")
Back to Journals » International Medical Case Reports Journal » Volume 14
Suspicion of Frasier’s Syndrome in the Nephrology Unit of the State University Hospital of Haiti: Case Study and Review of Literature
Authors Jean Paul A , Louis D, Desravines AJ , Jean RM, Jean Baptiste A, Buteau JH, Andre W
Received 19 June 2021
Accepted for publication 29 July 2021
Published 12 August 2021 Volume 2021:14 Pages 533—538
DOI https://doi.org/10.2147/IMCRJ.S325619
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Axler Jean Paul,1 Dieuguens Louis,2 Ansly Jefferson Desravines,1 Raema Mimrod Jean,1 Alfadler Jean Baptiste,1 Jean Henold Buteau,2 Wislet Andre2
1General Medicine, State University Hospital of Haiti, Port-au-Prince, West, Haiti; 2Internal Medicine, State University Hospital of Haiti, Port-au-Prince, West, Haiti
Correspondence: Axler Jean Paul 79, Impasse Dady, Juvenat, Petionville, West, Haiti
Tel +509 37349648
; +509 43776900
Email [email protected]
Objective: Frasier syndrome is a rare genetic nephropathy characterized by the presence of progressive glomerulopathy with proteinuria associated with male pseudo hermaphroditism. This case study described a picture of a young boy where the clinical suspicion context reminded the Frasier syndrome. To our knowledge, this case is the first described in Haiti.
Case Study: This is a 19-year-old young phenotypically male, born with a genital anomaly, was seen on referral at the nephrology/dialysis unit of the internal medicine department of the State University Hospital of Haiti for evaluation and follow-up. Insidious progression of symptoms had occurred over 3 years. Over three months of outpatient follow-up, he had four sets of renal labs drawn, and all showed impaired renal function. At the ultrasound, a bilateral cryptorchidism is described in the inguinal, and presence of functional ovaries with follicles of variable size scattered in the parenchyma. So, in the light of these anamnestic, clinical and paraclinical findings, we concluded to the diagnosis of end-stage renal failure by progressive glomerulopathy in a context of Frasier’s syndrome.
Conclusion: With any clinical picture consisting of genital anomalies at birth, renal symptomatology during childhood and the diagnosis of renal failure during adolescence, rare genetic nephropathies, such as Frasier syndrome must be considered.
Keywords: Frasier syndrome, end-stage renal failure, young, HUEH, Haiti
Background
Frasier syndrome is a rare genetic nephropathy caused by a point mutation in the WT 1 gene located on chromosome 11.1–4 First described in 1964 by Professor Frasier,5 it is characterized by the presence of progressive glomerulopathy with proteinuria associated with male pseudo hermaphroditism (presence of female external genitalia in a male phenotype).2,3,6–8 Initial manifestations of patients with Frasier syndrome include morning edema (facial puffiness), elevated blood pressure, proteinuria, with hypospadias and/or ambiguity of the external genitalia accompanied by cryptorchidism at birth.2,3,9 The symptomatology evolves into a treatment-resistant nephrotic syndrome that progressively leads to end-stage renal failure during the first two decades of the child’s life.2,3,6,9–11 We present a case of Frasier syndrome that was seen at the nephrology/dialysis unit of HUEH.
Case Study
This is a young phenotypically male (presence of secondary male characteristics) who is 19 years old and born with a genital malformation in a family without history of consanguinity. They were seen on referral at the nephrology/dialysis unit of the internal medicine department of HUEH for evaluation and follow-up. Insidious progression of symptoms had occurred over 3 years. However, the patient developed new symptoms over the 21 days preceding this consultation at the hospital. The patient was experiencing morning puffiness of the face and postprandial vomiting. He had episodes of dyspnea with effort that evolved into dyspnea at rest, orthopnea requiring two pillows, and paroxysmal nocturnal dyspnea. He also developed ascending edema of the lower limbs with malleolar onset. At an outside hospital, urine examination showed proteinuria (+++), urinary tract infection with severe leukocyturia (30–35 white blood cells per field) and hematuria (6–8 red blood cells per field). He had a severe anemia (7.8 g/dl), normocytic normochromic (MCV: 84.1; MCHC: 34.2), a predominantly neutrophilic hyperleukocytosis (14,000; 86%), an altered ionogram with mild hyperkalemia (5.71 mEq/L) and an altered renal balance with creatininemia (25.25 mg/dl); BUN (90 mg/dl). In the light of these assessments, he was referred to the emergency department and followed up with nephrology for appropriate care.
In the emergency room, he was noted to have a puffy face, pale palpebral conjunctiva, and crackling rales at the base of the left lung. His scrotum was empty to palpation and enlarged, though nontender. His external genitalia were characterized by a micropenis with the presence of a rough vaginal orifice attached to the perineum.
