")
Back to Journals » Cancer Management and Research » Volume 13
Synergistic Effects of TW-37 and ABT-263 on Renal Cell Carcinoma Cells
Authors Yu R, Lu Y, Yu R, Xie J, Zhou S
Received 6 July 2020
Accepted for publication 8 January 2021
Published 3 February 2021 Volume 2021:13 Pages 953—963
DOI https://doi.org/10.2147/CMAR.S265788
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Rui Yu,1 Yefen Lu,2 Ren Yu,3 Jianjun Xie,4 Shoujun Zhou4
1Department of Biochemistry and Molecular Biology, Zhejiang Key Laboratory of Pathophysiology, Medical School of Ningbo University, Ningbo, People’s Republic of China; 2Department of Neurology, The Fifth Affiliated Hospital of Wenzhou Medical University, Lishui, People’s Republic of China; 3Department of Urology, Ningbo Urology and Nephrology Hospital, Ningbo, People’s Republic of China; 4Suzhou Science & Technology Town Hospital, The Affiliated Suzhou Science & Technology Town Hospital of Nanjing Medical University, Suzhou, People’s Republic of China
Correspondence: Shoujun Zhou Email [email protected]
Background: Renal cell carcinoma (RCC) is a common urological system malignancy lack of effective therapeutic options. Upregulation of the Bcl-2 proteins was correlated with poor prognosis of RCC, suggesting that BH-3 mimetics may be a promising treatment option. ABT-263 is a BH3 mimetic that possesses anti-tumor effects. TW-37 is another inhibitor of Bcl-2 family protein with potential anti-tumor activities. However, since their effect as single agent is limited, combination treatment represents a strategy to improve the efficiency. We studied the ABT-263 in combination with TW-37 and analyzed the molecular mechanisms of action in RCC cells.
Methods: MTT and colony formation assays were used to measure the proliferation of RCC cells. Transwell assay was used to assay the migration and invasion of RCC cells. Cell cycle distribution and apoptosis were measured using the flow cytometry and apoptotic nucleosome assay, respectively. Western blotting was performed to measure the change of proteins. The anti-tumor effects of ABT-263, TW-37 and their combination were also evaluated in vivo.
Results: Cotreatment of TW-37 and ABT-263 synergistically repressed the proliferation of RCC cells. TW-37 and ABT-263 also synergistically inhibited the migration and invasion of RCC cells It was also showed that TW-37 and ABT-263 synergistically induced cell cycle arrest at the G2/M phase. Furthermore, increased apoptosis was observed after exposure to TW-37 and ABT-263. Mechanism investigation showed that TW-37 and ABT-263 synergistically induced apoptosis via the mitochondrial pathway and relied on the activation of Bax and caspases. Furthermore, ERK signaling pathway activation was detected after treated with TW-37 and ABT-263. Finally, TW-37 and ABT-263 also synergistically repressed the growth of RCC cells in xenograft mice.
Conclusion: In summary, our data demonstrated that combined treatment with TW-37 and ABT-263 exhibited synergistic RCC cell death and this combination may be applied as an effective therapeutic strategy against RCC.
Keywords: renal cell carcinoma; RCC, TW-37, ABT-263, apoptosis, ERK signaling
Introduction
Renal cell carcinoma (RCC) is a common malignancy of the urinary system with an increasing incidence worldwide.1 Till now, the major treatment for early RCC patients is radical nephrectomy.2 However, more than 30% of RCC patients had signs of metastasis at the initial diagnosis and missed the opportunity for surgery.3 Currently, the survival of RCC patients is still dismal and the overall 5-year survival rate is less than 10%.4 In the past decades, enormous progress have been made in understanding the pathogenesis and progression mechanisms underlying the RCC and multiple targeting agents have been developed such as lapatinib, sunitinib and sorafenib.5 Nevertheless, application of those drugs was limited by various factors such as high-cost, side effects and resistance to drug.6 Therefore, its necessary to identify new and more efficient therapeutic agents for RCC.
TW-37 is a second-generation Bcl-2 homology 3 mimetic derivative of gossypol that initially purified from cotton seeds and roots.7 TW-37 can bind to the Bcl-2 homology domain 3 (BH3) groove of Bcl-2 to intervene the heterodimerization of Bcl-2 with proapoptotic proteins and thereby inducing the apoptosis.8 TW-37 has similar affinity to Mcl-1 and Bcl-2 while has a lower affinity to Bcl-xl.9 It was found that TW-37 had anti-cancer activities against various cancers such as oral cancer, neuroblastoma, non-small cell lung cancer, colorectal cancer, nasopharyngeal carcinoma and pancreatic cancer.7,10–14 Previous studies have found that Bcl-2 proteins were frequently overexpressed in RCC.15 Hence targeting Bcl-2 proteins might be a promising strategy for inhibiting RCC.
However, the effects of TW-37 alone or in combination with other chemotherapeutic agents on RCC are still elusive.
ABT-263, also known as Navitoclax, is an orally available BH-3-mimetic agent which was tested in Phase I/II for various cancers.16 ABT-263 has been reported to possess high affinity to Bcl-2, Bcl-xl but low affinity to Bcl-w and Mcl-1.16,17 ABT-263 has shown promising anti-tumor activity toward chronic lymphocytic leukemia (CLL) either as a single agent or in combination with other therapeutic agents.18 However, the application of ABT-263 in solid tumors is limited by high levels of Mcl-1. Given that TW-37 could bind and inhibit Mcl-1, it would be intriguing to test the combination effects of TW-37 and ABT-263 against tumors.
In the present study, we found that a combination of TW-37 and ABT-263 showed synergistic anti-tumor effects against RCC. Mechanisms investigation revealed that this TW-37 acts together with ABT-263 to induce apoptosis via the mitochondrial pathway and relied on the activation of caspase and ERK signaling pathway. Moreover, TW-37 also synergize with ABT-263 to inhibit the growth of RCC in a xenograft mice mode.
Materials and Methods
Cell Culture and Chemicals
Human RCC cells (786-O and Caki-1) were obtained from Shanghai Bank for Cell Culture (Shanghai, China). All cells were cultured in RPMI-1640 medium (Life Technologies, USA) supplemented with 10% FBS (Gibco, USA). Cells were maintained in humidified atmosphere with 5% CO2 at 37°C. TW-37, ABT-263 and all other chemicals were obtained from Sigma-Aldrich (St Louis, USA). TW-37 and ABT-263 were dissolved in DMSO at a stock solution of 100 mM and diluted with FBS-free medium to achieve desired concentrations.
