")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Swimming Differentially Affects T2DM-Induced Skeletal Muscle ER Stress and Mitochondrial Dysfunction Related to MAM
Authors Zhang Z , Cui D, Zhang T, Sun Y, Ding S
Received 19 December 2019
Accepted for publication 2 April 2020
Published 30 April 2020 Volume 2020:13 Pages 1417—1428
DOI https://doi.org/10.2147/DMSO.S243024
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Zhe Zhang,1,2 Di Cui,1,2 Tan Zhang,1,2 Yi Sun,1,2 Shuzhe Ding1,2
1Key Laboratory of Adolescent Health Assessment and Exercise Intervention of Ministry of Education, East China Normal University, Shanghai 200241, People’s Republic of China; 2College of Physical Education and Health, East China Normal University, Shanghai 200241, People’s Republic of China
Correspondence: Shuzhe Ding
Key Laboratory of Adolescent Health Assessment and Exercise Intervention of Ministry of Education, East China Normal University, Shanghai 200241, People’s Republic of China
Tel +86 13601798505
Email [email protected]
Purpose: Mitochondrial dysfunction and endoplasmic reticulum stress (ERS) are associated with metabolic diseases such as obesity and Type 2 diabetes mellitus (T2DM). Mitochondria and ER are connected via mitochondria-associated membranes (MAM) that are involved in glucose homeostasis and insulin resistance. We postulated that exercise might positively benefit T2DM-induced ER and mitochondrial dysfunction that might be associated with MAM.
Materials and Methods: Mice were fed a high-fat diet and injected with streptozotocin (STZ) to create T2DM models. Glucose tolerance, mitochondrial quality, MAM quality, and ERS were investigated in diabetic mice after six weeks of swimming.
Results: Type 2 DM induced decreased MAM quantity, impaired mitochondrial quality, and deteriorated ERS in skeletal muscle that led to endoplasmic reticulum-associated degradation (ERAD). Swimming alleviated strong ERS caused by T2DM. Importantly, MAM quantity was positively associated with mitochondrial function and tended to negatively correlate with the ERS branch, ATF6. Moreover, both ATF6 branches of ERS and ERAD were positively associated with the pIRE1α branch of ERS.
Conclusion: Type 2 DM induced glucose intolerance, powerful ERS, and mitochondrial dysfunction associated with decreased amounts of MAM. Swimming improved glucose intolerance and selectively mitigated the ERS in skeletal muscle. Therefore, MAM quality and ATF6 might be novel and important targets for T2DM treatment. Endoplasmic reticulum stress might be an effective target of swimming to improve diabetes.
Keywords: T2DM, skeletal muscle, mitochondrial quality, ERS, MAM
Introduction
Type 2 DM is a complex metabolic disorder and its incidence has dramatically increased over the past few decades. Consequently, a better understanding of the mechanisms underlying this pathology is crucial for proposing new therapeutic approaches.1 Type 2 DM is often associated with the development of secondary complications in various organs, such as the eyes, kidneys, heart, brain, and skeletal muscle.2 Among these organs, skeletal muscle is crucial for glucose homeostasis as it is the primary site of insulin-stimulated glucose uptake. Therefore, it is the main target of changes in insulin resistant states and T2DM.3 Type 2 DM is associated with reduced mitochondrial function, dynamics and membrane potential, (MMP),1,4–10 and aberrant ERS,11 all of which could impair energy metabolism in skeletal muscle. Recent structural and functional studies have revealed interactions between mitochondria and ER networks, and zones of close contact between these organelles are called MAM (mitochondria associated membranes)12 (11). These MAM allow mitochondria and ER to share some important cellular functions that are critical for insulin production and insulin-stimulated glucose disposal.1 Given the structural and functional link between ER and mitochondria, a potential emerging mechanism of T2DM might involve interaction between mitochondria and ER (MAM).1,13,14
Molecular chaperones such as calnexin, calreticulin, and the sigma-1 receptor (sigma-1R) at MAM regulate associations between the ER and mitochondria.12 The sigma-1R cloned in 199615,16 represents an integral membrane protein that is localized at the ER (and the ER–mitochondria interface) suggesting that it functions as an ER chaperone. Thus, Sigma-1R is considered as the gold standard for quantifying MAM. This receptor also modulates several signaling pathways including ion channels, lipid rafts, autophagy, and the ER stress response.17,18
Endoplasmic reticulum stress (ERS) refers to a subcellular organelle process in which ER homeostasis is disrupted by physiological factors such as heat shock, mitochondrial calcium overload,19 or a high-fat diet.20 These perturbations can lead to the accumulation of unfolded proteins and protein aggregates in the lumen of the ER which is called the unfolded protein response (UPR). The occurrence of a UPR suggests ERS.21 The UPR consists of three main branches, each of which is activated by the specific stress transducers, protein kinase R-like ER protein kinase (PERK), activating transcription factor 6 (ATF6) and inositol requiring enzyme 1 (IRE1). In the basal state, these are all associated with the chaperone Bip (also called glucose-regulated protein 78; GRP78). When unfolded proteins accumulate, each transducer can disassociate from Bip, which results in self-activation of the transducer.22 Both PERK and IRE1 are activated by auto-phosphorylation oligomerization, whereas ATF6 is translocated to the Golgi apparatus for activation.23 When the ERS exceeds the processing capacity of a cell, ER-associated degradation (ERAD) can result.24 Caspase 12 is a specific indicator of ERAD because it is active during ER deterioration to ERAD.25
Physical exercise plays a role in regulating various bodily functions, such as cellular metabolism, as well as cardiovascular and muscular functions. Endurance exercise increases mitochondrial function in skeletal muscle26,27 and protects against ERS, as this kind of exercise increases the expression of several chaperones in mice fed with a high-fat diet.28 Therefore, we questioned whether exercise intervention would also be associated with interactions between mitochondria and ER in patients with diabetes. Mitochondria interact with the sarcoplasmic endoplasmic reticulum (SR/ER) in skeletal muscle. However, a correlation between MAM and insulin resistance in skeletal muscle has not been investigated in detail. To further understand the relationship between MAM in skeletal muscle and diabetes, and to confirm whether crosstalk between mitochondria and ER is a mechanism of positive outcomes of exercise intervention for T2DM, we generated T2DM animal models by feeding mice with a high-fat diet and injecting them intraperitoneally (ip) with STZ. We then trained them to swim, and analyzed the effects of swimming on blood glucose, mitochondrial function in skeletal muscle, Sigma-1R and the ERS signaling pathway. We found that T2DM impaired glucose tolerance, induced ERS that led to ERAD in skeletal muscle, and caused mitochondrial dysfunction associated with reduced MAM. Swimming alleviated deleterious ERS in diabetic mice and thus mitigated ERAD.
