")
Back to Journals » International Journal of Nanomedicine » Volume 11
Sustained-release liquisolid compact tablets containing artemether-lumefantrine as alternate-day regimen for malaria treatment to improve patient compliance
Authors Nnamani PO, Ugwu AA, Ibezim EC, Kenechukwu FC, Akpa PA, Ogbonna JD, Obitte NC, Odo AN, Windbergs M, Lehr CM, Attama AA
Received 20 July 2015
Accepted for publication 22 January 2016
Published 28 November 2016 Volume 2016:11 Pages 6365—6378
DOI https://doi.org/10.2147/IJN.S92755
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Petra Obioma Nnamani,1,2 Agatha Adaora Ugwu,1 Emmanuel Chinedu Ibezim,1 Franklin Chimaobi Kenechukwu,1 Paul Achile Akpa,1 John-Dike Nwabueze Ogbonna,1 Nicholas Chinedu Obitte,3 Amelia Ngozi Odo,4 Maike Windbergs,2 Claus-Michael Lehr,2,5,6 Anthony Amaechi Attama1
1Drug Delivery and Nanomedicines Research Group, Department of Pharmaceutics, Faculty of Pharmaceutical Sciences, University of Nigeria, Nsukka, Nigeria; 2Department of Drug Delivery, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research, Saarland University, Saarbrücken, Germany; 3Department of Pharmaceutical Technology and Industrial Pharmacy, Faculty of Pharmaceutical Sciences, 4Department of Human Kinetics and Health Education, University of Nigeria, Nsukka, Nigeria; 5PharmBioTec GmbH, 6Department of Biopharmaceutics and Pharmaceutical Technology, Saarland University, Saarbrücken, Germany
Abstract: The present study aimed to develop low-dose liquisolid tablets of two antimalarial drugs artemether–lumefantrine (AL) from a nanostructured lipid carrier (NLC) of lumefantrine (LUM) and estimate the potential of AL as an oral delivery system in malariogenic Wistar mice. LUM-NLCs were prepared by hot homogenization using Precirol® ATO 5/Transcutol® HP and tallow fat/Transcutol® HP optimized systems containing 3:1 ratios of the lipids, respectively, as the matrices. LUM-NLC characteristics, including morphology, particle size, zeta potential, encapsulation efficiency, yield, pH-dependent stability, and interaction studies, were investigated. Optimized LUM-NLCs were mixed with artemether powder and other dry ingredients and the resultant powder evaluated for micromeritics. Subsequent AL liquisolid tablets were tested for in vitro drug release and in vivo antiplasmodial activity in mice infected with Plasmodium berghei berghei (NK 65). Results showed that optimized LUM-NLC were stable, spherical, polydispersed but nanometric. Percentage yield and encapsulation efficiency were ~92% and 93% for Precirol® ATO 5/Transcutol® HP batch, then 81% and 95% for tallow fat/Transcutol® HP batch while LUM was amorphous in NLC matrix. In vitro AL release from liquisolid compacts revealed initial burst release and subsequent sustained release. Liquisolid tablet compacts formulated with Precirol® ATO 5/Transcutol® HP-AL4 achieved higher LUM release in simulated intestinal fluid (84.32%) than tallow fat/Transcutol® HP-BL3 (77.9%). Non-Fickian (anomalous) diffusion and super case II transport were the predominant mechanisms of drug release. Equal parasitemia reduction was observed for both batches of tablet compacts (~92%), superior to the reduction obtained with commercial antimalarial formulations: Coartem® tablets (86%) and chloroquine phosphate tablets (66%). No significant difference (P<0.05) in parasite reduction between double (4/24 mg/kg) and single (2/12 mg/kg) strength doses of AL compacts was observed. Our result highlights that AL could be formulated in much lower doses (4/24 mg/kg), for once-in-two days oral administration to improve patient compliance, which is currently not obtainable with conventional AL dosage forms.
Keywords: malaria, artemisinin-based combination therapy, antiplasmodial activity, liquisolid compacts, nanostructured lipid carriers
Introduction
Malaria remains one of the most serious vector-borne diseases, affecting millions of people, especially in the warmer and wetter regions of the tropics.1,2 The artemisinin drugs remain the most important and effective against parasites resistant to almost all other classes of drugs. After oral administration, artemether (ART) is absorbed fairly rapidly with peak plasma concentrations reached in ~2 hours.3 This invariably implies frequent dosing with associated numerous side effects, especially a nausea–vomiting tendency that makes people discontinue treatment or comply poorly, in addition to many other serious ones. Intramuscularly, absorption is very variable especially in children with poor peripheral perfusion besides the pain of injection.4,5 The elimination half-life is ~1 hour and there is no dose modification in renal or hepatic impairment, despite the drug’s ability to cause neurotoxicity especially following sustained blood concentration from intramuscular administration.3 With a novel drug delivery system, such as the nanostructured lipid carrier (NLC), which has been credited as more advantageous over all other colloidal systems (liposomes, nanoemulsions, solid lipid nanoparticles and microemulsions, etc)6–9 due to higher drug solubility in liquid lipids (oils) than solid lipids, we aimed to achieve the following: higher encapsulation efficiency (EE), loading capacity, controlled drug release from the payload in the matrix core as well as lower crystallinity of particles.3,10,11 This will reduce dose-related side effects (ie, improved patient friendliness), facilitate more reproducible absorption than conventional dosage forms as well as protect the drug against degradation in order to achieve site-specific drug delivery.12
Artemether–lumefantrine (AL) remains a first-line drug of choice among all other artemisinin-combination therapies in malaria treatment. It is available as an oral tablet of different fixed combinations (80/480, 40/240, and 20/120 mg) for the treatment of uncomplicated falciparum malaria, including multidrug resistant ones. Though AL is challenged with twice daily dosing and degradation in stomach acidic condition,13,14 the complementarities rest on the fact that while ART peak plasma concentration is reached ~2 hours after dosing, lumefantrine (LUM), a highly lipophilic drug, starts after a lag period of up to 2 hours, with peak plasma concentration ~6–8 hours after dosing. Also, the relative bioavailability of ART has been observed to increase more than two-fold and that of LUM 16-fold after a high fat meal than in fasted conditions.15,16 This informed our choice for the use of lipid excipients to propose a built-in fat-supplemented regimen for better absorption. In light of the above, the present study therefore demonstrates the potential of an extremely low-dose liquisolid tablet of AL obtained from NLC involving two solid homolipids (Precirol® ATO 5 and tallow fat) and an oil (Transcutol® HP) to reduce parasitemia in malariogenic Wistar mice following an alternate-day regimen. NLCs are more advantageous than any other nanoparticle preparation because of their oil-rich core, which makes for greater drug entrapment.3 More so, fatty food gives 16× and 2× more absorption of LUM and ART, respectively, a condition already built into the design of this novel delivery system. The formulation also combined surfactants of both oil- (Solutol® HS 15 and Tween® 80) and water-soluble (Poloxamer® 188) natures to ensure higher retention of LUM within particles.
In the present investigation, only NLC of LUM was prepared, while ART was incorporated as a crystalline drug alongside other direct compression excipients to produce the liquisolid tablet compacts, as sustained blood circulation of ART has been shown to have a neurological effect following intramuscular injections.16–18 Preparation of liquisolid systems is based on the principles of conversion of drug(s) in the liquid state into a free flowing, readily compressible, and apparently dry powder by simple physical blending with selected excipients, excipient-carriers, and coating materials.19 Liquid drug could also be sprayed into the carrier material in fluid-bed equipment for homogeneous distribution of the active substance. Liquid drug is incorporated into the porous structure of a carrier material due to adsorption and absorption.20–22 AL at two fixed combinations of 2/12 mg (od and/or bid) and 4/24 mg (od and/or once-in-two days) was investigated for antiplasmodial potentials in Plasmodium berghei berghei-infected (NK 65) adult male albino Wistar mice alongside commercial AL formulation (Coartem®) and chloroquine phosphate (CQ-PO4) oral tablets.
