")
Back to Journals » International Journal of Nanomedicine » Volume 14
Synthesis and characterization of a long-acting emtricitabine prodrug nanoformulation
Authors Ibrahim IM, Bade AN, Lin Z, Soni D, Wojtkiewicz M, Dyavar Shetty BL, Gautam N , McMillan JM, Alnouti Y, Edagwa BJ, Gendelman HE
Received 12 May 2019
Accepted for publication 10 July 2019
Published 7 August 2019 Volume 2019:14 Pages 6231—6247
DOI https://doi.org/10.2147/IJN.S215447
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Ibrahim M Ibrahim,1,2 Aditya N Bade,1 Zhiyi Lin,3 Dhruvkumar Soni,3 Melinda Wojtkiewicz,1 Bhagya Laxmi Dyavar Shetty,1 Nagsen Gautam,3 JoEllyn M McMillan,1 Yazen Alnouti,3 Benson J Edagwa,1 Howard E Gendelman1,3
1Department of Pharmacology and Experimental Neuroscience, College of Medicine, University of Nebraska Medical Center, Omaha, NE, USA; 2Department of Pharmacology, College of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia; 3Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, NE, USA
Purpose: A palmitoylated prodrug of emtricitabine (FTC) was synthesized to extend the drug’s half-life, antiretroviral activities and biodistribution.
Methods: A modified FTC prodrug (MFTC) was synthesized by palmitoyl chloride esterification. MFTC’s chemical structure was evaluated by nuclear magnetic resonance. The created hydrophobic prodrug nanocrystals were encased into a poloxamer surfactant and the pharmacokinetics (PK), biodistribution and antiretroviral activities of the nanoformulation (NMFTC) were assessed. The conversion of MFTC to FTC triphosphates was evaluated.
Results: MFTC coated with poloxamer formed stable nanocrystals (NMFTC). NMFTC demonstrated an average particle size, polydispersity index and zeta potential of 350 nm, 0.24 and −20 mV, respectively. Drug encapsulation efficiency was 90%. NMFTC was readily taken up by human monocyte-derived macrophages yielding readily detected intracellular FTC triphosphates and an extended PK profile.
Conclusion: NMFTC shows improved antiretroviral activities over native FTC. This is coordinate with its extended apparent half-life. The work represents an incremental advance in the development of a long-acting FTC formulation.
Keywords: palmitoyl chloride, viral reservoirs, long-acting antiretrovirals, human immunodeficiency virus type 1, monocyte-derived macrophage
Introduction
UNAIDS has recently highlighted the significant toll seen by human immunodeficiency virus (HIV) infections worldwide.1 In 2017, approximately 37 million people worldwide were infected of which 1.8 million were children under the age of 15. Despite global efforts to treat and prevent viral infection, up to a million people have died of infection. Attempts to reduce the spread of HIV infection centers around improved access to antiretroviral therapy (ART). Currently 21.7 million infected people have access to these medicines and it is estimated, in the developing world, that $26.2 billion will be required to control and prevent HIV infection by 2020.1 Thus, both improved access and continuous adherence to ART regimens are needed immediately.2
Effective antiretroviral treatment of infection requires early, daily and lifelong therapy.3 While currently available ART regimens have demonstrated remarkable reductions in disease morbidity and mortality, treatment failures abound.4 These are due, in part, to patient non-adherence to what is daily combination drug regimens and the stigma of the infection itself.4–6 The regimens are required to ensure reductions in viral load, maintenance of CD4+ T cell counts, prevention of the emergence of drug-resistant viral strains and elimination of viral transmission.2
ART regimens have also shown limitations in their pharmacokinetic (PK) and biodistribution profiles that center on the drugs' abilities to target viral reservoirs (including the lymph nodes, gut, central nervous system, genitourinary system and spleen) and inherent toxicities.7,8 These can result in reduced therapeutic success.9 To address each of these apparent limitations, long-acting (LA) ART regimens were developed in order to improve patient adherence, decrease drug toxicities and maintain viral suppression while also diminishing the stigma of antiretroviral drug (ARV) use.10 LA ART produced by poloxamer drug encasements can facilitate intracellular drug depots.11 These include the site of injection and within the reticuloendothelial system.10 To extend the apparent half-life of the drug, high protein binding and slow metabolic and release rates are required.12 Research in our own laboratory has extended this drug platform by the use of monocyte-derived macrophages (MDMs) as cell-based drug depots and as vehicles for drug delivery to viral reservoir tissue sites.1,13,14 Each of these serve to improve ARV PK and pharmacodynamic profiles.1
Despite active research into LA ARVs, only two antiretrovirals, cabotegravir and rilpivirine (CAB and RPV) are now being developed and will soon be available for human use.15 The strong sustained interest in LA ARV formulations by patients at large makes research in this area of immediate value.16 A focus of work has now been set in LA transformation of the nucleoside reverse transcriptase inhibitors (NRTIs) as they represent the backbone of current ART regimens.17 The standard of care for HIV therapy is two NRTIs and an integrase inhibitor or non-NRTI.17 Amongst these, the most potent and widely used NRTIs are emtricitabine (FTC, 2`-deoxy-5-fluoro-3`thiacytidine), tenofovir disoproxil fumarate (TDF) and lamivudine (3TC).18–20 Truvada, an FTC and TDF combination, is also effective in pre-exposure prophylaxis.21 Thus, expanding available LA formulations for each of these medicines would have a significant impact on patient care, HIV treatment and prevention.10
In efforts to achieve once-a-month or even longer dosing frequencies, our laboratory created first-generation LA NRTI prodrug formulations.11,22–25 To build upon this library we chemically modified FTC with the goal of extending its apparent half-life. FTC is noted for its bioavailability and limited systemic toxicities.20 In the current report, a first-generation hydrophobic FTC prodrug (MFTC) was created amenable to nanoparticle encasements. Drug was conjugated to a long chain palmitoyl fatty acid. The hydrophobic nanocrystal prodrugs (NMFTC) were stabilized into an aqueous solution by poloxamer 407 (P407). The nanosuspension offered increased dissolution rates, LA profiles with high encapsulation efficiency and physicochemical stability.26 The developed nanoparticles were readily taken up by MDMs and reside inside subcellular compartments with stable FTC-triphosphate (FTC-TP) conversions. PK profiles were improved over the native drug. As such, this nanoformulated prodrug could be employed toward further formulation developments.
Materials and methods
Materials
FTC was purchased from HBCChem (Union City, CA, USA). Chemicals, reagents, media and buffers used for chemical reactions or in vitro studies were purchased from either Sigma Aldrich (St. Louis, MO, USA) or Fisher Scientific (Waltham, MA, USA). Thin-layer chromatography was performed on precoated F-254 silica plates (250 µm) from SiliCycle, Inc. (Quebec City, QC, Canada). Monoclonal mouse anti-human HIV-1 p24 (clone Kal-1), and the polymer-based HRP-conjugated anti-mouse EnVision+ secondary antibodies were purchased from Dako (Carpinteria, CA, USA). MTT was purchased from Sigma Aldrich. FTC-TP was purchased from Toronto Research Chemicals (North York, ON, Canada). Sep-Pak QMA anion-exchange cartridges and OASIS HLB reverse-phase cartridges were purchased from Waters Corp. (Milford, MA, USA).
MFTC synthesis
FTC (2.0 g, 8.089 mmol, 1 equivalent) was dried by azeotroping from anhydrous pyridine (15 mL) and resuspended in anhydrous tetrahydrofuran (THF) (30 mL) under an argon atmosphere. The reaction flask was then cooled to −78°C followed by addition of tert-butylmagnesium chloride (8.089 mmol, 1 equivalent, 1 M in THF) and allowed to react for 15 mins to deprotonate the alcohol group in FTC. A solution of palmitoyl chloride (2.26 g, 8.089 mmol, 1 equivalent) was then added dropwise to the FTC anion solution at −78°C. The reaction mixture was gradually warmed to room temperature and stirred for 16 hrs, quenched using saturated ammonium chloride solution, concentrated and subjected to flash chromatography purification to yield MFTC as a colorless powder. Proton (1H)- and carbon (13C)- nuclear magnetic resonance (NMR) spectra were recorded on a Varian Unity/Inova-500 NB (500 MHz; Varian Medical Systems Inc., Palo Alto, CA, USA). Fourier transform infrared spectroscopy (FTIR) was performed on a PerkinElmer universal attenuated total reflectance Spectrum Two (Waltham, MA, USA). The aqueous solubility of FTC and MFTC were determined by adding excess drug to water followed by mixing for 24 hrs at room temperature. Samples were centrifuged at 20,000× g for 10 mins to remove undissolved drug. Solubilized drug in the supernatant was quantified by reversed phase HPLC using a Waters ACQUITY H-class ultra-performance liquid chromatography-TUV system (UPLC) and Empower 3 software (Milford, MA, USA). FTC and MFTC solutions were separated on a Phenomenex Kinetex 5 µm C18 column 100Å (Torrance, CA, USA) using either 95% 20 mM ammonium acetate buffer in 5% ACN, pH 3.9 (FTC), or 90% methanol/10% water (MFTC) with a flow rate of 1.0 mL/min and detection at 272 and 280 nm, respectively. Drug content was quantified by comparison of peak areas to those of known standards (0.05–50 µg/mL FTC or MFTC in methanol).
