also provide promising evidence of the flexibility and the consequent shape remodeling of VP6.
The interest in developing and refining new approaches to manipulate the mechanical properties of low-cost VP6 nanoparticles is rapidly increasing, in order to improve their suitability for biomedical applications. An evident difference in the macroscopic properties, which are varied according to the 3-D organization, is reported in this work. Indeed, the potential of protein NTs to induce strong immune responses and an adjuvant effect has been demonstrated by others.8,34,35 Immunogenicity of tubular structures is strongly linked to the exclusive framework organization. Epitopes displayed may be differentially more spaced out than in spherical particles36 with a higher expression ratio, thus affecting IgG and IgA response.37 Furthermore, tubular structures administered to mice may have longer circulation times than spherical particles. Therefore, a deeper knowledge of their physicochemical properties may be helpful to obtain the best compromise between stability and immunogenicity. We point out that a better understanding of VLP mechanics may be relevant to understanding virus biology. Indeed, mechanical features of virus capsids have evolved by natural selection for as long as relevant adaptive features involving interaction forces between cells and biomolecules have developed.38
On the other hand, functionalization of VP6 (VLP or NT construct) would allow the enhancement and spread of many different VP6 applications. Chemical conjugation with specific drugs and with a targeting molecule to specific receptors will enable the setup of a drug-delivery assay to be tested in selected cell cultures.
Acknowledgments
Financial support by the Italian Ministry of University and Research (Linea D1 Università Cattolica del Sacro Cuore) is gratefully acknowledged. We thank Professor Andrea Brancaccio from the Consiglio Nazionale delle Richerche for providing the pHis-Trx vector.
Disclosure
The authors report no conflicts of interest in this work.
References
Crisci E, Bárcena J, Montoya M. Virus-like particles: the new frontier of vaccines for animal viral infections. Vet Immunol Immunopathol. 2012;148(3–4):211–225. | ||
Ma Y, Nolte RJ, Cornelissen JJ. Virus-based nanocarriers for drug delivery. Adv Drug Deliv Rev. 2012;64(9):811–825. | ||
Shang W, Liu J, Yang J, Hu Z, Rao X. Dengue virus-like particles: construction and application. Appl Microbiol Biotechnol. 2012;94(1):39–46. | ||
Chandran A, Fitzwater S, Zhen A, Santosham M. Prevention of rotavirus gastroenteritis in infants and children: rotavirus vaccine safety, efficacy, and potential impact of vaccines. Biologics. 2010;4:213–229. | ||
Lepault J, Petitpas I, Erk I, et al. Structural polymorphism of the major capsid protein of rotavirus. EMBO J. 2001;20(7):1498–1507. | ||
Mukhopadhya I, Anbu D, Iturriza-Gomara M, et al. Anti-VP6 IgG antibodies against group A and group C rotaviruses in South India. Epidemiol Infect. 2010;138(3):442–447. | ||
Richardson V, Hernandez-Pichardo J, Quintanar-Solares M, et al. Effect of rotavirus vaccination on death from childhood diarrhea in Mexico. N Engl J Med. 2010;362(4):299–305. | ||
Blazevic V, Lappalainen S, Nurminen K, Huhti L, Vesikari T. Norovirus VLPs and rotavirus VP6 protein as combined vaccine for childhood gastroenteritis. Vaccine. 2011;29(45):8126–8133. | ||
Clarke BE, Newton SE, Carroll AR, et al. Improved immunogenicity of a peptide epitope after fusion to hepatitis B core protein. Nature. 1987;330(6146):381–384. | ||
Olsson SE, Villa LL, Costa RL, et al. Induction of immune memory following administration of a prophylactic quadrivalent human papillomavirus (HPV) types 6/11/16/18 L1 virus-like particle (VLP) vaccine. Vaccine. 2007;25(26):4931–4939. | ||
Pattenden LK, Middelberg AP, Niebert M, Lipin DI. Towards the preparative and large-scale precision manufacture of virus-like particles. Trends Biotechnol. 2005;23(10):523–529. | ||
Mena JA, Ramírez OT, Palomares LA. Intracellular distribution of rotavirus structural proteins and virus-like particles expressed in the insect cell-baculovirus system. J Biotechnol. 2006;122(4):443–452. | ||
Zhao Q, Chen W, Chen Y, Zhang L, Zhang J, Zhang Z. Self-assembled virus-like particles from rotavirus structural protein VP6 for targeted drug delivery. Bioconjug Chem. 2011;22(3):346–352. | ||
Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189(1):113–130. | ||