Materials and methods
Materials
LUM (Figure 1A) was obtained from Hangzhou Dayangchem. Co. Limited, (Hangzhou, People’s Republic of China), whereas ART (Figure 1B) was a gift from Ipca Laboratories Ltd. (Charkop, India). Precirol® ATO 5 and Transcutol® HP (PT) were donated by Gattefossé, St Priest, France. Phospholipon® 90G (P90G) was a gift from Phospholipid GmbH, Köln, Germany, while Poloxamer® 188 and Solutol® HS 15 (BASF, Ludwigshafen, Germany) were also received as donations. Other ingredients used were Polysorbate® 80 (Merck KGaA, Darmstadt, Germany), colloidal silicon dioxide (Aeroperl® 300, Degussa, Hanau, Germany), microcrystalline cellulose (Avicel® PH 102, FMC Biopolymer, PA, USA), and magnesium stearate (Peter Greven Fett Chemie, Muenstereifel, Germany). Tallow fat was obtained from a batch processed at the Department of Pharmaceutics, University of Nigeria Nsukka. The biorelevant media, simulated gastric fluid (SGF) (pH 1.2) and simulated intestinal fluid (SIF) (pH 6.8), were prepared without pepsin and pancreatin, respectively. Distilled water was used throughout the study.
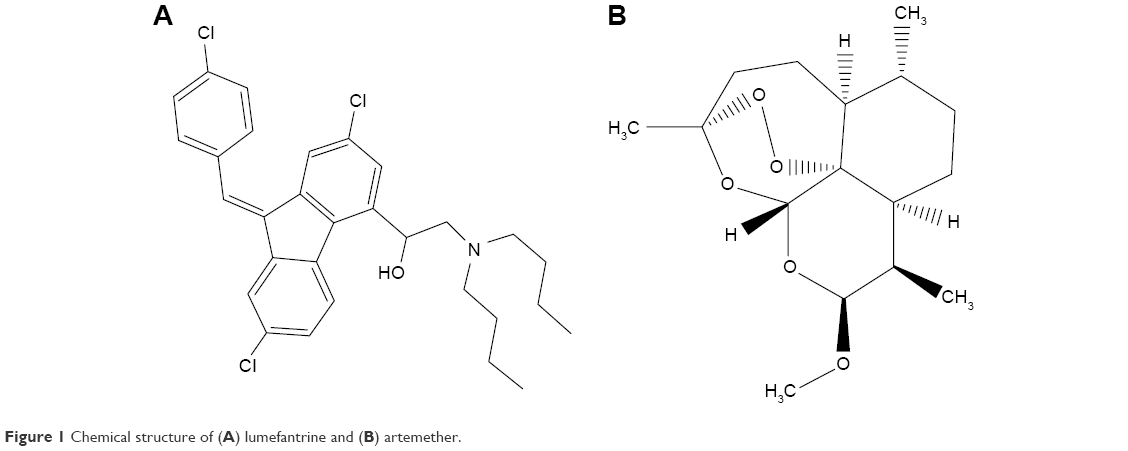 | Figure 1 Chemical structure of (A) lumefantrine and (B) artemether. |
Animals
All animal experiments were performed in accordance with the National Institute of Health guideline on principles of laboratory animal care (National Institute of Health publication 85–23, revised 1996) and were approved by the Institutional Animal Care and Use Committee of the University of Nigeria, Nsukka. Firstly, 40 adult male albino Wistar mice were procured, housed, and fed normally to acclimatize to the laboratory environment of the Department of Pharmacology and Toxicology, University of Nigeria, Nsukka. P. berghei (NK 65) malaria parasite employed in the study was sourced from the National Institute of Medical Research (Yaba Lagos, Nigeria), and the parasite was hosted by donor mice.
Lipid selection
Tallow fat was extracted following standard methods.23 Thermal properties of the bulk lipids, including tallow fat, Precirol® ATO 5, P90G and their mixtures, Transcutol® HP, and drugs (AL), were investigated by differential scanning calorimetry using a differential scanning calorimeter (DSC) (NETZSCH DSC 204 FI, NETZSCH, Selb, Germany). Binary and ternary lipid mixtures were also tested. The lipids were weighed on an electronic balance (HH.W21–Cr42II, Adventurer Ohaus, Changzhou, People’s Republic of China), mixed in a crucible, and melted together on a hot plate (SR1 UM 52188, Remi Equipment, Maharashtra, India) at 70°C and thereafter stirred until solidification. The DSC program used for all the lipids and matrices was 35–190°C at 10°C/min under a 20 mL/min nitrogen flux with a sample size of 3–5 mg. An empty standard aluminum pan was used as reference. Optimized binary mixtures of Precirol® ATO 5/Transcutol® (PT) at 3:1 ratios were thereafter selected.
Preparation of nanostructured lipid carriers
Hot homogenization technique was adopted. NLC matrices of 15% (PT or TT) were melted at 90°C and loaded with graded concentration of LUM (0.2%, 0.5%, 0.8%, and 1% w/w). Solutol® HS 15 (3% w/w) and polysorbate 80 (Tween® 80, 1% w/w) were added to the lipid phase to enhance solubility of LUM in the lipid medium. Aqueous phase containing Poloxamer® 188 (1% w/w) was heated to the same temperature and added to the molten lipid matrix. This was immediately subjected to high shear homogenization (Ultra-Turrax, T18 basic, IKA, Staufen, Germany) at 25,000 rpm for 15 minutes to produce an oil/water emulsion. The emulsion was allowed to cool at room temperature. NLCs containing no drug were also prepared to serve as a negative control.
Determination of percentage yield and EE
NLC from each batch was weighed after formulation to get the yield. Percentage (%) yield was calculated using the formula:
|
|
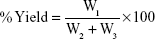
where W1=weight of the NLC formulated (g), W2=weight of the drug added (g), and W3=weight of the lipid matrix and surfactant (g).
Microconcentrator tubes, Vivaspin 6 (MWCO 10,000 Da) (Vivascience, Hanover, Germany), were used for the determination of EE%. Undiluted LUM-NLC formulation (5 mL) was placed in the upper chamber and sample recovery chamber was fitted below the membrane in the lower compartment. The unit was assembled and subjected to centrifugation in a centrifuge (TDL-4 B. Bran Scientific and Instrument Co., Sheffield, UK) at 8,000× g for 2 hours. A 1 mL volume of the filtrate was appropriately diluted with 0.1 M methanolic HCl and absorbance readings were obtained using UV-VIS spectrophotometer (6405 Jenway, Dunmow, UK) at 335 nm. Drug content was estimated by reference to a standard Beer’s plot, while EE of all batches of NLC was calculated using Equation 2:
|
|
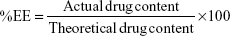
Determination of particle size, surface charge, and physical stability
Mean diameter (z-average, nm) and polydispersity index of the NLC particles were measured by photon correlation spectroscopy using a zetasizer nano-ZS (Malvern Instruments, Malvern, UK) equipped with a 10 mw He-NE laser employing the wavelength of 633 nm and a backscattering angle of 173° at 25°C. All samples were diluted with double-distilled water to obtain a suitable scattering intensity, before photon correlation spectroscopy analysis. The zeta potential (ZP) of NLC formulations (n=3) was determined via electrophoretic mobility measurements using a zetasizer nano-ZS (Malvern Instruments) and applying the Helmholtz–Smoluchowski equation.
Differential scanning calorimetry
Thermal properties of formulations were determined using a DSC (NETZSCH DSC 204 FI, Germany) with an empty standard aluminum pan as reference after baseline correction. Measurement was done according to the earlier described program. Briefly, the DSC program used for all the lipids and matrices was 35–190°C at 10°C/min under a 20 mL/min nitrogen flux with a sample size of 3–5 mg. An empty standard aluminum pan was used as reference.3
Fourier transform infrared spectroscopy
The compatibility among the pure drug, lipid matrices, and NLC was studied using a Shimadzu FTIR 8300 spectrophotometer (Shimadzu, Tokyo, Japan). Spectra were recorded in the wavelength region of 4,000–400 cm−1 with a threshold of 1.303, sensitivity of 50, and resolution of 2 cm−1. In each case, the sample was dispersed in KBr and then compressed into discs by applying a pressure of 5 tons for 5 minutes in a hydraulic press. The pellet was placed in the light path and the spectrum obtained.
pH variation during storage
The NLCs were subjected to time-dependent pH analysis to determine the short-term stability. Dispersion pH of each batch (A = PT-LUM-NLC and B = TT-LUM-NLC) was determined using a pH meter (pHep® Hana Instrument, Limena, Italy) after 24 hours of preparation and upon storage at room temperature for 1, 3, and 6 months.