Nanoformulation preparation and characterization
For preparation of a MFTC nanosuspension (NMFTC), a pre-suspension of 1% (w/v) MFTC and 0.5% (w/v) P407 in 10 mM HEPES (pH 5.5±0.5) was made and processed by high-pressure homogenization (Avestin EmulsiFlexC3; Avestin Inc., ON, Canada) at 20,000 psi until the desired particle size of 300–400 nm was achieved. Particle size, polydispersity index (PDI) and zeta potential were assessed by dynamic light scattering (DLS) using a Malvern Zetasizer Nano Series Nano-ZS (Westborough, MA, USA). Three independent experiments were performed each containing three replicates. The stability of NMFTC was assessed at room temperature and 4°C for up to 10 weeks by monitoring particle size, PDI, and zeta potential by DLS. The crystalline structure of MFTC was determined by X-ray diffraction (XRD) that was performed in the 2θ range of 2–40° using a PANalytical Empyrean diffractometer (Westborough, MA, USA) with Cu-Kα radiation (1.5418 Å) at 40 kV and 45 mA setting. A mask of 20 mm and a divergence slit of 1/8° were used on the incident beam path. The diffraction data were collected in steps of 0.04 degrees by continuously scanning at the rate of 0.013°/s.
Nanoparticle macrophage uptake, retention, and cytotoxicity
Human peripheral blood monocytes were obtained from HIV- and hepatitis-seronegative donors and purified by centrifugal elutriation at the University of Nebraska Medical Center (UNMC) elutriation core facility. The monocytes were cultured and allowed to differentiate into macrophages as described previously.25 Cell uptake was assessed at 1, 2, 4, 8 and 24 hrs after incubation with 100 µM NMFTC or FTC. At each time point, adherent MDMs were washed three times with 1 mL PBS, scraped into 1 mL PBS and pelleted by centrifugation at 1000× g for 8 mins at 4°C. The cell pellet was reconstituted in 200 µL of methanol and probe-sonicated at 15% amplitude (QSonica Q55; Qsonica, LLC, Newtown, CT, USA), followed by centrifugation at 20,000× g for 10 mins at 4°C to pellet cell debris. FTC or MFTC concentration in the supernatant fluid was quantified by UPLC as described above. To determine the extent and duration of retention, cells were incubated with 100 µM NMFTC or FTC for 8 hrs. Then, cells were washed with PBS and fresh drug-free medium was added. At days 1, 5, 10 and 15 following treatment, cells were washed with PBS, collected and analyzed for drug concentrations as described for the uptake study. For assessment of cytotoxicity, the MTT assay was performed. Monocytes were cultured on a 96-well plate at a density of 80,000 cells per well and allowed to differentiate into macrophages as described previously.25 MDMs were treated with various concentrations (10–400 µM) of FTC, MFTC or NMFTC for 4 days. After drug treatment, cells were washed and incubated with 100 μL/well of MTT solution, 5 mg/mL for 45 mins at 37°C. After incubation, MTT was removed. Then, 200 μL/well of DMSO was added and mixed thoroughly. Absorbance was measured at 490 nm on a Molecular Devices SpectraMax M3 plate reader with SoftMax Pro 6.2 software (Sunnyvale, CA, USA).
Antiretroviral activities
Antiretroviral efficacy was determined by measuring HIV reverse transcriptase (RT) activity in culture medium. For determination of the EC50 of MFTC and of FTC, MDMs were treated with 0.01–1000 nM FTC or MFTC for 1 hr followed by challenge with HIV-1ADA at a multiplicity of infection (MOI) of 0.1 infectious particles per cell for 4 hrs. Media were collected and HIV RT activity was determined as previously described.27,28 Concentration response curves were generated by GraphPad Prism 6.0 software (La Jolla, CA, USA) using nonlinear regression fit and the 50% inhibitory concentration determined. To evaluate antiretroviral efficacy, MDMs were treated with 100 μM FTC or NMFTC for 8 hrs. The cells were then washed with PBS to remove extracellular drug nanoparticles. At various time points (0 hr to 15 days following treatment duration), MDMs were challenged with HIV-1ADA (MOI 0.1) for 18 hrs. Cells were cultured for an additional 10 days with medium changes every other day. At day 10, viral activity was determined by measuring RT activity in the culture medium and HIV-1 p24 antigen expression in adherent cells fixed with 4% paraformaldehyde by immunocytochemistry as previously described.25 Uninfected and HIV-1ADA infected cells were controls.
Particle morphology and intracellular distribution
Particle morphologies and intracellular distribution of nanoparticles in MDM were assessed using transmission electron microscopy (TEM). For nanoparticle morphology, the nanosuspension was dried on a copper grid at room temperature and bright field images were taken with exposure times of 2 s. For intracellular distribution, MDMs were treated with 100 µM NMFTC for 4 hrs. Cells were collected in PBS, centrifuged at 1000× g for 8 mins at room temperature. The cell pellet was fixed and processed for TEM as described previously.29 Both copper grids, for nanoparticles and macrophages, were examined on FEI Tecnai G2 Spirit TWIN electron microscope (Hillsboro, OR, USA) operating at 80 kV. Images were acquired digitally with AMT imaging system (Woburn, MA, USA).29
MFTC plasma stability
To determine species differences in MFTC plasma stability, 100 µL mouse, rat, rabbit, dog, monkey or human plasma was incubated with 1 µM MFTC at 37°C. At different time points (0, 30 and 90 mins), 1 mL methanol was added to each sample and vortexed for 3 mins to stop the reaction. For the 0 timepoint, 100 µL ice-cold plasma was spiked with 100-x prodrug spiking solution in 20% DMSO/80% methanol and immediately 1 mL of ice-cold methanol was added. Heat-inactivated plasma was incubated at the same conditions and used as a negative control to differentiate chemical vs biological instability. Following the addition of methanol, samples were centrifuged at 15,000× g for 10 mins, 10 µL supernatant was mixed with 80% methanol containing IS, and then 10 µL was used for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.30 MFTC and FTC concentrations were determined in the supernatants by UPLC-tandem mass spectrometry (UPLC-MS/MS).30
PK studies and biodistribution
Male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA, USA) maintained on normal diet were injected intramuscularly (IM; caudal thigh muscle) with FTC or NMFTC at doses of 45, 75 or 100 mg/kg FTC-equivalents in a volume of 200 µL/200 g rat with FTC or 170–262 µl/200 g rat with NMFTC. Blood was collected at 2 hrs, and days 1, 7 and 14; tissues, including liver, spleen, lymph nodes and brain, were collected at days 1, 7 and 14. FTC was quantified by LC-MS/MS using a modification of an established method.30 Briefly, 25 μL of plasma was mixed with 1 mL ice-cold methanol (Optima grade, Fisher Scientific, Waltham, MA, USA), and spiked with 10 μL of internal standard (IS; 15N213C- lamivudine at 80 ng/mL, final concentration of 8 ng/mL). Samples were then vortexed for 3 mins and centrifuged at 17,000x g for 10 mins. Supernatants were dried, reconstituted in 100 µL 25% methanol and injected for LC/MS/MS analysis. Plasma standards were extracted at a final concentration of 0.05–500 ng/mL. For tissue sample preparation, approximately 20–100 mg of tissue was weighed and homogenized at 5–20x dilutions in 90% methanol. A quantity of 100 μL of the homogenate was mixed with 280 μL of methanol and 10 μL of IS. Samples were vortexed for 3 mins and centrifuged at 17,000x g for 10 mins. A quantity of 30 μL of supernatant was collected and mixed with 70 μL water (Optima grade, Fisher) before LC/MS/MS analysis. Tissue standards were extracted at a final concentration of 0.05–500 ng/mL. FTC was quantified using a Waters ACQUITY H-class UPLC coupled to a Waters Xevo TQ-S micro mass spectrometer (Waters Corp., Milford, MA, USA) with an ESI source in positive mode. An ACQUITY UPLC® CSH C18 1.7 μm analytical column (2.1 mm×100 mm, Waters Corp.) with an ACQUITY CSH C18 Vanguard column (2.1 mm×5 mm, Waters Corp.) were used for analyte separation.30 The transitions of 247.98<129.80 m/z and 233.23<114.97 m/z were used for FTC and IS quantification. Compounds were eluted using a gradient of Mobile phase A and B as follows. The gradient was held at 90% A for 1.5 mins and then linearly decreased to 5% A over 2 mins. The percentage was held for another minute to wash and then ramped back for equilibration to 90% A for 2 mins. The flow rate was 0.25 mL/min. Mobile phase A consisted of 7.5 mM ammonium bicarbonate in water (Optima grade, Fisher), pH adjusted to 7.0 with glacial acetic acid (ACS grade, Sigma). Mobile phase B was 100% methanol (Optima grade, Fisher).30
FTC-TP quantification
On each sacrifice day during the PK study in rats (days 1, 7 and 14), cells from fresh blood, spleen and lymph nodes were isolated and stored in 70% methanol as previously described.30 After isolation, all cell counting was performed using an Invitrogen Countess Automated Cell Counter (Carlsbad, CA, USA). FTC-TP extraction was performed as described previously with some modifications.30 Briefly, Sep-Pak QMA cartridges were used to separate FTC-TP from its mono- and di-phosphate forms. The triphosphate fractions were eluted and dephosphorylated using type XA sweet potato acid phosphatase. The 15N213C-3TC internal standard was added following the incubation (final concentration of 0.5 ng/mL). Dephosphorylated samples were then subjected to a second SPE extraction step using Waters OASIS HLB cartridges.30 The FTC fraction was eluted with 1.4 mL of methanol and evaporated under vacuum. Once dry, the residues were stored at −20°C then reconstituted with 100 µL 25% methanol before performing LC-MS/MS analyses as described above.