Cell Transfection
siRNA against Bax (si-Bax) or negative control (si-NC) were ordered from GenePharm (Shanghai, China). RCC cells were transiently transfected with siRNA using the Lipofectamine 2000 (Life Technologies, USA) according to the manufacturer’s guide.
Cell Viability Assay
Cell viabilities were performed as described.19 In short, cells were seeded at a density of 2×104 cells/well in 96-well plates. 10 μL of 5 mg/mL MTT solution (Beyotime, Beijing, China) was added into each well and the absorbance was read by a microplate reader (BioTek, USA). Data were presented as the mean ± SD of triplicate experiments.
Cellular Apoptosis Assay
Cellular apoptosis was measured by Apoptotic Nucleosome ELISA kit (Calbiochem, USA) according to the manufacturer’s guide. Briefly, cells were seeded at a density of 1×105 cells/well in 96-well plates, after different treatment cell lysates were incubated with biotinylated antibody according to the manufacturer’s guide and the results were read by a microplate reader (BioTek, USA). The experiments were performed in triplicate.
Cell Cycle Distribution Assay
Cells were seeded into 24-well plate at a density of 5×104 cells/well and treated with agents. Then, cells were harvested and washed with PBS twice and fixed with 20% ethanol for 4 hours avoiding the light. Then cells were collected after centrifuging for 5 min at 1500 rpm. Next, cells were stained with propidium iodide (PI) for 20 mins in the dark. The cell cycle distribution was analyzed by flow cytometry (BD Bioscience, USA).
Colony Formation Assay
For colony formation assay, cells were seeded into 6-well plates at a density of 200 cells/well. Then cells were exposed to different treatment for two weeks, cells were fixed with methanol and staining with 0.1% crystal violet and the number of colonies were recorded under inverted microscope (Olympus, Tokyo, Japan).
Cellular Migration and Invasion Assay
For migration/invasion assay, around 1×104 cells were cultured in serum-free medium and seeded into the upper chambers of transwell plate (8 μm pore size, Costar, USA) that were coated with or without Matrigel (BD Biosciences, USA) for migration and invasion assay, respectively. Bottom chamber was filled with full medium with 10% FBS. Then cells were cultured in a cell incubator for 48 h. Next, cells in the top chamber were removed with cotton swabs and cells on the lower surface were fixed with methanol and stained with 0.1% crystal violet, the number of cells was visualized and recorded under the microscope (Olympus, Tokyo, Japan).
Caspase-3 Activity Assay
Caspase-3 activity was measured using a caspase-3 activity assay kit (Abcam, USA) according to the manufacturer’s guide. Briefly, after different treatments, cellular lysates were mixed with equal amounts of colorimetric substrate and incubated at room temperature for 4 h. The results were measured using a microplate reader (BioTek, USA).
Western Blot and Immunoprecipitation Assay
After different treatment, cells were harvested and lysed with CHAPS lysis buffer. The purification of cytosol was performed as described earlier.20 The concentration of total protein was calculated using the BCA protein assay kit (Beyotime, Beijing, China). The protein was separated by 12% SDS-PAGE and then transferred to PVDF membrane (Millipore, USA). The PVDF membrane was blocked with 5% skimmed milk at room temperature for 1 h and then incubated with various primary antibodies (Cell Signaling Technologies, USA) overnight at 4 °C. Next, the membrane was incubated with corresponding secondary antibodies for 1 h at room temperature. For detection of activation of Bax, immunoprecipitation was conducted as described before.20
Xenograft Models
Male BALB/c nude mice (4- to 6-weeks-old) were randomly divided into four groups (Control group, TW-37 treated group, ABT-263 treated group, TW-37 and ABT-263 treated group). 786-O and Caki-1 cells (1×107) were implanted into mice. When the tumour size reached about 100 mm3, mice were intravenous injected with different agents. Tumors size were calculated using a caliper every 3 days, and two perpendicular diameters of each tumour were recorded. The tumour volume was measured using the following formula: volume = (width2 × length)/2.
Immunohistochemistry
Immunohistochemistry was performed in paraffin-embedded (5 μm) tissue sections. Sections were immunostained with anti-Ki67 (Abcam, USA) and anti-cleaved caspase-3 (Abcam, USA) afterwards detected by MACH3 HRP-Polymer kit (CST, USA) according to the manufacturer’s guide. The images were visualized using a microscope (Olympus, Tokyo, Japan).
Statistical Analysis
The data are presented as the mean ± S.D. for at least three independent experiments. Statistical analysis of two means was performed with Student’s t-test. Three or more means were calculated with one-way ANOVA and Bonferroni’s post hoc test. A probability value of less than 0.05 was considered to be statistically significant. The combined treatment (combination index [CI]) effects were analyzed using the CalcuSyn software program according to the Chou–Talalay equation,21 and the value of the CI for the combination treatment was quantitatively used to define the effect with CI < 1 (synergism), CI = 1 (additivity), and CI > 1 (antagonism).
Results
The Combination of TW-37 and ABT-263 Synergistically Repressed the Proliferation of RCC Cells
Firstly, we measured the viabilities of RCC cells (786-O, Caki-1) under the treatment of various doses of TW-37, ABT-263 and their combinations for 24 h. As indicated in Figure 1A, TW-37 and ABT-263 alone decreased the viability of RCC cells in a dose-dependent manner. Meanwhile, co-treatment of TW-37 and ABT-263 repressed the viability of RCC cells in a higher extent when compared to the treatment with TW-37 or ABT-263 alone (Figure 1A). Then we tested the inhibitory effects of combination of TW-37 and ABT-263. It was showed that the combination of various doses of TW-37 and ABT-263 decreased the viabilities of RCC cells in a time-dependent manner (Figure 1B). Furthermore, the CI calculation revealed that CI values were less than 0.8 at almost all fraction-affected points, suggesting strong synergistic effect between TW-37 and ABT-263 in RCC cells (Figure 1B). Because the synergistic effect is the most potent while the TW-37 dose is 20 μM and ABT-263 is 40 μM, so this concentration is chosen in the following experiments.