Materials and Methods
The Committee for the Care of Laboratory Animal Resources, East China Normal University, approved all experimental protocols, and the study proceeded in accordance with the National Institutes of Health Guide For The Care And Use Of Laboratory Animals (NIH Publications No. 8023, revised 1978).
Animals
Thirty-nine clean-grade male C57BL/6 mice (age, 4 weeks; weight, 16.49 ± 1.50 g; Shanghai SLAC Laboratory Animals Co., Ltd., Shanghai China, Animal Production License No. SCXK (Shanghai) 2007–0005, animal use identification certificate: SYXK (Shanghai) 2004–0001) were randomly divided into control (n = 16) and diabetes (n = 23) groups. Clean grade means that the animals are free of pathogens that are harmful to humans and other animals, or that could interfere with scientific investigations. The mice are quarantined, and have specific documentation including quarantine and lineage certificates. All mice were housed in cages maintained at 22 ± 1°C and 50% ± 5% humidity under a 12 h light/dark cycle. Fodder and water were available ad libitum.
The control group was fed with standard chow. The group with diabetes was fed with a high-fat diet (54.6% basic feed, lard 16.9%, sucrose 14%, casein 10.2%, premix feed 2.1%, maltodextrin 2.2%) for four weeks, then were injected with STZ (50 mg/kg/day) for 5 days to induce T2DM. Control mice were injected with citrate buffer. Fasting blood glucose >11.1 mmol/L was taken as the criterion for diabetes in the mice.29 The mice were randomly assigned to control (C), exercised (E), diabetic sedentary (D), and diabetic exercised (DE) groups (n = 8 each).
Blood Glucose Evaluation and Intraperitoneal Glucose Tolerance Test (IPGTT)
Mice fasted overnight before blood glucose tests. Blood samples were obtained from the tail vein, and blood glucose was measured using an ACCU-CHEK Active glucometer (Roche Holdings AG, Basel, Switzerland) with glucose test strips (Roche Holdings AG).
After an overnight fast, venous blood was collected from the tail to measure baseline glucose before, and 15, 30, 45, 60, 75, 90 and 120 min after an intraperitoneal injection of glucose (2 g/kg body weight).
Swimming
A swimming pool was set up as described.30 Water to a depth of 50 cm in a glass tank was kept at a thermostatically controlled temperature of 30°C ± 2°C. During the first week, the mice of Groups E and DE swam for short periods every day for five days to get acclimated to swimming. Thereafter, the mice swam for 1 h per day (4:30–5:30 pm), 5 days per week for 6 weeks.
Tissue Harvesting
After 6 weeks, all of the mice were fasted for 12 h then sacrificed. The left gastrocnemius muscles were collected to extract mitochondria and the right gastrocnemius muscles were snap-frozen in liquid nitrogen and stored at −80°C.
Mitochondrial Membrane Potential
Mitochondria were extracted as described.31,32 The left gastrocnemius was ground in a mortar and homogenized in ice-cold buffer (100 mM KCl, 5 mM MgSO4, 5 mM EDTA, 50 mM Tris-Base, 1 mM ATP, pH 7.4). Mitochondria were fractionated by differential centrifugation. We used the fluorescent probe JC-1 to detect the mitochondrial membrane potential (MMP) Δψ. The probe was diluted to prepare the dye solution according to the specifications recommended by the manufacturer of Beyotime C2006 (Beyotime Biotechnology, Shanghai, China), and fluorescent signals were detected using a Tecan2000 microplate reader (Tecan Group Ltd., Männedorf, Switzerland). Protein was quantified using the bicinchoninic acid (BCA) method.
RNA Extraction and Quantitative Real-Time PCR
We extracted RNA as described.30 Total RNA extracted from gastrocnemius muscles (50 µg) using Trizol assay kits was quantified by spectrophotometry (260 nm). Complementary DNA was prepared from 5 mL of total RNA using ReverTra Ace qPCR RT kits (FSQ-101; Toyobo Co., Ltd., Osaka, Japan). Real-time PCR proceeded using a Step One instrument (Applied Biosystems, Waltham, MA, USA), software (Applied Biosystems) a SYBR and Green PCR Master Mix (QPK201; Toyobo Co. Ltd.) for detection. The quantity of transcripts was normalized to the internal control, GAPDH. Specific primers for Fis1, Mfn1, Mfn2, OPA1 and GAPDH were designed based on the findings of a PubMed search. Topic-related publications were included without date of publication or study design limits. We selected credible publications in which the primers were described and cited when gene target-specific, then specific conditions were set to prepare the primers33–37 (Table 1). All primers were reverse validated (http://genome.ucsc.edu/cigi-bin/hgPcr?hgsid=687005089_cq47CErF0NZA0acwDe6fl0tV6e7i).
 |
Table 1 Primer Sequences for Real-Time PCR |
Preparation of Tissue Lysates for Western Blotting
Gastrocnemius muscles were homogenized in lysis buffer (50 mM Tris HCl, 150 mM NaCl, 1 mM EDTA, 0.2 mM PMSF, and 1% NP-40, pH 7.4), then separated by centrifugation at 8000g for 10 min. Protein concentrations in the supernatant were measured using DC protein assay kits (Bio-Rad Laboratories Inc., Hercules, CA, USA). Proteins were resolved by 8–10% SDS-PAGE, then blotted onto PVDF membrane (Bio-Rad Laboratories Inc). Nonspecific binding was blocked with 5% skim milk then the membranes were incubated overnight with primary antibodies diluted in 3% BSA in PBS-T,38 followed by secondary antibodies for 2 h. Proteins on the membranes were probed using the high sensitivity ECL chemiluminescence kit, ECL Plus (Thermo Fisher Scientific Inc., Waltham, MA, USA), then quantified using a gel imager (Alphatech Inc., Burlington, MA, USA).
