")
Back to Journals » OncoTargets and Therapy » Volume 9
Synergistic activity of vorinostat combined with gefitinib but not with sorafenib in mutant KRAS human non-small cell lung cancers and hepatocarcinoma
Authors Jeannot V, Busser B, Vanwonterghem L, Michallet S, Ferroudj S, Cokol M, Coll JL, Ozturk M, Hurbin A
Received 20 July 2016
Accepted for publication 29 August 2016
Published 9 November 2016 Volume 2016:9 Pages 6843—6855
DOI https://doi.org/10.2147/OTT.S117743
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Victor Jeannot,1,2 Benoit Busser,1–3 Laetitia Vanwonterghem,1,2 Sophie Michallet,1,2 Sana Ferroudj,1,2 Murat Cokol,4 Jean-Luc Coll,1,2 Mehmet Ozturk,1,2,5 Amandine Hurbin1,2
1INSERM U1209, Department Cancer Targets and Experimental Therapeutics, Grenoble, France; 2University Grenoble Alpes, Institute for Advanced Biosciences, Grenoble, France; 3Department of Biochemistry, Toxicology and Pharmacology, Grenoble University Hospital, Grenoble, France; 4Faculty of Engineering and Natural Sciences, Sabanci University, Istanbul, Turkey; 5Faculty of Medicine, Dokuz Eyul University, Izmir Biomedicine and Genome Center, Izmir, Turkey
Abstract: Development of drug resistance limits the efficacy of targeted therapies. Alternative approaches using different combinations of therapeutic agents to inhibit several pathways could be a more effective strategy for treating cancer. The effects of the approved epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor (gefitinib) or a multi-targeted kinase inhibitor (sorafenib) in combination with a histone deacetylase inhibitor (vorinostat) on cell proliferation, cell cycle distribution, apoptosis, and signaling pathway activation in human lung adenocarcinoma and hepatocarcinoma cells with wild-type EGFR and mutant KRAS were investigated. The effects of the synergistic drug combinations were also studied in human lung adenocarcinoma and hepatocarcinoma cells in vivo. The combination of gefitinib and vorinostat synergistically reduced cell growth and strongly induced apoptosis through inhibition of the insulin-like growth factor-1 receptor/protein kinase B (IGF-1R/AKT)-dependent signaling pathway. Moreover, the gefitinib and vorinostat combination strongly inhibited tumor growth in mice with lung adenocarcinoma or hepatocarcinoma tumor xenografts. In contrast, the combination of sorafenib and vorinostat did not inhibit cell proliferation compared to a single treatment and induced G2/M cell cycle arrest without apoptosis. The sorafenib and vorinostat combination sustained the IGF-1R-, AKT-, and mitogen-activated protein kinase-dependent signaling pathways. These results showed that there was synergistic cytotoxicity when vorinostat was combined with gefitinib for both lung adenocarcinoma and hepatocarcinoma with mutant KRAS in vitro and in vivo but that the combination of vorinostat with sorafenib did not show any benefit. These findings highlight the important role of the IGF-1R/AKT pathway in the resistance to targeted therapies and support the use of histone deacetylase inhibitors in combination with EGFR-tyrosine kinase inhibitors, especially for treating patients with mutant KRAS resistant to other treatments.
Keywords: targeted therapy, combined treatments, non-small cell lung cancer, hepatocarcinoma
Introduction
Although the use of new therapeutic agents has improved the treatment of cancer patients in recent years, the efficacy of targeted therapies is limited by the development of drug resistance. Thus, the identification of alternative approaches that further disrupt tumor cell growth is essential, and these strategies could have a significant clinical impact. In particular, combinations of targeted agents could be exploited to inhibit more than one pathway and could be significantly more effective for achieving tumor regression than single therapeutic agents.
Gefitinib and erlotinib are epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors (TKIs) that improve the survival of patients with EGFR-mutated non-small cell lung cancer (NSCLC).1 For patients with EGFR wild-type tumors, first-line chemotherapy is still the standard of care.2 EGFR-TKIs are approved for use in second- and third-line treatments of advanced NSCLC or as a maintenance therapy. However, the limited response to EGFR-TKIs observed in patients with wild-type EGFR NSCLC showed that there were intrinsic resistance mechanisms to EGFR-TKIs including the KRAS mutation.3
Histone deacetylase (HDAC) inhibitors induce a range of anticancer effects, including tumor cell apoptosis, cell cycle arrest, differentiation, senescence, modulation of immune responses, and altered angiogenesis.4 Vorinostat and romidepsin are the most advanced HDAC inhibitors and are currently approved for treating cutaneous T-cell lymphomas.4–6 Belinostat is approved for the treatment of peripheral T-cell lymphoma, and panobinostat is approved for use in combination treatments for multiple myeloma.7,8 Several studies support the use of HDAC inhibitors in combination with EGFR-TKIs in NSCLC cells to restore EGFR-TKI sensitivity.9–14 In this context, we recently showed the role of HDAC in the EGFR-TKI resistance of mutant KRAS adenocarcinoma.15,16
Sorafenib is a small-molecule TKI that targets vascular endothelial growth factor receptors, Raf kinases, and platelet-derived growth factor receptor. It was the first inhibitor to produce a survival benefit for advanced hepatocellular carcinoma (HCC).17 However, the majority of HCC patients do not respond to sorafenib, and most, if not all, patients who initially respond to sorafenib develop tumor resistance after a few months of treatment.18 Preclinical studies have also shown that combining HDAC inhibitors with sorafenib can have antiproliferative, antiangiogenic, and proapoptotic effects on epithelial tumor cells including HCC cells.19–22
Based on these data, we hypothesized that a combination treatment with HDAC inhibitors and TKIs could overcome the intrinsic resistance of epithelial tumor cells to TKI, and lead to more effective treatment, especially for both HCC and NSCLC with wild-type EGFR and mutant KRAS. Therefore, we investigated the effects of combinations of the HDAC inhibitor vorinostat with either multi-targeted kinase inhibitor sorafenib or EGFR-TKI gefitinib on antiproliferative and survival pathways in NSCLC and HCC cells with wild-type EGFR and mutant KRAS in vitro and in vivo.
Materials and methods
Cell lines
NSCLC (A549, H1299, H358, H322, and H1719) and HCC (HepG2, Hep3B, HuH7, Hep40, and PLC/PRF5) cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA), and further authentication was not performed. These cells were maintained in RPMI 1640 (Gibco, Cergy Pontoise, France) supplemented with 10% fetal bovine serum in a humidified atmosphere with 5% CO2. We routinely carried out morphology checks on all cell lines, and we only passaged the cell lines for 3 months. All cell lines were routinely tested for the presence of mycoplasma (MycoAlert® Mycoplasma Detection Kit, Lonza, France).