Microscopy
Following the results of particle characterization, optimized samples were selected for microstructure analysis. Particle morphology of NLCs loaded with LUM was observed using a scanning electron microscope (Hitachi S-4000 Microscope, Schaumburg, USA). Samples were diluted with double-distilled water and deposited on film-coated copper grids to dry overnight at room temperature. The dried samples were visualized under the scanning electron microscope.
Preparation of powder blend for tablets
Optimized NLCs loaded with LUM equivalent to 240 mg from each of PT (batch A) and TT (batch B) were mixed with crystalline ART (40 mg), colloidal silicone dioxide (0.5%), magnesium stearate (2 mg), and microcrystalline cellulose in sufficient quantity to produce 300 mg for 5 minutes in a planetary mixer (B-30 Guangdong, People’s Republic of China). Different batches (A and B) of liquisolid tablets were produced.
Micromeritics of powder
Bulk and tapped densities of powder were separately determined using 10 mL measuring cylinder and after 500 tappings on horizontal surface, respectively. Hausner’s quotient and compressibility index were calculated from the two densities. Fixed-height funnel method was used to determine flow rate and angle of repose. Briefly, time taken for ~20 g powder mass to flow through a funnel orifice height of 7.5 cm was recorded in g/s. Mean height of the powder peak and base diameter were calculated to obtain the angle of repose as ratio of powder height to radius of powder base. Triplicate measurements were carried out to ensure validity of result.
Production and evaluation of liquisolid tablet compacts of AL
Direct compression method was used for tablet production. Powder blend/mixture was compressed for 60 seconds at 50 kgf in a 10 mm diameter die to produce 300 mg liquisolid tablet compacts using a single punch tableting machine (Manesty F3, West Molesey, UK). Tablets were stored over silica gel in a desiccator for 24 hours to allow for elastic recovery and hardening before evaluation.
Weight uniformity test
Twenty tablets were randomly selected from each batch (A and B) and individual weight was determined using an electronic balance. Coefficient of variation, mean, and standard deviation of the tablets were noted.
Tablet friability test
Ten tablets were randomly selected from each batch (A and B). The tablets were dedusted and accurately weighed together using an electronic balance (HH.W21–Cr42II, Adventurer Ohaus) and the initial weight was recorded. The tablets were then placed in a friabilator (CS-1 Henan, People’s Republic of China) set to rotate at 25 rpm for 4 minutes. Afterwards, they were removed, dedusted, and the final weight was determined. The percentage friability was therefore determined using Equation 3.
|
|
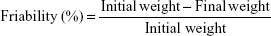
Crushing strength (hardness)
The crushing strength of the tablets was analyzed by a diametric compression test using a hardness tester (Lab Hosp. Tablet hardness tester, Tamil Nadu, India) with a constant speed of 0.1 mm/s. Ten tablets from each batch were tested and the average reported.
Erosion time of tablets
Six tablets were randomly selected from each batch. The biorelevant media, SGF (pH 1.2) and SIF (pH 6.8) without enzymes (500 mL), were used as erosion media separately at 37±2°C using Erweka apparatus following the British Pharmacopoeia method.24 Briefly, one tablet was placed into each of the six tubes of the disintegration unit and the time taken for complete erosion of each tablet was determined. The mean and standard deviation of all the six tablets were appropriately calculated.
Assay of drugs in the formulated liquisolid tablets
An amount of the tablet theoretically containing 20 mg of ART was weighed and dissolved in sufficient methanol to produce 50 mL. The resulting solution was allowed to stand for ~5 minutes and then filtered through a filter paper while discarding the first 10 mL. Some 2 mL of the resulting solution was transferred into a test tube and 2 mL of concentrated HCl added. The test tube was securely stoppered and allowed to stand in a water bath set at 80°C for 30 minutes. The resulting solution was diluted with sufficient methanol to make 25 mL. Absorbance of this solution was taken at 345 nm in a spectrophotometer (6405 Jenway) against a blank solution (concentrated HCl [2 mL] made up to 25 mL with methanol). The content of ART was calculated by reference to the calibration curve. The assay of lumefantrine in the liquisolid compact tablets followed the method earlier described under EE.
Dissolution rate and release kinetics of tablets of tablets
Dissolution tests were performed on tablets using the USP apparatus II (Paddle) method. The revolution speed of the paddle and volume of the dissolution medium were set at 75 rpm and 900 mL, respectively. SGF (pH 1.2) and SIF (pH 6.8) without enzymes were used as the dissolution media. As soon as the paddle was rotated, one tablet from each batch of formulation was introduced into the dissolution medium. Samples (5 mL) were withdrawn at predetermined sampling points (0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 16, 18, 20, 22, and 24 hours) and filtered through 0.45 μm filter membrane. Sample replacement was done with fresh 5 mL of dissolution medium after each withdrawal to maintain sink condition. For ART assay, 2 mL of each withdrawn sample was added to 2 mL of concentrated HCl in a test tube, securely stoppered, and stood in a water bath at 80°C for 30 minutes and afterwards cooled, diluted with distilled water (20 mL), and filtered. Absorbance of this solution was taken at 345 nm against a blank solution of HCl (2 mL) made up to 25 mL with distilled water. For LUM assay, another 2 mL of withdrawn solution was diluted with sufficient methanol to 25 mL volume. Absorbance of this solution was taken at 335 nm against a blank solution of methanol in a spectrophotometer (Jasco V-630UV/VIS Double Beam, Tokyo, Japan). Contents of AL in liquisolid tablets were separately calculated by reference to relevant calibration curves.
Kinetics of release from liquisolid tablets
Different release models were applied to study the kinetics and mechanisms of AL release from liquisolid tablets, including zero order, first order, Higuchi, and Ritger–Peppas models. Zero order was done by fitting cumulative % drug release versus time, while first order assessed log cumulative of % drug remaining versus time.25 The cumulative % drug release versus square root of time described the Higuchi model, whereas Ritger–Peppas and Korsmeyer–Peppas models assessed both Fickian and non-Fickian release of drug from swelling as well as nonswelling systems by Log (Mt/M∝) = Log k + n Log t. The best fit model depended on the linearity of the plots as represented by the R2 values.
In vitro release of AL from liquisolid tablet compacts
In vitro drug release studies for the different batches of formulation were studied in 900 mL each of SGF and SIF placed on a magnetic stirrer hot plate with temperature and speed of rotation maintained at 37±2°C and 100 rpm, respectively. One tablet from each batch of formulation was introduced in a basket clamped on a tripod stand. The basket was then deepened into the medium at zero time “t0” and samples were withdrawn at different time intervals while replacing each withdrawal with equal volume of fresh dissolution medium at the same temperature. Absorbance of the withdrawn samples was determined using UV-VIS spectrophotometer (6405 Jenway) at different wavelengths (345 nm for ART and 335 nm for LUM) after appropriate treatments as described earlier and the concentrations were calculated. The amount of drug released at each time point was calculated with reference to the relevant Beer’s plot.
In vivo schizonticidal activity of the liquisolid tablet compacts
Evaluation of the curative potential of the formulated tablets against established plasmodium infection was carried out according to standard protocols.15,26 Briefly, the mice were divided into seven groups of five mice each in clearly painted colors to avoid any mix-up. Blood of the donor mice was collected by cardiac puncture and diluted with physiological saline (normal saline) to give a concentration of 108 parasitized erythrocytes per mL. A 0.2 mL volume of the donor mouse erythrocyte equivalent to 2×107 parasitized erythrocytes was injected intraperitoneally into each of the experimental mice on day 1 (D1). All 35 mice were inoculated with chloroquine sensitive strain of P. berghei (NK 65) and left untreated until the fourth day (D4) post inoculation.