Statistics and data analyses
All data are presented as mean±standard deviation or standard error of the mean for in vitro and in vivo studies, respectively. In vitro studies were performed using a minimum of three biological replicates. All experiments were repeated three times. In vivo studies included a minimum of three animals per treatment group. For comparisons of two groups, Student’s t-test (two-tailed) was used. For comparison between multiple groups, one-way ANOVA with Bonferroni correction were performed. For studies with multiple time points, two-way ANOVA and Bonferroni’s post hoc tests for multiple comparisons were performed. Results with P<0.05 were considered significant. All data were graphed and analyzed using GraphPad Prism 6.0 software (La Jolla, CA, USA).31,32
Study approvals
All experimental protocols involving the use of laboratory animals were approved by the UNMC Institutional Animal Care and Use Committee ensuring the ethical care and use of laboratory animals in experimental research. All animal studies were performed in compliance with UNMC institutional policies and NIH guidelines for laboratory animal housing and care. Human blood cells were isolated by leukapheresis from HIV-1/2 and hepatitis seronegative donors and were deemed exempt from approval by the Institutional Review Board (IRB) of UNMC, as IRB of UNMC has determined that this experimental procedure does not constitute human subject research.
Results
The principal focus of the current work rested on developing an “injectable” FTC prodrug formulation through changing the chemical and physical nature of the agent from a highly hydrophilic drug to nanocrystals of lipophilic and hydrophobic prodrug. The FTC prodrug and nanoformulation were thoroughly characterized including NMR, FTIR, XRD, aqueous solubility, plasma stability and morphology. The biological characterization of the FTC formulation included human macrophage uptake, retention, antiretroviral potency and cytotoxicity measurements. Antiretroviral activities were monitored and found to be sustained with readily detectable intracellular FTC triphosphate (FTC-TP, the active metabolite of FTC), levels for up to 15 days after a single treatment of cell culture. Following the administration of NMFTC at 45, 75 and 100 mg/kg FTC equivalent doses in rats, sustained levels of FTC-TP levels were evident in peripheral blood mononuclear cells (PBMCs) and lymphatic tissues, and sustained FTC levels were measured in plasma with targeting of lymphatic tissues, liver and brain for 14 days.
MFTC chemical analyses
To overcome challenges associated with the short half-life of hydrophilic water-soluble FTC, we created poloxamer-encased hydrophobic drug nanosuspensions with enhanced cellular drug uptake and improved antiretroviral efficacy.33 To perform this task, a 16-carbon fatty acid palmitoyl chloride was conjugated to FTC to form a hydrophobic ester prodrug, MFTC, with a final yield of 90% (Figure 1A). The chemical modifications of FTC were confirmed by (1H) and carbon (13C) NMR spectra (Figure S1A and B, respectively), and FTIR spectroscopy. The 1H-NMR spectrum showed peaks corresponding to terminal methyl group (CH3R) and repeating methylene (RCH2R) protons of the fatty acid chain. 1H NMR (500 MHz, CDCl3): 7.94 (d, J=6.5 Hz 1H), 6.27–6.31 (m, 1H), 5.35 (q, J=4.3 Hz 1H), 4.43 (dd, J=12.5, 2.9 Hz, 1H), 3.56 (dd, J=12.4, 5.3 Hz, 1H), 3.18 (dd, J=12.4, 3.1 Hz, 1H), 2.40 (t, J=7.5 Hz 1H), 1.63–1.70 (m, 4H), 1.25 (br, 24H), 0.87 (t, J =6.9 Hz 1H). 13C NMR (125 MHz, CDCl3): δ 173.1, 157.9, 157.8, 153.5, 137.1, 135.2, 125.8, 125.6, 87.4, 84.6, 63.2, 38.9, 34.0, 31.9, 29.8, 29.7, 29.6, 29.4, 29.3, 29.2, 29.1, 24.8, 22.7, 14.1. MS-ES+ (m/z): calculated for C24H40FN3O4S, 485.25 (100%), 486.28 (26.0%), 487.27 (4.5%); and found, 486.21[M+H+]. The FTIR spectrum of MFTC (Figure 1B) showed the appearance of bands at 2920 and 2860 cm−1 that correspond to C-H stretches of the fatty acid chain. Additionally, both palmitoyl chloride and MFTC spectra exhibited absorption bands corresponding to the carbonyl group at 1805 and 1745 cm−1. XRD analysis revealed distinct diffraction patterns for FTC and MFTC suggesting different arrangement of atoms within the crystal lattice of each (Figure 1C). Esterification of FTC decreased the aqueous solubility of the parent drug by 1200 folds (Figure 1D). Measurement of HIV-1 RT activity in MDMs demonstrated that MFTC did not significantly change the EC50 of MFTC (0.033 µM) as compared to FTC (0.079 µM). These results indicated that the prodrug is efficiently hydrolyzed into the parent drug (Figure 1E).
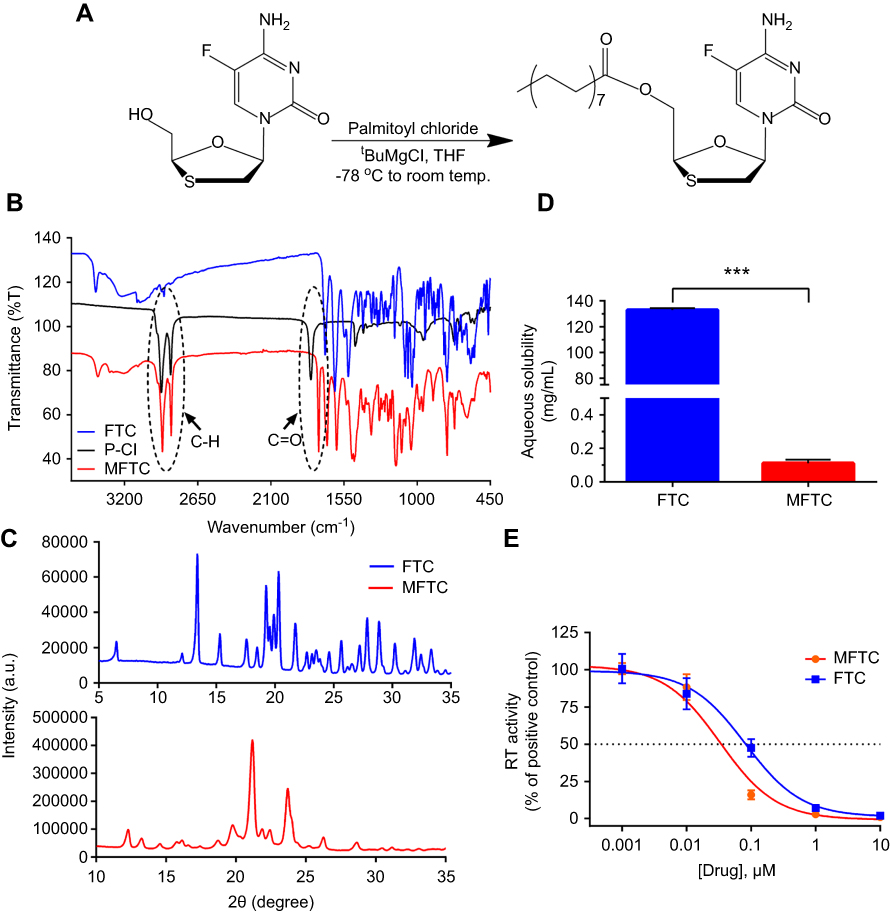 |
Figure 1 Synthesis and characterization of MFTC. (A) A 16-carbon fatty acid modified MFTC was synthesized with a final yield of 90%. (B) FTIR spectrum of MFTC showing, highlighted in dashed circles, absorption peaks at 2920 and 2860 cm−1 that correspond to CH2-CH2 stretches of the fatty acid chain and at 1745 cm−1 that corresponds to the carbonyl functional group that is part of the formed ester bond. (C) XRD analysis of FTC and MFTC demonstrates the crystalline nature of both drugs. (D) Aqueous solubility of FTC and MFTC demonstrates decrease in solubility of MFTC (mean±SD, n=3; ***P≤0.001). (E) EC50 (dashed line) was determined in vitro by HIV-1 RT activity assay (0.079 and 0.033 µM for FTC and MFTC, respectively) demonstrating the chemical modification did not significantly change the antiretroviral activity of FTC. Results were analyzed by nonlinear regression fit (mean±SD, n=3). Abbreviations: FTC, emtricitabine; MFTC, modified FTC prodrug; FTIR, Fourier transform infrared spectroscopy; XRD, X-ray diffraction; RT, reverse transcriptase; EC50, half maximum effective concentration. |
NMFTC preparation and characterization
NMFTC was prepared by high-pressure homogenization using P407 as the stabilizing surfactant. The resultant nanoparticle size, PDI and zeta potential were 350±10 nm, 0.24±0.02 and −20±2 mV, respectively, with an encapsulation efficiency of 90%. These data were an average of three experiments each performed with three replicates. A fourth experiment was performed by a different scientist and the results were as follows: nanoparticle size of 279.2±98.09 nm, PDI of 0.226 and zeta potential of −22±6.323 mV. Long-term stability of NMFTC was monitored at 25°C and 4°C. Nanoparticle size, PDI and zeta potential remained reproducible amongst three independent experiments with three replicates each and ±10% variability for at least 10 weeks (Figure 2A, B). NMFTC particle morphology was predominantly rod-shaped as shown by TEM (Figure 2C). It needs to be noted that there were differences between the analytical values of size distribution between the TEM and DLS tests. This was linked to the fact that TEM measures the diameter of dehydrated and immobilized nanoparticles on solid support while DLS measures the diameter in a hydrated state.34 Therefore, nanoparticles have larger hydrodynamic volume due to the solvent effect when hydrated.