Drugs
Sorafenib tosylate, vorinostat (SAHA, MK0683), and linsitinib (OSI-906) were obtained from Selleckchem (Munich, Germany). Gefitinib (ZD1839) was provided by AstraZeneca (Paris, France). All drugs were dissolved in sterile dimethyl sulfoxide at 10 mmol/L stock solution.
Cell proliferation assay
Cells that were growing exponentially were seeded in 96-well plates and exposed to serial dilutions of gefitinib, sorafenib, and vorinostat in regular growth medium containing 10% fetal bovine serum for 96 hours. Cell proliferation was measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Growth inhibition was expressed as the percentage of surviving drug-treated cells compared to untreated control cells. The drug concentrations required to inhibit cell growth by 50% (IC50) were determined by interpolation from the dose to response curves. Combinations of treatments were performed in 96-well plates using serial dilutions ranging from 0 to IC50 (0; 0.2 IC50; 0.4 IC50; 0.6 IC50; 0.8 IC50; and IC50). The combination effect of treatments was evaluated using the method described by Chou et al23 using the CompuSyn program (ComboSyn Inc., Paramus, NJ, USA). Interactions between drugs were expressed as the combination index (CI) determined with CompuSyn software: CI <0.9 represented synergistic cytotoxicity; 0.9< CI <1.1 represented additive cytotoxicity; and CI >1.1 represented antagonistic cytotoxicity. The dose reduction index (DRI) values represented the amount that the dose of each drug in a synergistic combination could be reduced to achieve a given effect level compared to a dose of each drug alone. Fa–CI and Fa–DRI plots were built and showed CI or DRI on the y-axis as a function of the effect level (Fa) on the x-axis.
Cell cycle analysis
Cells were harvested, pooled, fixed with 70% ethanol, and incubated in phosphate-buffered saline (PBS) at 37°C for 30 minutes before being stained with 20 μg/mL propidium iodide. The percentage of cells in specific cell cycle phases (G0/G1, S, and G2/M) was determined using a flow cytometer (Accuri C6, Becton Dickinson, Le Pont-de-Claix, France).
Apoptosis assays
The morphological changes related to apoptosis were assessed by fluorescence microscopy after staining the cells with Hoechst 33342 (5 μg/mL, Sigma). The percentage of apoptotic cells was scored after counting at least 500 cells. Active caspase-3 and poly(adenosine diphosphate ribose) polymerase (PARP) cleavage were detected with immunoblotting. Apoptotic cells with hypodiploid DNA staining were counted in the “sub-G1” peak using flow cytometry.
Immunoblotting
Cells were lysed and immunoblotting was performed as previously described15,16 using antibodies against cleaved caspase-3 (Asp175), actin, phospho-EGFR-Y1068, EGFR, phospho-protein kinase B (AKT)-S473, pan-AKT, phospho-signal transducer and activator of transcription (STAT)3-Y705, STAT3, phospho-extracellular signal-regulated kinase (ERK)1/2-T202/Y204, and ERK1/2 (Cell Signaling Technology, St Quentin en Yvelines, France), PARP (SantaCruz Biotechnology, Heidelberg, Germany), acetylated α-tubulin and α-tubulin (Abcam, Paris, France), and p21WAF1 (Merck Millipore, Molsheim, France).
In vivo models
The effect of the combination of gefitinib and vorinostat was measured in established subcutaneous tumor-bearing mice. All the animal experiments were performed in agreement with the European Economic Community guidelines and the “Principles of Laboratory Animal Care” (NIH publication N 86-23 revised 1985). Animal experiment studies were approved through institutional guidelines and by the European Community for the use of experimental animals (authorization to experiment 2015031115126706). Female NMRI nude mice (6–8 weeks old, Janvier, Le Genest-Saint Isle, France) were injected subcutaneously in the flank with 20×106 H358 cells suspended in PBS or 5×106 PLC/PRF5 cells suspended in 50% Matrigel (BD Biosciences, San Jose, CA, USA). Tumor size was measured twice a week using a caliper, and the tumor volume was calculated as follows: length × (width)2 ×0.4. When tumors of ~250 mm3 in size were detected, the mice were randomized into four groups (8–11 mice per group) and were orally treated with 5 (H358) or 50 mg/kg/day (PLC/PRF5) gefitinib and/or vorinostat (100 mg/kg/day) for either 5 (H358) or 3 (PLC/PRF5) days a week. Control mice received an oral vehicle (Tween/dimethyl sulfoxide). Mice bearing tumors ≥2 cm3 were euthanized immediately. At the end of the experiment, tumor samples were excised and frozen for Western blot and immunohistochemical analyses.
Immunohistochemical staining
Tumor sections of a 7-μm thickness were fixed with 4% paraformaldehyde and incubated overnight at 4°C with antibodies. Immunohistochemical staining of Ki67 and cleaved caspase-3 was performed as previously described.15 Terminal deoxynucleotidyl transferase dUTP nick end labeling was performed according to the manufacturer’s instructions (Roche Diagnostics, Mannheim, Germany).
Statistical analysis
A comparison of mean values was performed using the Mann–Whitney U-test or an analysis of variance test. A P<0.05 was considered to be significant. Univariate analyses were conducted using the Kaplan–Meier method and the log-rank test for tumor-bearing mice. All analyses were performed using Statview 4.1 software (Abacus Concept, Berkeley, CA, USA).
Results
Single-drug inhibition of proliferation in NSCLC and HCC cell lines
We first evaluated the ability of gefitinib, sorafenib, or vorinostat to inhibit growth in human NSCLC (A549, H1299, H358, H1719, and H322) and HCC (Hep3B, Huh7, HepG2, Hep40, and PLC/PRF5) cell lines harboring wild-type EGFR, and wild-type or mutant KRAS (Table 1). Cancer cells were treated with increasing concentrations of gefitinib, sorafenib, or vorinostat. There was a wide range of gefitinib sensitivity for NSCLC cell lines, and HCC cells were resistant to gefitinib. The IC50 concentrations for sorafenib had the same range in NSCLC and HCC cells. The IC50 concentrations indicated that the HCC cell lines were slightly more sensitive to vorinostat than the NSCLC cell lines.
 | Table 1 The sensitivity of human NSCLC and HCC cell lines to gefitinib, sorafenib, and vorinostat |
Effects of drug combinations on NSCLC and HCC cell growth
H358, A549, and PLC/PRF5 mutant KRAS cell lines were selected to be treated with combinations of vorinostat and either gefitinib or sorafenib. Compared to single treatments, H358 and PLC/PRF5 cells treated with increasing concentrations of gefitinib and vorinostat exhibited significantly decreased viability (Figure 1A and B). This effect was confirmed in A549 cells (Figure 1) and in wild-type KRAS H322 cells (Supplementary material S1), which harbored an amplification of KRAS.24 The CI values measuring the degree of drug interaction indicated a synergistic interaction between gefitinib and vorinostat (Figure 1A and B, Supplementary material S2). Synergism was confirmed by measuring how many folds the dose of each drug in a synergistic combination may be reduced using the DRI. In contrast, the effect of the combination of vorinostat with sorafenib was not significantly different from the effect of each drug alone for each of the concentrations studied in H358, PLC/PRF5, and A549 cells (Figure 1C, D, and F) and in wild-type KRAS cell lines (Supplementary material S1). Vorinostat and sorafenib treatment resulted in an antagonistic interaction in KRAS mutant cells and H322 cells (Figure 1C and D, Supplementary material S2). Taken together, these findings indicate that vorinostat synergized with gefitinib, but not with sorafenib, especially in the mutant or amplified KRAS cells.