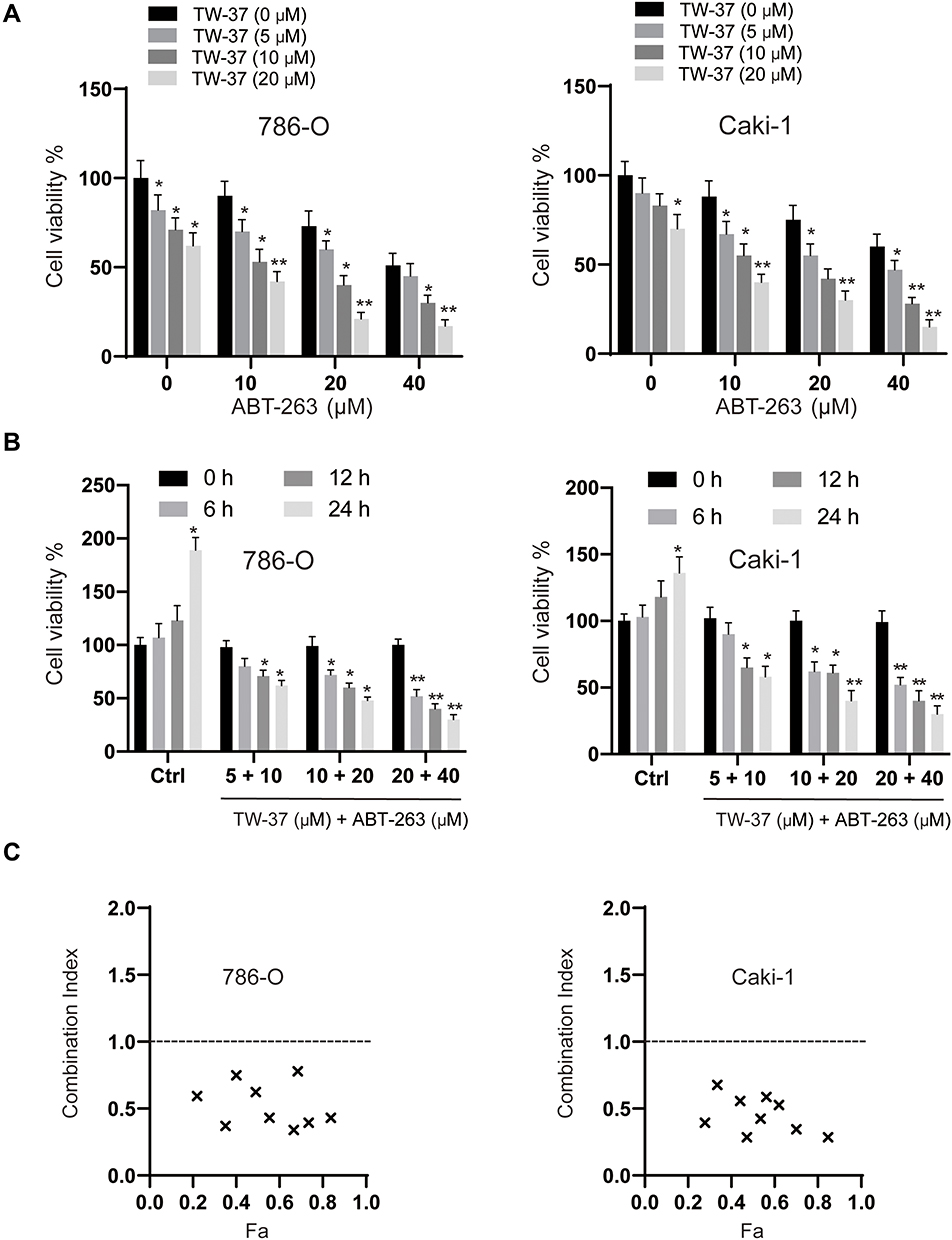 |
Figure 1 TW-37 and ABT-263 cotreatment synergistically inhibited the viability of RCC cells. (A) RCC cell lines (786-O and Caki-1) were treated with various doses of TW-37 (0 μM, 5 μM, 10 μM, 20 μM) and ABT-263 (0 μM, 10 μM, 20 μM, 40 μM) for 24 h, then cell viability was measured by MTT assay. (B) RCC cell lines (786-O and Caki-1) were treated with different combinations of TW-37 and ABT-263 (5 μM + 10 μM; 10 μM + 20 μM; 20 μM + 40 μM), then cell viabilities were assayed at various time points (6 h, 12 h, 24 h). (C) Combination index (CI) of TW-37 and ABT-263 in RCC cells were calculated using the Chou-Talalay method (CompuSyn). Fa: Fraction affected. Data was presented as mean±SD. Experiments were performed at least three times. *p<0.05; **p<0.01 vs. control group. |
Synergistic Effects of TW-37 and ABT-263 Co-Treatment on the Colony Formation, Migration, Invasion and Cell Cycle Distribution of RCC Cells
Next, we assayed the effects of TW-37, ABT-263 and their combination on the colony formation ability of RCC cells. As indicated in Figure 2A, compared to the groups treated with TW-37 or ABT-263 alone, the colony numbers in the TW-37 and ABT-263 co-treated groups were marked decreased. Furthermore, co-treatment of TW-37 and ABT-263 also significantly inhibited the migration (Figure 2B) and invasion (Figure 2C) of RCC cells. Western blot assays showed that TW-37 and ABT-263 treatment decreased the protein levels of MMP-2/-7, and EMT transition markers such as Vimentin and N-cadherin while increased the expression of epithelial marker (E-cadherin) and this effect is more obvious in the group co-treated with TW-37 and ABT-263 (Figure 2D). Cell cycle distribution analysis showed that TW-37 and ABT-263 co-treatment synergistically decreased the number of cells in the G1 phase and increased the number of cells in the G2/M phase (Figure 2E). Western blot analysis showed that TW-37 and ABT-263 synergistically increased the expression of p21 while decreased the expression of Cyclin B1, D1 and E (Figure 2F).
