")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Synergistic roles of p53 and HIF1α in human renal cell carcinoma-cell apoptosis responding to the inhibition of mTOR and MDM2 signaling pathways
Authors Liu Q, Shen H, Lin J, Xu X, Ji Z, Han X, Shang D, Yang P
Received 18 May 2015
Accepted for publication 6 September 2015
Published 18 February 2016 Volume 2016:10 Pages 745—755
DOI https://doi.org/10.2147/DDDT.S88779
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Qing-jun Liu,* Hong-liang Shen,* Jun Lin, Xiu-hong Xu, Zheng-guo Ji, Xiao Han, Dong-hao Shang, Pei-qian Yang
Department of Urology Surgery, Beijing Friendship Hospital, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Introduction: mTOR and MDM2 signaling pathways are frequently deregulated in cancer development, and inhibition of mTOR or MDM2 independently enhances carcinoma-cell apoptosis. However, responses to mTOR and MDM2 antagonists in renal cell carcinoma (RCC) remain unknown.
Materials and methods: A498 cells treated with MDM2 antagonist MI-319 and/or mTOR inhibitor rapamycin were employed in the present study. Cell apoptosis and Western blot analysis were performed.
Results and conclusion: We found that the MDM2 inhibitor MI-319 induced RCC cell apoptosis mainly dependent on p53 overexpression, while the mTOR antagonist rapamycin promoted RCC cell apoptosis primarily through upregulation of HIF1α expression. Importantly, strong synergistic effects of MI-319 and rapamycin combinations at relatively low concentrations on RCC cell apoptosis were observed. Depletion of p53 or HIF1α impaired both antagonist-elicited apoptoses to differential extents, corresponding to their expression changes responding to chemical treatments, and double knockdown of p53 and HIF1α remarkably hindered MI-319- or rapamycin-induced apoptosis, suggesting that both p53 and HIF1α are involved in MDM2 or mTOR antagonist-induced apoptosis. Collectively, we propose that concurrent activation of p53 and HIF1α may effectively result in cancer-cell apoptosis, and that combined MDM2 antagonists and mTOR inhibitors may be useful in RCC therapy.
Keywords: renal cell carcinoma, mTOR, MDM2, p53, HIF1α, apoptosis
Introduction
Renal cell carcinoma (RCC) leads to approximately 2% of all adult malignancies, and accounts for over 90% of the kidney neoplasms. Recent statistics report that there are predicted to be 65,000 new cases and 14,000 deaths in 2013 from RCC,1 highlighting the harm of this cancer. However, the pathogenesis of RCC and the signaling pathways and genes associated with RCC remain unclear. The most common histologic subtype of RCC is clear-cell RCC, and the majority of sporadic RCC patients have mutations or epigenetic silencing of the VHL tumor-suppressor gene.2 The best-characterized VHL functions negatively regulate the HIFα family of transcription factors (HIF1α, HIF2α, HIF3α) in an oxygen-dependent manner via its E3 ubiquitin ligase activity.3,4
EGFR overexpression has frequently been identified in RCC, and EGFR signaling is essential for malignant renal tubular cells.5,6 Downstream effectors of EGFR signaling include the Ras/Raf/MAP kinase and PI3K/AKT pathways. PI3K signaling leads to phosphorylation of AKT and the activation of protein translation initiation via mTOR,7 which have been widely investigated in RCC and other aggressive diseases.8–10 The mTOR protein kinase interacts with several proteins to form two distinct complexes – mTORC1 and mTORC2 – to regulate downstream-factor activity. mTOR integrates extracellular growth signals with such cellular responses as proliferation, autophagy, metabolism, cell growth, and survival.11 mTOR activates HIF1α and inhibits neuronal apoptosis in the developing rat brain.12,13 In contrast, inhibition of mTOR activity suppresses the expression of HIF1α, which determines sensitivity to mTOR inhibitors in cancers.14,15 Deregulation of HIF1α appears to be essential to the disease process. These observations demonstrate that HIF1α is tightly interacted with the mTOR signaling pathway in cell survival and apoptosis. Blocking mTOR signaling activity by rapamycin and its derivatives, which are serine/threonine kinase inhibitors, has been revealed to inhibit tumor-cell growth and induce cancer-cell apoptosis in various cancers, including RCC.9,16,17 Two rapamycin derivatives – RAD001 and CCI-779 – have demonstrated promising antitumor activity with relatively minor toxicity in early clinical trials in advanced cancer patients.18–20 Notably, CCI-779 has been examined as a single agent in RCC, although with a low response rate (7%).21 Rapamycin has shown additive or synergistic activity when combined with other tyrosine-kinase inhibitors.22 Rapamycin also enhances chemotherapy responses in breast cancer cells23 and overcomes EGFR resistance in non-small-cell lung cancer cells.24 Therefore, the combination of mTOR inhibitors and other antagonists may be more active than either agent alone.
The tumor suppressor p53 is the most frequently mutated gene in human cancers, and mediates cell-cycle arrest and apoptosis in RCC.25–27 p53 Transactivation is inhibited by MDM2, a nuclear protein induced by p53, and MDM2 also mediates p53 stabilization and degradation in p53-mediated apoptosis.28–30 A large percentage of human tumors have amplified MDM2, leading to p53 loss and tumorigenesis.31 These observations implicate the importance of p53 and MDM2 feedback-loop interactions in regulating cancer development. Inhibition of MDM2 by antagonists or depletion of MDM2 expression induces apoptosis in leukemia cells32 and other kinds of carcinoma cells.33–35 Although MDM2 has been suggested to be a biomarker and potential therapy target in RCC prognosis and treatment,36–38 but the role of MDM2 in RCC cells is not well understood. Moreover, dual inhibition of PI3K/mTOR could inhibit MDM2 to enhance p53-mediated apoptosis in leukemia,39 but the cross talk between MDM2 and mTOR signaling pathways in RCC is not clear either.
We hypothesized that the combination of an MDM2 inhibitor plus rapamycin might induce apoptosis in RCC cell lines in a synergistic manner, which was proved at low drug concentrations. We also explored the role of p53 and HIF1α, which act downstream of the MDM2 and mTOR signaling pathways, to mediate antagonist-induced apoptosis in a cooperative cross-regulated manner.