Antibodies
The following rabbit primary antibodies were diluted 1:1000 for immunoblotting: monoclonal antibody to phospho-PERK (MA5-15033) and polyclonal antibody to pIRE1α (PA1-16927) (Thermo Fisher), polyclonal antibodies to ATF6 (ab62576), caspase 12 (ab62484) and Sigma-1R (ab151288) (ABCAM, Cambridge, MA, USA), and combined with HRP-conjugated secondary antibodies diluted 1:500 (Santa Cruz Biotechnology Inc., Dallas, TX, USA).
Statistical Analysis
Blood glucose levels in the C and D groups before swimming were analyzed using independent Student t-tests. Multiple comparison data were analyzed by a one-way ANOVA and correlations were determined by Pearson correlations, both using GraphPad Prism version 5.0 software. Data are presented as means ± SD. P values are shown as *P < 0.05, #P < 0.05, **P < 0.01 and ***P < 0.001, unless indicated otherwise.
Results
Swimming Improved Glucose Intolerance in Diabetic Mice
We fed 3–4-week-old mice with a high-fat diet and injected them ip with STZ to induce T2DM. These mice had significantly increased fasting blood glucose levels (>11.1mmol/L) (Figure 1A). Glucose tolerance was further assessed in C and D mice using the IPGTT. Blood glucose levels were significantly higher in the D, than the C mice throughout the test, and did not return to basal levels in the D mice until 120 min after the test (P < 0.001) (Figure 1B). The area under curve (AUC) of the IPGTT graph was significantly increased in the D, compared with the C group (P < 0.001; Figure 1D). Since exercise can improve glucose tolerance,39 we applied the IPGTT to the mice after swimming for 1 h/day, 5 days/week for 6 weeks to further assess glucose tolerance. Swimming improved glucose tolerance in both groups (Figure 1C). Blood glucose levels declined earlier (P < 0.05) and were significantly lower at 60 min of the test in the E than the C group. The results were similar between the DE and D groups, with blood glucose levels at 120 min after the glucose injection being lower (P < 0.05) in the DE, than the D group (Figure 1C). The AUC was significantly reduced in the DE, compared with the D group (P < 0.05; Figure 1E), indicating that swimming improved glucose tolerance in control and diabetic mice.
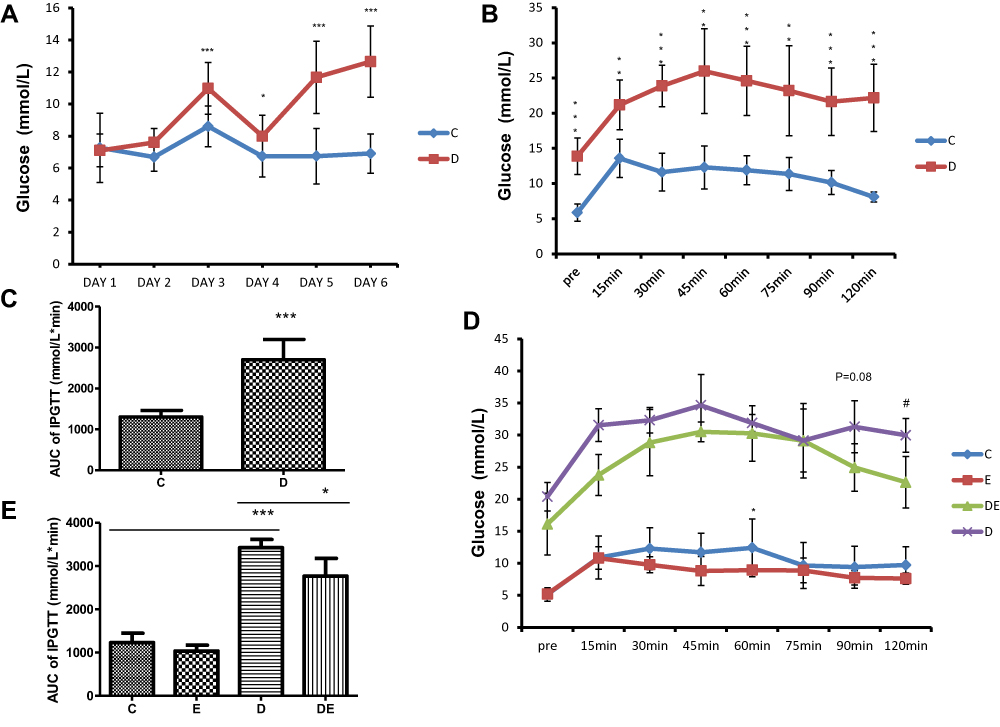 |
Figure 1 Swimming improves T2DM-induced glucose intolerance. (A) Changes in fasting blood glucose after STZ injection. *P < 0.05, ***P < 0.001 (n = 16–23). (B) Blood glucose levels in groups C and D in IPGTT tests. **P < 0.01, ***P < 0.001 (n = 6). (C) Area under curve of IPGTT graph of (B). ***P < 0.001 (n = 6). (D) Blood glucose levels in IPGTT test of groups C, D, E and DE after swimming exercise. *P < 0.05 group C vs E; #P < 0.05 group D vs DE (n = 6). (E) Area under curve of IPGTT graph (D). *P < 0.05, ***P < 0.001 (n = 6). |
Diabetes Reduced Mitochondrial Membrane Potential (MMP) of Skeletal Muscle
The onset of obesity and T2DM are associated with abnormal mitochondrial function1,8,13,14,40 and exercise can alter mitochondrial quality. Given that MMP is considered the most sensitive indicator of mitochondrial quality, we examined MMP in mitochondria extracted from gastrocnemius muscles after swimming. The MMP was significantly decreased in the D, compared with the C group (**P < 0.01), and significantly increased in the E, compared with the C group (*P < 0.05), whereas those between the D and DE groups did not significantly differ (Figure 2).