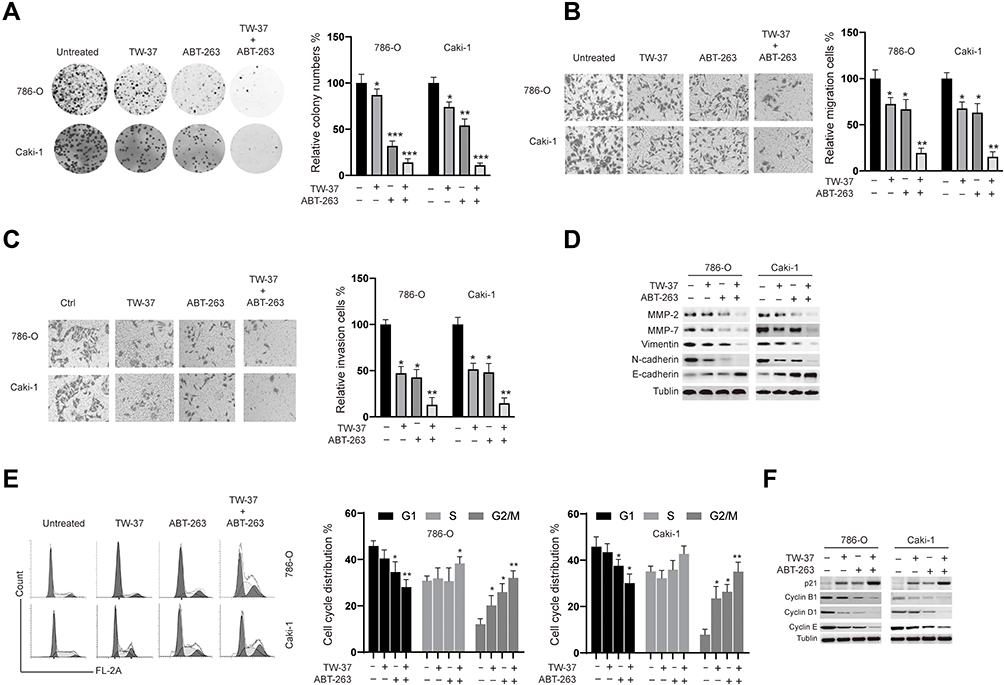 |
Figure 2 The effects of TW-37 and ABT-263 cotreatment on the colony formation, migration, invasion and cell cycle distribution of RCC cells. RCC cells were treated TW-37 (20 μM), ABT-263 (40 μM) or their combination. (A) Two weeks later, the colony formation ability was assayed. (B) 48 h later, migration of RCC cells was measured. (C) 48 h later, invasion of RCC cells was measured. (D) 48 h later, cellular lysates were subjected to Western blot analysis with indicated antibodies. (E) 48 h later, cell cycle distribution was assayed. (F) 48 h later, cellular lysates were subjected to Western blot analysis with indicated antibodies. Data was presented as mean±SD. Experiments were performed at least three times. *p<0.05; **p<0.01; ***p<0.001 vs. control group. |
TW-37/ABT-263 Co-Treatment Triggered Apoptosis in a Caspase-Dependent Manner in RCC Cells
To reveal the molecular mechanisms underlying the synergy of TW-37/ABT-263, we evaluated the apoptosis after the treatment of TW-37/ABT-263. As indicated in Figure 3A, single treatment with either TW-37 or ABT-263 resulted in an increase of apoptosis. Co-treatment of TW-37 and ABT-263 triggered the apoptosis in a more extent (Figure 3A). Meanwhile, caspase-3 activity assay showed that treatment of TW-37 or ABT-263 alone induced enhancement of activity of caspase-3 while co-treatment of TW-37 and ABT-263 induced more activation of caspase-3 (Figure 3B). We then measured expression levels of proteins involved in the apoptosis by Western blotting. It was found that cotreatment of TW-37 and ABT-263 caused a significantly decrease of anti-apoptotic Bcl-2 proteins while increase the expression of pro-apoptotic protein Bax and cleavage of caspase-3 (Figure 3C). We then examined whether co-treatment with TW-37 and ABT-263 results in Bax activation by immunoprecipitation. As indicated, TW-37 or ABT-263 alone induced activation of Bax which was further increased by cotreatment of TW-37 and ABT-263 (Figure 3D, top). Furthermore, it was also observed that the combination of TW-37 and ABT-263 resulted in the release of mitochondrial proteins cytochrome c and Smac/DIABLO in a greater extent than the treatment of TW-37 or ABT-263 alone (Figure 3D, bottom). To explore whether or not activation of caspase is essential for the apoptosis caused by TW-37/ABT-263, a pan caspase-inhibitor zVAD.fmk was added. As shown in Figure 3E, z.VAD.fmk almost blocked apoptosis after the cotreatment of TW-37 and ABT-263 in RCC cells. Moreover, in order to further evaluate the molecular mechanisms underlying the apoptosis caused by TW-37 and ABT-263, we silenced the expression of Bax in RCC cells (Figure 3F, left). It was found that downregulation of Bax inhibited the release of mitochondrial proteins (Figure 3F, right) and cleavage of caspase-3 (Figure 3G) caused by TW-37/ABT-263. Meanwhile, knockdown of Bax also inhibited the apoptosis (Figure 3H) and increase of caspase-3 activity (Figure 3I) triggered by TW-37/ABT-263 in RCC cells. These experiments indicate that TW-37/ABT-263 induced apoptosis of RCC cells via the mitochondrial pathway in a caspase-dependent manner.