Materials and methods
Cell culture and treatment
The RCC cell line A498 (VHL-mutant) was originally obtained from the American Type Culture Collection (Manassas, VA, USA). A498 cells were grown in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal calf serum. Cells were washed once with phosphate-buffered saline and treated with indicated concentrations of the MDM2 antagonist MI-319 (synthesized using previously published methods)40 and/or the mTOR inhibitor rapamycin (Sigma-Aldrich Co, St Louis, MO, USA). The protocol in the present study was reviewed and approved by the Human Clinical and Research Ethics Committees.
Cell-apoptosis assay
Cells were plated in duplicate and treated with the indicated drug for 48 hours. Percentages of apoptotic cells were determined by staining with propidium iodide, and the percentages of sub-G1 cells were considered apoptotic cells. Flow cytometry was performed on a Beckman cytometer, and data were analyzed using FlowJo software.
Western blot
A498 cells were washed and lysed in buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% docetaxel, 0.1% sodium dodecyl sulfate, 1 μg/mL aprotinin, and 100 μg/mL phenylmethylsulfonyl fluoride supplemented with phosphatase-inhibitor cocktails (Sigma-Aldrich). Samples were separated by sodium dodecyl sulfate polyacrylamide-gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were blocked in 10% nonfat milk phosphate-buffered saline buffer and incubated with primary antibodies overnight at 4°C. ECL Lightning Plus reagent (GE Healthcare, Little Chalfont, UK) was used for protein detection. The primary antibodies used in the experiments were p53 (Cell Signaling Technology Inc, Danvers, MA, USA), HIF1α (Novus Biologicals, Littleton, CO, USA), p21 (Santa Cruz Biotechnology Inc, Dallas, TX, USA), MDM2 (Santa Cruz Biotechnology), Bax (BD Biosciences, San Jose, CA, USA), p-mTOR (Ser2448; Cell Signaling Technology), p-S6K (Ser389; Santa Cruz Biotechnology), and β-actin (Sigma-Aldrich).
siRNA
A498 cells were plated at 1×105 cells in 60 mm dishes. Chemically synthesized small interfering RNAs (siRNAs) were purchased from GeneChem (Montreal, Canada) and transfected into A498 cells with Lipofectamine 2000 according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA). The siRNA target sequences were: human p53, 5′-GCATCTTATCCGAGTGGAA-3′; human HIF1α, 5′-CCTATATCCCAATGGATGATG-3′; and control siRNA, 5′-CGTACGCGGAATACTTCGA-3′.
Statistical analysis
Data are presented as means ± standard deviation, and statistical evaluations were carried out using GraphPad Prism 5.0 (GraphPad Software Inc, La Jolla, CA, USA). For all tests, three independent experiments were performed and similar results obtained. Statistical significance was measured by Student’s t-test, and levels of statistical significance was set at *P<0.05 and **P<0.01.
Results
Suppression of MDM2 induces cell apoptosis and p53 upregulation in human RCC cells