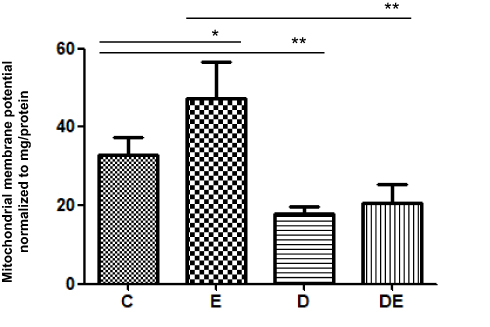 |
Figure 2 T2DM significantly impairs MMP of skeletal muscle. Groups C, E, D and DE. *P < 0.05, **P < 0.01 (n = 6). |
Diabetes Upregulated and Downregulated Mitochondrial Fission and Fusion, Respectively, in Skeletal Muscle
Mitochondrial fission and fusion (mitochondrial dynamics) play critical roles in maintaining functional mitochondria when cells are exposed to metabolic or environmental stresses.38,44 Mitochondrial fission is needed to create healthy mitochondria with a lower MMP.45 Since we found that diabetes reduced MMP, we postulated that lower MMP is associated with mitochondrial fission. We measured mRNA levels of Fis1, Mfn1, Mfn2 and OPA1 to determine mitochondrial dynamics. The Fis1 mRNA levels were significantly increased in the E (P < 0.01,) and D (P < 0.001) groups compared with group C (Figure 3A). The, mRNA levels of OPA1 were significantly decreased in the D (P < 0.05) compared with the C group (Figure 3D), whereas Mfn1 and Mfn2 differ did not significantly differ between them (Figure 3B and C). These results suggest that swimming increased mitochondrial fission compared with control mice, and that diabetes excessively upregulated mitochondrial fission, but downregulated mitochondrial fusion.
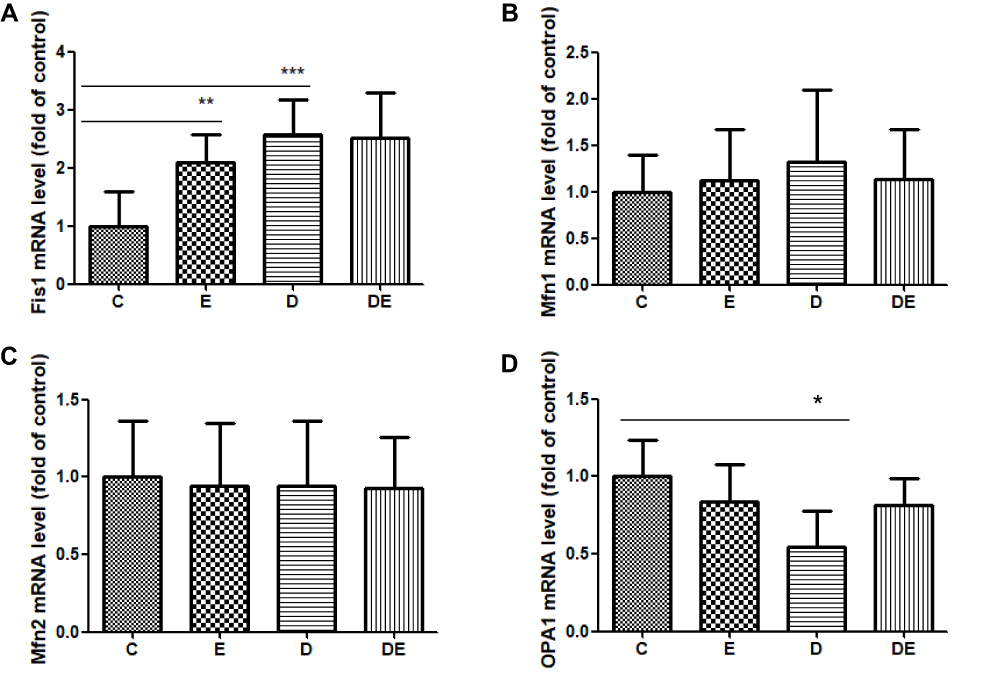 |
Figure 3 Swimming and T2DM increase mitochondrial fission, but T2DM decreases mitochondrial fusion. (A–D) Levels of mRNA in groups C, E, D and DE. (A) Fis1 mRNA. **P < 0.01, ***P < 0.001 (n = 8). (B) Mfn1 mRNA (n = 8). (C) Mfn2 mRNA (n = 8). (D) OPA1 mRNA. *P < 0.05 (n = 6). |
Swimming Ameliorated MAM Impaired by Diabetes
Interactions between mitochondria and ER comprise a potential emerging mechanism of T2DM pathogenesis,1 and Sigma-1R is considered as the gold standard for quantifying MAM. We assessed Sigma-1R expression in lysates of gastrocnemius muscles prepared from groups C, E, D, and DE by immunoblotting. Diabetes significantly reduced (P < 0.01), whereas swimming increased (P = 0.07) Sig-1R protein levels in diabetic mice (Figure 4), suggesting that diabetes decreased, whereas swimming ameliorated the amount of MAM.
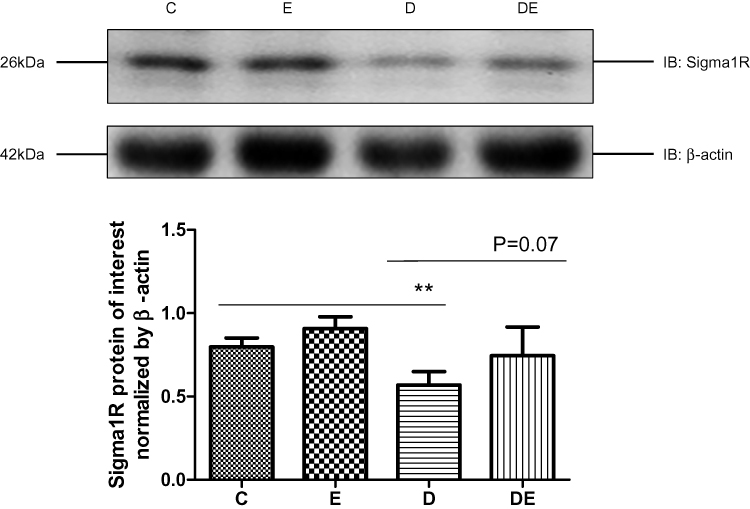 |
Figure 4 T2DM reduces sigma-1R protein expression. Western blots of Sigma-1R expression in gastrocnemius muscles from groups C, E, D and Group DE. **P < 0.01 (n = 6). |
Swimming Alleviated ERS in Skeletal Muscle of Diabetic Mice
Given the changes in mitochondrial quality and MAM along with the ER-mitochondrial crosstalk, we speculated that diabetes aggravates, whereas exercise helps to alleviate ER stress. Therefore, we examined the expression of p-PERK, p-IRE1α and ATF6 proteins, which are critical components of the ERS pathway, in immunoblots of gastrocnemius muscle lysates prepared from the four groups of mice. We also measured the level of caspase 12 protein to assess ERAD. Diabetes resulted in significantly increased levels of pIRE1α (P < 0.01,), ATF6 (P < 0.05) and caspase 12 (P < 0.001) expression (Figure 5). However, swimming significantly reduced the protein levels of pIRE1, ATF6, and caspase 12 (P<0.05 for all) in the gastrocnemius of the DE compared with the D group (Figure 5). The level of p-PERK expression in the gastrocnemius did not change in either group.