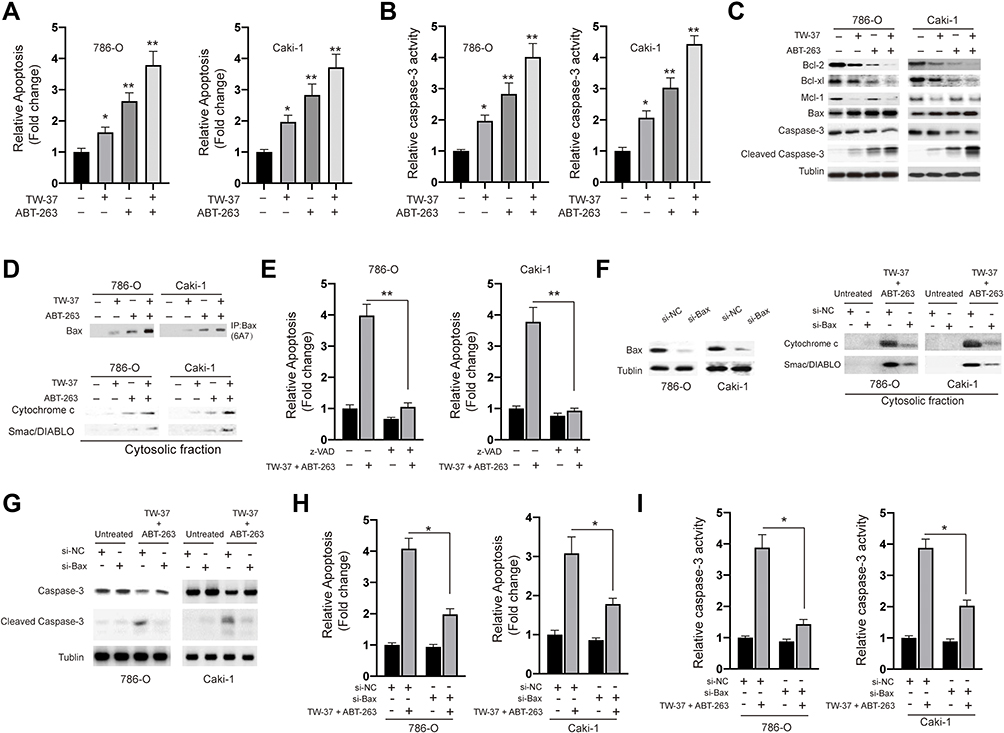 |
Figure 3 TW-37 and ABT-263 cotreatment synergistically induced apoptosis in RCC cells. RCC cells were treated with TW-37 (20 μM), ABT-263 (40 μM) or their combination for 24 h. (A) Cellular apoptosis rates were measured using the apoptosis nucleosome ELISA assay. (B) Caspase-3 activity was assayed. (C) the expression of indicated proteins were measured using the Western blotting assay. (D) the activation of Bax (top) and release of mitochondrial proteins (bottom) were assayed. (E) cells were co-treated with TW-37 and ABT-263 in the presence of zVAD.fmk or not for 24 h, then cellular apoptosis was measured. (F) RCC cells were transfected with siRNAs for 24 h, the level of Bax was measured by Western blotting (left). Transfected cells were co-treated with TW-37 and ABT-263, the release of mitochondrial proteins was assayed (right). (G) RCC cells were transfected with siRNAs for 24 h, then cells were co-treated with TW-37 and ABT-263 for another 24 h and the cleavage of caspase-3 was measured by Western blotting. (H) RCC cells were transfected with siRNAs for 24 h, then cells were co-treated with TW-37 and ABT-263 for another 24 h and cellular apoptosis was assayed. (I) RCC cells were transfected with siRNAs for 24 h, then cells were co-treated with TW-37 and ABT-263 for another 24 h and activity of caspase-3 was assayed. Data was presented as mean±SD. Experiments were performed at least three times. *p<0.05; **p<0.01. |
TW-37/ABT-263 Co-Treatment Triggered Apoptosis Relied on the Activation of ERK Signaling Pathway in RCC Cells
Next, we investigated whether there are any signaling pathways involved in the apoptosis induced by TW-37/ABT-263. As indicated in Figure 4A, treatment of TW-37/ABT-263 resulted in the activation of ERK, JNK but not p38 signaling pathway. In order to confirm the role of MAPKs, we applied various specific inhibitors. Interestingly, it was found that ERK inhibitor SB220024 but not JNK inhibitor (SP600125) and p38 inhibitor (SB202190) could successfully blocked the apoptosis induced by combination of TW-37 and ABT-263 (Figure 4B). Moreover, it was also found that SB220024 but not SP600125 and SB202190 could repress the cleavage of caspase-3 (Figure 4C), activation of Bax (Figure 4D) and release of mitochondrial proteins into cytosol (Figure 4D). Taken together, those data suggested that TW-37 and ABT-263 cooperates to induce apoptosis relied on the activation of ERK pathway in RCC cells.
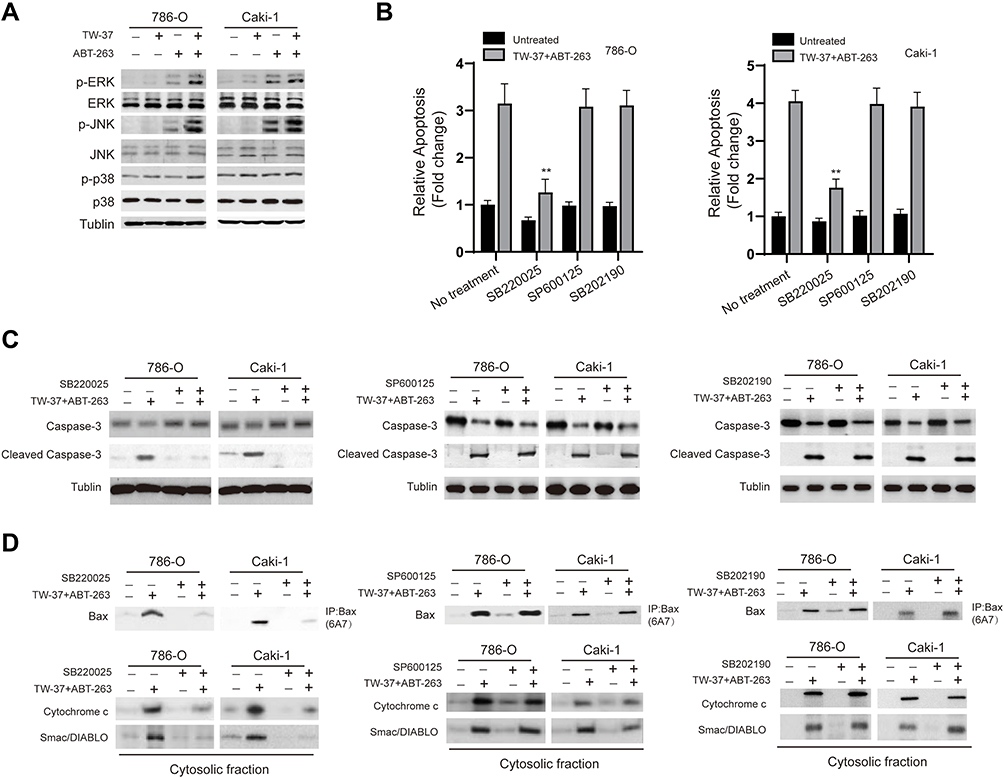 |
Figure 4 TW-37 and ABT-263 cotreatment lead to the activation of ERK signaling pathway. (A) RCC cells were treated with TW-37 (20 μM), ABT-263 (40 μM) or their combination for 24 h, then total cellular lysates were subjected to Western blotting analysis with indicated antibodies. (B) RCC cells were pretreated with various specific inhibitors for 4 h, then cells were incubated with TW-37 in combination with ABT-263 for another 24 h, cellular apoptosis was measured. (C) RCC cells were pretreated with various specific inhibitors for 4 h, then cells were incubated with TW-37 in combination with ABT-263 for another 24 h, the cleavage of caspase-3 was measured by Western blotting. (D) RCC cells were pretreated with various specific inhibitors for 4 h, then cells were incubated with TW-37 in combination with ABT-263 for another 24 h, the activation of Bax and release of mitochondrial proteins were assayed. Data was presented as mean±SD. Experiments were performed at least three times. **p<0.01 vs. control group. |
TW-37/ABT-263 Synergistically Inhibited the Growth of RCC Cells in Mouse Xenograft
Finally, we evaluated the effects of TW-37/ABT-263 in a xenograft mouse model. As shown in Figure 5A, TW-37 (10 mg/kg) or ABT-263 (10 mg/kg) alone reduced the growth of xenograft tumors when compared to the control group. In line with in vitro experiments results, cotreatment of TW-37 and ABT-263 show synergistic effects against the growth of tumor in vivo (Figure 5A). Meanwhile, we also observed that cotreatment of TW-37 and ABT-263 reduced the tumor weight more than the treatment of TW-37 and ABT-263 alone (Figure 5B). Further study showed that combination treatment repressed the expression of Ki-67 and induced the upregulation of cleaved caspase-3 more than treatment with single agent (Figure 5C). Western blotting results also confirmed that cotreatment of TW-37 and ABT-263 induced more cleaved caspase-3 in xenograft tumors (Figure 5D). Taken together, those data suggested that TW-37 and ABT-263 synergistically repressed the growth of RCC cells in vivo.