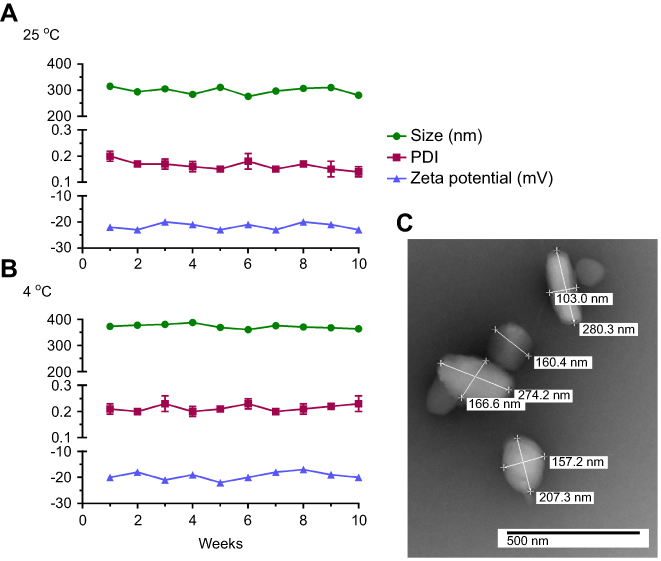 |
Figure 2 Nanoformulation characteristics. NMFTC was synthesized by high-pressure homogenization using P407 as excipient. NMFTC stability was assessed for up to 10 weeks at (A) 25°C, and (B) 4°C in terms of particle size, PDI, and zeta potential (mean±SD, n=3). (C) The size and morphology of NMFTC was assessed by TEM. Abbreviations: NMFTC, nanoparticle of modified FTC prodrug; PDI, polydispersity index; TEM, transmission electron microscopy. |
NMFTC-macrophage interactions
The rationale behind the use of macrophages in these assays is based on their role as drug depots and vehicles for drug delivery. Treatment with up to 100 µM NMFTC or FTC did not affect MDM viability, as determined by the MTT assay, after 4 days of exposure (Figure 3A). This concentration was thus selected for subsequent in vitro studies. Quantitation of intracellular MFTC levels showed that NMFTC was readily taken up by MDMs, reaching the highest concentration of 25 nmol/106 cells after 4 hrs followed by a rapid prodrug decay over 24 hrs (Figure 3B). In contrast, exposure to FTC resulted in minimal detectable intracellular drug levels. Surprisingly, FTC was not detected in cells treated with NMFTC. These results led us to quantify intracellular levels of the active triphosphate metabolite, FTC-TP. Quantitation of FTC-TP demonstrated enhanced intracellular FTC-TP levels after exposure to NMFTC, reaching the highest concentration of 75 pmol/106 cells after 8 hrs, compared to only 10 pmol/106 cells for FTC exposure (Figure 3C). After 8 hrs of NMFTC treatment , intracellular FTC-TP levels were detected for up to 10 days (1.5 pmol/106 cells). NMFTC treatment resulted in threefold higher FTC-TP levels at day 5 (3.2 pmol/106 cells) compared to FTC treatments (Figure 3D). TEM showed MDM cytoplasm loaded with nanoparticles after 4 hrs exposure to NMFTC (Figure 3E).
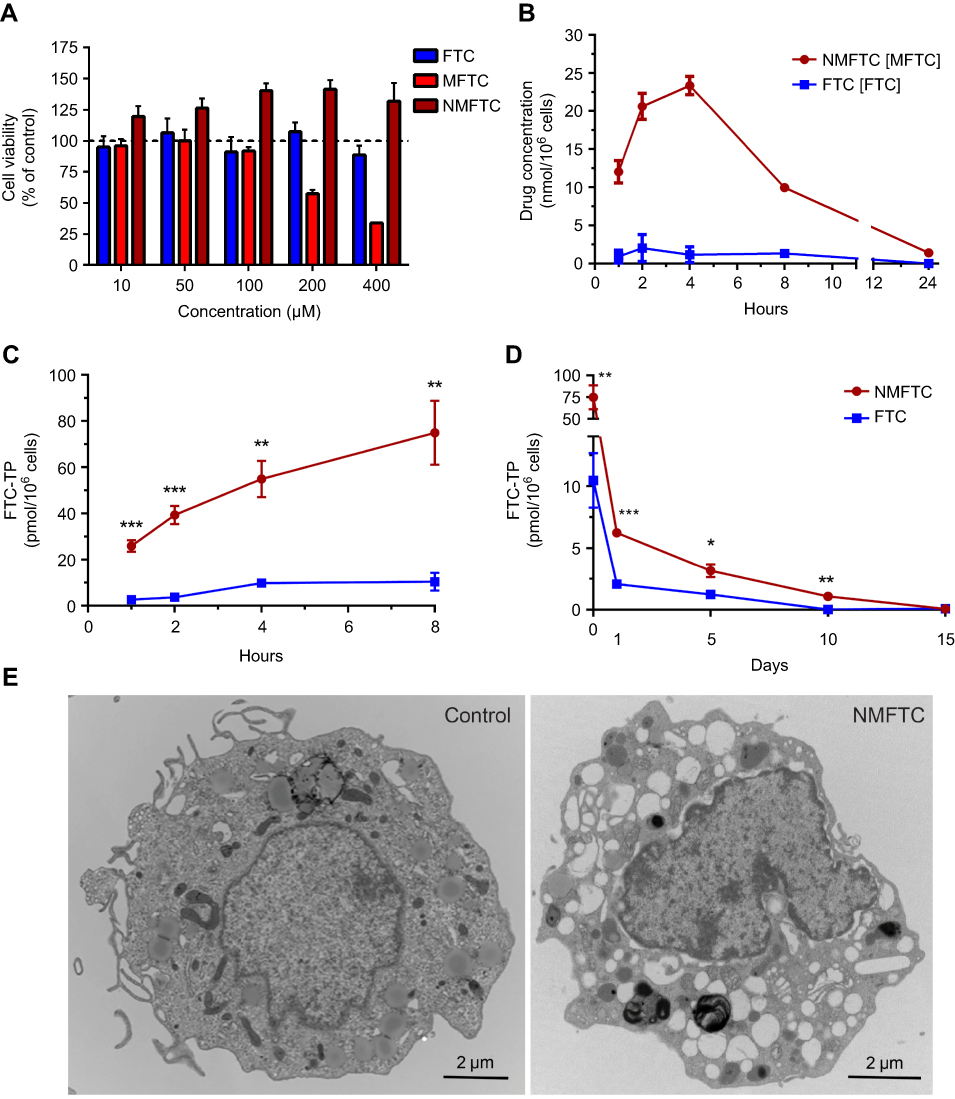 |
Figure 3 NMFTC in vitro characteristics. (A) Cytotoxicity was assessed in MDMs by MTT assay after 4 days of treatment with FTC or MFTC or NMFTC over a range of concentrations (10–400 µM). Results were normalized to untreated control cells. (B) Drug uptake in MDMs was measured over 24 hrs showing higher NMFTC uptake compared to FTC reaching high peak after 4 hrs. Intracellular FTC-TP concentrations demonstrate higher (C) uptake of NMFTC over 8-hr period, and longer (D) retention after 8-hr exposure for up to 10 days as compared to FTC. (E) TEM showing an MDM loaded with NMFTC (white clusters) after 4-hr exposure. Results in (A), (B), (C) and (D) are shown as mean±SD, n=3; *P≤0.05, **P≤0.01, ***P≤0.001. Abbreviations: MDM, monocyte-derived macrophage; FTC, emtricitabine; NMFTC, nanoparticle of modified FTC prodrug; TEM, transmission electron microscopy; CCK-8: cell counting kit-8. |
Antiretroviral activities
MDMs incubated with 100 µM FTC or NMFTC for 8 hrs were challenged with HIV-1ADA. Prior exposure to NMFTC completely inhibited viral RT activity analyzed in the cell culture medium when cells were challenged sequentially up to day 10 (Figure 4A). This effect was reduced to 25% inhibition with NMFTC when cells were challenged 15 days after drug preloading and to 30% at 24 hrs with FTC. Results were confirmed in adherent cells stained for HIV-1 p24 antigen. No HIV-1 positive (brown) cells were observed in cells challenged with HIV-1ADA 10 days after NMFTC loading; however, HIV-1 p24+ cells were observed after a 15-day challenge. In contrast, HIV-1 p24+ cells were observed by 12 hrs following native FTC treatments (Figure 4B).
 |
Figure 4 NMFTC antiretroviral activity. (A) HIV-1 RT activity in FTC and NMFTC pre-exposed MDMs showing complete inhibition with NMFTC for up to 10 days as compared with FTC that did not have effect beyond day 1 (mean±SD, n=9; ***P≤0.001). (B) HIV-1 p24 antigen staining showing results similar to RT assay demonstrated by the absence of p24 staining (brown color) for up to 10 days after NMFTC pre-exposure whereas FTC had no effect by day 1. Abbreviations: RT, reverse transcriptase; FTC, emtricitabine; NMFTC, nanoparticle of modified FTC prodrug; MDM, monocyte-derived macrophage. |
MFTC plasma stability
To assess the metabolic stability of MFTC in plasma, we incubated MFTC for 90 mins in plasma from six species (mouse, rat, rabbit, monkey, dog and human). MFTC rapidly declined in mouse and rat plasma, and within 30 mins no detectable prodrug was observed. MFTC disappearance was slower in the plasma of other species with % decline compared to 0 min at 92% in rabbit, 33% in monkey, 20% in human and 11% in dog after 90 mins of incubation (Figure 5A). Prodrug decay was accounted for with formation of the parent drug (FTC) as a result of ester bond hydrolysis (Figure 5B). The differences in conversion rates were based on known higher metabolic rates and species difference of esterase expression in plasma. We also have shown in our cell-based experiments that NMFTC was persistent within the cells due to lowered but sustained hydrolysis by cellular carboxylesterase activities.