Over three months of outpatient follow-up, he had four sets of renal labs drawn, and all showed impaired renal function (Figure 1). The last one revealed a creatinine level of 10.5 mg/dl. The glomerular filtration rate was calculated: MDRD eGFR = 8.21 mL/min/1.73. His phosphocalcic assessment showed hypocalcemia at 8.1 mg/dl and a 24-hour proteinuria at 578 mg. During the same period, he had four altered blood counts showing severe anemia that did not correct (Figure 1), and the last of these revealed a severe microcytic hypochromic anemia with a hemoglobin level of 7.4 gr/dl, MCV at 79.2 fl. and a MCHC at 26.6 pg, and hyperleukocytosis of 15,800 with neutrophilic predominance (76%). His hormonal tests showed a decrease in testosterone level to 242 ng/dl, an increase in hydroxyprogesterone level to 67.9, and an increased LH level to 26 mlU/mL.
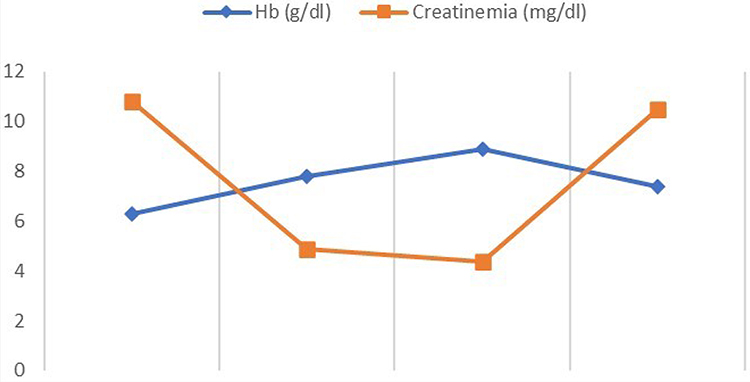 |
Figure 1 Variation of hemoglobin and creatininemia over the last 3 months. |
He also had an abdominal pelvic ultrasound showing poor cortico-medullary differentiation with small kidneys (right kidney 9.7*5.02 cm; left kidney 9.7*5.6 cm) and dilatation of the pyelocaliceal system and proximal ureter (Figure 2). Bilateral cryptorchidism is also described in the inguinal region (right testicles 2.7*1.1 cm and left 1.37*1.04 cm), and presence of functional ovaries (1.6*1.1 cm on the right and 1.5*1.7 cm on the left) with follicles of variable size scattered in the parenchyma (Figure 3). He also has a prostate gland (4.83cm3). Doppler ultrasound of the kidneys described a significant alteration of the renal vascularization and an absence of opacification of the cortical arteries in the periphery. He had performed an echocardiogram concluding a dilated cardiomyopathy with mild alteration of the systolic ejection fraction (46.4%) associated with a pericardial effusion without signs of tamponade. Chest radiography also described congestive cardiomegaly with signs of vessel cephalization and impaction of the left hilum.
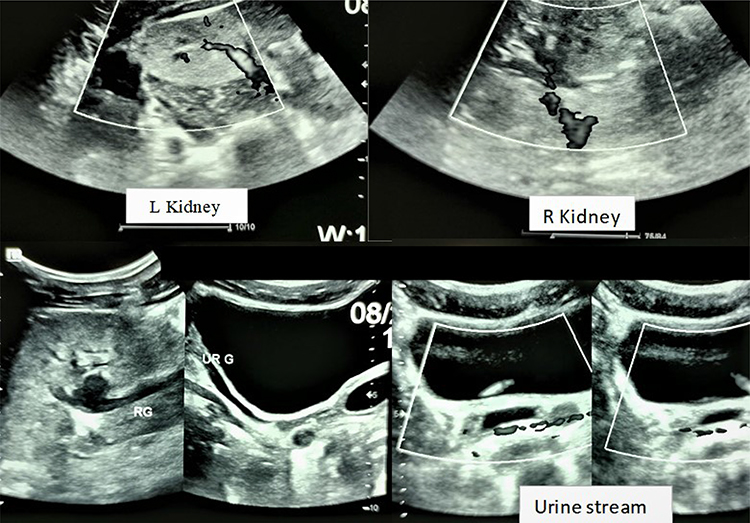 |
Figure 2 Ultrasound of the kidneys (L: left; R: right). |
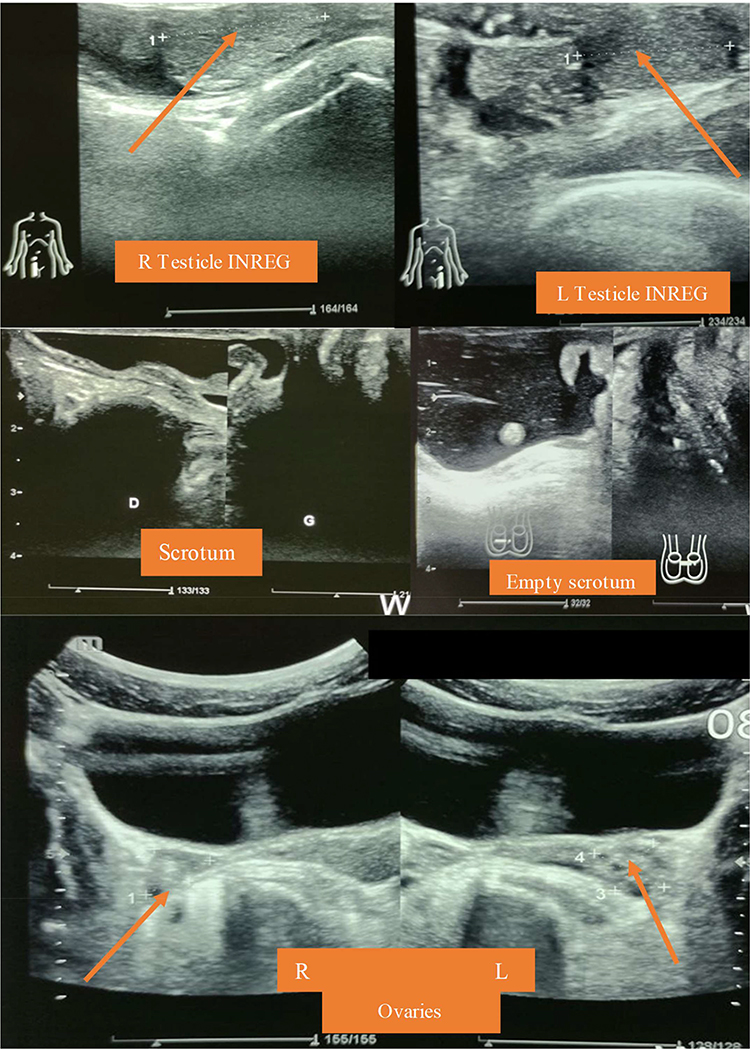 |
Figure 3 Identification of the testicles (in the inguinal region), ovaries and the empty srotum on abdominal-pelvic ultrasound. |
In the light of these anamnestic, clinical and paraclinical findings, we concluded to the diagnosis of end-stage renal failure by progressive glomerulopathy in a context of Frasier’s syndrome complicated by a global cardiac decompensation class IV.