MDM2 overexpression has been associated with increased metastasis and advanced disease in several cancers.36 MDM2 is considered a risk factor and has clinical significance combining with the p53 and BCL2 genes in RCC.37,41 However, the role of MDM2 in RCC remains unknown. We used an MDM2 antagonist (MI-319) to treat RCC A498 cells for 48 hours, and then cell apoptosis was examined. As expected, MDM2 inhibition resulted in A498 cell apoptosis in a dose-dependent manner (Figure 1A and B). The potentially lethal activities of p53 are regulated by the proto-oncogene MDM2.36 To investigate the correlation between MDM2–p53 signaling and the mTOR–HIF1α cascade, the protein levels of these pathway-related proteins and apoptosis-related Bax were determined. We found that p53, p21, MDM2, and Bax were remarkably upregulated, while p-mTOR and p-S6K (Ser389) levels, which represent mTOR signaling activity, were suppressed upon MI-319 treatment (Figure 1C and D). However, the expression of HIF1α was only slightly increased by MI-319 stimulation. Therefore, blocking MDM2 activity results in strong accumulation of p53 proteins and efficient RCC cell apoptosis.
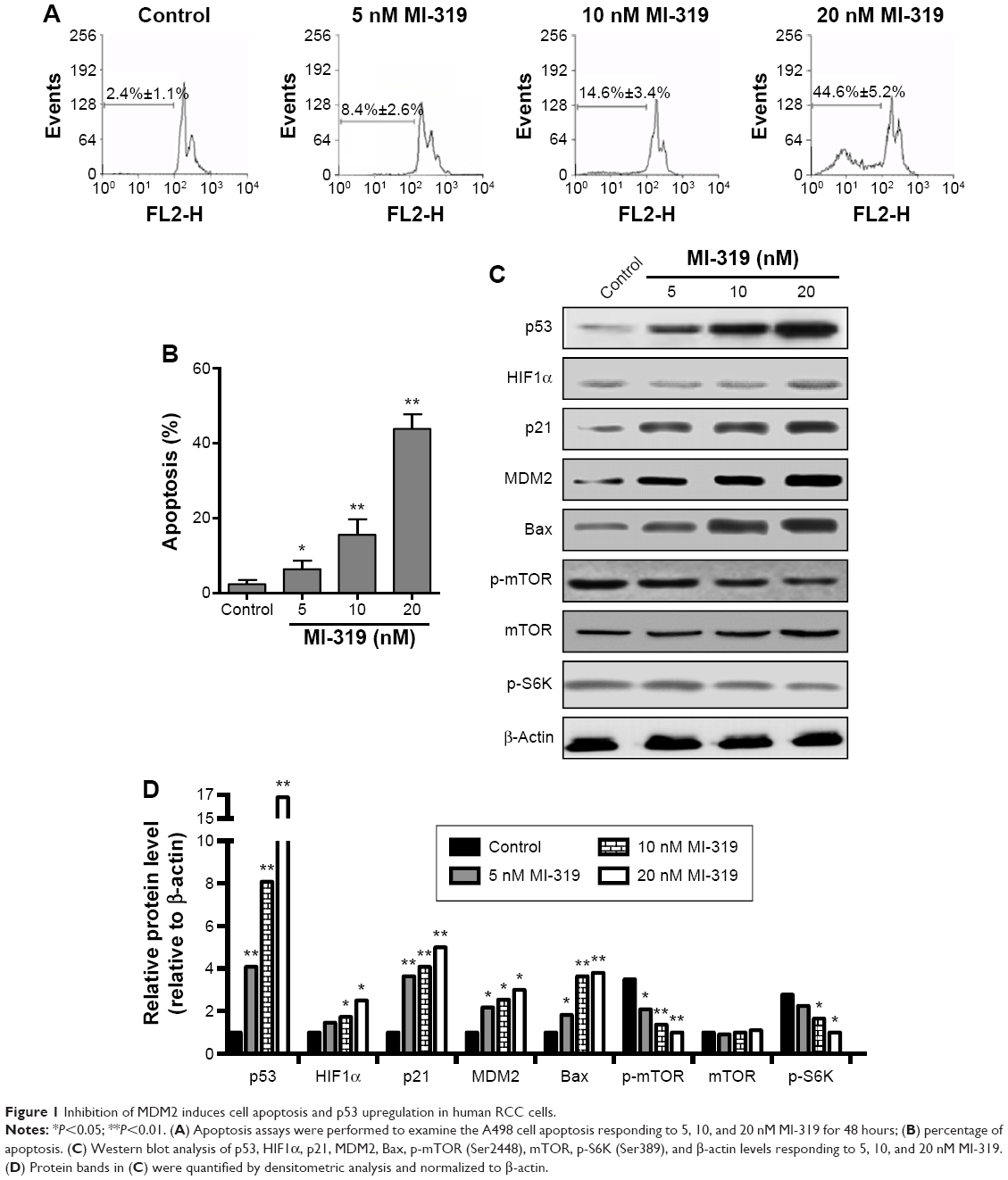 | Figure 1 Inhibition of MDM2 induces cell apoptosis and p53 upregulation in human RCC cells. |
mTOR inhibition leads to apoptosis and remarkable HIF1α upregulation in RCC cells
mTOR signaling is overactivated in RCC, and inhibition of mTOR leads to multiple kinds of cancer-cell apoptosis.42,43 Therefore, the effects of mTOR inhibitors on RCC cell survival, as well as its downstream signaling, were tested. Similar to MI-319 treatment, the mTOR inhibitor rapamycin induced cell apoptosis in a dose-dependent manner, and both antagonists induced only <10% of apoptotic cells at 5 nM (Figure 2A and B). Moreover, HIF1α, p21, and Bax were strongly induced upon increased concentrations of rapamycin treatment, and MDM2, p-mTOR, and p-S6K levels were concurrently downregulated (Figure 2C and D). Interestingly, p53 exhibited minor increased expression upon rapamycin stimulation, and mTOR signaling activity downregulation was more obvious than MI-319-elicited effects (Figures 1C and 2C, D). Therefore, we concluded that mTOR-HIF1α might be the major target of rapamycin-induced apoptosis.
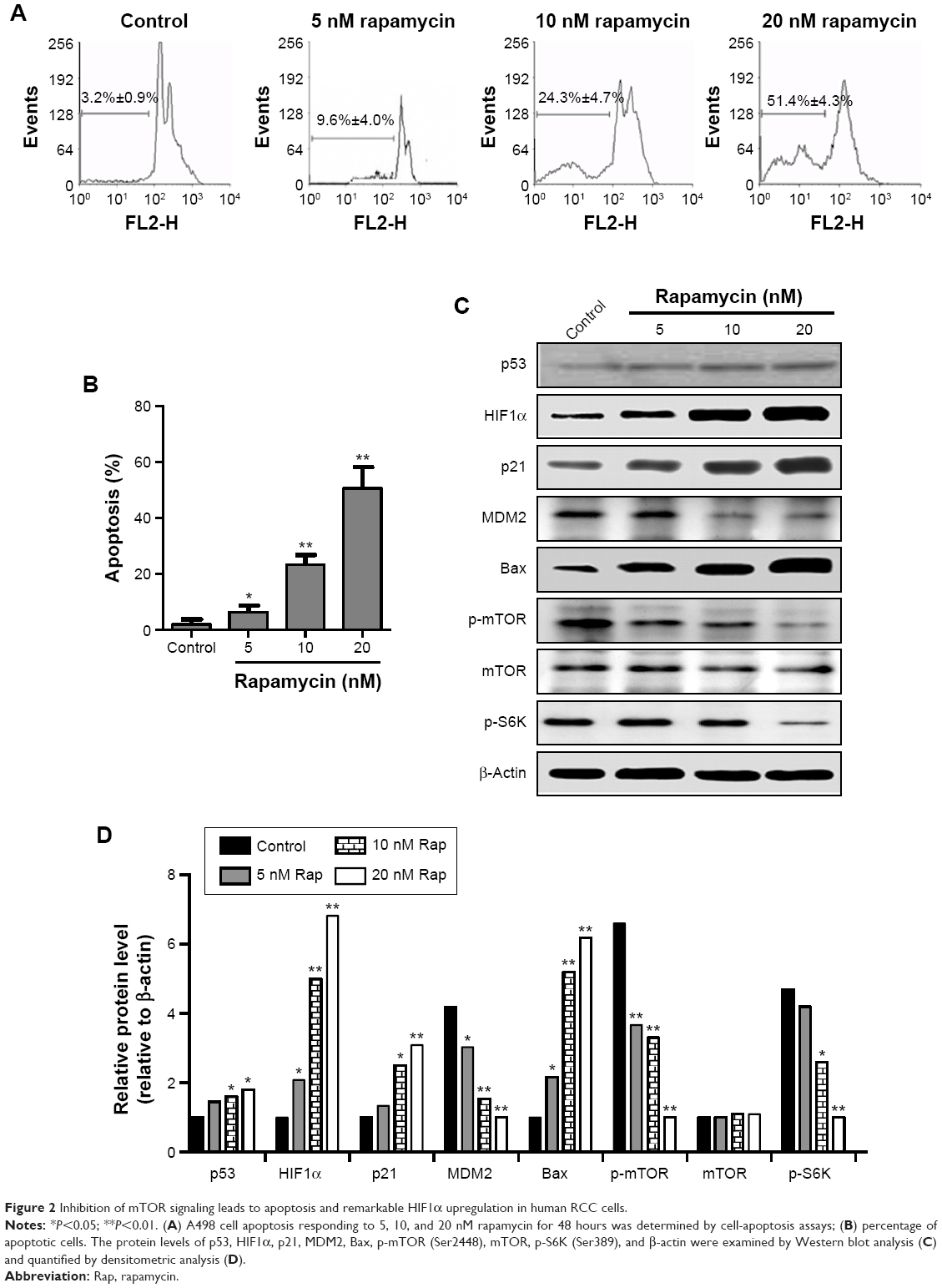 | Figure 2 Inhibition of mTOR signaling leads to apoptosis and remarkable HIF1α upregulation in human RCC cells. |
P53 and HIF1α mediate antagonist-induced apoptosis
To simply define the induction sensitivity of p53 and HIF1α expression by MDM2 and mTOR antagonists, we calculated the p53- and HIF1α-expression response to MI-319 or rapamycin (Figures 1D and 2D), and found that both p53 and HIF1α were enhanced by MI-319 or rapamycin, while p53 was more responsive to MI-319, HIF1α was more responsive to rapamycin treatment. Therefore, we hypothesized that both p53 and HIF1α account for both antagonist-induced apoptoses with division of labor. To test this, p53 and HIF1α were knocked down by their specific siRNAs (Figure 3B and E). Then, the apoptosis of siRNA-transfected A498 cells responding to MI-319 or rapamycin treatment was determined. p53 depletion antagonized MI-319- or rapamycin-induced (20 nM for both inhibitors) apoptosis, while p53 siRNA displayed much stronger effect in antagonizing MI-319-induced effects than rapamycin-elicited apoptosis (Figure 3C and D). In contrast, HIF1α depletion inhibited rapamycin-induced apoptosis to a more apparent extent than MI-319-elicited effects (Figure 3E and F). Combining these results with p53- and HIF1α-expression changes upon MI-319 or rapamycin treatment (Figures 1C and 2C), we concluded that both p53 and HIF1α may play essential roles in the division of labor in MDM2 or mTOR inhibition-induced apoptosis, depending on their expression-change response to different stimulations in RCC cells.