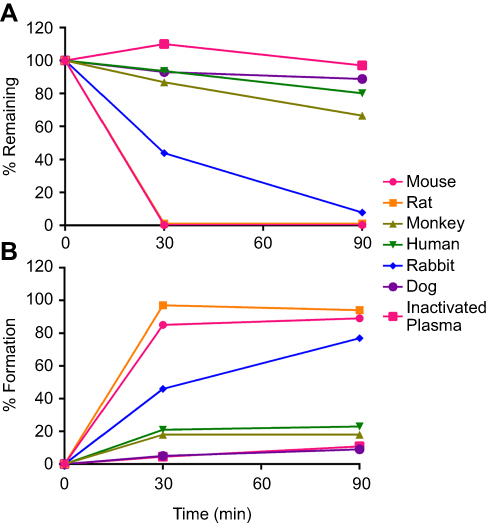 |
Figure 5 MFTC enzymatic cleavage kinetics in plasmas of six different species demonstrate species difference in (A) prodrug hydrolysis; rate of hydrolysis: mouse, rat>rabbit>monkey>dog and human. (B) The hydrolyzed prodrug reverted back to the parent drug as indicated by increased formation of FTC. Abbreviations: FTC, emtricitabine; MFTC, modified FTC prodrug. |
PK assessments
Sprague-Dawley rats were injected IM with either FTC or NMFTC at 45, 75 or 100 mg/kg FTC-equivalent doses to determine the effect of dose escalation on the PK profile. At 2 hrs, FTC plasma levels (Figure 6A) were several fold higher following FTC treatment compared to NMFTC treatment; however, by day 1 FTC concentrations had dropped markedly in the FTC treated animals, regardless of dose. On day 1, NMFTC provided 20-fold higher FTC plasma levels at all three doses compared with parent FTC (25, 34 and 52 ng/mL with FTC; 552, 696 and 1150 ng/mL with NMFTC for 45, 75 and 100 mg/kg treatments, respectively), indicating a slow release of FTC from the nanoformulation versus immediate release of the parent FTC. At day 7, NMFTC showed 11-, 14- and 40-fold higher FTC levels with respect to the three doses as compared with parent FTC (0, 3, and 1.4 ng/mL with FTC; 11, 42 and 55 ng/mL with NMFTC). At day 14, plasma FTC concentrations were fivefold lower than the reported 50% inhibitory concentration (IC50; ie, 8 ng/mL) and eightfold lower than the IC90 (ie, 14 ng/mL) in all NMFTC treatment groups. Tissue FTC levels in liver, spleen, lymph nodes and brain are shown in Figure 6B–E, respectively. At day 1, NMFTC showed 11- to 28-fold higher FTC levels in all tissues compared to parent FTC. In the liver at day 7, NMFTC showed 5-, 2- and 127-fold higher FTC levels compared to parent FTC at the respective doses (2.6, 14 and 0.6 ng/g with FTC; 13, 27 and 76 ng/g with NMFTC). In the spleen at day 7, NMFTC showed 10-, 15- and 10-fold higher FTC levels compared to parent FTC at the respective doses (0, 1.6 and 6 ng/g with FTC; 10, 25 and 59 ng/g with NMFTC). In lymph nodes at day 7, NMFTC showed 2-, 6- and 13-fold higher FTC levels as compared with parent FTC at the respective doses (17, 6 and 8 ng/g with FTC; 25, 35 and 104 ng/g with NMFTC). In the brain at day 7, NMFTC showed 2-, 18- and 10-fold higher FTC levels as compared with parent FTC at the respective doses (1.3, 1, and 0.7 ng/g with FTC; 2.7, 18, and 7 ng/g with NMFTC). At day 14, tissue FTC levels were detectable only in lymph nodes from the highest NMFTC dosed treatment group (36.9 ng/g).
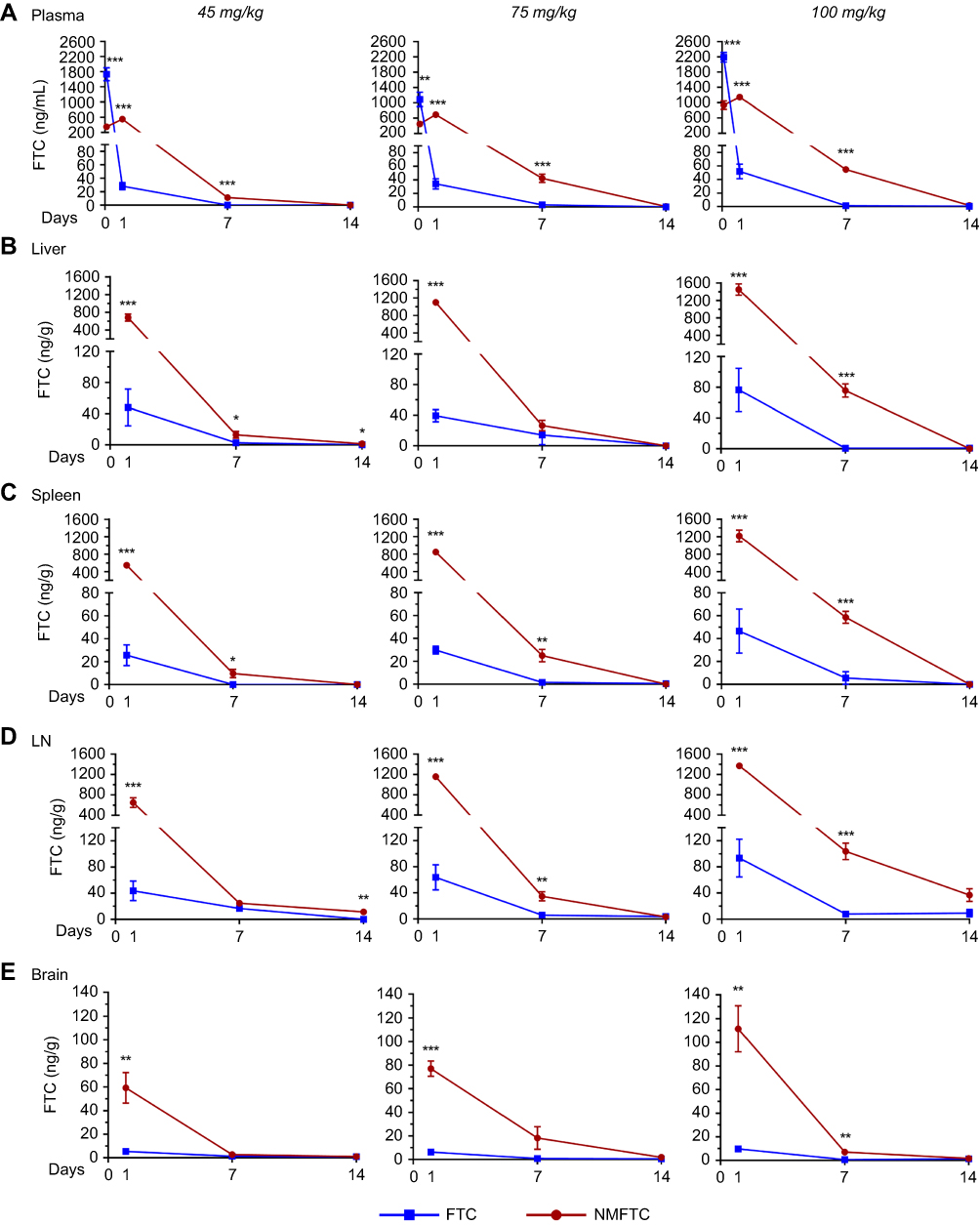 |
Figure 6 NMFTC pharmacokinetics. Sprague-Dawley rats were injected IM with 45, 75 or 100 mg/kg FTC or FTC-equivalent dose of NMFTC. NMFTC resulted in significantly higher FTC concentrations, as compared to FTC, in rat (A) plasma, (B) liver, (C) spleen, (D) LNs and (E) brain. Results are shown as mean±SEM, n=3–5; *P≤0.05, **P≤0.01, ***P≤0.001.Abbreviations: FTC, emtricitabine; NMFTC, nanoparticle of modified FTC prodrug; LN, lymph node; SEM, standard error of the mean. |
To determine whether the active triphosphate concentrations in lymphoid cells would be improved in vivo by treatment of rats with NMFTC compared to FTC, FTC-TP levels in PBMCs, splenocytes and lymph node cells were determined. Intracellular FTC-TP levels in PBMCs mirrored the FTC concentrations in plasma. At 2 hrs, PBMC FTC-TP levels were 2–3-fold higher with parent FTC treatment compared to NMFTC treatment (Figure 7A). However, on day 1, this was reversed and NMFTC treatment provided up to twofold higher FTC-TP levels compared to parent FTC (100, 133 and 163 fmol/106 cells with FTC; 116, 267 and 407 fmol/106 cells with NMFTC). At day 7, FTC-TP levels were eightfold higher with NMFTC compared to parent FTC (6, 7 and 8 fmol/106 cells with FTC; 43, 60 and 64 fmol/106 cells with NMFTC). Intracellular FTC-TP levels in spleen and lymph nodes are shown in Figure 7B and C, respectively. As compared to parent FTC, NMFTC treatment resulted in 2- to 4-fold higher FTC-TP levels in splenocytes and lymph node cells at days 1 and 7, respectively, with all doses. FTC-TP levels in spleen at day 1 were 6–13 fmol/106 cells with FTC and 27–36 fmol/106 cells with NMFTC. These levels decreased by day 7 to 2–3 fmol/106 cells with FTC and 6–8 fmol/106 cells with NMFTC, regardless of dose. FTC-TP levels in lymph node cells at day 1 were 4–7 fmol/106 cells with FTC and 8–13 fmol/106 cells with NMFTC. FTC-TP levels in lymph node cells dropped by day 7 to 1–2 fmol/106 cells with FTC and 3–4 fmol/106 cells with NMFTC regardless of dose. At day 14, detectable concentrations of FTC-TP in PBMCs, spleen and lymph node cells were observed following NMFTC treatment, with the highest concentration in lymph node cells of animals treated with the highest dose (up to 2 fmol/106 cells). Thus, NMFTC provided slower release of the drug in plasma and significantly higher drug levels in plasma and tissues for at least 7 days compared to the parent drug. Increasing the dose resulted in higher drug levels in plasma and tissues, however, no dose-dependent differences in intracellular FTC-TP were observed.