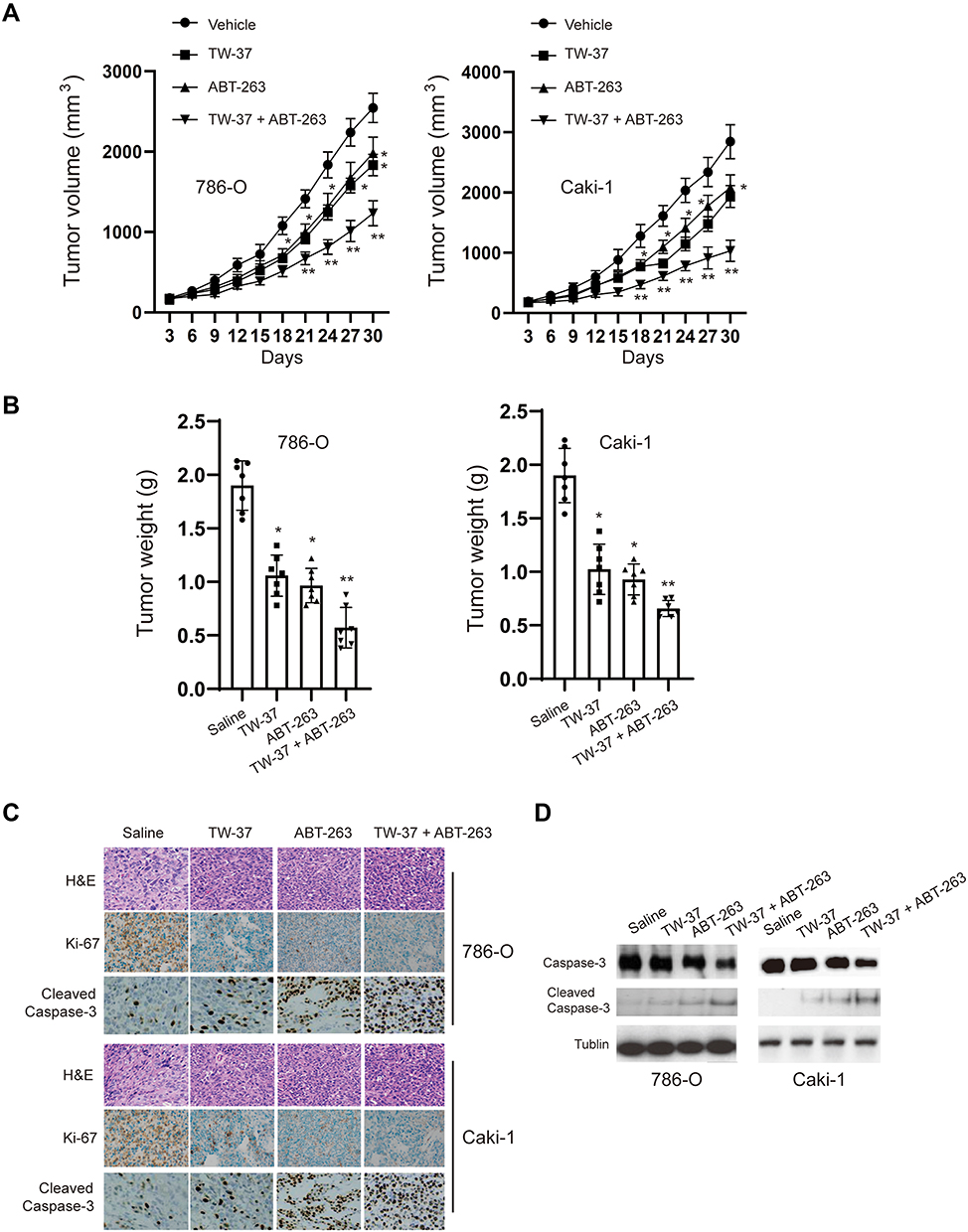 |
Figure 5 TW-37 and ABT-263 synergistically inhibited the growth of tumor in xenograft mice model. (A) Tumor-bearing nude mice were treated with vehicle, TW-37 (10 mg/kg), and/or ABT-263 (10 mg/kg) for 30 days, and tumor sizes were measured every 3 days. (B) Mice were sacrificed 30 days after treatment, tumor weights were measured. (C) H&E staining and Immunohistochemical (IHC) analysis for changes of Ki-67 and cleaved-caspase 3 expression in harvested tumors after different treatments as indicated. Representative images are shown (magnification: ×40). (D) Western blotting analysis of cleavage of caspase-3 in collected tumors from different groups. *p<0.05; **p<0.01 vs. control group. |
Discussion
To date, there’s lack of effective treatment for the RCC and better therapeutic options are urgently needed. A group of small-molecule compounds, known as “BH3-mimetics,” can bind to anti-apoptotic Bcl-2 proteins to directly activate apoptosis of tumor cells. Considering that evaluated expression of Bcl-2 proteins was correlated with poor prognosis of RCC patients.22 Therefore, BH3-mimetics might be promising in the treatment of RCC, especially those with high levels of Bcl-2 proteins. However, the anti-tumor effects of BH3-mimetics are limited by single agent. Thus, BH3-mimetics treatment of cancers with other agents is an essential strategy to improve the outcome. For instance, BH3-mimetic ABT-737 could significantly enhance the anti-tumor effects of PI3K inhibitor LY294002 in RCC.23 ABT-737 in combination with Chloroquine also showed synergistic anti-tumor effects against RCC.24 In the current study, the effects of TW-37 and ABT-263 were evaluated on RCC cells. We found that TW-37 and ABT-263 showed strong synergistic anti-tumor effects against RCC cells. The potency of this synergism is proved by CI values. Besides the in vitro experiments, TW-37 and ABT-263 also synergistically inhibited the growth of tumor in xenograft mice models.