During his stay on the unit, he received 31 sessions of hemodialysis and 9-unit bags of compatible packed red blood cells that did little to correct his anemia (Figure 1). He was managed for cardiac decompensation and was placed on furosemide, enalapril, carvedilol, iron, thiamine, erythropoietin and omeprazole. After 12 days, he developed a compensated heart disease and his clinical condition improved, so he was discharged. He continued hemodialysis sessions in the unit and genetic tests were planned to investigate possible mutations that would be diagnostic of Frasier syndrome. However, he suddenly succumbed to cardiac arrest in post-dialysis.
Discussion
Mutations in the WT1 gene result in a spectrum of very rare congenital diseases characterized by abnormalities in gonadal and urinary tract development and are the cause of WAGR (Wilms-Aniridia-Genitourinary-mental Retardation) syndromes, Denys-Drash syndrome and Frasier syndrome.3,7,12,13 Initial manifestations of patients with nephropathy due to the WT1 gene mutation include edema, proteinuria, pseudo hermaphroditism, hypospadias or cryptorchidism at birth.9 Frasier syndrome and Denys-Drash syndrome (DDS) share common features and are both characterized by a steroid-resistant nephrotic syndrome, which leads to renal failure, gonadal tumor (most often gonadoblastoma or dysgerminoma), and male pseudo hermaphroditism.2,12,14 The main differences lie in the more prominent manifestation of renal symptomatology during childhood in DDS, contrasting FS which mostly manifests during the second decade, particularly during adolescence or young adulthood.3,7,9,12,15,16 Additionally, the risk of development Wilms’ tumors is higher in DDS.3,6,7,12,13,16,17 In our case, we eliminated DDS because the renal failure occurs mainly in the second decade (16–19 years), as mentioned in the literature.6,7,18,19
Frasier syndrome is defined by the presence of progressive glomerulopathy with proteinuria associated with male pseudo hermaphroditism (presence of female external genitalia in a male phenotype).2,3,6–8 To our knowledge, this case is the first described in Haiti. Taking into account the appearance of the external genitalia, phenotype and sex chromosomes, Frasier syndrome is classified into 3 types:2
- Type 1: characterized by the presence of external female genitalia with sex chromosome 46, XY; the most common.
- Type 2 by the presence of male external genitalia with sex chromosome 46, XY.