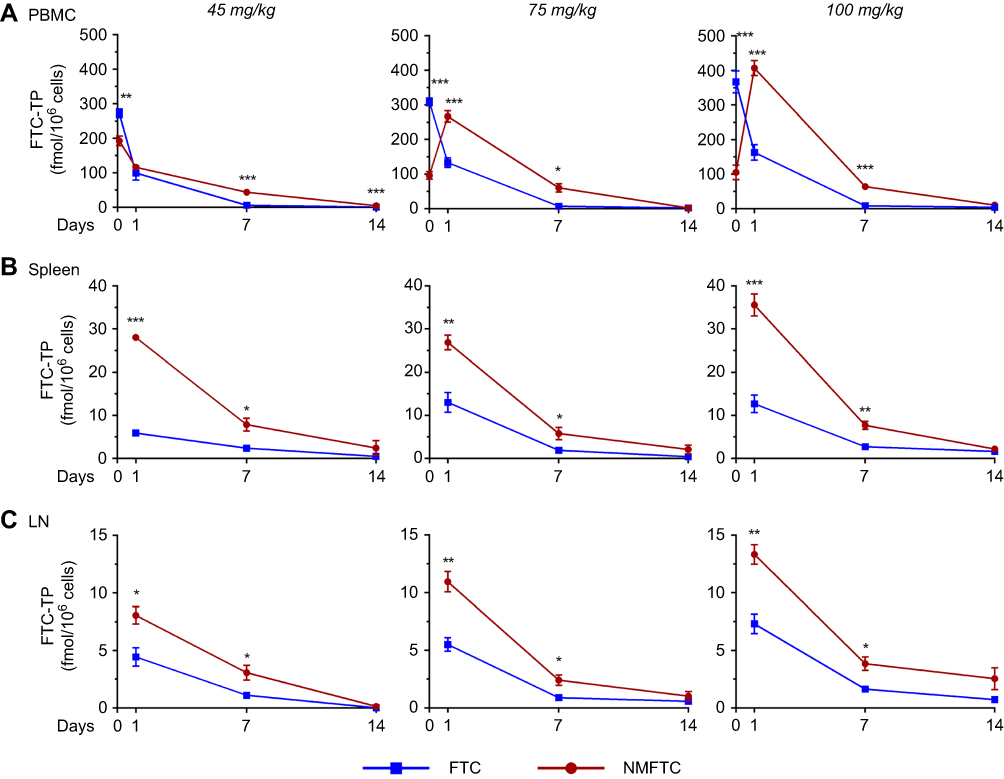 |
Figure 7 Intracellular FTC-TP. Sprague-Dawley rats were injected IM with 45, 75 or 100 mg/kg FTC or FTC-equivalent dose of NMFTC. NMFTC resulted in significantly higher intracellular FTC-TP concentrations, as compared to FTC, in rat (A) PBMCs, (B) spleen and (C) LNs. Results are shown as mean±SEM, n=3–5; *P≤0.05, **P≤0.01, ***P≤0.001.Abbreviations: FTC, emtricitabine; FTC-TP, FTC triphosphate; NMFTC, nanoparticle of modified FTC prodrug; PBMC, peripheral blood mononuclear cell; LN, lymph node; SEM, standard error of the mean. |
Discussion
While prior studies had attempted to generate a longer acting FTC prodrug, none were fully characterized or developed as a LA ARV strategy.35 In addition, critical measurement of FTC TP formation was lacking as well as in vivo and drug tissue biodistribution assays.35 In the current report, evaluation of drug potency in human MDMs demonstrated improved EC50 values for MFTC at 33 nM compared to 79 nM for FTC. Additionally, NMFTC showed enhanced lymphatic (spleen, lymph node and brain) tissue penetrance as compared to native FTC. The effective dose of MFTC required to show equipotent activity to FTC was reduced due to tissue targeting and sustained bioavailability of FTC that was cleaved from MFTC in plasma. This was compared to a rapid decline in FTC levels after native drug administrations. The preparation of poloxamer-coated nanocrystals can improve the surface area of the prepared structure and as such facilitate the interactions between the nanomaterial and cell and tissue targets. Additionally, crystallinity provides ease in formulation processing, scaleup, stability and solubility. Since MFTC is hydrophobic, its uniform dispersal was assisted by crystallization and subsequent homogenization, forming nanocrystals with physical properties that allow its dispersal in water and used as a nanosuspension for injection.
FTC is a potent cytidine analog that is transformed into its active triphosphate metabolite in cells where it competes with endogenous cytidine triphosphates for incorporation into the viral DNA chain during viral reverse transcription.32 It is a potential candidate for conversion into a LA ARV. However, a major obstacle in transforming FTC into an LA agent centers on its hydrophilicity. LA ART has taken several forms toward its development.36,37 One is controlled release microparticle carrier systems that rely on drug depot formation at the injection site.38 However, the created systems exhibit a tendency to aggregate due to lack of particle homogeneity leading to injection site reactions limiting their usage.39 Another is nanotechnology which comes into play by offering properties that facilitate creation of particles capable of crossing biological barriers and forming intracellular drug depots.40,41 These encased drug particles access into tissues where conventional formulations cannot readily penetrate.9 Yet another is enabling slow release which can be furthered by creation of prodrug nanocrystals.42 This is opposed to macroparticles that tend to form injection site depots where local adverse reactions are quite common.39
In the current study, a prodrug nanoformulation of FTC was developed to extend its apparent half-life and improve distribution to viral reservoir cells and tissues. FTC is a pivotal component of initial HIV therapy as per recommendations of the International Antiviral Society-USA Panel.17 Furthermore, FTC and TDF in combination have been shown to be effective as pre-exposure prophylaxis prevention in high-risk populations.21 Therapeutic limitations of current FTC include a short plasma half-life, which requires once-daily administration, and restriction of drug penetrance to viral reservoirs. These limitations emphasize the need for a LA parenteral nanoformulated FTC. LA therapeutics utilizing hydrophobic prodrugs have been very successful in the clinic for management of psychotic disorders.43,44 Our group has used similar strategies to develop hydrophobic prodrug nanoformulations of the HIV integrase inhibitors CAB and dolutegravir that extended their antiretroviral efficacy for more than a month.11,25 Others reported improvements in antiretroviral activity and cellular uptake with other FTC prodrugs.35,45 However, in contrast to our current study, some prodrugs were less hydrophobic and not encased in nanoformulations.46 Herein, modification of FTC was made by conjugating a 16-carbon fatty acid chain, palmitoyl chloride, markedly increased the drug’s hydrophobicity bringing the aqueous solubility from 134 to 0.12 mg/mL. This chemical modification allowed for synthesis of stable nanocrystals with high encapsulation efficiency.47 Furthermore, our method formed homogenous rod-shaped nanoparticles. Such nanoparticles have been shown to be readily taken up by macrophages as opposed to globular-shaped or spherical particles.48–50 Our prodrugs are designed to be both lipophilic and hydrophobic and as such requires a delivery strategy for sufficient dispersal in aqueous media or equivalent biological fluids. Thus, nanoformulating prodrug crystals allow their use in isotonic solutions while avoiding aggregation. This occurs at or following injection precluding emboli formation.