Ab initio, on day 0 of the test (D0), percentage parasitemia and red blood cell count of the donor mice were determined by Giemsa-stained thin blood smear of the donor mice and improved Neubauer Counting Chamber, respectively. Post inoculation, all treatments were given orally per day for 3 days (D4–D6). On day 1 (D1), mice from Group A were administered with 0.2 mL/kg body weight of normal saline, while Group B mice received 10 mg/kg body weight of CQ-PO4 tablet dispersed in distilled water, then 5 mg/kg on day 2 and 3 (D2 and D3). Group C mice received 4 and 24 mg/kg of commercial fixed combination dose of AL (Coartem®) once daily. Groups D and E received 4 and 24 mg/kg of AL liquisolid tablets once daily from each batch of parent NLC (PT and TT) formulation. For convenience, the optimized AL liquisolid tablets originating from PT matrix NLC (batch A) was referred to as AL4 whereas that of TT (batch B) was called BL3. Mice in Groups F and G were administered with 2 and 12 mg/kg of AL liquisolid tablets twice daily, respectively, for AL4 and BL3. The once-in-two days dosage regimen of the liquisolid compact was tested for Groups D and E as well as Groups F and G. On day 7 (D7), each mouse was tail-bled and a thin blood film was made on a microscope slide. The films were stained with 10% Giemsa solution and examined microscopically to monitor the parasitemia level. The antimalarial activity was determined by the equation:
|
|

Statistical analysis
All the data generated were expressed as mean ± standard deviation. For group comparisons, one-way analysis of variance with duplication was applied. Statistical significance was determined using Student’s t-test, with P<0.05 considered to be statistically significant.
Results and discussion
Lipid selection
Lipid selection was based on low enthalpy value as an indicator of low crystallinity and subsequent high propensity for drug entrapment. Table 1 shows that NLCs from PT (or batch A) as well as TT (or batch B) combinations, respectively, had the lowest enthalpy values and, hence, were selected for further studies.
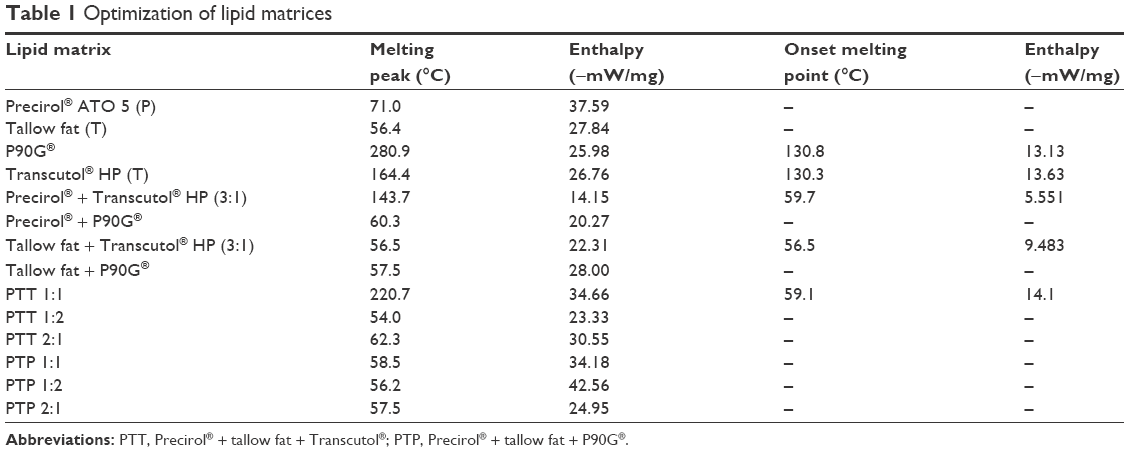 | Table 1 Optimization of lipid matrices |
Melting endotherm of tallow fat was 56.4°C with enthalpy of −27.84 mW/mg, while its combination with Transcutol® at 3:1 yielded nanostructured matrix with melting peak at 56.5°C and enthalpy of −22.31 mW/mg. Addition of Phospholipon® 90G to tallow fat resulted in matrices with higher enthalpy values, and hence high crystallinity in much the same way as with Precirol®. DSC trace of Precirol® was 71.0°C with an enthalpy of −37.59 mW/mg, while its combination with Transcutol® at 3:1 exhibited a melting peak at 59.7°C with enthalpy of −14.15 mW/mg. These NLCs had better thermal properties than the ternary systems. Meanwhile, higher enthalpy values imply more crystallinity with subsequent higher probability of low drug entrapment.27
Determination of yield (%), EE, and particle properties of LUM-NLCs
Table 2 shows general particle properties of NLC whereas Figure 2 shows the percentage yield of LUM-NLCs. The EE of the formulation shows that two formulations (one from each batch) had the highest drug payload (Table 2). These corresponded to LUM-NLC-A4 (PT batch) and LUM-NLC-B3 (TT batch) with EE of 93.2% and 94.8%, respectively. LUM-NLC-A4 had a particle size of 900 nm, polydispersity index of 0.7, and ZP of −27.4 mV, whereas LUM-NLC-B3 had z-average of ~899 nm, polydispersity index of 0.7, and ZP of −17.3 mV, among many other formulations (Table 2). Percentage yield was highest (~92%) in LUM-NLC-A4 whereas LUM-NLC-B3 had a yield of 81.16% lower than ~86% of LUM-NLC-B5, which was not stable after 6 months of storage as the ZP value was the lowest (−6.8 mV). Generally, increase in drug concentration from 0.2% to 1.0% caused instability of the resultant NLC formulation. In other words, optimum stability of LUM-loaded NLC was observed at 0.5% and/or 0.8% concentrations depending on type of matrix. By implication, LUM-NLC-A4 was stable when loaded with up to 0.8% of LUM (93.2% EE), whereas LUM-NLC-B3 was stable when loaded with up to 0.5% of LUM (EE of ~95%). It was observed that EE (%) increased with particle size only within the nanometer size range. In other words, a decrease in EE (%) was observed in particles existing in the micrometer ranges as seen in LUM-NLC-A3 (1,491 μm) and LUM-NLC-B4 (1,134 μm) having an EE of ~78% and 83%, respectively, though nanoformulations in these size ranges do not have a problem if intended for oral use as compared to parenteral and/or transdermal uses.3,27,28
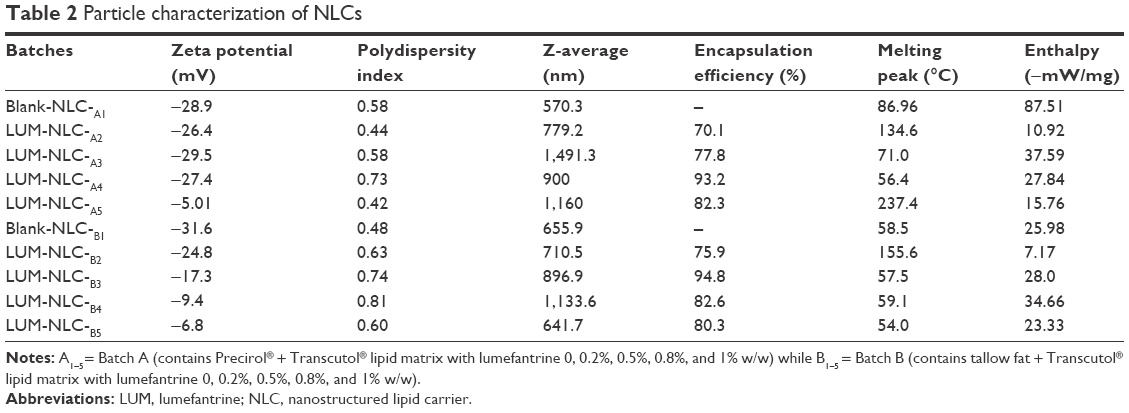 | Table 2 Particle characterization of NLCs |
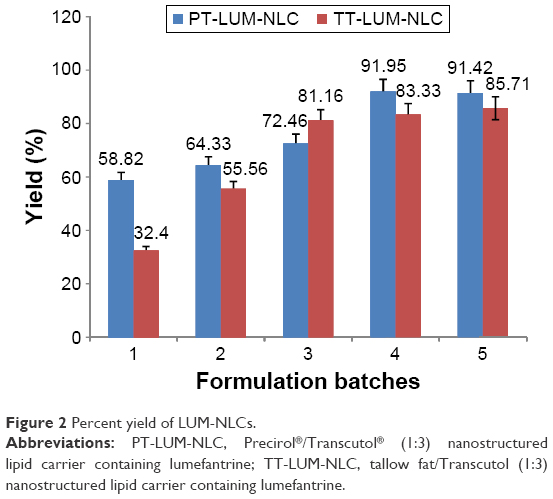 | Figure 2 Percent yield of LUM-NLCs. |
Interaction study by DSC
Table 2 depicts the thermal properties of NLC formulations. It could be seen that LUM-NLC-A4 and LUM-NLC-B3 had the least enthalpy values among all formulations. LUM had a melting peak at 134.6°C with an enthalpy of −78.62 mW/mg, whereas LUM-NLC-A4 and LUM-NLC-B3 melted differently at 58.5°C and 54.0°C with respective enthalpies of −25.98 and −23.33 mW/mg. There was no peak due to the drug in the thermogram traces. The complete absence of drug peak implies that LUM was molecularly dispersed in the NLC matrices and as such existed in an amorphous form, which would make for high drug encapsulation. This supports the fact provided by EE (%) as well as ZP (stability index). As a sufficient amount of drug was contained in the matrices used for DSC tests, we therefore rule out low drug concentration as a factor contributing to the disappearance of the drug peak in the thermograms.