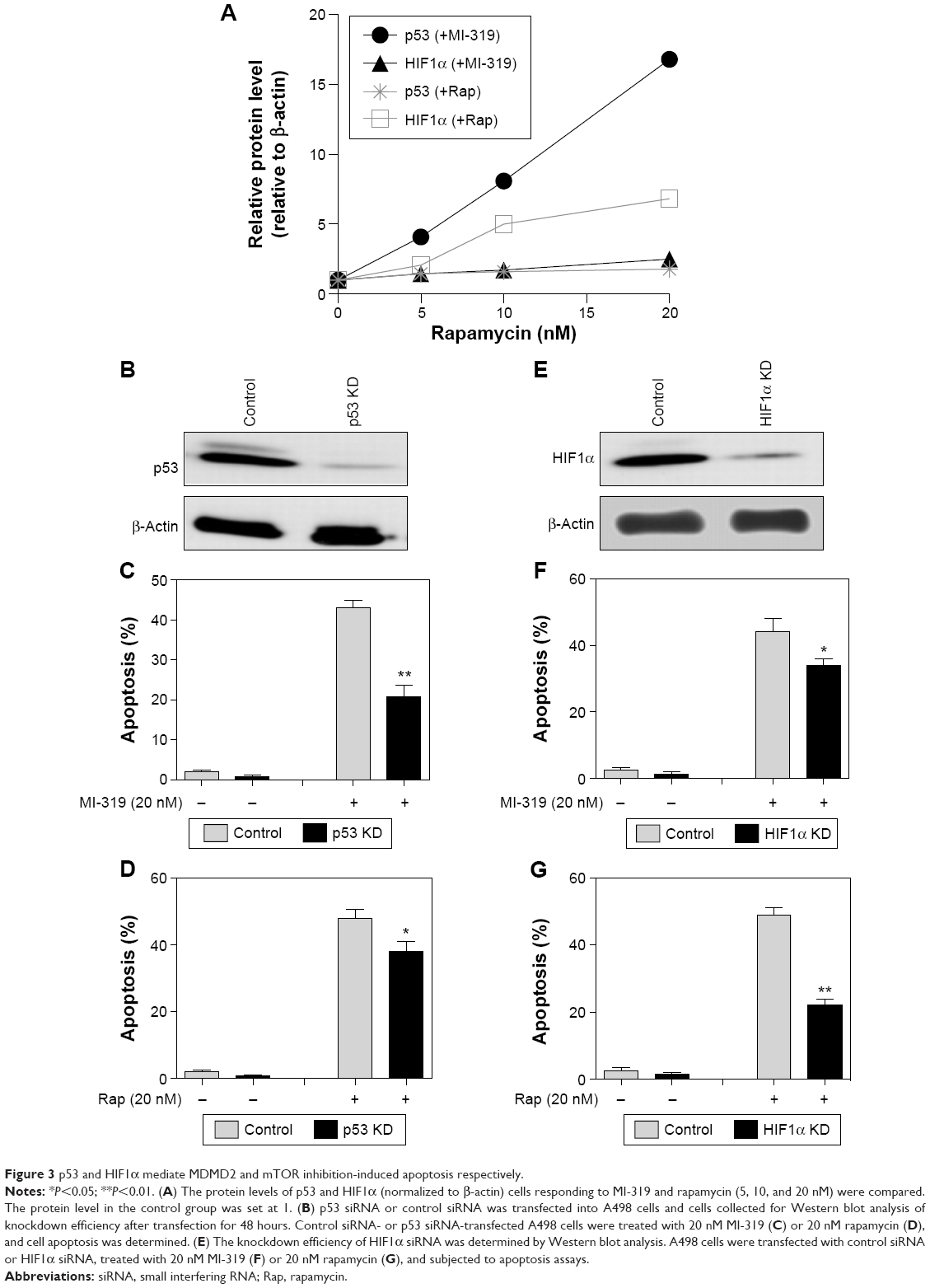 | Figure 3 p53 and HIF1α mediate MDMD2 and mTOR inhibition-induced apoptosis respectively. |
Synergistic roles of MDM2 and mTOR inhibition in RCC cell apoptosis
Inhibition of MDM2 or mTOR by their antagonists resulted in strong RCC cell apoptosis at 20 nM, while the effect was minor at lower concentrations. We asked whether the mTOR inhibitor could cooperate with MDM2 antagonists to generate stronger apoptosis effects at lower concentrations. We treated A498 cells with 5 nM MI-319, 5 nM rapamycin, or combinations of both inhibitors. Surprisingly, synergistic effects of MDM2 and mTOR inhibitors were observed, and the combination-induced apoptosis was much more than the summation of the percentages of single agent-elicited apoptosis (Figure 4A). Accordingly, p53 and HIF1α expression was markedly enhanced by the combination treatment accompanied by the upregulation of Bax and p21 expression (Figure 4B and C). Therefore, MDM2 and mTOR inhibition produces synergistic effects in RCC cell apoptosis and upregulates both p53 and HIF1α expression.