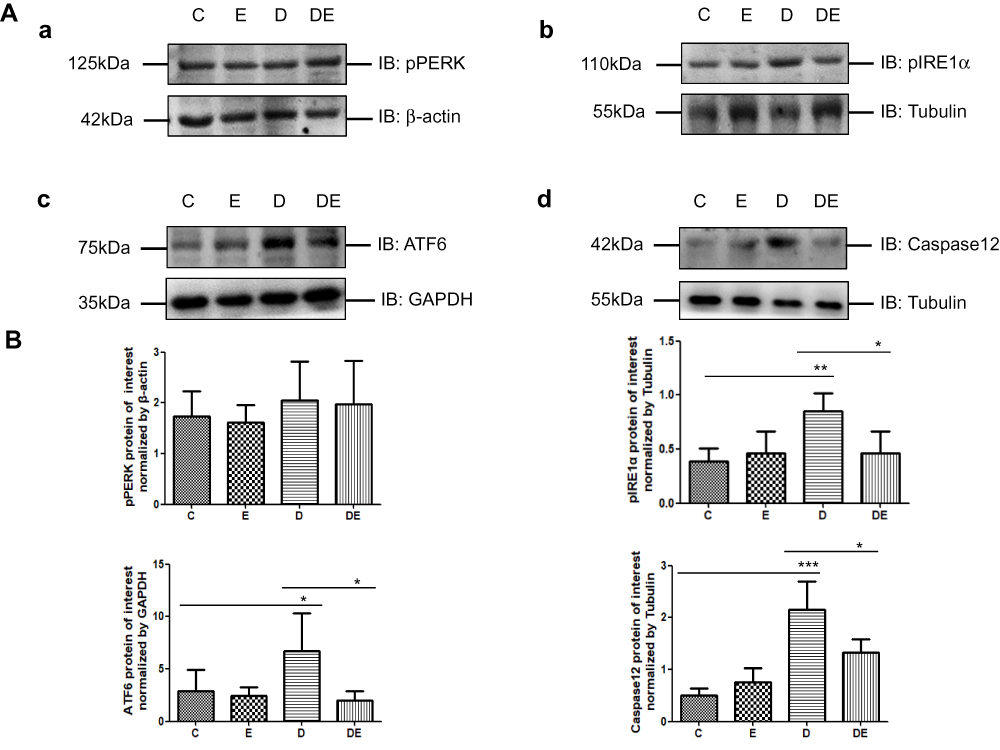 |
Figure 5 Type 2 diabetes exacerbates ER stress leading to ERAD, which is mitigated by swimming (A,B). (a–d) Western blots of p-PERK, p-IRE1α, ATF6 and caspase 12 in gastrocnemius muscles of mice in groups C, E, D, and DE (n = 6 per group. *P < 0.05, **P < 0.01, and ***P < 0.001. |
Changes in MMP and ERS are Associated with MAM
To further verify whether mitochondrial quality and ERS are linked to MAM, we investigated correlations between the amounts of MMP/ERS signaling and of Sigma-1R expression. Levels of Sigma-1R expression correlated positively with MMP (R2 = 0.4901, P < 0.05; Figure 6A), but inversely with those of ATF6 from gastrocnemius muscle (R2 = 0.2658, P = 0.06; Figure 6A). These results suggested a positive correlation between MAM and quantitative as well as mitochondrial function, and a negative correlation between MAM and the UPR branch, which is activated by ATF6. Since two branches (ATF6 and pIRE1) of the UPR and ERAD (caspase 12) were activated by diabetes, we also assessed correlations among protein levels of ATF6, caspase 12 and pIRE1α. We found positive correlations between the levels of pIRE1α and ATF6 proteins (R2 = 0.2283, P < 0.05), and between those of pIRE1α and caspase 12 (R2 = 0.4653, P < 0.01; Figure 6B).
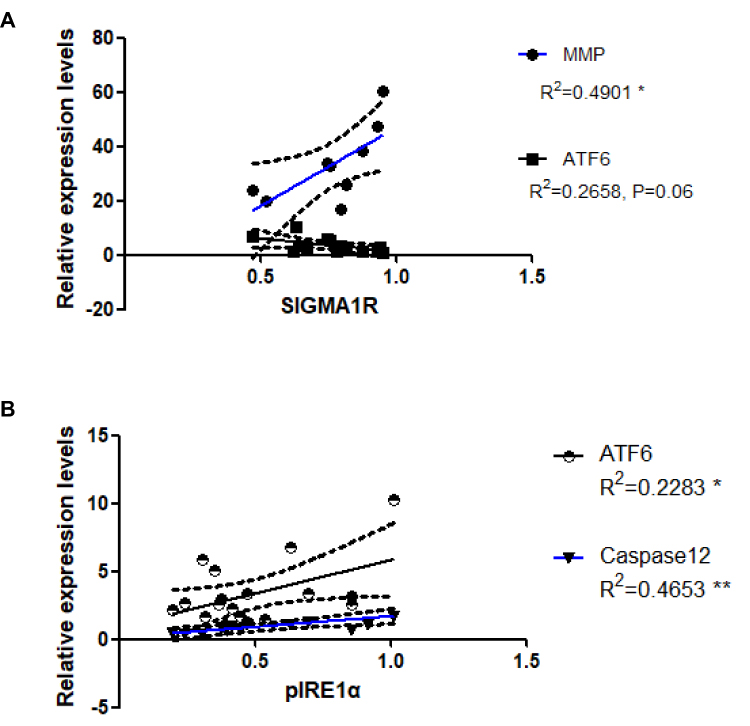 |
Figure 6 Associations between expression levels of Sigma-1R and MMP, and that of pIRE1α and ATF6 and caspase 12. (A) Correlations between Sigma-1R and MMP and ATF6. *P < 0.05. (B) Correlations between the pIRE1α and ATF6 and caspase12. *P < 0.05, **P 0.01. |
Discussion
The present study provides new insights into the role of MAM in mitochondrial quality and ERS in T2DM and the effects of swimming as intervention. We found that STZ-induced diabetes results in impaired glucose tolerance, ER stress and abnormal mitochondrial quality associated with reduced MAM. We also found that swimming significantly improved glucose tolerance, and alleviated the ERS. Swimming tended to alleviate mitochondrial dysfunction and the decreased MAM caused by type 2 diabetes.