Waugh DS. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif. 2011;80(2):283–293. | ||
Loughran ST, Walls D. Purification of poly-histidine-tagged proteins. Methods Mol Biol. 2011;681:311–335. | ||
Sun P, Tropea JE, Waugh DS. Enhancing the solubility of recombinant proteins in Escherichia coli by using hexahistidine-tagged maltose-binding protein as a fusion partner. Methods Mol Biol. 2011;705:259–274. | ||
Walls D, Loughran ST. Tagging recombinant proteins to enhance solubility and aid purification. Methods Mol Biol. 2011;681:151–175. | ||
Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics. 2004;5(1–2):75–86. | ||
Zuo X, Li S, Hall J, et al. Enhanced expression and purification of membrane proteins by SUMO fusion in Escherichia coli. J Struct Funct Genomics. 2005;6(2–3):103–111. | ||
Zuo X, Mattern MR, Tan R, et al. Expression and purification of SARS coronavirus proteins using SUMO-fusions. Protein Expr Purif. 2005;42(1):100–110. | ||
Jenny RJ, Mann KG, Lundblad RL. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr Purif. 2003;31(1):1–11. | ||
Butt TR, Edavettal SC, Hall JP, Mattern MR. SUMO fusion technology for difficult-to-express proteins. Protein Expr Purif. 2005;43(1):1–9. | ||
Ushijima H, Koike H, Mukoyama A, Hasegawa A, Nishimura S, Gentsch J. Detection and serotyping of rotaviruses in stool specimens by using reverse transcription and polymerase chain reaction amplification. J Med Virol. 1992;38(4):292–297. | ||
Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 3rd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1989. | ||
Reichelt P, Schwarz C, Donzeau M. Single step protocol to purify recombinant proteins with low endotoxin contents. Protein Expr Purif. 2006;46(2):483–488. | ||
Bugli F, Graffeo R, Paroni Sterbini F, et al. Monoclonal antibody fragment from combinatorial phage display library neutralizes alpha-latrotoxin activity and abolishes black widow spider venom lethality, in mice. Toxicon. 2008;51(4):547–554. | ||
Missori M, Mondelli C, De Spirito M, et al. Modifications of the mesoscopic structure of cellulose in paper degradation. Phys Rev Lett. 2006;97(23):238001. | ||
Papi M, Maulucci G, De Spirito M, et al. Ristocetin-induced self-aggregation of von Willebrand factor. Eur Biophys J. 2010;39(12):1597–1603. | ||
Brunelli R, Papi M, Arcovito G, et al. Globular structure of human ovulatory cervical mucus. FASEB J. 2007;21(14):3872–3876. | ||
Molinari P, Peralta A, Taboga O. Production of rotavirus-like particles in Spodoptera frugiperda larvae. J Virol Methods. 2008;147(2):364–367. | ||
Clark KB, Lin SC, Humphrey C, et al. Expression and characterization of human group C rotavirus virus-like particles in insect cells. Virology. 2009;387(2):267–272. | ||
Kanno T, Yamada T, Iwabuki H, et al. Size distribution measurement of vesicles by atomic force microscopy. Anal Biochem. 2002;309(2):196–199. | ||
Ghosh MK, Borca MV, Roy P. Virus-derived tubular structure displaying foreign sequences on the surface elicit CD4+ Th cell and protective humoral responses. Virology. 2002;302(2):383–392. | ||
Ward RL, McNeal MM. VP6: A candidate rotavirus vaccine. J Infect Dis. 2010;202 Suppl:S101–S107. | ||
Mathieu M, Petitpas I, Navaza J, et al. Atomic structure of the major capsid protein of rotavirus: implications for the architecture of the virion. EMBO J. 2001;20(7):1485–1497. | ||
Geng Y, Dalhaimer P, Cai S, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2(4):249–255. | ||
Mateu MG. Mechanical properties of viruses analyzed by atomic force microscopy: a virological perspective. Virus Res. 2012;168(1–2):1–22. |
Supplementary material
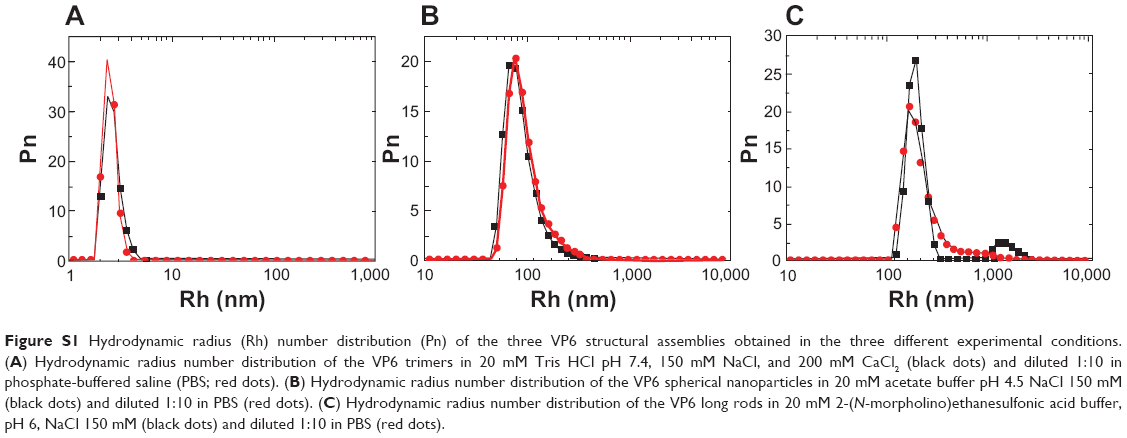 | Figure S1 Hydrodynamic radius (Rh) number distribution (Pn) of the three VP6 structural assemblies obtained in the three different experimental conditions. (a) Hydrodynamic radius number distribution of the VP6 trimers in 20 mM Tris HCl pH 7.4, 150 mM NaCl, and 200 mM CaCl2 (black dots) and diluted 1:10 in phosphate-buffered saline (PBS; red dots). (B) Hydrodynamic radius number distribution of the VP6 spherical nanoparticles in 20 mM acetate buffer pH 4.5 NaCl 150 mM (black dots) and diluted 1:10 in PBS (red dots). (C) Hydrodynamic radius number distribution of the VP6 long rods in 20 mM 2-(N-morpholino)ethanesulfonic acid buffer, pH 6, NaCl 150 mM (black dots) and diluted 1:10 in PBS (red dots). |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.