Fourier transform infrared spectroscopy
To demonstrate that it was possible to incorporate LUM into NLCs, infrared spectra of free LUM, LUM-loaded NLCs (LUM-NLC-A4 and LUM-NLC-B3) were obtained alongside individual lipid matrices, and presented in Figure 3. The bands at 3,325, 2,858, and 1,139 cm−1 were due to the vibration of the functional groups [(–OH, CH and C-O-C (asym)-)] present in the structure of LUM and can be seen in the Fourier transform infrared spectroscopy spectra of the free LUM, PT, and TT. Spectra from NLCs containing no drug did not show these peaks, indicating successful incorporation of LUM in the NLC formulations.
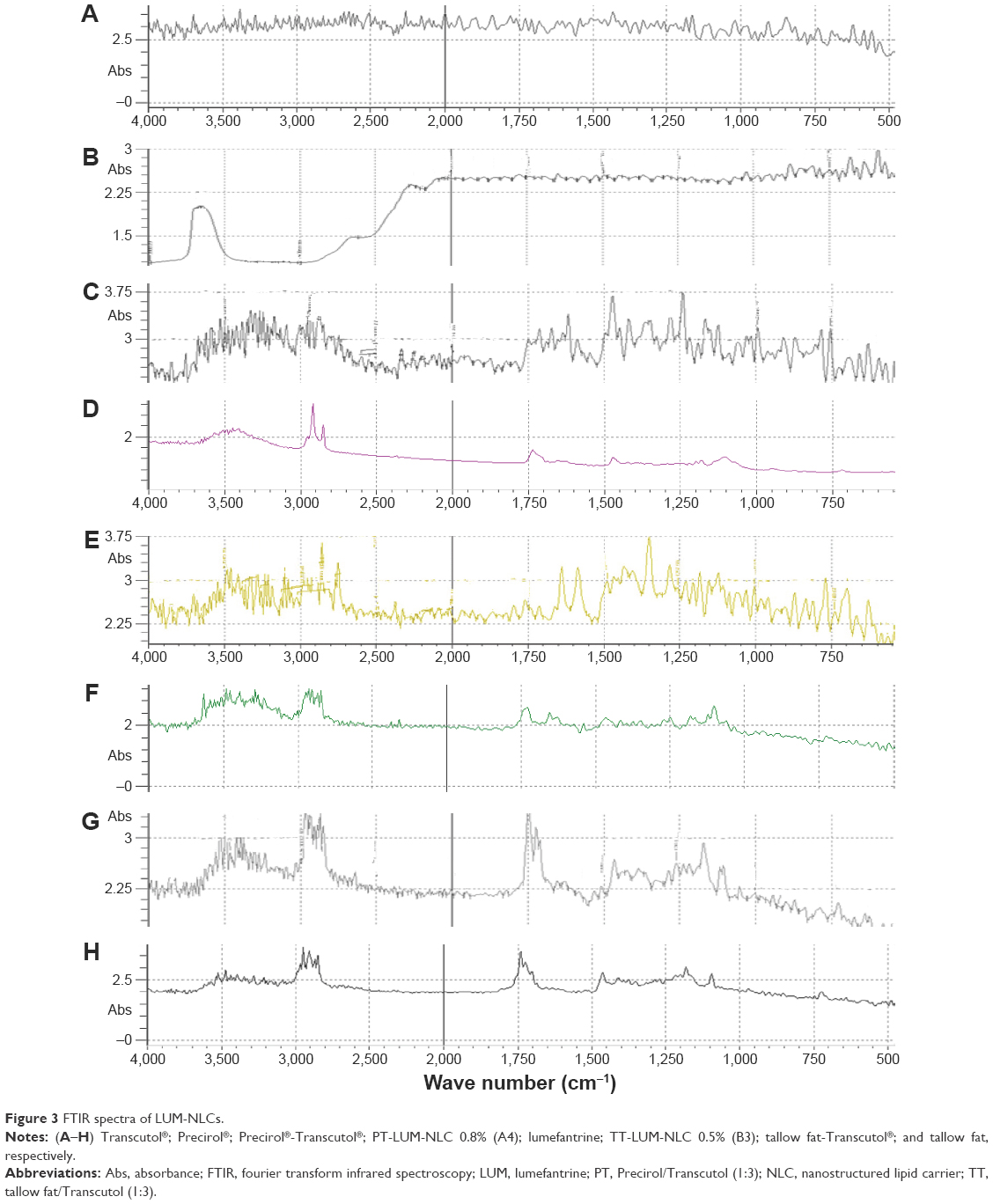 | Figure 3 FTIR spectra of LUM-NLCs. |
pH variation on storage
Figure 4 shows the result of pH stability of LUM-NLC formulations after 24 hours post preparation and at room temperature storage for 3 and 6 months. Figure 4A and B represents NLC formulations produced from PT and TT matrices at 3:1 ratios, respectively. In (A), the most stable formulation for the graded concentration of LUM in NLC formulations was LUM-NLC-A4 because it showed minimal variation in storage pH compared to other batches. This observation agreed with the stability result of ZP analysis where the formulation had −27.4 mV and incorporated as high as 93.2% of LUM. However, in (B), LUM-NLC-B3 was the most stable formulation having maintained negligible fluctuations in pH during storage in agreement with its ZP value of −17.3 mV and EE of ~95%. In light of all the above results, LUM-NLC-A4 and LUM-NLC-B3 were chosen as the optimized formulations for further investigations.