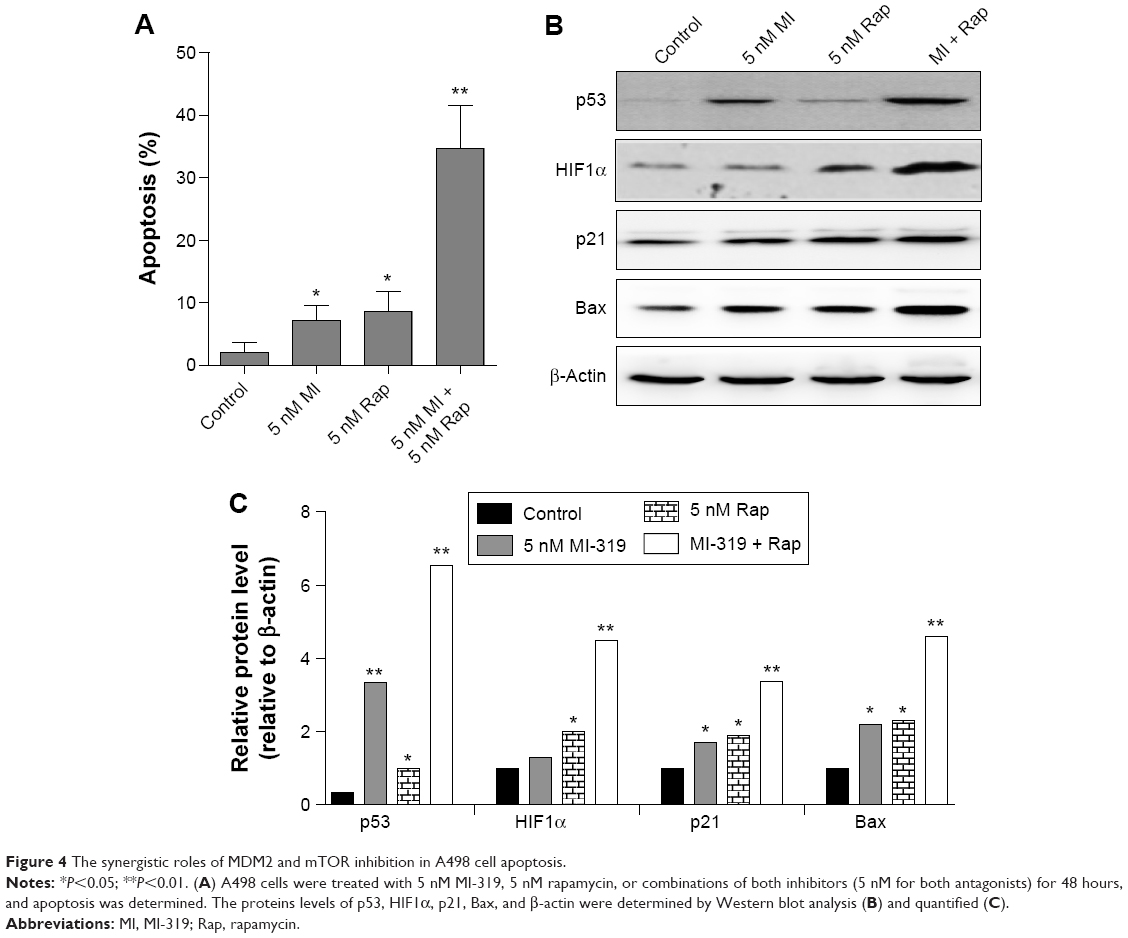 | Figure 4 The synergistic roles of MDM2 and mTOR inhibition in A498 cell apoptosis. |
The cooperative roles of p53 and HIF1α in mediating antagonist-induced apoptosis
In view of the strong response of p53 and HIF1α to concurrent inhibition of MDM2 and mTOR, the hypothesis that p53 and HIF1α may play cooperative roles in MDM2 or mTOR inhibition-induced apoptosis was tested. p53 siRNA and HIF1α siRNA were transfected into A498 cells simultaneously, leading to the downregulation of both proteins (Figure 5A). Then, the double-transfected cells were treated with MI-319 or rapamycin (20 nM for both inhibitors), and cell apoptosis was examined by apoptosis assays. We found that either MI-319- or rapamycin-induced apoptosis was substantially decreased by concurrent depletion of p53 and HIF1α (Figure 5B). We also calculated the percentage of p53- or HIF1α-mediated apoptosis upon MI-319 or rapamycin treatment. It was found that p53 and HIF1α are responsible for MI-319- or rapamycin-induced apoptosis with more or less division of labor, whereas p53 and HIF1α cooperatively account for over 70% of the both antagonist-induced apoptosis. Collectively, p53 and HIF1α play independent and cooperative roles in mediating MDMs and mTOR antagonist-induced apoptosis.