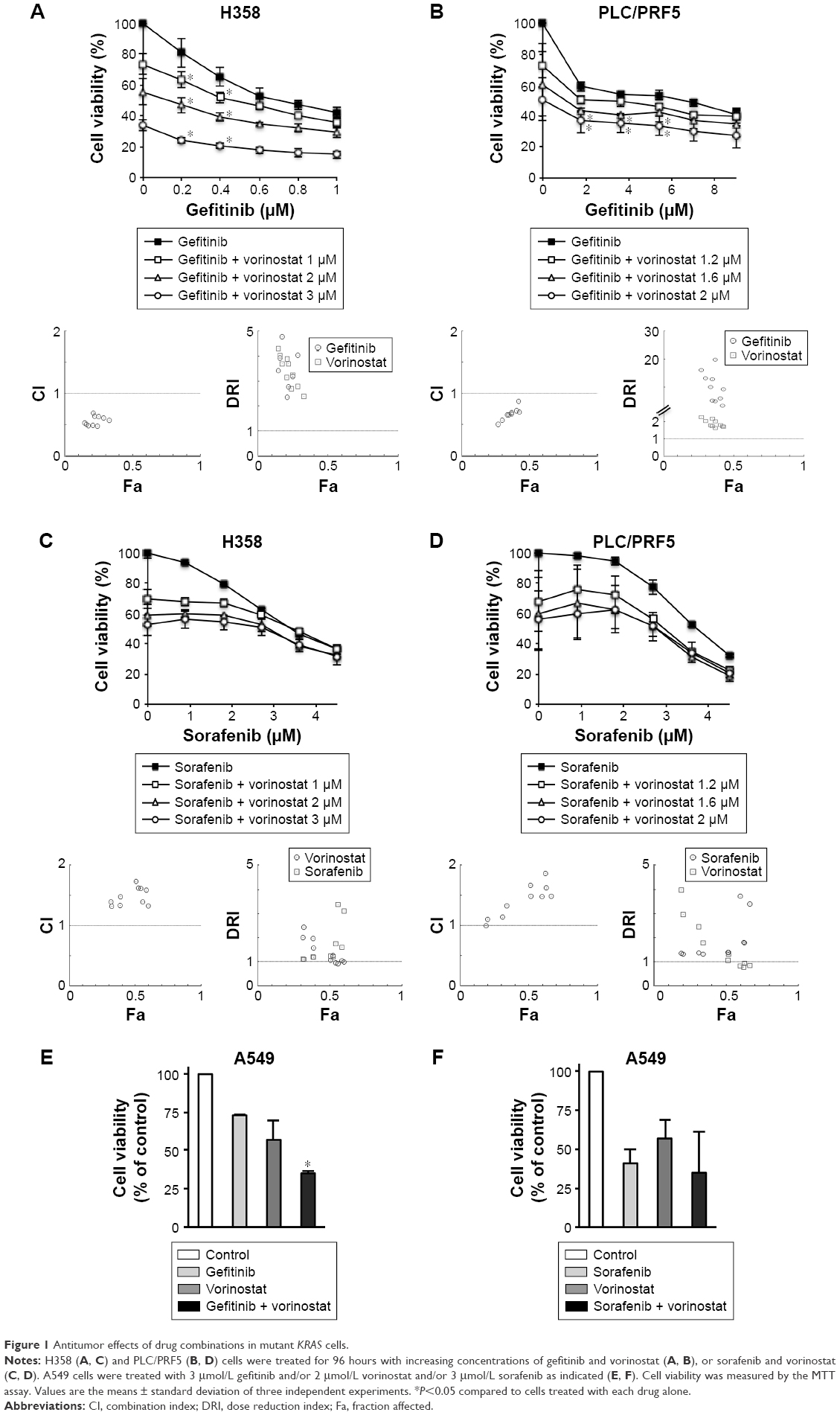 | Figure 1 Antitumor effects of drug combinations in mutant KRAS cells. |
Effects of drug combinations on NSCLC and HCC cell cycle distribution
The effect of vorinostat combined with either gefitinib or sorafenib on cell cycle distribution was analyzed by flow cytometry in H358 and PLC/PRF5 cells (Supplementary material S3). None of the three single agent treatments resulted in a marked change in the cell cycle distribution (Figure 2). However, the combination of gefitinib with vorinostat induced a strong and statistically significant accumulation of sub-G1 cells together with a decrease in the G0/G1 and/or G2/M phases (Figure 2A and B). In contrast, the combination of sorafenib and vorinostat induced a strong accumulation of cells in the G2/M phases in H358 cells (Figure 2C) and had no significant effect in PLC/PRF5 cells (Figure 2D). These cell cycle aberrations strongly suggest that vorinostat, when combined with gefitinib, “induced” apoptotic in both cell lines, whereas when combined with sorafenib induced a G2/M arrest.