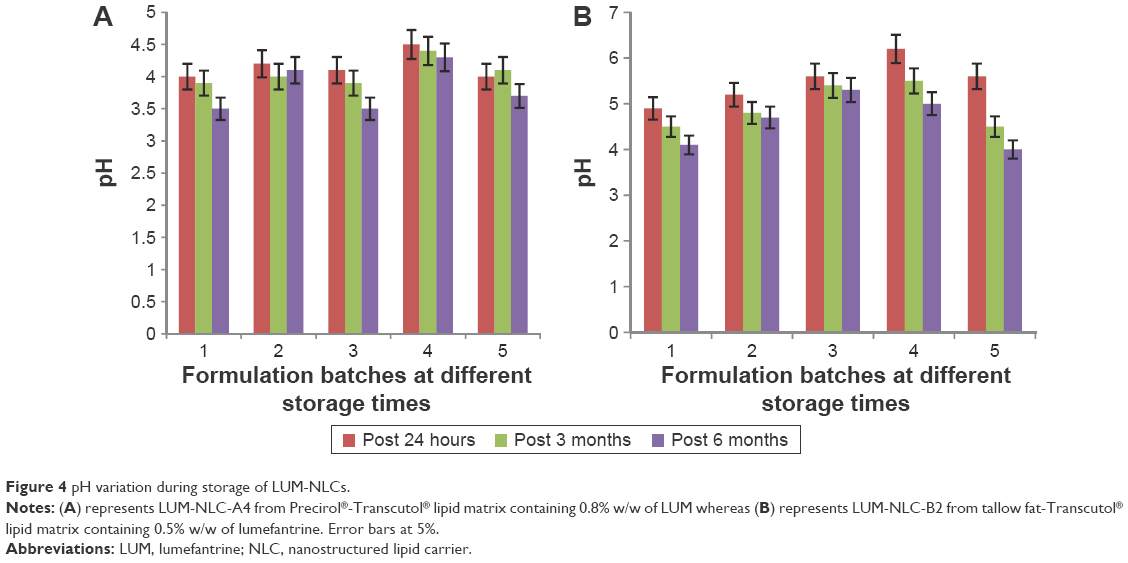 | Figure 4 pH variation during storage of LUM-NLCs. |
Microscopy
The microstructures of optimized sample formulations of LUM-NLC-A4 and LUM-NLC-B3 are shown in Figure 5. It could be seen that the pure drugs were crystalline and different in shape from those of optimized formulations. Generally, LUM-NLCs were smooth, nonporous, and spherical in shape. This implies that the drug existed in an amorphous form in the NLC matrices, in agreement with earlier results.3,23,28
 | Figure 5 SEM images of optimized formulations prepared from Precirol®/Transcutol® (LUM-NLC-A4) and tallow fat/Transcutol® (LUM-NLC-B3). |
Micromeritics and liquisolid compact characteristics
The flow properties of powder blend of drug-free NLC and LUM-NLCs with crystalline ART and other direct compressible excipients (Table 3) to produce AL fixed-dose combination liquisolid tablets are presented in Table 4. Generally, all batches had good flow properties, while Hausner’s quotient was ~1.2; angle of repose was majorly 25° and Carr’s compressibility index was ~15%, in addition to low resistance to flow observed across all batches. Table 5 details the tablet properties studied, such as weight uniformity, hardness, friability, thickness, diameter, and erosion times of AL liquisolid compacts. Variation in weight was within official acceptable ranges.24,29,30 AL tablets originating from NLCs prepared from Precirol/Transcutol, PT (AL1 and AL4) were harder than those of tallow/Transcutol, TT (BL1 and BL3) despite having fast erosion time (but erosion time was faster than the latter). By implication, AL1 and AL4 both had complete drug erosion at 3.10 and 6.30 hours, respectively, faster than BL1 and BL3, which showed complete drug erosion at 25.30 and 25.60 hours, respectively, despite having lower hardness and friability. This perhaps may be due to the presence of both water- and oil-soluble surfactants, which allowed more passage of water through the drug-free compacts (AL1) much more than the AL4. In other words, there were more hydro-channels in the tablets of AL1 than in AL4. This is reasonable as the oil-soluble surfactants had enabled much drug incorporation as seen in the result of EE to the tune of 93.2%, thereby reducing available spaces created by water-soluble surfactants. In contrast to this, it was observed that the BL1 and BL3 both had longer erosion times despite having lower hardness values and friability. This could imply limited availability of hydro-channels to facilitate water penetration into compacts to disentangle the particles as the particle had earlier entrapped up to ~95% of LUM, which is a practically water-insoluble drug, in coexistence with a similar drug, ART.3,13,14
 | Table 3 Micromeritics of powder blends |
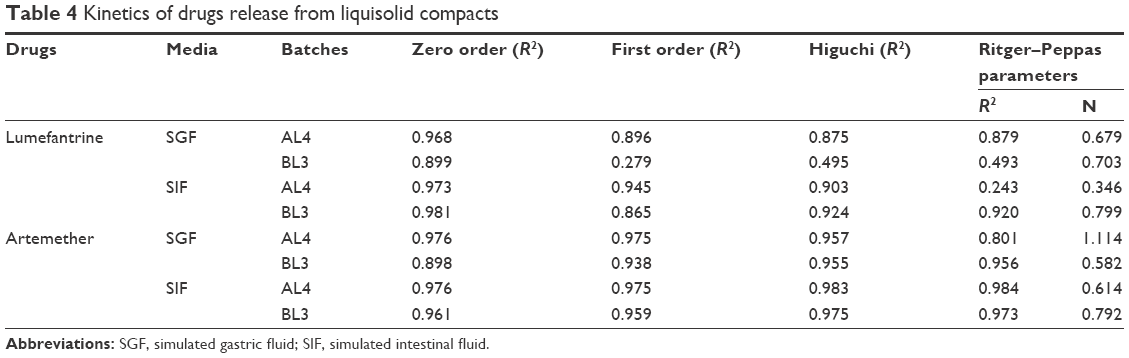 | Table 4 Kinetics of drugs release from liquisolid compacts |
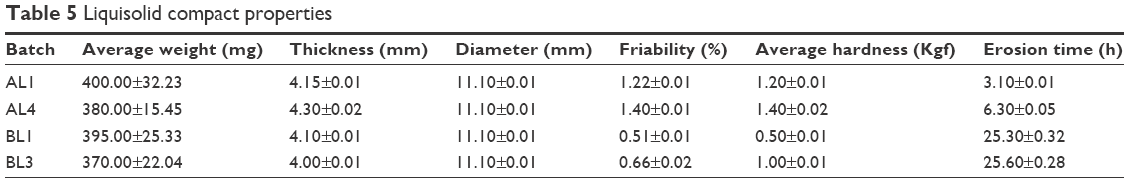 | Table 5 Liquisolid compact properties |
In vitro drug release
Drugs incorporated into lipid nanoparticles are either dissolved or dispersed.27,31 As a matter of fact, solubility of drug in the lipid matrix is a crucial factor for drug release from NLCs. To evaluate the release of AL from fixed-dose combination liquisolid compacts, in vitro drug release were performed separately for each drug using the basket-mesh method. Biorelevant media of SGF (pH 1.2) and SIF (pH 6.8) were used at 37°C. Comparative studies were carried out for free-AL compacts (data not shown), AL4 and BL3 and results are shown in Figure 6. Both AL4 and BL3 compacts showed slightly fast initial phase of 10.44±0.2% and 9.8±0.06% of LUM in the first 1 hour of study in SIF and SGF, respectively whereas ART release was 17.1±0.9% in both SIF and SGF. It is understandable why ART should have a higher initial release in 1 hour over LUM. This is because ART was used as a crystalline drug ab initio not incorporated into NLC as was the case with LUM. In other words, the initial fast release was because of superficial entrapment of LUM at the outer layers of the NLC particles as the formulation cooled from high temperature of production to room temperature.32,33 However, slower and more prolonged release patterns were observed later. Summarily, LUM release was lower than ART in both media. The highest amount of LUM release in 24 hours from AL4 (84.32±10.85) occurred in SIF followed by 77.97±9.6% in BL3 (better than 71.84% and 61.4% in SGF), respectively. ART release from compacts in both SIF and SGF were equal amounting to 91.25±12% and 86.59±10.68%, respectively. As ART is a fast-acting schizonticidal drug for malaria treatment, its rapid initial but gradual release would reduce parasite load and quickly resolve clinical symptoms while sustained release of LUM would prevent recrudescence.3
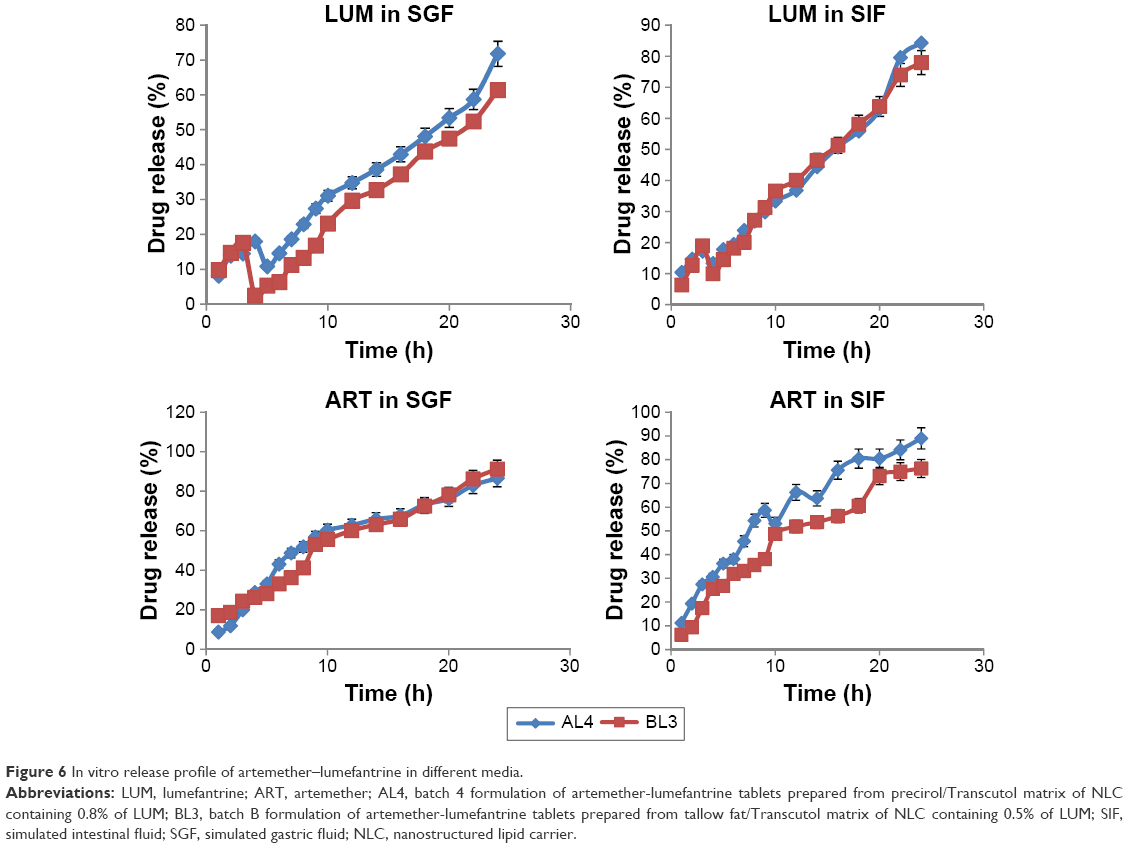 | Figure 6 In vitro release profile of artemether–lumefantrine in different media. |
Kinetics and mechanism of AL release from liquisolid compacts