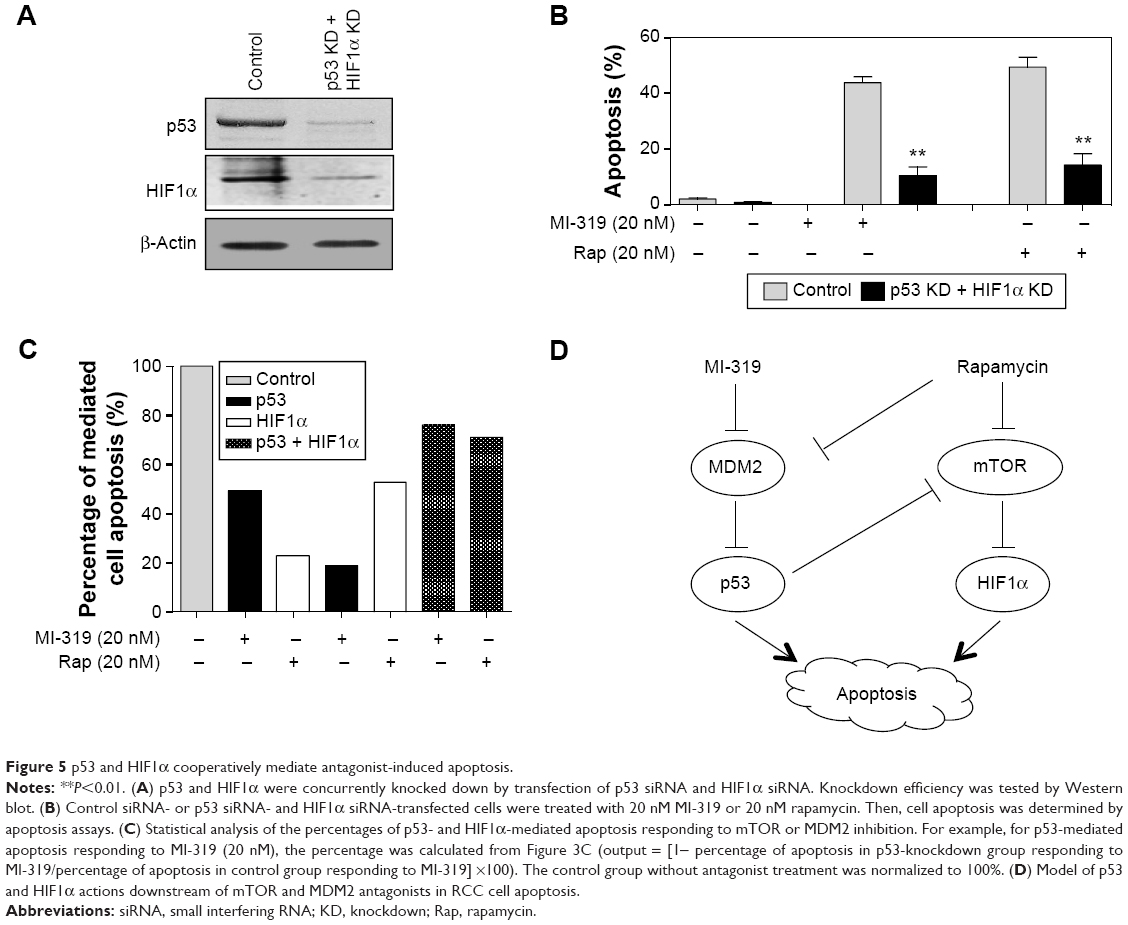 | Figure 5 p53 and HIF1α cooperatively mediate antagonist-induced apoptosis. |
Discussion
The present study demonstrates that combined inhibition of MDM2 and mTOR provides more effective induced apoptosis in RCC cell lines than either agent alone, and that p53 and HIF1α cooperatively mediate the synergistic effects elicited by MDM2 and mTOR inhibitors. More importantly, the proportions of p53- or HIF1α-mediated apoptosis depend on their response sensitivity to MDM2-antagonist or mTOR-inhibitor treatment. These results indicate that multiple signaling pathways, such as MDM2 and mTOR signaling, are involved in RCC cell survival and apoptosis through activating different downstream effectors, including p53 and HIF1α.
We showed that mTOR inhibition resulted in efficient RCC cell apoptosis, mainly through upregulation of HIF1α. Consistently, it has been reported that catalytic mTOR inhibition decreases cellular proliferation and induces apoptosis in RCC cell lines.9 It has also been demonstrated that mTOR inhibition induces a more robust induction of compensatory MEK/ERK signaling.9 Nonetheless, MEK/ERK signaling is considered a cell survival-signaling pathway and can be targeted with small-molecule kinase inhibition, resulting in enhanced therapeutic efficacy.9 However, we found that mTOR inhibition strongly enhanced HIF1α expression (Figure 2), and the upregulated HIF1α always acts as an apoptosis inducer.44 Elevated HIF1α may disrupt the downstream metabolic pathways and activate the apoptotic pathway.44 Depletion of HIF1α significantly antagonized mTOR inhibitor-induced apoptosis, suggesting that HIF1α is a negative regulator of the mTOR pathway in cell survival. Although it has been reported that inhibition of mTOR activity suppresses the expression of HIFα,14,15 this may result from a difference in cell types, especially for normal somatic cells responding to hypoxia or cancer cells.14,44 The oncogenic or tumor-suppressive effects of HIFα may also depend on other signaling activities.44 Here, at least in RCC cells, anti-mTOR-induced HIFα showed antitumor activity to induce RCC cell apoptosis. When mTOR signaling activity is inhibited by rapamycin, inhibition of HIF1α expression is released, resulting in HIF1α upregulation and consequent apoptosis. Moreover, rapamycin can also increase p53-dependent apoptosis by inhibition of MDM2 in cancer cells.45 Consistently, rapamycin treatment results in MDM2 downregulation and p53 upregulation in RCC cells (Figure 2), and p53 also mediates a minority of rapamycin-induced apoptosis (Figure 3). This demonstrates that there is a cross-link between MDM2 and mTOR signaling, emerging at key proapoptotic factors, such as p53 and HIF1α. Therefore, rapamycin may induce cell apoptosis or cell-cycle arrest, indicated by p21 increase through induction of HIF1α, and Bax upregulation is the outcome of HIF1α-induced apoptosis.
Although MDM2 has been found to be associated with RCC disease,36–38 the specific role of MDM2 inhibition in anti-RCC remains unknown. Here, we first showed that an MDM2 antagonist promotes RCC cell apoptosis in a dose-dependent manner (Figure 1). As a classical downstream factor of MDM2, p53 stabilization and degradation is regulated by MDM2 in p53-mediated apoptosis.28–30 To explore the mechanism underlying MDM2 inhibitor-induced apoptosis, p53 was preferentially considered. We found that p53 indeed mediated large proportions of MI-319-induced apoptosis. MI-319 induced the upregulation of p53, p21, MDM2, as well as Bax, and a slight increase in HIF1α. Interestingly, HIF1α also mediated a minority of MI-319-elicited apoptosis (Figure 3). It is possible that MI-319 triggers the apoptotic pathway by upregulation of p53 and HIF1α, and MDM2, Bax, and p21 upregulation is the molecular consequence of cell-status alterations undergoing apoptosis. As for downregulation of the mTOR pathway, this could be explained by the possibility that MI-319-induced p53 activation/accumulation inhibits the activity of the mTOR–HIF1α signaling cascade.46,47 These results further confirm the cross talk between MDM2 and mTOR signaling pathways at distinct emerging signaling cascades.