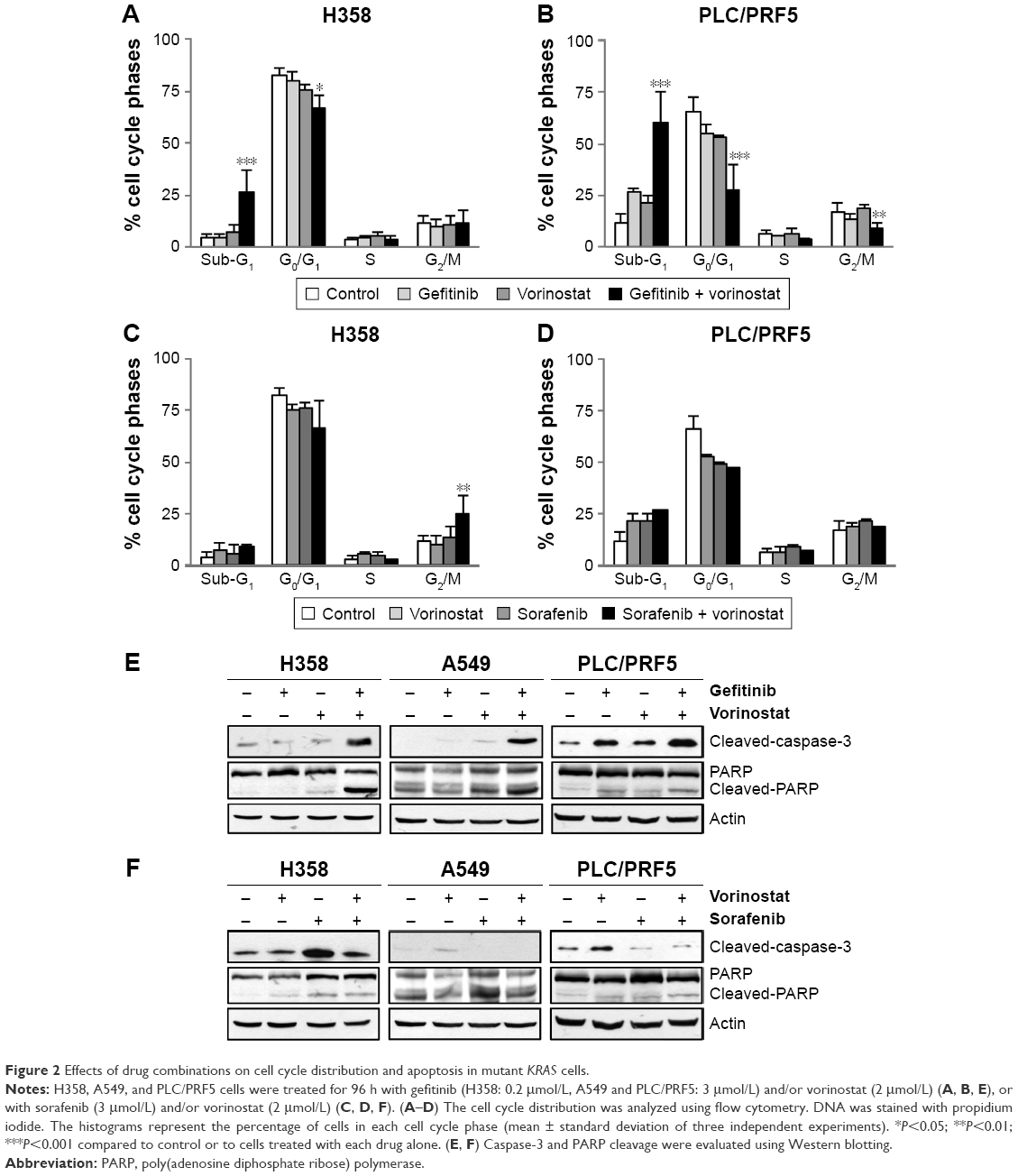 | Figure 2 Effects of drug combinations on cell cycle distribution and apoptosis in mutant KRAS cells. |
Apoptotic effects of drug combinations in NSCLC and HCC cell lines
We further investigated the apoptotic effects of vorinostat combined with the TKIs with Western blots. Similar to the increase in the apoptotic sub-G1 population, a combination of gefitinib with vorinostat was more effective than each drug alone for inducing caspase-3 and PARP cleavage in KRAS mutant cells (Figure 2E). This combination also strongly induced apoptosis in amplified KRAS H322 cells and wild-type KRAS H1719 cells (Supplementary material S3). In contrast, the combination of vorinostat with sorafenib did not enhance the level of cleaved caspase-3 and PARP compared to single treatments in all the cells that were tested (Figure 2F, Supplementary material S3). Taken together, these findings indicate that the synergistic interaction between gefitinib and vorinostat is associated with the induction of apoptosis. In contrast, the combination of vorinostat and sorafenib failed to induce apoptosis, which showed that there was no beneficial interaction.
Effects of drug combinations on signaling pathways in NSCLC and HCC cell lines
We measured the activity of the combined treatments on key intracellular pathways in cell survival and proliferation. As expected, vorinostat enhanced the acetylation of α-tubulin and induced the expression of p21WAF1. The combination of vorinostat and gefitinib slightly enhanced α-tubulin acetylation but attenuated the induction of p21WAF1 by vorinostat (Figure 3A). These effects were also observed in wild-type KRAS cells (Supplementary material S3). Sorafenib alone and in combination with vorinostat enhanced the acetylation of α-tubulin in an additive manner but markedly inhibited p21WAF1 and its induction by vorinostat (Figure 3B).
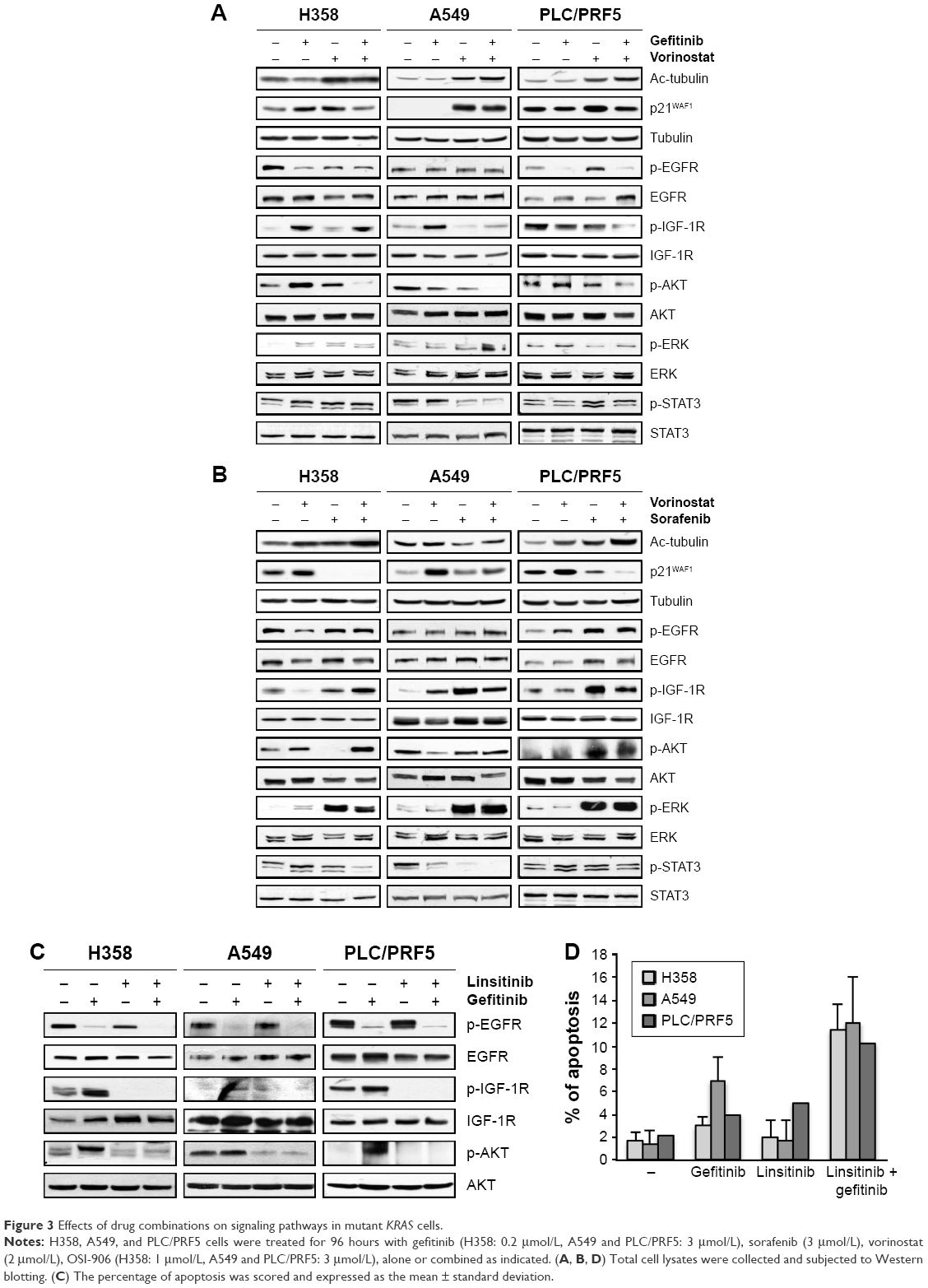 | Figure 3 Effects of drug combinations on signaling pathways in mutant KRAS cells. |
Gefitinib alone markedly inhibited phospho-EGFR (p-EGFR) but enhanced or maintained phosphor-insulin-like growth factor-1 receptor (p-IGF-1R) in resistant KRAS mutant cells (Figure 3A). Vorinostat alone had no significant effect on the activation of EGFR or IGF-1R. When gefitinib was combined with vorinostat, a marked reduction of p-IGF-1R, except in H358 cells, was observed compared to each drug alone in the KRAS mutant cell lines (Figure 3A). No effect of a single or combined treatment was observed in EGFR or IGF-1R levels. Downstream of these receptors, we showed that gefitinib or vorinostat alone had no significant effect on p-AKT, p-ERK, and p-STAT3. The exception was H358 cells in which p-AKT and p-ERK were slightly enhanced, and this effect was previously observed.25 The drug combination markedly reduced p-AKT in the three KRAS mutant cell lines (Figure 3A) in correlation with apoptosis induction.