Physical exercise is an effective intervention for treating metabolic diseases,14,28,43 such as obesity, hypoglycemia and diabetes. Indeed, our IPGTT findings showed that swimming improves glucose tolerance in diabetic mice, which was similar to previous results.39,46
Both swimming and diabetes resulted in mitochondrial fission, with which Fis1 is thought to be related. Fission molecules are upregulated in the skeletal muscles of mice with obesity induced by a high-fat diet.47 Notably, moderate mitochondrial fission is beneficial for exercise adaptation,43 whereas excessive mitochondrial fission disrupts mitochondrial pool health, which consequently damages skeletal muscle homeostasis.38,42 The present study found that swimming induced, whereas diabetes significantly increased mitochondrial fission (group E vs D: P < 0.01 vs P < 0.001). We speculated that swimming regularly for six weeks induced moderate mitochondrial fission which was advantageous for exercise adaption, whereas diabetes induced excessive mitochondrial fission that is detrimental to the mitochondrial network.
Fission is needed to create new mitochondria and regulate the mitochondrial network through a selective autophagic process called mitophagy.44 This process includes the removal of depolarized mitochondria with a lower MMP than healthy mitochondria during cellular stress. Therefore, mitochondrial fission is closely associated with changes in MMP. Here, we showed that diabetes significantly decreased, whereas swimming obviously increased MMP, which was consistent with changes in Fis1 mRNA levels. We recently showed that a Fis1 deficiency results in abnormal mitochondrial dynamics and delayed mitophagy execution in skeletal muscle at rest and under exercise stress, indicating that Fis1 not only mediates fission, but also maintains normal mitophagy.38,48,49 Therefore, we speculate that the decreased MMP induced by diabetes arises from excessively elevated Fis1 transcription, and that both of these processes work in concert to affect mitophagy. However, this awaits confirmation. By contrast, diabetes reduced the level of OPA1 mRNA suggesting that diabetes downregulates mitochondrial fusion. These findings agreed with the fact that OPA1 expression is decreased in the skeletal muscles of patients with T2DM.50 Furthermore, findings in vitro have shown that OPA1 disruption in mammal cells caused by RNA interference (RNAi) blocks mitochondrial fusion, which leads to poor cell growth and mitochondrial dysfunction, such as decreased mitochondrial membrane potential and reduced cellular respiration.51 We suppose that a decreased OPA1 mRNA level is also associated with the decreased MMP in diabetic mice. Together, these finding suggest that the significantly reduced MMP arises from elevated mitochondrial fission and decreased mitochondrial fusion in the skeletal muscles of diabetic mice.
Both mitochondrial dysfunction and ER stress could be common changes in insulin-sensitive tissues that play key roles in glucose homeostasis that is altered in the context of obesity and T2DM, and the link between mitochondria and ER is MAM.52,53 We found that the expression of Sigma-1R protein, which is considered a specific quantitative component of MAM, was significantly decreased, whereas swimming increased its expression in the skeletal muscle of diabetic mice. These findings suggested that swimming can somewhat attenuate MAM impairment induced by diabetes. Notably, MAM disruption is an essential subcellular alteration associated with muscle insulin resistance in mice and humans, indicating that reduced ER-mitochondria coupling could be a common change in several insulin sensitive tissues that play key roles in altered glucose homeostasis in the context of obesity and T2DM.52 We also identified a positive correlation between levels of Sigma-1R protein and MMP, suggesting that mitochondrial function is positively associated with the quantity of MAM in the pathogenesis of diabetes.
High-fat diets, as well as excessive or insufficient exercise can activate ERS in skeletal muscle. In contrast, repeated moderate endurance exercise that attenuates the activation of various chaperone molecules associated with ERS protects skeletal muscle from excessive ERS.11 We similarly showed that diabetes significantly increased pIRE1α and ATF6 expression in skeletal muscle and that swimming alleviated the ERS in diabetic mice, whereas p-PERK remained unchanged in both groups. These findings indicated that diabetes activated ERS, whereas swimming attenuated the ERS through ATF6 and pIRE1α branches in skeletal muscle. Degrees of activation tended to differ among the three branches of UPR (p-PERK, pIRE1α, ATF6) under the same conditions. Our findings are similar to those of a previous study54 in which the UPR-specific ATF6/IRE1α channel in the skeletal muscles of 18 untrained persons was activated after resistance training, whereas PERK was not involved.
However, recovery might not be achieved when ER stress is prolonged or severe,55 and the UPR will generate pro-apoptotic signals to remove affected cells, that is, ERAD. The pro-apoptotic signals associated with the UPR involve the activation of caspase 12 by IRE1α.56 Our findings suggested that the remarkably elevated expression of both pIRE1α and caspase 12 was caused by diabetes and that the amounts of pIRE1α and caspase 12 proteins in diabetic mice were significantly reduced in response to swimming. Importantly, the correlation between pIRE1α and caspase 12 expression was significant and positive (Figure 6B), which is consistent with the pro-apoptotic pathway associated with ER referred to as IRE1α-caspase 12. These findings indicated that diabetes activates IRE1α-caspase 12, which results in ERAD, and that swimming can alleviate ERAD through pIRE1α.
Mitochondrial function is associated with MAM, and we identified a negative correlation between levels of Sig-1R and ATF6 expression. The latter was also positively associated with that of pIRE1α (Figure 6B), indicating that the ATF6 branch is associated with the pIRE1α branch during the pathogenesis of diabetes and exercise intervention. We speculate that impaired MAM, mitochondrial dysfunction and ERS are all related. However, compared with ERS, mitochondrial function is more closely related to MAM quantity in diabetic mice, and ERS might be a more effective target for swimming intervention, which would probably contribute to improving glucose intolerance. However, the specific mechanisms of these processes need to be further clarified during the pathogenesis of diabetes and exercise intervention.