Macrophages are a major HIV reservoir. Thus targeting macrophages for effective treatment of HIV infection has received considerable attention.51–54 Moreover, macrophages are responsible for sustaining viral load and for spreading virus to other tissues.14,55 NMFTC macrophage uptake was sustained for up to 15 days. This supports the idea that macrophages could be harnessed as a vehicle for drug delivery to tissues where current ARVs have limited access.56 Interestingly, NMFTC resulted in inhibition of viral replication after viral challenge 10 days after drug loading. These observations were extended to in vivo studies where we observed several fold higher FTC and FTC-TP for at least 7 days in plasma, tissues and lymphoid cells after a single NMFTC injection compared to treatment with FTC. Importantly, the rate of hydrolysis of the prodrug and release of parent FTC was much slower in human plasma compared to rat plasma. These results suggest that this prodrug nanoformulation could prolong drug residence time and efficacy in humans due to differences in plasma esterase activities among species.57
Overall, and since their introduction in 1996, combination ART has changed the HIV clinical care landscape. The ARV regimens have effectively decreased HIV viral load and substantially reduced disease morbidity and mortality.58 However, due to the unique viral characteristics and disease course of HIV, its management requires strict regimen adherence.59 Moreover, since rapid viral replication can result in the emergence of mutations and drug resistance,60 new classes of drugs and alternative drug formulation delivery platforms are needed that allow infrequent dosing. To these ends, we created an LA FTC nanoformulation and demonstrated its abilities to inhibit HIV replication over extended time periods. We also note that the volume used in each of the studies can reflect values for human equivalent doses. All together, higher drug levels and distribution in tissue viral reservoirs for longer periods indicate that LA ARVs hold great promise towards prevention and treatment of HIV infections.61 Creation of hydrophobic prodrugs and their incorporation into nanoparticles provide one effective pathway towards improving current ART regimens.
Conclusion
We now demonstrate that a palmitoylated ester prodrug of FTC can extend in vivo drug concentrations to provide improved antiretroviral activity. The increased drug hydrophobicity facilitated the formation of a P407-stabilized lipophilic nanoformulation with high drug loading efficiency. This enhanced drug uptake and antiretroviral activity in MDMs. Retention of intracellular FTC-TP provided extended antiretroviral activity of NMFTC in MDMs. Interestingly, following a single IM injection, FTC concentrations were maintained in blood, reticuloendothelial tissues, and brain for at least 7 days at significantly higher levels with NMFTC as compared with the parent drug. These results paralleled intracellular FTC-TP levels in PBMCs and in cells isolated from spleen and lymph nodes.
Acknowledgements
This research was supported by the University of Nebraska Foundation, which includes donations from the Carol Swarts, M.D. Emerging Neuroscience Research Laboratory, the Margaret R. Larson Professorship, and the Frances, and Louie Blumkin, and Harriet Singer Endowment, the Vice Chancellor’s Office of the University of Nebraska Medical Center for Core Facility Developments, and National Institutes of Health grants R01 MH104147, P01 DA028555, R01 NS36126, P01 NS31492, 2R01 NS034239, P01 MH64570, P30 MH062261, P30 AI078498, R01 AG043540 and 1 R56 AI138613-01A1.
Author contributions
I.M.I prepared the work plan, contributed to the data acquisition, analysis, and interpretation of experimental results, and wrote the manuscript. H.E.G. conceived and supervised the project, designed and supervised the pharmacokinetic and virological experiments and directed the data interpretation. B.E. conceived the design of prodrug synthesis and formulations used, supervised the experiments, data analysis and interpretation. J.M.M, M.W. and B.L.D.S. assisted in tissue drug extraction, sample preparation, LC-MS/MS data acquisition and analysis. Y.A. and N.G. assisted in developing the method for FTC-TP extraction and contributed to LC-MS/MS data acquisition and analysis. A.N.B. planned, oversaw and implemented the animal studies and performed data analyses. Z.L. and D.S. assisted in drug injection, blood and tissue collection during the animal studies. H.E.G., B.E. and A.N.B. edited the manuscript. All authors contributed to data analysis, drafting or revising of the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dou H, Destache CJ, Morehead JR, et al. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood. 2006;108(8):2827–2835. doi:10.1182/blood-2006-03-013334
2. Chesney M. Adherence to HAART regimens. AIDS Patient Care STDS. 2003;17(4):169–177. doi:10.1089/108729103322494320
3. Pirrone V, Thakkar N, Jacobson JM, Wigdahl B, Krebs FC. Combinatorial approaches to the prevention and treatment of HIV-1 infection. Antimicrob Agents Chemother. 2011;55(5):1831–1842. doi:10.1128/AAC.01370-10
4. Li JZ, Etemad B, Ahmed H, et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS. 2016;30(3):343.
5. Bangsberg DR, Perry S, Charlebois ED, et al. Non-adherence to highly active antiretroviral therapy predicts progression to AIDS. AIDS. 2001;15(9):1181–1183.
6. Rueda S, Park‐Wyllie LY, Bayoumi A, et al. Patient support and education for promoting adherence to highly active antiretroviral therapy for HIV/AIDS. Cochrane Database Syst Rev. 2006;(3):CD001442.
7. Wong JK, Yukl SA. Tissue reservoirs of HIV. Curr Opin HIV AIDS. 2016;11(4):362. doi:10.1097/COH.0000000000000293
8. Carr A. Toxicity of antiretroviral therapy and implications for drug development. Nat Rev Drug Discov. 2003;2(8):624. doi:10.1038/nrd1151
9. Chun TW, Moir S, Fauci AS. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat Immunol. 2015;16(6):584–589. doi:10.1038/ni.3152
10. Barnhart M. Long-acting HIV treatment and prevention: closer to the threshold. Global Health Sci Pract. 2017;5(2):182–187. doi:10.9745/GHSP-D-17-00206
11. Zhou T, Su H, Dash P, et al. Creation of a nanoformulated cabotegravir prodrug with improved antiretroviral profiles. Biomaterials. 2018;151:53–65. doi:10.1016/j.biomaterials.2017.10.023
12. Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–1822. doi:10.1126/science.1095833
13. Meltzer MS, Skillman DR, Hoover DL, et al. Macrophages and the human immunodeficiency virus. Immunol Today. 1990;11:217–223. doi:10.1016/0167-5699(90)90086-O
14. Sattentau QJ, Stevenson M. Macrophages and HIV-1: an Unhealthy Constellation. Cell Host Microbe. 2016;19(3):304–310. doi:10.1016/j.chom.2016.02.013
15. Margolis DA, Gonzalez-Garcia J, Stellbrink H-J, et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet. 2017;390(10101):1499–1510. doi:10.1016/S0140-6736(17)31917-7
16. Williams J, Sayles HR, Meza JL, et al. Long-acting parenteral nanoformulated antiretroviral therapy: interest and attitudes of HIV-infected patients. Nanomedicine. 2013;8(11):1807–1813. doi:10.2217/nnm.12.214
17. Saag MS, Benson CA, Gandhi RT, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2018 recommendations of the International Antiviral Society-USA panel. JAMA. 2018;320(4):379–396. doi:10.1001/jama.2018.8431
18. Bruno R, Regazzi MB, Ciappina V, et al. Comparison of the plasma pharmacokinetics of lamivudine during twice and once daily administration in patients with HIV. Clin Pharmacokinet. 2001;40(9):695–700. doi:10.2165/00003088-200140090-00005
19. Ruane PJ, DeJesus E, Berger D, et al. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of tenofovir alafenamide as 10-day monotherapy in HIV-1–positive adults. J Acquir Immune Defic Syndr. 2013;63(4):449–455. doi:10.1097/QAI.0b013e3182965d45
20. Nelson M, Schiavone M. Emtricitabine (FTC) for the treatment of HIV infection. Int J Clin Pract. 2004;58(5):504–510. doi:10.1111/ijcp.2004.58.issue-5
21. McCormack S, Dunn DT, Desai M, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2016;387(10013):53–60. doi:10.1016/S0140-6736(15)00056-2
22. Guo D, Zhou T, Araínga M, et al. Creation of a long-acting nanoformulated 2′, 3′-dideoxy-3′-thiacytidine. J Acquir Immune Defic Syndr. 2017;74(3):e75.
23. Singh D, McMillan J, Hilaire J, et al. Development and characterization of a long-acting nanoformulated abacavir prodrug. Nanomedicine (Lond). 2016;11(15):1913–1927. doi:10.2217/nnm-2016-0233
24. Puligujja P, Balkundi SS, Kendrick LM, et al. Pharmacodynamics of long-acting folic acid-receptor targeted ritonavir-boosted atazanavir nanoformulations. Biomaterials. 2015;41:141–150. doi:10.1016/j.biomaterials.2014.11.012
25. Sillman B, Bade AN, Dash PK, et al. Creation of a long-acting nanoformulated dolutegravir. Nat Commun. 2018;9(1):443. doi:10.1038/s41467-018-02885-x
26. Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3(9):785. doi:10.1038/nrd1494
27. Arainga M, Guo D, Wiederin J, Ciborowski P, McMillan J, Gendelman HE. Opposing regulation of endolysosomal pathways by long-acting nanoformulated antiretroviral therapy and HIV-1 in human macrophages. Retrovirology. 2015;12::
28. Gendelman HE, Orenstein JM, Martin MA, et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167(4):1428–1441. doi:10.1084/jem.167.4.1428
29. Kevadiya BD, Woldstad C, Ottemann BM, et al. Multimodal theranostic nanoformulations permit magnetic resonance bioimaging of antiretroviral drug particle tissue-cell biodistribution. Theranostics. 2018;8(1):256. doi:10.7150/thno.22764
30. Gautam N, Lin Z, Banoub MG, et al. Simultaneous quantification of intracellular lamivudine and abacavir triphosphate metabolites by LC–MS/MS. J Pharm Biomed Anal. 2018;153:248–259. doi:10.1016/j.jpba.2018.02.036
31. Ottemann BM, Helmink AJ, Zhang W, et al. Bioimaging predictors of rilpivirine biodistribution and antiretroviral activities. Biomaterials. 2018;185:174–193. doi:10.1016/j.biomaterials.2018.09.018
32. Piliero PJ. Pharmacokinetic properties of nucleoside/nucleotide reverse transcriptase inhibitors. J Acquir Immune Defic Syndr. 2004;37(Suppl 1):S2–S12.