Sorafenib induced a paradoxical increase of p-IGF-1R and p-ERK (Figure 3B). The combination of sorafenib with vorinostat maintained or enhanced p-EGFR, p-IGF-1R, p-AKT, and p-ERK in comparison with each drug alone and in correlation with the absence of apoptosis, which suggested that sorafenib induced survival pathways that were not inhibited by vorinostat.
Moreover, the inhibition of IGF-1R using an IGF-1R-TKI (linsitinib) decreased the gefitinib-induced activation of AKT (Figure 3C) and restored apoptosis in the three KRAS mutant cell lines (Figure 3D). These data strongly suggest that the IGF-1R-dependent AKT signaling pathway mediated resistance to gefitinib in cells with mutant KRAS and is controlled by vorinostat.
Together, these findings show that vorinostat combined with gefitinib potently repressed IGF-1R/AKT-dependent survival signaling in the NSCLC and HCC KRAS mutant cells, but a similar effect was not observed when vorinostat was combined with sorafenib.
Antitumor efficacy of a gefitinib and vorinostat combination in NSCLC and HCC cells in vivo
Because the gefitinib and vorinostat combination showed synergistic effects in vitro, we examined their in vivo cooperativity using H358 or PLC/PRF5 subcutaneous xenografts in mice. Oral treatments with gefitinib (5 mg/kg) and vorinostat (100 mg/kg) were well-tolerated, and no significant weight loss was observed in the H358 tumor-bearing mice (Figure 4A). Single-drug treatment did not inhibit H358 tumor growth and weight compared with the control group. The combination of gefitinib and vorinostat had a strong significant inhibitory effect on both H358 tumor growth and weight (Figure 4A). This combination of treatments reduced the tumor volume more effectively than each drug alone, and the growth inhibition index calculations showed synergistic effects of the gefitinib/vorinostat combination in H358 tumors (Table 2). This effect was also observed in the PLC/PRF5 xenograft model. According to the IC50 concentrations (Table 1), we used 50 mg/kg gefitinib for treating PLC/PRF5 tumor-bearing mice in combination with 100 mg/kg vorinostat. Because this dose of gefitinib induced 8%–12% weight loss 1 week after beginning the treatment in PLC/PRF5 tumor-bearing mice (not shown), we reduced the frequency of treatment administration to three times a week. This adaptation was better tolerated and was continued until the end of the experiment (Figure 4B). Gefitinib alone or in combination with vorinostat inhibited PLC/PRF5 tumor growth with a synergistic interaction compared with the control group (Figure 4B, Table 2). In addition, because of the rapid growth of PLC/PRF5 tumors and the reduced frequency of the treatment, some animals in the control and vorinostat-treated groups presented rapidly growing tumors that reached 2 cm3. These animals were removed from the experiment for ethical reasons, which reduced the population of these two groups. This phenomenon significantly decreased when the mice received gefitinib or gefitinib/vorinostat, which highlighted the benefit of these two treatments (Figure 4C). This effect was not observed in H358 tumor-bearing mice because of the growth rate of H358 tumors and the frequency of the treatments.