The kinetics of release showed mixed zero order and Higuchi release patterns in all media as shown in Table 4. Briefly, only ART release in SIF from both AL4 and BL3 showed an R2 value >0.9 for all four studied models, while release of LUM in SIF and ART in SGF both from BL3 had R2 values >0.9 for only three models. Kinetics of release of ART from AL4 (R2=0.98) fitted into all models, while release form BL3 in SIF has R2 values lower than 0.98 for all models. Zero order best described the kinetics of LUM release in SIF and SGF from both AL4 and BL3 whereas Higuchi model best described ART release from BL3 in SGF (R2=0.96) and SIF (R2=0.98), respectively. This shows that mechanism of release was Fickian which might have depended on the porosity of the compact as Higuchi model deals with drug release from porous systems.34 This is in consonance with the result of in vitro release, which showed fast onset release of ART due to hydro-channels already created by the water-soluble surfactants (amphipathy) in the NLC particles making for fast erosion and release of ART followed by gradual prolonged release of both drugs. However, mechanism of drug release in terms of Peppas could be described by release exponent “n” value and kinetic constant “k,” which incorporates the structural and geometric characteristics of the release device. In this study, the n values were generally greater than 0.45 but less than 0.81, which suggests a non-Fickian (anomalous) type. LUM release from AL4 in SIF gave an n value of 0.35, suggesting Fickian diffusion, whereas ART release from AL4 in SGF gave n value of 1.11, typical of super case II transport (matrix erosion) and Fickian diffusion.
In vivo antiplasmodial activity of liquisolid compacts
All the liquisolid tablet compacts tested exhibited much greater plasmodial growth inhibition than the commercially available fixed combination tablet of AL (Coartem®) and CQ-PO4 tablets as well as the placebo (drug-free liquisolid compact tablets). From Figure 7, it is clear that liquisolid compacts had better plasmodial growth inhibition than the conventional market samples probably due to the nature of their compositions. As fatty meals improve the bioavailability of AL, the study result is therefore in consonance with this observation as the NLC matrices stand in this gap.13,16 In other words, the enhanced activity of AL liquisolid compacts over other conventional options was due to its lipid-based particle carrier, which provided high drug solubility and entrapment thereby improving drug absorption in much smaller doses (4/24 and/or 2/24 mg) compared to the large doses of the conventional samples (80/480, 40/240, and/or 20/120 mg). On the other hand, the particles would have also protected AL from first pass effect by enhancing lymphatic uptake as the component homolipids of Precirol® and tallow fat were primarily composed of palmitic, lauric, myristic, and other similar fatty acids, which have been shown to possess penetration enhancing properties, and hence facilitated lymphatic transport of AL.33 Additionally, the high solubility of LUM in the lipid matrices might have facilitated drug absorption in agreement with earlier works, through a number of ways, including lipolysis and cellular transport3,13,14,33 as well as opening of tight junctions due to fluidization of intestinal cell membranes by the action of surfactants.34–37 Therefore, liquisolid compacts of AL4 and AL3 exhibited higher reduction in parasitemia than the commercial fixed-dose combination (Coartem®) and CQ-PO4 tablets as well as individual LUM or ART tablets (data not shown). Compacts of AL4 and BL3 containing 4/24 mg of AL and given once-in-two days reduced parasitemia by 91% and 92%, respectively, whereas AL4 and BL3 containing 2/12 mg of AL and given twice daily reduced parasitemia by 88.4% and 91%, respectively. By implication, there was no significant difference (P<0.05) in parasitemia reduction across all AL liquisolid compacts. Yet, as the current problem with conventional fixed-dose combination of AL is noncompliance due to dose frequency (twice daily) of large amounts (80/480, 40/240, and/or 20/120 mg) and untoward development of resistance, we hereby report that the present investigation could stand in this gap. Meanwhile, AL4 and BL3 containing 4/24 mg of AL and taken once in 2 days could provide more use convenience than the counterpart regimen containing 2/12 mg of AL and taken twice daily or even once daily. This would certainly increase compliance and reduce development of resistance due to noncompliance, in addition to reduction of numerous side effects due to large doses with no option of dose modification in hepatic and/or renal impairment,3 as inherent in conventional regimens.
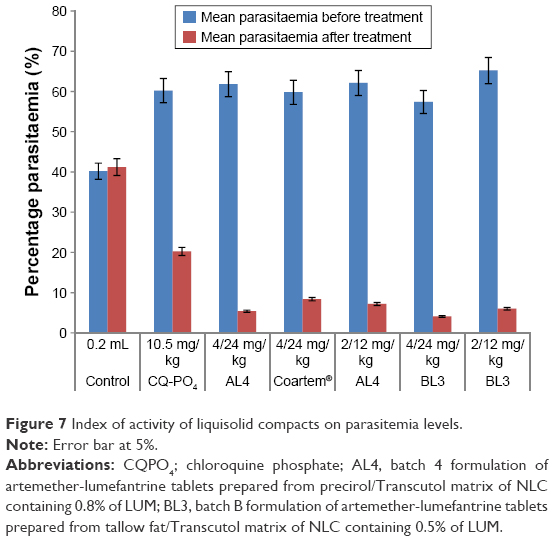 | Figure 7 Index of activity of liquisolid compacts on parasitemia levels. |
Conclusion
In this study, stable NLCs with mean particle size of 900 nm and high LUM EE (~95%) were successfully prepared from homolipid systems of PT (batch A) and TT (batch B) at 3:1 combinations, respectively, by the hot homogenization method. These NLCs were converted to liquisolid tablet compacts by direct compression with ART and other excipients to attain the World Health Organization current recommendation for artemisinin-combination therapy for malaria treatment. The AL compacts (AL4 and BL3) containing two fixed-dose combinations of 4/24 and 2/12 mg of AL, respectively, showed superior plasmodial growth inhibition (~92%) compared to conventional tablets of AL (Coartem®) and another antimalarial drug, CQ-PO4, which showed 85% and 66% inhibition, respectively, in malariogenic mice. The most important finding here was that AL could be reformulated in smaller, more tolerable doses with moderately fast ART release (17% in 1 hour), yet gradually sustained over 24 hours (91%) as well as LUM (84%) both in SIF, for resolution of malaria symptoms (>92% activity), while providing more use convenience of once-in-two days dosing of 4/24 mg of AL to improve compliance and reduce development of resistance from noncompliance, hence, proffering solution to the current problem encountered with the available marketed AL regimens.
Acknowledgments
The authors appreciate the assistance of Dr Chiara Rossi for SEM analysis while thanking Professor Dr CM Lehr for making his laboratory available for particle characterization. The authors are thankful to Phospholipid GmbH, Köln, Germany, BASF Ludwigshafen, Germany, and Gattefossé, St Priest, France, for gift samples of Phospholipon® 90G; Poloxamer® 188 and Solutol® HS 15; and Precirol® ATO 5 and Transcutol® HP, respectively.