Notably, the synergistic effects of RCC apoptosis upon combined MDM2-antagonist and mTOR-inhibitor treatment were observed (Figure 4). It is widely known that the combination of an mTOR antagonist with other kinase inhibitors produces much stronger effects in cell-proliferation suppression and proapoptosis and overcomes chemical resistance.48–50 The synergistic role of MDM2 and mTOR inhibitors was observed at low concentrations, which may produce minor toxicity and be convenient for therapy applications. The combined use of an MDM2 antagonist and mTOR inhibitor may prevent RCC cell survival and cancer progression. Concurrent depletion of p53 and HIF1α strikingly antagonized MI-319- or rapamycin-induced apoptosis, demonstrating the synergistic roles of p53 and HIF1α in mediating the two antagonist-elicited apoptoses. However, depletion of p53 and HIF1α did not fully block the antagonist-induced apoptosis (Figure 5A). This might have been because of the incomplete knockdown efficiency and other apoptosis-related pathways involved in this process. Although there have been implications indicating a correlation between p53 and HIF1α regulation in apoptosis,51,52 here we first established the synergistic linkage between p53 and HIF1α downstream of the MDM2 and mTOR pathways, which has clinical significance for understanding the mechanism of potential anticancer agents. Recently, mTOR has emerged as an attractive cancer-therapy target, while resistance to mTOR inhibitors in RCC cells severely impairs the efficiency for cancer therapy. Several potential mechanisms leading to resistance to mTOR inhibitors have been proposed, including activation of the HIF pathway.53 According to our current observations, the combination of MDM2 inhibitors with mTOR inhibitors may overcome or impair resistance to mTOR inhibitors in RCC chemotherapy.
Previously, it has been revealed that the effects of mTOR inhibition or combination with other kinase inhibitors are influenced by the mutational states of VHL. Wild-type or VHL-mutant RCC cells display different apoptotic sensitivity to these antagonists.54 Here we found that combination of an mTOR inhibitor and MDM2 antagonist efficiently induced VHL-mutant A498 RCC cell apoptosis at relatively low concentrations (Figure 4). We also tested this combination effect in wild-type RCC7 cells, and synergistic effects were also observed (data not shown). We primarily suggest that the combination of mTOR and MDM2 inhibition induces RCC apoptosis independently of VHL-mutation states, which needs to be fully elucidated in future. Besides, both p53 and mTOR are involved in the autophagy pathway, favoring the adaptation and survival of cells,55,56 but here both MI-319 and rapamycin treatment elicited a large percentage of cell death, accompanied by cell disassembly (data not shown). Therefore, MI-319 and rapamycin treatment is not likely to activate the autophagy pathway.
In summary, our study demonstrates that inhibition of MDM2 or mTOR signaling leads to effective RCC cell apoptosis, and combination of MDM2 and mTOR antagonists’ results in more cell apoptosis at low concentrations. MDM2 inhibition acts mainly through upregulation of p53 to promote apoptosis, and the increased p53 may also inhibit the mTOR pathway to promote HIF1α expression and apoptosis. Alternatively, mTOR inhibition promotes apoptosis through enhancing HIF1α expression, and the mTOR inhibitor rapamycin may also repress MDM2 signaling to promote apoptosis (Figure 5D). Suppression of the mTOR and MDM2 signaling pathways promotes RCC cell apoptosis through concurrent activation of p53 and HIF1α through direct or indirect pathways, and the integrated output signaling activity synergistically results in cancer-cell apoptosis. The striking effects of combined MDM2 antagonists and mTOR inhibitors provide a novel potential therapeutic strategy for RCC treatment.
Acknowledgment
This study was supported by the National Natural Science Foundation of China (81202029).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | ||
Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994;91(21):9700–9704. | ||
Hanna SC, Heathcote SA, Kim WY. mTOR pathway in renal cell carcinoma. Expert Rev Anticancer Ther. 2008;8(2):283–292. | ||
Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8(11):865–873. | ||
Dorđević G, Matušan Ilijaš K, Hadžisejdić I, Maričić A, Grahovac B, Jonjić N. EGFR protein overexpression correlates with chromosome 7 polysomy and poor prognostic parameters in clear cell renal cell carcinoma. J Biomed Sci. 2012;19:40. | ||
Szymańska K, Moore LE, Rothman N, et al. TP53, EGFR, and KRAS mutations in relation to VHL inactivation and lifestyle risk factors in renal-cell carcinoma from central and eastern Europe. Cancer Lett. 2010;293(1):92–98. | ||
Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. | ||
Pal SK, He M, Tong T, et al. RNA-seq reveals aurora kinase-driven mTOR Pathway activation in patients with sarcomatoid metastatic renal cell carcinoma. Mol Cancer Res. 2015;13(1):130–137. | ||
Bailey ST, Zhou B, Damrauer JS, et al. mTOR inhibition induces compensatory, therapeutically targetable MEK activation in renal cell carcinoma. PloS One. 2014;9(9):e104413. | ||
Okazaki H, Matsunaga N, Fujioka T, et al. Circadian regulation of mTOR by the ubiquitin pathway in renal cell carcinoma. Cancer Res. 2014;74(2):543–551. | ||
Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22. | ||
Chen H, Xiong T, Qu Y, Zhao F, Ferriero D, Mu D. mTOR activates hypoxia-inducible factor-1α and inhibits neuronal apoptosis in the developing rat brain during the early phase after hypoxia-ischemia. Neurosci Lett. 2012;507(2):118–123. | ||
Land SC, Tee AR. Hypoxia-inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282(28):20534–20543. | ||
Thomas GV, Tran C, Mellinghoff IK, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12(1):122–127. | ||
Karar J, Cerniglia GJ, Lindsten T, Koumenis C, Maity A. Dual PI3K/mTOR inhibitor NVP-BEZ235 suppresses hypoxia-inducible factor (HIF)-1α expression by blocking protein translation and increases cell death under hypoxia. Cancer Biol Ther. 2012;13(11):1102–1111. | ||
Daste A, Gross-Goupil M, Roca S, Bernhard JC, Ravaud A. Prolonged efficacy of mTOR inhibitors in papillary renal cell carcinoma: progression-free survival lasting for over 3 years, a case report and review of the literature. Target Oncol. 2014;9(1):81–84. | ||