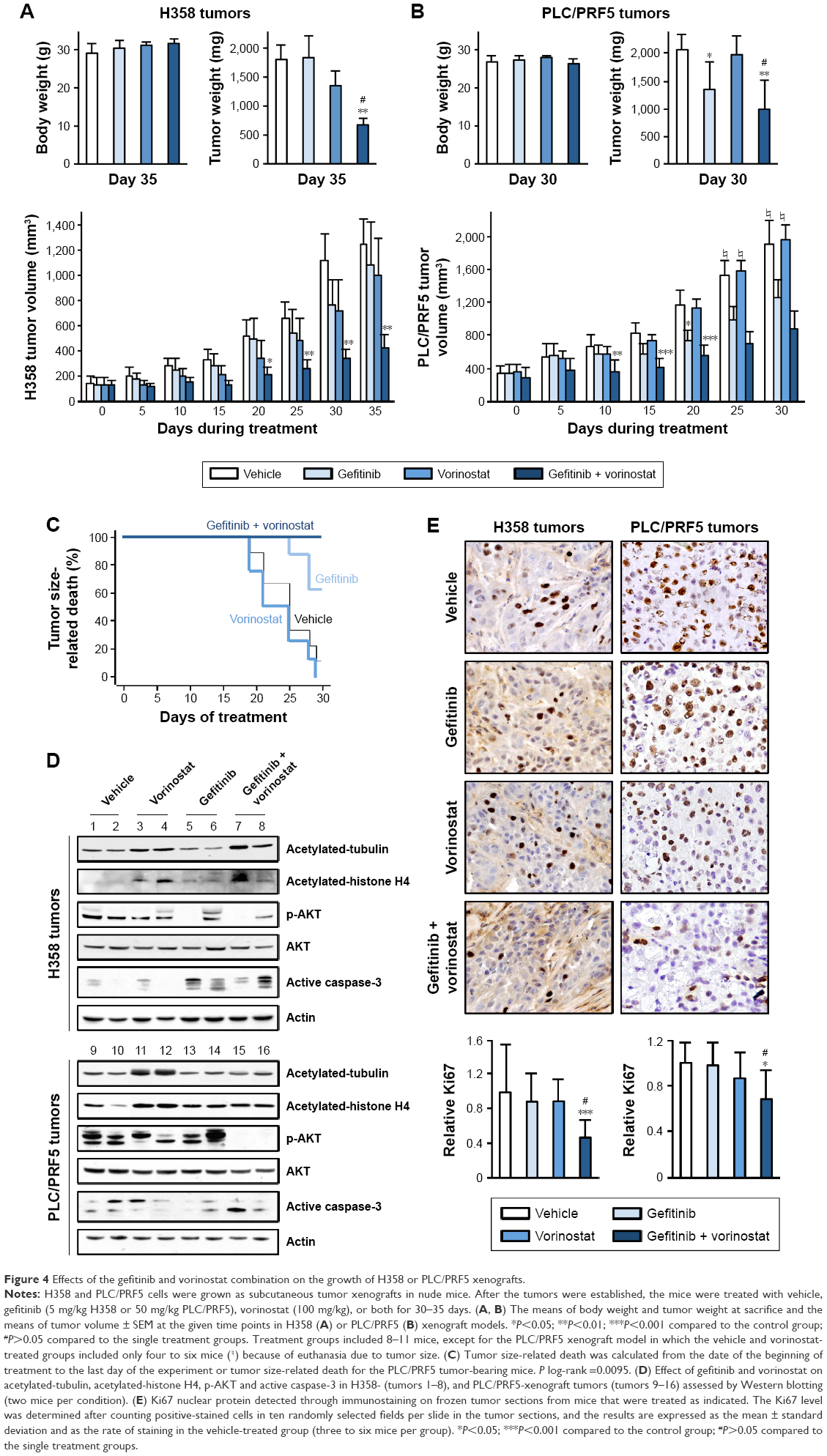 | Figure 4 Effects of the gefitinib and vorinostat combination on the growth of H358 or PLC/PRF5 xenografts. |
 | Table 2 Synergistic indexes of combination treatment with gefitinib and vorinostat in vivo |
Intratumoral biomarkers were assessed with Western blot and immunohistochemical analyses. Consistent with the in vitro data, vorinostat enhanced the acetylation of tubulin and histone H4, and the combined treatments decreased p-AKT and slightly enhanced active-caspase three levels (Figure 4D). Ki67 immunostaining showed that the gefitinib and vorinostat combination significantly decreased cell proliferation by approximately twofold in H358 tumors and 1.5-fold in PLC/PRF5 tumors compared to the control- or single-treatment groups (Figure 4E). Active-caspase-3 and terminal deoxynucleotidyl transferase dUTP nick end labeling in the tumors showed that single and combined treatments enhanced apoptosis compared to the control group (Supplementary material S4). Thus, the gefitinib and vorinostat combined treatments enhanced the antitumor activity of each drug in vivo.
Discussion
Although the development of selective molecular targeted therapies has led to advances in cancer treatment, several relevant issues for the optimal and effective use of targeted therapies remain unsolved. Clinical responses to a single agent EGFR-TKI or the multi-kinase inhibitor sorafenib have rarely been observed in NSCLC and HCC patients, respectively,1,17 and these patients will eventually develop resistance.2,18,26 There is an urgent need for the identification of alternative therapeutic strategies. In this respect, combinations of targeted drugs could be key factors for increasing therapeutic efficacy, especially in patients with mutant KRAS NSCLC or HCC.
Despite the demonstrated benefits of EGFR-TKI in NSCLC patients with EGFR-activating mutations, not all patients with NSCLC respond to treatment and they eventually develop resistance.1,2 Unlike NSCLC, very few studies have shown the effects of single agent EGFR-TKI treatment in HCC cells.27–29 The clinical efficacy of EGFR inhibitors as single agents in HCC is modest.30 Likewise, a limited clinical benefit was observed with HDAC inhibitors for patients with solid tumors, including NSCLC and HCC.11 In lung cancer, phase II trials of vorinostat showed encouraging results.31 However, the subsequent phase III randomized trial failed to demonstrate improvements in the response rate and survival with vorinostat.11
The results demonstrated that in comparison to single agents, the combined inhibition of EGFR and HDAC provided a stronger inhibition of cell growth and induction of cell death in NSCLC and HCC cell lines. These effects were demonstrated in vitro and in vivo. We observed that the synergistic interaction of gefitinib and vorinostat triggered apoptosis in NSCLC and HCC cell lines with wild-type EGFR and mutant KRAS and that these therapies showed inhibition of NSCLC and HCC tumor growth in vivo. Other preclinical studies reported enhanced efficacy of EGFR and HDAC co-inhibition in NSCLC cells9,12–16,32 and in head and neck carcinoma cells.33 Interestingly, we were not able to find reports on the combined effects of EGFR-TKI and HDAC inhibitors in HCC. The sub-optimal doses of gefitinib and vorinostat that were used in H358 tumor-bearing mice, as well as the lower frequency of administration used in PLC/PRF5 tumor-bearing mice, were not associated with any documented side effects, and no signs of toxicity were observed in the co-treated animals. Few clinical trials investigated EGFR-TKI and HDAC inhibitor combination in NSCLC patients, but they did not improve the outcomes in unselected patients or in patients with EGFR mutations.34–36 In contrast, the results strongly support the value of associating HDAC inhibitors with EGFR-TKI treatments in drug-resistant mutant KRAS patients.