It was reported that ABT-263 could efficiently inhibit Bcl-xl, Bcl-2 and Bcl-w while it fails to bind Mcl-1.25 Thus, Mcl-1 drives resistance toward BH3-mimetics and therefore strategies that can counteract Mcl-1 are essential to take full advantage of ABT-263 based therapies. It was well recognized that TW-37, a non-peptidic small molecular, has a great affinity for Mcl-1.26 In the present study, we revealed that combination of TW-37 and ABT-263 significantly repressed cell viability of RCC cells when compared to the treatment of single agent alone. It was also found that TW-37 and ABT-263 synergistically inhibited the migration and invasion of RCC cells. Furthermore, the current study showed that treatment of TW-37 and ABT-263 led to the downregulation of mesenchymal makers N-cadherin and Vimentin and to the upregulation of epithelial marker E-cadherin. Hence, the process of EMT process was repressed. Considering EMT process played an essential role in metastasis of RCC.27 Thus, cotreatment with TW-37 and ABT-37 might have impact on metastasis of RCC and it would be intriguing to test it. Furthermore, it was also found that TW-37 and ABT-263 triggered cell cycle arrest. Our finding is in line with previous studies which also reported that TW-37 or ABT-263 could trigger cell cycle arrest in various cancer cells.28–30
In addition, we also investigated the underlying molecular mechanisms.
We showed that TW-37 and ABT-263 synergistically induced apoptosis via the mitochondrial pathway. This conclusion is supported by various pieces of evidence. For instance, downregulation of anti-apoptotic Bcl-2 proteins and upregulation of pro-apoptotic Bcl-2 protein. In addition, activation of Bax was observed after treatment of TW-37/ABT-263. The relevance of activation of Bax in determining the sensitivity to TW-37/ABT-263 co-treatment is verified by knockdown studies showing that silencing of Bax significantly protects cells from TW-37/ABT-263 triggered apoptosis. Noteworthy, it was recognized that blockage of Mcl-1 is essential for augment the anti-tumor effects of ABT-263.31 In our scenario, it was found that TW-37 treatment led to the downregulation of Mcl-1 while ABT-263 alone had no effects on the expression of Mcl-1. It may explain the synergistic effects between TW-37 and ABT-263 and further investigation is required to verify it. Activation of caspase-3 was also detected after treatment of TW-37/ABT-263. We also found that pan caspase inhibitor zVAD.fmk blocked the apoptosis induced by TW-37/ABT-263. Therefore, TW-37/ABT-263 triggered apoptosis in a caspase-dependent manner via the mitochondrial pathway in RCC cells.
Furthermore, we also observed activation of ERK after treatment of TW-37/ABT-263. Our study is in line with previous study which also found that ABT-263 induced activation of ERK pathway in hepatocellular carcinoma cells.32 However, another study reported that TW-37 inhibited the activation of ERK pathway in melanoma cell.33 This discrepancy might be caused by different cells lines and more investigation regarding the effects of TW-37/ABT-263 on MAPKs is required.
In addition, in vivo experiments were conducted on a xenograft mice model to test the synergistic effects between TW-37 and ABT-263. The dosage we used is lower than previous studies to avoid any possible side effects.10,34,35 Our data indicated that TW-37/ABT-263 treatment significantly inhibited RCC growth in vivo in the xenograft model, which could in part be caused by decreased proliferation and increased apoptosis as evidence by reduced Ki-67 and enhanced cleavage caspase-3 immunoreactivity in the xenograft tumors.
Some limitations of our study are, firstly, the central mechanism that accounts for the entire synergistic effect of TW-37 and ABT-263 was not established in our study. Secondly, it remains unclear whether any other pathways are also involved in the combination effects of TW-37 and ABT-263. Thirdly, the drugs dosages are relatively high in our study. Therefore, its meaningful to find strategies to decrease the concentrations of agents. Such as modification of agents to improve its efficiency or in combination with other chemotherapeutics.
In summary, our data suggests that combination of TW-37 and ABT-263 exhibited synergistic effects against RCC cells. The synergistic anti-tumor effects of TW-37 and ABT-263 is relied on the activation of mitochondrial pathway in a caspase-dependent manner. In addition, activation of ERK is essential for the anti-tumor effects of TW-37/ABT-263. Furthermore, in vivo data showed that the combination of TW-37 and ABT-263 leads to significant tumor regression in RCC xenografts. Our study reveals for the first time that combination of TW-37 and ABT-263 is a novel potential strategy for the treatment of RCC.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gerlinger M, Horswell S, Larkin J. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46(3):225–233. doi:10.1038/ng.2891