33. Li F, Maag H, Alfredson T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J Pharm Sci. 2008;97(3):1109–1134. doi:10.1002/jps.21047
34. Ito T, Sun L, Bevan MA, Crooks RM. Comparison of nanoparticle size and electrophoretic mobility measurements using a carbon-nanotube-based coulter counter, dynamic light scattering, transmission electron microscopy, and phase analysis light scattering. Langmuir. 2004;20(16):6940–6945. doi:10.1021/la0490184
35. Hobson JJ, Al-Khouja A, Curley P, et al. Semi-solid prodrug nanoparticles for long-acting delivery of water-soluble antiretroviral drugs within combination HIV therapies. Nat Commun. 2019;10(1):1413. doi:10.1038/s41467-019-09354-z
36. Rothstein SN, Huber KD, Sluis-Cremer N, Little SR. In vitro characterization of a sustained-release formulation for enfuvirtide. Antimicrob Agents Chemother. 2014;58(3):1797–1799. doi:10.1128/AAC.02045-12
37. Boffito M, Jackson A, Owen A, Becker S. New approaches to antiretroviral drug delivery: challenges and opportunities associated with the use of long-acting injectable agents. Drugs. 2014;74(1):7–13. doi:10.1007/s40265-013-0163-7
38. Li Q, Xie L, Caridha D, et al. Long-term prophylaxis and pharmacokinetic evaluation of intramuscular Nano-and microparticle decoquinate in mice infected with P. berghei sporozoites. Malar Res Treat. 2017;7508291. doi: 10.1155/2017/7508291.
39. Kohane DS. Microparticles and nanoparticles for drug delivery. Biotechnol Bioeng. 2007;96(2):203–209. doi:10.1002/bit.21193
40. Juillerat-Jeanneret L. The targeted delivery of cancer drugs across the blood–brain barrier: chemical modifications of drugs or drug-nanoparticles? Drug Discov Today. 2008;13(23–24):1099–1106. doi:10.1016/j.drudis.2008.09.005
41. Edagwa BJ, Guo D, Puligujja P, et al. Long-acting antituberculous therapeutic nanoparticles target macrophage endosomes. FASEB J. 2014;28(12):5071–5082. doi:10.1096/fj.14-255786
42. Sedky K, Nazir R, Lindenmayer J-P, Lippmann S. Paliperidone palmitate: once-monthly treatment option for schizophrenia. Curr Psychiatr. 2010;9(3):48–50.
43. Ravenstijn P, Remmerie B, Savitz A, et al. Pharmacokinetics, safety, and tolerability of paliperidone palmitate 3‐month formulation in patients with schizophrenia: a phase‐1, single‐dose, randomized, open‐label study. J Clin Pharmacol. 2016;56(3):330–339. doi:10.1002/jcph.578
44. Raedler LA. Aripiprazole lauroxil (Aristada): long-acting atypical antipsychotic injection approved for the treatment of patients with schizophrenia. Am Health Drug Benefits. 2016;9(Spec Feature):40.
45. Mandal S, Belshan M, Holec A, Zhou Y, Destache CJ. An enhanced emtricitabine-loaded long-acting nanoformulation for prevention or treatment of HIV infection. Antimicrob Agents Chemother. 2017;61(1):e01475–01416. doi:10.1128/AAC.01475-16
46. Agarwal HK, Chhikara BS, Bhavaraju S, Mandal D, Doncel GF, Parang K. Emtricitabine prodrugs with improved anti-HIV activity and cellular uptake. Mol Pharm. 2013;10(2):467–476. doi:10.1021/mp300361a
47. Dumortier G, Grossiord JL, Agnely F, Chaumeil JC. A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm Res. 2006;23(12):2709–2728. doi:10.1007/s11095-006-0283-9
48. Doshi N, Mitragotri S. Macrophages recognize size and shape of their targets. PLoS One. 2010;5(4):e10051. doi:10.1371/journal.pone.0010051
49. Nowacek AS, Balkundi S, McMillan J, et al. Analyses of nanoformulated antiretroviral drug charge, size, shape and content for uptake, drug release and antiviral activities in human monocyte-derived macrophages. J Control Release. 2011;150(2):204–211. doi:10.1016/j.jconrel.2010.11.019
50. Gratton SE, Ropp PA, Pohlhaus PD, et al. The effect of particle design on cellular internalization pathways. Proc National Acad Sci. 2008;105(33):11613–11618. doi:10.1073/pnas.0801763105
51. Barr SD, Ciuffi A, Leipzig J, Shinn P, Ecker JR, Bushman FD. HIV integration site selection: targeting in macrophages and the effects of different routes of viral entry. Mol Ther. 2006;14(2):218–225. doi:10.1016/j.ymthe.2006.03.012
52. Duncan CJ, Sattentau QJ. Viral determinants of HIV-1 macrophage tropism. Viruses. 2011;3(11):2255–2279. doi:10.3390/v3112255
53. Genis P, Jett M, Bernton EW, et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176(6):1703–1718. doi:10.1084/jem.176.6.1703
54. Unwalla H, Banerjea AC. Inhibition of HIV-1 gene expression by novel macrophage-tropic DNA enzymes targeted to cleave HIV-1 TAT/Rev RNA. Biochem J. 2001;357(1):147–155. doi:10.1042/bj3570147
55. Kumar A, Abbas W, Herbein G. HIV-1 latency in monocytes/macrophages. Viruses. 2014;6(4):1837–1860. doi:10.3390/v6041837
56. Ikehara Y, Niwa T, Biao L, et al. A carbohydrate recognition–based drug delivery and controlled release system using intraperitoneal macrophages as a cellular vehicle. Cancer Res. 2006;66(17):8740–8748. doi:10.1158/0008-5472.CAN-06-0470
57. Bahar FG, Ohura K, Ogihara T, Imai T. Species difference of esterase expression and hydrolase activity in plasma. J Pharm Sci. 2012;101(10):3979–3988. doi:10.1002/jps.23258
58. Carpenter CC, Fischl MA, Hammer SM, et al. Antiretroviral therapy for HIV infection in 1997: updated recommendations of the International AIDS Society—USA Panel. JAMA. 1997;277(24):1962–1969.
59. Friedland GH, Williams A. Attaining higher goals in HIV treatment: the central importance of adherence. AIDS. 1999;13:S61–72.
60. Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350(10):1023–1035. doi:10.1056/NEJMra025195
61. Edagwa JB, Zhou T, et al. Development of HIV reservoir targeted long acting nanoformulated antiretroviral therapies. Curr Med Chem. 2014;21(36):4186–4198. doi:10.2174/0929867321666140826114135
Supplementary material
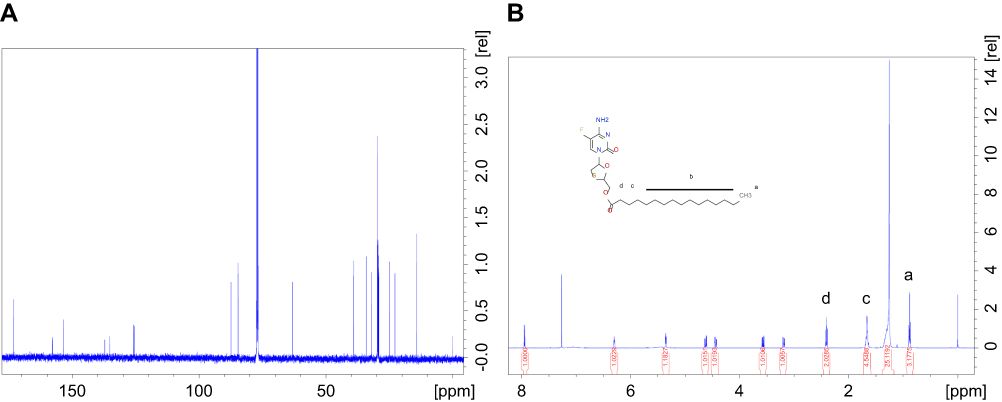 |
Figure S1 (A) 1H and (B) 13C- NMR spectra of MFTC in CDCl3. Additional peaks corresponding to the protons and carbon atoms of the fatty acid chain confirmed derivatization of FTC.Abbreviations: 1H-NMR, proton nuclear magnetic resonance; 13C-NMR, carbon nuclear magnetic resonance; MFTC, modifies FTC prodrug. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.