The mechanistic analysis suggested that the synergism between gefitinib and vorinostat involved the inhibition of the IGF-1R and AKT pro-survival pathways and that apoptosis was activated in both NSCLC and HCC cells with mutant or amplified KRAS.24 The combined treatment maintained p-IGF-1R only in H358 cells but still inhibited p-AKT, which suggested that it was acting downstream of the IGF-1R via a mechanism that still needed to be elucidated. Accordingly, IGF-1R was involved in resistance to EGFR-TKI in mutant KRAS NSCLC cells.16,25,37 Moreover, acetylation mechanisms regulated IGF-1R- and PI3K/AKT signaling,38,39 and the combination of an HDAC inhibitor with an EGFR-TKI inhibited AKT signaling in lung and head and neck cancer cells.9,14,16,33 It has also been reported that HDAC and EGFR co-inhibition modulated ErbB receptor levels but that these effects were independent of the EGFR or KRAS status.9,33 We also observed that gefitinib slightly downregulated vorinostat-induced p21WAF1 expression. P21WAF1 is a cyclin-dependent kinase inhibitor that played an important role in the regulation of the G1/S cell cycle and that may have protected cells from undergoing apoptosis.40 Therefore, the apoptotic effects of a gefitinib and vorinostat combination may have occurred through p21WAF1 inhibition.
Sorafenib is the standard of care for the treatment of advanced HCC.17 However, its clinical benefits remain modest and most often consist of temporary tumor stabilization.18 In NSCLC, sorafenib did not show clinical activity in a randomized phase III trial in NSCLC patients with a KRAS mutation.41 The first completed prospective, biopsy-mandated, biomarker-based, adaptively randomized study in pretreated lung cancer patients (Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination, BATTLE trial) reported a benefit of sorafenib in patients with mutated or wild-type KRAS.42
Preclinical studies have demonstrated antiproliferative, antiangiogenic, and proapoptotic effects from combining HDAC inhibitors with sorafenib in epithelial tumor cells,19–21 including HCC cells.22 Several phase I studies investigated HDAC inhibitors in combination with sorafenib, and these studies either support future clinical development of an entinostat/sorafenib combination in patients with advanced solid tumors43 or show that the combined sorafenib and vorinostat treatment was not tolerated and had no confirmed response in patients with renal cell carcinoma and NSCLC.44 We observed that an antagonistic combination of vorinostat with sorafenib failed to enhance apoptosis in both NSCLC and HCC cells. This combination of treatments induced G2/M cell cycle arrest, maintained IGF-1R, AKT, and ERK activation, but failed to induce apoptosis. Accordingly, IGF-1R/AKT pathway inhibition has been shown to improve the efficacy of molecular-targeted therapies such as sunitinib for HCC.45 Although sorafenib targets B-Raf and C-Raf in the ERK signaling pathway, we observed that sorafenib did not inhibit ERK phosphorylation in cells with mutant KRAS, as previously shown.46 In addition, the sorafenib and vorinostat combination strongly downregulated p21WAF1 expression. Accordingly, the p21WAF1 knockdown has been shown to decrease the anti-proliferative activity of vorinostat.47 The antagonistic effects of a vorinostat and sorafenib combination may result from the inhibition of the expression of the tumor-suppressor gene p21WAF1. In addition, other cyclin-dependent kinases may also be involved. After labeling the senescent cells using acid β-galactosidase staining, we did not observe any significant signals in response to a sorafenib and vorinostat combination (data not shown), which suggested that this treatment did not induce senescence. Interestingly, autophagy played a compensatory role during treatment with sorafenib, vorinostat, or their combination in HCC cells.48 This result could explain the absence of apoptosis and should be investigated further.
Conclusion
In summary, HDAC inhibitors have the potential to restore tumor cell sensitivity to EGFR-TKIs and dramatically reduce the concentration of EGFR-TKI necessary to induce cell death in mutant KRAS NSCLC and HCC cells in vitro and in vivo through IGF-1R-dependent AKT signaling downregulation. In contrast, sorafenib exerts an antagonistic effect when combined with vorinostat both in NSCLC and HCC cell lines through IGF-1R-dependent AKT signaling activation. Many clinical trials have been completed without prospectively evaluating or targeting the specific subpopulation of drug-resistant patients; the addition of a new agent could be clinically effective when based on preclinical data. The observations, which demonstrated the synergistic antitumor activity of gefitinib combined with vorinostat, should be considered when designing future clinical trials based on the association of these inhibitors in NSCLC or HCC patients with mutant KRAS.
Acknowledgments
We thank Anthony Lucas for immunohistochemical studies. This study was supported by grants from “La Ligue Contre le Cancer” (“comités Isère” and “Puy de Dôme” – AH), “La Région Rhône-Alpes” (VJ), the “ANR projet blanc” (Agence Nationale pour la Recherche, grant ANR 11-BSV5-018-02 – AH), and TÜBITAK (grant no 113S389 – MO).