Santoni M, Berardi R, Amantini C, et al. Role of natural and adaptive immunity in renal cell carcinoma response to VEGFR-TKIs and mTOR inhibitor. Int J Cancer. 2014;134(12):2772–2777. | ||
Panwalkar A, Verstovsek S, Giles FJ. Mammalian target of rapamycin inhibition as therapy for hematologic malignancies. Cancer. 2004;100(4):657–666. | ||
Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27(13):2278–2287. | ||
Huang S, Houghton PJ. Targeting mTOR signaling for cancer therapy. Curr Opin Pharmacol. 2003;3(4):371–377. | ||
Atkins MB, Hidalgo M, Stadler WM, et al. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol. 2004;22(5):909–918. | ||
Mohi MG, Boulton C, Gu TL, et al. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004;101(9):3130–3135. | ||
Mondesire WH, Jian W, Zhang H, et al. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10(20):7031–7042. | ||
Fei SJ, Zhang XC, Dong S, et al. Targeting mTOR to overcome epidermal growth factor receptor tyrosine kinase inhibitor resistance in non-small cell lung cancer cells. PloS One. 2013;8(7):e69104. | ||
Ku BM, Kim DS, Kim KH, et al. Transglutaminase 2 inhibition found to induce p53 mediated apoptosis in renal cell carcinoma. FASEB J. 2013;27(9):3487–3495. | ||
Zhu Z, Xing S, Cheng P, et al. The relationship of expression of Bcl-2, p53, and proliferating cell nuclear antigen (PCNA) to cell proliferation and apoptosis in renal cell carcinoma. J Huazhong Univ Sci Technolog Med Sci. 2004;24(4):354–357. | ||
Miyake H, Hara I, Gohji K, Arakawa S, Kamidono S. p53 Modulation of Fas/Apo-1 mediated apoptosis in a human renal cell carcinoma cell line. Int J Oncol. 1998;12(2):469–473. | ||
de Rozieres S, Maya R, Oren M, Lozano G. The loss of mdm2 induces p53-mediated apoptosis. Oncogene. 2000;19(13):1691–1697. | ||
Grossman SR, Perez M, Kung AL, et al. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2(4):405–415. | ||
Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69(7):1237–1245. | ||
Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362(6423):857–860. | ||
Jin L, Tabe Y, Kojima K, et al. MDM2 antagonist Nutlin-3 enhances bortezomib-mediated mitochondrial apoptosis in TP53-mutated mantle cell lymphoma. Cancer Lett. 2010;299(2):161–170. | ||
Yu H, Zou Y, Jiang L, et al. Induction of apoptosis in non-small cell lung cancer by downregulation of MDM2 using pH-responsive PMPC-b-PDPA/siRNA complex nanoparticles. Biomaterials. 2013;34(11):2738–2747. | ||
Mir R, Tortosa A, Martinez-Soler F, et al. Mdm2 antagonists induce apoptosis and synergize with cisplatin overcoming chemoresistance in TP53 wild-type ovarian cancer cells. Int J Cancer. 2013;132(7):1525–1536. | ||
Kong L, Yuan Q, Zhu H, et al. The suppression of prostate LNCaP cancer cells growth by selenium nanoparticles through Akt/Mdm2/AR controlled apoptosis. Biomaterials. 2011;32(27):6515–6522. | ||
Noon AP, Vlatkovic N, Polanski R, et al. P53 and MDM2 in renal cell carcinoma: biomarkers for disease progression and future therapeutic targets? Cancer. 2010;116(4):780–790. | ||
Uchida T, Gao JP, Wang C, et al. Clinical significance of p53, mdm2, and Bcl-2 proteins in renal cell carcinoma. Urology. 2002;59(4): 615–620. | ||
Haitel A, Wiener HG, Baethge U, Marberger M, Susani M. Mdm2 expression as a prognostic indicator in clear cell renal cell carcinoma: comparison with p53 overexpression and clinicopathological parameters. Clin Cancer Res. 2000;6(5):1840–1844. | ||
Kojima K, Shimanuki M, Shikami M, et al. The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53 wild-type AML. Leukemia. 2008;22(9):1728–1736. | ||
Mohammad RM, Wu J, Azmi AS, et al. An MDM2 antagonist (MI-319) restores p53 functions and increases the life span of orally treated follicular lymphoma bearing animals. Mol Cancer. 2009;8:115. | ||
Hirata H, Hinoda Y, Kikuno N, et al. MDM2 SNP309 polymorphism as risk factor for susceptibility and poor prognosis in renal cell carcinoma. Clin Cancer Res. 2007;13(14):4123–4129. | ||
Lin F, Zhang PL, Yang XJ, Prichard JW, Lun M, Brown RE. Morphoproteomic and molecular concomitants of an overexpressed and activated mTOR pathway in renal cell carcinomas. Ann Clin Lab Sci. 2006;36(3):283–293. | ||
Fulda S. Synthetic lethality by co-targeting mitochondrial apoptosis and PI3K/Akt/mTOR signaling. Mitochondrion. 2014;19 Pt A:85–87. | ||
Minet E, Michel G, Remacle J, Michiels C. Role of HIF-1 as a transcription factor involved in embryonic development, cancer progression and apoptosis (review). Int J Mol Med. 2000;5(3):253–259. | ||
Kao CL, Hsu HS, Chen HW, Cheng TH. Rapamycin increases the p53/MDM2 protein ratio and p53-dependent apoptosis by translational inhibition of mdm2 in cancer cells. Cancer Lett. 2009;286(2):250–259. | ||
Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102(23):8204–8209. | ||
Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010;20(7):427–434. | ||
von Roemeling CA, Marlow LA, Kennedy WP, Kennedy GT, Copland JA, Menefee ME. Preclinical evaluation of the mTOR inhibitor, temsirolimus, in combination with the epothilone B analog, ixabepilone in renal cell carcinoma. Am J Cancer Res. 2013;3(4):390–401. | ||
Roulin D, Waselle L, Dormond-Meuwly A, Dufour M, Demartines N, Dormond O. Targeting renal cell carcinoma with NVP-BEZ235, a dual PI3K/mTOR inhibitor, in combination with sorafenib. Mol Cancer. 2011;10:90. | ||
Holland WS, Tepper CG, Pietri JE, et al. Evaluating rational non-cross-resistant combination therapy in advanced clear cell renal cell carcinoma: combined mTOR and AKT inhibitor therapy. Cancer Chemother Pharmacol. 2012;69(1):185–194. | ||
Choy MK, Movassagh M, Bennett MR, Foo RS. PKB/Akt activation inhibits p53-mediated HIF1A degradation that is independent of MDM2. J Cell Physiol. 2010;222(3):635–639. | ||
Zuckerman V, Wolyniec K, Sionov RV, Haupt S, Haupt Y. Tumour suppression by p53: the importance of apoptosis and cellular senescence. J Pathology. 2009;219(1):3–15. | ||
Santoni M, Pantano F, Amantini C, et al. Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochim Biophys Acta. 2014;1845(2):221–231. | ||
Gemmill RM, Zhou M, Costa L, Korch C, Bukowski RM, Drabkin HA. Synergistic growth inhibition by Iressa and rapamycin is modulated by VHL mutations in renal cell carcinoma. Br J Cancer. 2005;92(12):2266–2277. | ||
Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22(2):181–185. | ||
Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–687. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.