Data supporting these concepts are still preliminary, and further investigations are needed to elucidate the precise mechanism of MAM, mitochondrial function and ERS during T2DM and exercise intervention in vivo. Further study is also needed to define more mitochondrial aspects that could fully describe mitochondrial quality, such as mitochondrial biogenesis and mitophagy. Although we could show changes in sigma-1R and mitochondrial dynamics, electronic microscopy is necessary to assess ER-mitochondria contact and mitochondrial morphology. Moreover, glucose-regulated protein 75 (GRP75) is a protein localized in MAM and is reduced by long-term diabetes. Investigating GRP75 expression is also necessary for our future study. In summary, we found that T2DM induces a decrease in MAM quantity, impaired mitochondrial quality, and deleterious ERS that leads to ERAD. Swimming mitigated T2DM-induced glucose intolerance and ERS in mouse skeletal muscles. Importantly, MAM quantity was not only associated positively with mitochondrial function, but also tended to correlate negatively with an ERS branch (ATF6). Moreover, both the ATF6 branch and ERAD were positively associated with the pIRE1α branch of ERS. Therefore, MAM quality and ATF6 might be novel and important targets of T2DM treatment. Endoplasmic reticulum stress might be a more effective target for swimming to improve diabetes.
Abbreviations
ATF6, activating transcription factor 6; GRP78, Bip chaperone (also called glucose-regulated protein 78); ERAD, endoplasmic reticulum-associated degradation; ERS, endoplasmic reticulum stress; IPGTT, intraperitoneal glucose tolerance test; IRE1, inositol requiring enzyme 1; MAM, mitochondria-associated membranes; MMP, mitochondrial membrane potential; PERK, protein kinase R-like ER protein kinase; RNAi, RNA interference; RT-PCR, reverse transcription polymerase chain reaction; Sigma-1R, sigma-1 receptor; SR/ER, sarcoplasmic endoplasmic reticulum; STZ, streptozotocin; T2DM, Type 2 diabetes mellitus; UPR, unfolded protein response.
Acknowledgments
We thank all staff (Zhe Ge, Haixiu Jin, Qiang Zhang and Zhengtang Qi) at the Shuzhe Ding laboratory for critical comments and technical assistance.
Funding
This study was funded by the National Natural Science Foundation of China (Grant 31671241), National Natural Science Foundation of China (Grant 31600967) and Collaborative Innovation Center of POWER Project Adolescents (Grant 44801400). This work was also supported by Fundamental Research Funds for the Central Universities (2018ECNU-HLYT048).
Disclosure
The authors have no competing interests to declare.
References
1. Rieusset J. Mitochondria and endoplasmic reticulum: mitochondria-endoplasmic reticulum interplay in type 2 diabetes pathophysiology. Int J Biochem Cell Biol. 2011;43(9):1257–1262. doi:10.1016/j.biocel.2011.05.006
2. Gispen WH, Biessels GJ. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000;23(11):542–549. doi:10.1016/S0166-2236(00)01656-8
3. Karlsson HK, Zierath JR. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem Biophys. 2007;48(2–3):103–113. doi:10.1007/s12013-007-0030-9
4. Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100(14):8466–8471. doi:10.1073/pnas.1032913100
5. Greco AV, Tataranni PA, Mingrone G, et al. Daily energy metabolism in patients with type 1 diabetes mellitus. J Am Coll Nutr. 1995;14(3):286–291. doi:10.1080/07315724.1995.10718509
6. Brandenburg SL, Reusch JE, Bauer TA, et al. Effects of exercise training on oxygen uptake kinetic responses in women with type 2 diabetes. Diabetes Care. 1999;22(10):1640–1646. doi:10.2337/diacare.22.10.1640
7. Davidsen PK, Gallagher IJ, Hartman JW, et al. High responders to resistance exercise training demonstrate differential regulation of skeletal muscle microRNA expression. J Appl Physiol (1985). 2011;110(2):309–317. doi:10.1152/japplphysiol.00901.2010
8. Nishikawa T, Kukidome D, Sonoda K, et al. Impact of mitochondrial ROS production in the pathogenesis of insulin resistance. Diabetes Res Clin Pract. 2007;77(Suppl 1):S161–S164. doi:10.1016/j.diabres.2007.01.071
9. Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi:10.1038/35008121
10. Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444(7117):337–342. doi:10.1038/nature05354
11. Deldicque L. Endoplasmic reticulum stress in human skeletal muscle: any contribution to sarcopenia? Front Physiol. 2013;4:236. doi:10.3389/fphys.2013.00236
12. Giorgi C, De Stefani D, Bononi A, et al. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int J Biochem Cell Biol. 2009;41(10):1817–1827. doi:10.1016/j.biocel.2009.04.010
13. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–387. doi:10.1126/science.1104343
14. Bonnard C, Durand A, Peyrol S, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118(2):789–800. doi:10.1172/JCI32601
15. Kekuda R, Prasad PD, Fei YJ, et al. Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1). Biochem Biophys Res Commun. 1996;229(2):553–558. doi:10.1006/bbrc.1996.1842
16. Hanner M, Moebius FF, Flandorfer A, et al. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A. 1996;93(15):8072–8077. doi:10.1073/pnas.93.15.8072
17. Su TP, Hayashi T, Maurice T, et al. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci. 2010;31(12):557–566. doi:10.1016/j.tips.2010.08.007
18. Kourrich S, Su TP, Fujimoto M, et al. The sigma-1 receptor: roles in neuronal plasticity and disease[J]. Trends Neurosci. 2012;35(12):762–771. doi:10.1016/j.tins.2012.09.007
19. Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. 2002;51(Suppl 3):S455–S461. doi:10.2337/diabetes.51.2007.S455
20. Deldicque L, Cani PD, Philp A, et al. The unfolded protein response is activated in skeletal muscle by high-fat feeding: potential role in the downregulation of protein synthesis. Am J Physiol Endocrinol Metab. 2010;299(5):E695–E705. doi:10.1152/ajpendo.00038.2010