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
3. Jabaji RB. Trend of surgical treatment of localized renal cell carcinoma. Perm J. 2019;23:18–108.
4. Sanchez-Gastaldo A, Kempf E, González Del Alba A, et al. Systemic treatment of renal cell cancer: a comprehensive review. Cancer Treat Rev. 2017;60:77–89. doi:10.1016/j.ctrv.2017.08.010
5. Posadas EM, Limvorasak S, Figlin RA. Targeted therapies for renal cell carcinoma. Nat Rev Nephrol. 2017;13(8):496–511. doi:10.1038/nrneph.2017.82
6. Barata PC, Rini BI. Treatment of renal cell carcinoma: current status and future directions. CA Cancer J Clin. 2017;67(6):507–524. doi:10.3322/caac.21411
7. Lei S, Ding Y, Fu Y, et al. The preclinical analysis of TW-37 as a potential anti-colorectal cancer cell agent. PLoS One. 2017;12(10):e0184501. doi:10.1371/journal.pone.0184501
8. Mohammad RM, Goustin AS, Aboukameel A, et al. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res. 2007;13(7):2226–2235. doi:10.1158/1078-0432.CCR-06-1574
9. Wang G, Nikolovska-Coleska Z, Yang C-Y, et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem. 2006;49(21):6139–6142. doi:10.1021/jm060460o
10. Ahn CH, Lee WW, Jung YC, et al. Antitumor effect of TW-37, a BH3 mimetic in human oral cancer. Lab Anim Res. 2019;35(1):27. doi:10.1186/s42826-019-0028-7
11. Klenke S, Akdeli N, Stelmach P, et al. The small molecule Bcl-2/Mcl-1 inhibitor TW-37 shows single-agent cytotoxicity in neuroblastoma cell lines. BMC Cancer. 2019;19(1):243. doi:10.1186/s12885-019-5439-1
12. Chen X, Mao G, Chen H, et al. TW37 enhances the pro-apoptosis and anti-migration ability of gefitinib in non-small cell lung cancer. Cell Mol Biol (Noisy-Le-Grand). 2018;64(4):6–10. doi:10.14715/cmb/2018.64.4.2
13. Wang Z, Song W, Aboukameel A, et al. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and invasion in pancreatic cancer. Int J Cancer. 2008;123(4):958–966. doi:10.1002/ijc.23610
14. Lu Y, Huang H, Yang H, et al. Small molecule inhibitor TW-37 is tolerable and synergistic with chemotherapy in nasopharyngeal carcinoma. Cell Cycle. 2017;16(14):1376–1383. doi:10.1080/15384101.2017.1329066
15. Gobe G, Rubin M, Williams G, et al. Apoptosis and expression of Bcl-2, Bcl-X L, and bax in renal cell carcinomas. Cancer Invest. 2002;20(3):324–332. doi:10.1081/CNV-120001177
16. Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. doi:10.1158/0008-5472.CAN-07-5836
17. Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi:10.1038/nature03579
18. Kipps TJ, Eradat H, Grosicki S, et al. A Phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2015;56(10):2826–2833. doi:10.3109/10428194.2015.1030638
19. Yu R, Yao J, Ren Y. A novel circRNA, circNUP98, a potential biomarker, acted as an oncogene via the miR-567/PRDX3 axis in renal cell carcinoma. J Cell Mol Med. 2020;24(17):10177–10188. doi:10.1111/jcmm.15629
20. Yu R, Yu B-X, Chen J-F, et al. Anti-tumor effects of atractylenolide I on bladder cancer cells. J Exp Clin Cancer Res. 2016;35(1):40. doi:10.1186/s13046-016-0312-4
21. Chou T-C. Preclinical versus clinical drug combination studies. Leuk Lymphoma. 2008;49(11):2059–2080. doi:10.1080/10428190802353591
22. Itoi T, Yamana K, Bilim V, et al. Impact of frequent Bcl-2 expression on better prognosis in renal cell carcinoma patients. Br J Cancer. 2004;90(1):200–205. doi:10.1038/sj.bjc.6601454
23. Zhu S, Cohen MB, Bjorge JD, et al. PI3K inhibition potentiates Bcl-2-dependent apoptosis in renal carcinoma cells. J Cell Mol Med. 2013;17(3):377–385. doi:10.1111/jcmm.12019
24. Yin P, Jia J, Li J, et al. ABT-737, a Bcl-2 selective inhibitor, and chloroquine synergistically kill renal cancer cells. Oncol Res. 2016;24(1):65–72. doi:10.3727/096504016X14587366983838
25. Dai H, Meng XW, Kaufmann SH. Mitochondrial apoptosis and BH3 mimetics. F1000Res. 2016;5:2804. doi:10.12688/f1000research.9629.1
26. Brumatti G, Ekert PG. Seeking a MCL-1 inhibitor. Cell Death Differ. 2013;20(11):1440–1441. doi:10.1038/cdd.2013.114
27. Piva F, Giulietti M, Santoni M, et al. Epithelial to mesenchymal transition in renal cell carcinoma: implications for cancer therapy. Mol Diagn Ther. 2016;20(2):111–117. doi:10.1007/s40291-016-0192-5
28. Wang Z, Azmi AS, Ahmad A, et al. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and induces apoptosis in pancreatic cancer: involvement of notch-1 signaling pathway. Cancer Res. 2009;69(7):2757–2765. doi:10.1158/0008-5472.CAN-08-3060
29. Sale MJ, Cook SJ. The BH3 mimetic ABT-263 synergizes with the MEK1/2 inhibitor selumetinib/AZD6244 to promote BIM-dependent tumour cell death and inhibit acquired resistance. Biochem J. 2013;450(2):285–294. doi:10.1042/BJ20121212
30. Zhan Y, Wang Y, Qi M, et al. BH3 mimetic ABT-263 enhances the anticancer effects of apigenin in tumor cells with activating EGFR mutation. Cell Biosci. 2019;9(1):60. doi:10.1186/s13578-019-0322-y
31. Lian BSX, Yek AEH, Shuvas H, et al. Synergistic anti-proliferative effects of combination of ABT-263 and MCL-1 selective inhibitor A-1210477 on cervical cancer cell lines. BMC Res Notes. 2018;11(1):197. doi:10.1186/s13104-018-3302-0
32. Wang B, Ni Z, Dai X, et al. The Bcl-2/xL inhibitor ABT-263 increases the stability of Mcl-1 mRNA and protein in hepatocellular carcinoma cells. Mol Cancer. 2014;13(1):98. doi:10.1186/1476-4598-13-98
33. Verhaegen M, Bauer JA, Martín de la Vega C, et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006;66(23):11348–11359. doi:10.1158/0008-5472.CAN-06-1748
34. Yang IH, Jung J-Y, Kim S-H, et al. ABT-263 exhibits apoptosis-inducing potential in oral cancer cells by targeting C/EBP-homologous protein. Cell Oncol. 2019;42(3):357–368. doi:10.1007/s13402-019-00431-5
35. Zoeller JJ, Vagodny A, Taneja K, et al. Neutralization of BCL-2/X L enhances the cytotoxicity of T-DM1 in vivo. Mol Cancer Ther. 2019;18(6):1115–1126. doi:10.1158/1535-7163.MCT-18-0743
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.