Disclosure
The authors report no conflicts of interest in this work.
References
Guerra CA, Howes RE, Patil AP, et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010;4:e774. | ||
Hay SI, Okiro EA, Gething PW, et al. Estimating the global clinical burden of Plasmodium falciparum malaria in 2007. PLoS Med. 2010;7:e1000290. | ||
Nnamani PO, Hansen S, Windbergs M, Lehr CM. Development of artemether-loaded nanostructured lipid carrier (NLC) formulation for topical application. Int J Pharm. 2014;477:208–217. | ||
Nnamani PO, Scoles G, Kröl S. Preliminary characterization of N-trimethylchitosan as a nanocarrier for malaria vaccine. J Vector Borne Disease. 2011;48(4):224–230. | ||
Egunsola O, Oshikoya KA. Comparative safety of artemetherlumefantrine and other artemisinin-based combinations in children: a systematic review. Malar J. 2013;12:385. | ||
Varshosaz J, Eskandari S, Tabakhian M. Production and optimization of valproic acid nanostructured lipid carriers by the taguchi design. Pharm Dev Technol. 2010;15:89–96. | ||
Thatipamula RP, Palem CR, Gannu R, Mudragada S, Yamsani MR. Formulation and in vitro characterization of domperidone loaded solid lipid nanoparticles and nanostructured lipid carriers. Daru. 2011;19:23–32. | ||
Kasongo KW, Jansch M, Muller RH, Walker RB. Evaluation of the in vitro differential protein adsorption patterns of didanosine-loaded nano-structured lipid carriers (NLCs) for potential targeting to the brain. J Liposome Res. 2011;21:245–254. | ||
Mandawgade SD, Sharma S, Pathak S, Patravale VB. Development of SMEDDS using natural lipophile: application to β-artemether delivery. Int J Pharm. 2008;362:179–183. | ||
Varshosaz J, Eskandari S, Tabakhian M. Freeze-drying of nanostructure lipid carriers by different carbohydrate polymers used as cryoprotectants. Carbohyd Polym. 2012;88:1157–1163. | ||
Teeranachaideekul V, Souto EB, Müller RH, Junyaprasert V. Physico-chemical characterization and in vitro release studies of ascorbyl palmitate-loaded semi-solid nanostructured lipid carriers (NLC gels). J Microencapsul 2008;25:111–120. | ||
Yang B, Lin J, Chen Y, Liu Y. Artemether/hydroxypropyl-b-cyclodextrin host–guest system: characterization, phase-solubility and inclusion mode. Bioorg Med Chem. 2009;17:6311–6317. | ||
Parashar D, Aditya NP, Murthy RSR. Development of artemether and lumefantrine co-loaded nanostructured lipid carriers: physicochemical characterization and in vivo antimalarial activity. Drug Deliv. 2014;23(1):1–7. | ||
Aditya NP, Patankar S, Madhusudhan B, Murthy RS, Souto EB. Artemether loaded lipid nanoparticles produced by modified thin-film hydration: pharmacokinetics, toxicological and in vivo anti-malarial activity. Eur J Pharm Sci. 2010;40:448–455. | ||
Agubata CO, Nzekwe IT, Attama AA, Mueller-Goymann CC, Onunkwo GC. Formulation, characterization and anti-malarial activity of homolipid-based artemether microparticles. Int J Pharm. 2015;478:202–222. | ||
Lefèvre G, Thomsen MS. Clinical pharmacokinetics of artemether and lumefantrine (Riamet®). Clin Drug Invest. 1999;18:467–480. | ||
Ezzet F, Mull R, Karbwang J. Population pharmacokinetics and therapeutic response of CGP 56697 (artemether + benflumetol) in malaria patients. Br J Clin Pharmacol. 1998;46(6):553–561. | ||
Hien TT, Davis TM, Choung LV, et al. Comparative pharmacokinetics of intramuscular artesunate and artemether in patients with severe falciparum malaria. Antimicrob Agents Chemother. 2004;48(11):4234–4239. | ||
Vranikova B, Gajdziok J. Liquisolid systems and aspects influencing their research and development. Acta Pharm. 2013;63:447–465. | ||
Nagabandi VK, Ramarao T, Jayaveera KN. Liquisolid compacts: a novel approach to enhance bioavailability of poorly soluble drugs. Int J Pharm Biol Sci. 2011;1:89–102. | ||
Gonjari ID, Karmarkar AB, Hosmani AH. Evaluation of in vitro dissolution profile comparison methods of sustained release tramadol hydrochloride liquisolid compact formulations with marketed sustained release tablets. Dig J Nanomater Bios. 2009;4:651–661. | ||
Mahajan HS, Dhamne MR, Gattani SG, Rasal AD, Shaikh HT. Enhanced dissolution rate of glipizide by a liquisolid technique. Int J Pharm Sci Nanotech. 2011;3:1205–1213. | ||
Nnamani PO, Ibezim EC, Attama AA, Adikwu MU. Surface modified solid lipid microparticles based on homolipids and softisan® 142: preliminary characterization. Asian Pac J Trop Med. 2010;205–210. | ||
The British Pharmacopeia. London: Her Majesty’s Stationery office; 2004. Available from: https://www.pharmacopoeia.com/. Accessed June 10, 2006. | ||
Attama AA, Onugu CO, Nnamani PO, Adikwu M, Nzekwe IT. The use of self- emulsifying systems in the delivery of diclofenac. Int J Pharm. 2003;262:23–28. | ||
Ryley JF, Peters W. The antimalarial activity of some quinolone esters. Ann Trop Med Parasitol. 1970;84:209–222. | ||
Attama AA, Müller-Goymann CC. A critical study of novel physically structured lipid matrices composed of a homolipid from Capra hircus and theobroma oil. Int J Pharm. 2006;322:67–78. | ||
Muller RH, Radtke M, Wissing SA. Solid lipid nanoparticles (SLN) nanostructured lipid carriers (NLC) in cosmetics and dermatological preparations. Adv Drug Deliv Rev. 2002;54:S131–S155. | ||
Indian Pharmacopeia. The India Pharmacopoeia Commission Ghaziabad; 2007. Available from: http://www.ipc.gov.in. Accessed June 19, 2010. | ||
United States Pharmacopeia. In vitro and in vivo evaluation of dosage forms 2008. 27th Ed., Revision, Easton, PA, USA: Mack Publishing Co. | ||
Pinto MF, Moura CC, Nunes C, Segundo MA, Costa Lima SA, Reis S. A new topical formulation for psoriasis: development of methotrexate-loaded nanostructures lipid carriers. Int J Pharm. 2014;477:519–526. | ||
Humphreys GS, Merinopoulos I, Ahmed J, Whitty CJ, Mutabingwa TK, Sutherland CJ, Hallett RL. Amodiaquine and artemether-lumefantrine select distinct alleles of the Plasmodium falciparum mdr1 gene in Tanzanian children treated for uncomplicated malaria. Antimicrob Agents Chemother. 2007;51:991–997. | ||
Krugliak M, Deharo E, Shalmiev G, Sauvain M, Moretti C, Ginsburg H. Antimalarial effects of C18 fatty acids on Plasmodium falciparum in culture and on Plasmodium vinckei petteri and Plasmodium yoelii nigeriensis in vivo. Exp Parasitol. 1995;81:97–105. | ||
Koga K, Kusawake Y, Sugioka N, Shibata N. Enhancing mechanism of Labrasol on intestinal membrane permeability of the hydrophilic drug gentamicin sulfate. Eur J Pharm Biopharm. 2006;64:82–91. | ||
Rama Prasad YV, Eaimtrakarn S, Ishida M, et al. Evaluation of oral formulations of gentamicin containing Labrasol in beagle dogs. Int J Pharm. 2003;268:13–21. | ||
Sha X, Yan G, Wu Y, Li J, Fang X. Effect of self-microemulsifying drug delivery systems containing labrasol on tight junctions in Caco 2 cells. Eur J Pharm Sci. 2005;24:477–486. | ||
Chang RK, Shojaei AH. Effect of a lipoidic excipient on the absorption profile of compound UK 81252 in dogs after oral administration. J Pharm Pharm Sci. 2004;7:8–12. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.