Author contributions
VJ, LV, SM, and SF conducted the experiments. VJ, BB, J-LC, MO, and AH participated in study design. VJ, BB, MC, J-LC, MO, and AH analyzed the data. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. | ||
Hirsch FR, Janne PA, Eberhardt WE, et al. Epidermal growth factor receptor inhibition in lung cancer: status 2012. J Thorac Oncol. 2013;8(3):373–384. | ||
Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2(1):e17. | ||
West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30–39. | ||
Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–3115. | ||
Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–5417. | ||
O’Connor O, Masszi T, Savage K, Pinter-Brown L, Foss F, Popplewell L. Belinostat, a novel pan-histone deacetylase inhibitor (HDACi), in relapsed or refractory peripheral T-cell lymphoma (R/R PTCL): results from the BELIEF trial. J Clin Oncol. 2013;31(15):abstract 8507. | ||
Bailey H, Stenehjem DD, Sharma S. Panobinostat for the treatment of multiple myeloma: the evidence to date. J Blood Med. 2015;6:269–276. | ||
Chen MC, Chen CH, Wang JC, et al. The HDAC inhibitor, MPT0E028, enhances erlotinib-induced cell death in EGFR-TKI-resistant NSCLC cells. Cell Death Dis. 2013;4:e810. | ||
Lai CJ, Bao R, Tao X, et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010;70(9):3647–3656. | ||
Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011;7(2):263–283. | ||
Witta SE, Dziadziuszko R, Yoshida K, et al. ErbB-3 expression is associated with E-cadherin and their coexpression restores response to gefitinib in non-small-cell lung cancer (NSCLC). Ann Oncol. 2009;20(4):689–695. | ||
Leone A, Roca MS, Ciardiello C, et al. Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway. Free Radic Biol Med. 2015;89:287–299. | ||
Lin YC, Lin YC, Shih JY, et al. DUSP1 expression induced by HDAC1 inhibition mediates gefitinib sensitivity in non-small cell lung cancers. Clin Cancer Res. 2015;21(2):428–438. | ||
Busser B, Sancey L, Josserand V, et al. Amphiregulin promotes resistance to gefitinib in nonsmall cell lung cancer cells by regulating Ku70 acetylation. Mol Ther. 2010;18(3):536–543. | ||
Jeannot V, Busser B, Brambilla E, et al. The PI3K/AKT pathway promotes gefitinib resistance in mutant KRAS lung adenocarcinoma by a deacetylase-dependent mechanism. Int J Cancer. 2014;134(11):2560–2571. | ||
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | ||
Siegel AB, Olsen SK, Magun A, Brown RS Jr. Sorafenib: where do we go from here? Hepatology. 2010;52(1):360–369. | ||
Baradari V, Hopfner M, Huether A, Schuppan D, Scherubl H. Histone deacetylase inhibitor MS-275 alone or combined with bortezomib or sorafenib exhibits strong antiproliferative action in human cholangiocarcinoma cells. World J Gastroenterol. 2007;13(33):4458–4466. | ||
Park MA, Zhang G, Martin AP, et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther. 2008;7(10):1648–1662. | ||
Zhang G, Park MA, Mitchell C, et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res. 2008;14(17):5385–5399. | ||
Chen CH, Chen MC, Wang JC, et al. Synergistic interaction between the HDAC inhibitor, MPT0E028, and sorafenib in liver cancer cells in vitro and in vivo. Clin Cancer Res. 2014;20(5):1274–1287. | ||
Chou TC, Motzer RJ, Tong Y, Bosl GJ. Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: a rational approach to clinical protocol design. J Natl Cancer Inst. 1994;86(20):1517–1524. | ||
Sudhir PR, Hsu CL, Wang MJ, et al. Phosphoproteomics identifies oncogenic Ras signaling targets and their involvement in lung adenocarcinomas. PLoS One. 2011;6(5):e20199. | ||
Hurbin A, Wislez M, Busser B, et al. Insulin-like growth factor-1 receptor inhibition overcomes gefitinib resistance in mucinous lung adenocarcinoma. J Pathol. 2011;225(1):83–95. | ||
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Hopfner M, Sutter AP, Huether A, Schuppan D, Zeitz M, Scherubl H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J Hepatol. 2004;41(6):1008–1016. | ||
Okano J, Matsumoto K, Nagahara T, Murawaki Y. Gefitinib and the modulation of the signaling pathways downstream of epidermal growth factor receptor in human liver cancer cells. J Gastroenterol. 2006;41(2):166–176. | ||
Fuchs BC, Fujii T, Dorfman JD, et al. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008;68(7):2391–2399. | ||
Thomas MB, Chadha R, Glover K, et al. Phase 2 study of erlotinib in patients with unresectable hepatocellular carcinoma. Cancer. 2007;110(5):1059–1067. | ||
Ramalingam SS, Maitland ML, Frankel P, et al. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(1):56–62. | ||
Nakagawa T, Takeuchi S, Yamada T, et al. EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res. 2013;73(8):2428–2434. | ||
Bruzzese F, Leone A, Rocco M, et al. HDAC inhibitor vorinostat enhances the antitumor effect of gefitinib in squamous cell carcinoma of head and neck by modulating ErbB receptor expression and reverting EMT. J Cell Physiol. 2011;226(9):2378–2390. | ||
Han JY, Lee SH, Lee GK, et al. Phase I/II study of gefitinib (Iressa(®)) and vorinostat (IVORI) in previously treated patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2015;75(3):475–483. | ||
Reguart N, Rosell R, Cardenal F, et al. Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer. 2014;84(2):161–167. | ||
Witta SE, Jotte RM, Konduri K, et al. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J Clin Oncol. 2012;30(18):2248–2255. | ||
Suda K, Mizuuchi H, Sato K, Takemoto T, Iwasaki T, Mitsudomi T. The insulin-like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild-type epidermal growth factor receptor. Int J Cancer. 2014;135(4):1002–1006. | ||
Kim JS, Lee SC, Min HY, et al. Activation of insulin-like growth factor receptor signaling mediates resistance to histone deacetylase inhibitors. Cancer Lett. 2015;361(2):197–206. | ||
Pirola L, Zerzaihi O, Vidal H, Solari F. Protein acetylation mechanisms in the regulation of insulin and insulin-like growth factor 1 signalling. Mol Cell Endocrinol. 2012;362(1–2):1–10. | ||
Park C, Jeong NY, Kim GY, et al. Momilactone B induces apoptosis and G1 arrest of the cell cycle in human monocytic leukemia U937 cells through downregulation of pRB phosphorylation and induction of the cyclin-dependent kinase inhibitor p21Waf1/Cip1. Oncol Rep. 2014;31(4):1653–1660. | ||
Paz-Ares LG, Biesma B, Heigener D, et al. Phase III, randomized, double-blind, placebo-controlled trial of gemcitabine/cisplatin alone or with sorafenib for the first-line treatment of advanced, nonsquamous non-small-cell lung cancer. J Clin Oncol. 2012;30(25):3084–3092. | ||
Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1(1):44–53. | ||
Ngamphaiboon N, Dy GK, Ma WW, et al. A phase I study of the histone deacetylase (HDAC) inhibitor entinostat, in combination with sorafenib in patients with advanced solid tumors. Invest New Drugs. 2015;33(1):225–232. | ||
Dasari A, Gore L, Messersmith WA, et al. A phase I study of sorafenib and vorinostat in patients with advanced solid tumors with expanded cohorts in renal cell carcinoma and non-small cell lung cancer. Invest New Drugs. 2013;31(1):115–125. | ||
Ou DL, Lee BS, Chang YC, et al. Potentiating the efficacy of molecular targeted therapy for hepatocellular carcinoma by inhibiting the insulin-like growth factor pathway. PLoS One. 2013;8(6):e66589. | ||
Takezawa K, Okamoto I, Yonesaka K, et al. Sorafenib inhibits non-small cell lung cancer cell growth by targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant cells. Cancer Res. 2009;69(16):6515–6521. | ||
Arnold NB, Arkus N, Gunn J, Korc M. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances gemcitabine-induced cell death in pancreatic cancer. Clin Cancer Res. 2007;13(1):18–26. | ||
Yuan H, Li AJ, Ma SL, et al. Inhibition of autophagy significantly enhances combination therapy with sorafenib and HDAC inhibitors for human hepatoma cells. World J Gastroenterol. 2014;20(17):4953–4962. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.