21. Verfaillie T, Salazar M, Velasco G, et al. Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol. 2010;2010:930509. doi:10.1155/2010/930509
22. Gething MJ. Role and regulation of the ER chaperone BiP. Semin Cell Dev Biol. 1999;10(5):465–472. doi:10.1006/scdb.1999.0318
23. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi:10.1038/nrm2199
24. Araki E, Oyadomari S, Mori M. Endoplasmic reticulum stress and diabetes mellitus. Intern Med. 2003;42(1):7–14. doi:10.2169/internalmedicine.42.7
25. Rao RV, Peel A, Logvinova A, et al. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 2002;514(2–3):122–128. doi:10.1016/S0014-5793(02)02289-5
26. Joseph AM, Pilegaard H, Litvintsev A, et al. Control of gene expression and mitochondrial biogenesis in the muscular adaptation to endurance exercise. Essays Biochem. 2006;42:13–29. doi:10.1042/bse0420013
27. Coffey VG, Hawley JA. The molecular bases of training adaptation. Sports Med. 2007;37(9):737–763. doi:10.2165/00007256-200737090-00001
28. Deldicque L, Cani PD, Delzenne NM, et al. Endurance training in mice increases the unfolded protein response induced by a high-fat diet. J Physiol Biochem. 2013;69(2):215–225. doi:10.1007/s13105-012-0204-9
29. Lara-Riegos JC, Ortiz-Lopez MG, Pena-Espinoza BI, et al. Diabetes susceptibility in Mayas: evidence for the involvement of polymorphisms in HHEX, HNF4alpha, KCNJ11, PPARgamma, CDKN2A/2B, SLC30A8, CDC123/CAMK1D, TCF7L2, ABCA1 and SLC16A11 genes. Gene. 2015;565(1):68–75. doi:10.1016/j.gene.2015.03.065
30. Sun Y, Cui D, Zhang Z, et al. Attenuated oxidative stress following acute exhaustive swimming exercise was accompanied with modified gene expression profiles of apoptosis in the skeletal muscle of mice. Oxid Med Cell Longev. 2016;2016:8381242. doi:10.1155/2016/8381242
31. Rasmussen HN, Andersen AJ, Rasmussen UF. Optimization of preparation of mitochondria from 25–100 mg skeletal muscle. Anal Biochem. 1997;252(1):153–159. doi:10.1006/abio.1997.2304
32. Sun Y, Qi Z, He Q, et al. The effect of treadmill training and N-acetyl-l-cysteine intervention on biogenesis of cytochrome c oxidase (COX). Free Radic Biol Med. 2015;87:326–335. doi:10.1016/j.freeradbiomed.2015.06.035
33. Romanello V, Scalabrin M, Albiero M, et al. Inhibition of the fission machinery mitigates OPA1 Impairment in adult skeletal muscles. Cells. 2019;8(6):597. doi:10.3390/cells8060597
34. Silva RE, Motori E, Bruser C, et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 2019;15(6):e1008085. doi:10.1371/journal.pgen.1008085
35. Martinez J, Tarallo D, Martinez-Palma L, et al. Mitofusins modulate the increase in mitochondrial length, bioenergetics and secretory phenotype in therapy-induced senescent melanoma cells. Biochem J. 2019;476(17):2463–2486. doi:10.1042/BCJ20190405
36. Dhananjaya D, Hung KY, Tarn WY. RBM4 modulates radial migration via alternative splicing of Dab1 during cortex development. Mol Cell Biol. 2018;38(12):e00007–18.
37. Karbowski M, Norris KL, Cleland MM, et al. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443(7112):658–662. doi:10.1038/nature05111
38. Zhang Z, Sliter DA, Bleck C, et al. Fis1 deficiencies differentially affect mitochondrial quality in skeletal muscle. Mitochondrion. 2019;49:217–226. doi:10.1016/j.mito.2019.09.005
39. Sun Y, Cui D, Zhang Z, et al. Voluntary wheel exercise alters the levels of miR-494 and miR-696 in the skeletal muscle of C57BL/6 mice. Comp Biochem Physiol B Biochem Mol Biol. 2016;202:16–22. doi:10.1016/j.cbpb.2016.07.002
40. Fujimaki S, Kuwabara T. Diabetes-induced dysfunction of mitochondria and stem cells in skeletal muscle and the nervous system. Int J Mol Sci. 2017;18(10):2147.
41. Ju JS, Jeon SI, Park JY, et al. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J Physiol Sci. 2016;66(5):417–430. doi:10.1007/s12576-016-0440-9
42. Sliter DA, Martinez J, Hao L, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561(7722):258–262. doi:10.1038/s41586-018-0448-9
43. Ding H, Jiang N, Liu H, et al. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim Biophys Acta. 2010;1800(3):250–256. doi:10.1016/j.bbagen.2009.08.007
44. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. doi:10.1126/science.1219855
45. Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27(2):433–446. doi:10.1038/sj.emboj.7601963
46. Zhang T, Qi Z, Wang H, et al. Adeno-associated virus-mediated knockdown of SLC16A11 improves glucose tolerance and hepatic insulin signaling in high fat diet-fed mice. Exp Clin Endocrinol Diabetes. 2019
47. Liu R, Jin P, Yu L, et al. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS One. 2014;9(3):e92810. doi:10.1371/journal.pone.0092810
48. Yamano K, Fogel AI, Wang C, et al. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife. 2014;3:e1612. doi:10.7554/eLife.01612
49. Shen Q, Yamano K, Head BP, et al. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell. 2014;25(1):145–159. doi:10.1091/mbc.e13-09-0525
50. Joseph AM, Joanisse DR, Baillot RG, et al. Mitochondrial dysregulation in the pathogenesis of diabetes: potential for mitochondrial biogenesis-mediated interventions. Exp Diabetes Res. 2012;2012:642038. doi:10.1155/2012/642038
51. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280(28):26185–26192. doi:10.1074/jbc.M503062200
52. Tubbs E, Chanon S, Robert M, et al. Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes. 2018;67(4):636–650. doi:10.2337/db17-0316
53. Sasi U, Sindhu G, Raj PS, et al. Mitochondria associated membranes (MAMs): emerging drug targets for diabetes. Curr Med Chem. 2019
54. Ogborn DI, Mckay BR, Crane JD, et al. The unfolded protein response is triggered following a single, unaccustomed resistance-exercise bout. Am J Physiol Regul Integr Comp Physiol. 2014;307(6):R664–R669. doi:10.1152/ajpregu.00511.2013
55. Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18(1):59–68. doi:10.1016/j.molmed.2011.07.010
56. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. doi:10.1038/ncb0311-184
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.