- Type 3 presence of external female genitalia with sex chromosome 46, XX.
This case was closest to type 2 because of the male phenotype with predominant development of male sexual characteristics (no well-defined breasts or vagina). The mean age at diagnosis of Frasier syndrome was estimated to be 16.3±2.3 years,2,15,20 which is also the age at which the diagnosis of end-stage renal disease is often made, although the onset of renal symptoms of proteinuria, edema and hypertension typically begins between 2 and 10 years of age.1,2,6,9,10,19,20 This patient had a similar picture; since his childhood he was often in hospital for reasons of facial oedema and alteration of the renal function. This prompted the abdominal ultrasound revealing a decrease in kidney size of kidneys, and allowed the diagnosis of preterminal renal insufficiency at age 16.
Two elements are essential to confirm the diagnosis of Frasier syndrome: renal biopsy and cytogenetic study that identifies the WT1 gene mutation located on chromosome 11p13.7 Renal biopsy performed before the stage of end-stage failure describes segmental and focal glomerulosclerosis8,18,21 generally caused by the WT1 gene mutation leading to alterations in the podocytes with areas of proliferation.8,9,15 It is due to point mutations in intron 9 of the WT1 gene, resulting in the loss of the lysine-threonine-serine (KTS) containing isoforms of the WT1 protein and result in deficiency of the usually more abundant KTS positive isoforms and reversal of the normal KTS positive/negative ratio from 2:1 to 1:2.16 This has a great impact on tumor risk, because patients with Frasier syndrome have one normal copy of WT1 and one that can only produce the KTS negative isoform, this might explain why patients with Frasier syndrome do not develop Wilms’s tumor which also explains the observed external genitalia changes, because the KTS positive isoform might participate in another pathway required for normal male urogenital development.2,16,19 There are extrarenal manifestations of the WT1 gene mutation such as the presence of cardiomyopathies,21 a finding that was identified in our patient at echocardiography showing dilated cardiomyopathy with slight alteration of the systolic ejection fraction (46, 4%). However, this could also be a complication of his chronic end-stage renal failure.
In our case, the renal biopsy was no longer appropriate since he had arrived at the nephrology unit very late and already had end-stage renal failure with hypotrophic kidneys. During his stay, we conducted genetic tests to confirm the presence of the mutations. Unfortunately, he died suddenly due to cardiac arrest after a dialysis session. As a result, the genetic tests were not performed to confirm mutations that would explain the clinical and paraclinical findings characteristic of Frasier syndrome.
Conclusion
With any clinical picture consisting of genital anomalies at birth, renal symptomatology (facial edema, proteinuria and high blood pressure) during childhood and the diagnosis of renal failure during adolescence, rare genetic nephropathies, such as Frasier syndrome and Denys Drash syndrome, must be considered.7,12,20,23 In low-income settings such as ours, where genetic technology is not always available, the diagnosis of Frasier syndrome should be considered in any child with developmental abnormalities of the external genitalia presenting with isolated proteinuria and/or signs of impaired renal function during childhood or adolescence.
Statement of Ethics Approval
This study has been approved for publication by the head directory of the Internal Medicine of the State University Hospital of Haiti as it is a teaching hospital.
Statement on Participant Consent
The patient’s mother consented to participate and publish the relevant information about the case, and the information about the identity was kept confidential.
Acknowledgments
Special thanks to Dr. Nelle-Ange Mele, Dr. Joseph Daniel Perez de Corcho, and Dr. Rolvix Patterson, for their invaluable help in translating the manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Yang Y, Zhao F, Tu X, Yu Z. Mutations in WT1 in boys with sporadic isolated steroid-resistant nephrotic syndrome. Genet Mol Res. 2016;15(1):15017559. doi:10.4238/gmr.15017559
2. Ezaki J, Hashimoto K, Asano T, et al. Gonadal tumor in Frasier syndrome: a review and classification. Cancer Prev Res. 2015;8(4):271–276. doi:10.1158/1940-6207.CAPR-14-0415
3. Peco-Antić A, Ozaltin F, Parezanović V, Miloševski-Lomić G, Zdravković V. Proteinuria in Frasier syndrome. Srp Arh Celok Lek. 2013;141(9–10):685–688. doi:10.2298/SARH1310685P
4. Hildebrandt F. Genetic kidney diseases. Lancet. 2010;375(9722):1287–1295. doi:10.1016/S0140-6736(10)60236-X
5. Frasier SD, Bashore RA, Mosier HD. Gonadoblastoma associated with pure gonadal dysgenesis in monozygous twins. J Pediatr. 1964;64(5):740–745. doi:10.1016/S0022-3476(64)80622-3
6. Jalanko H, Kääriäinen H. Nephrotic Disorders.
7. Miller-Hodges E. Clinical aspects of WT1 and the kidney. Methods Mol Biol. 2016;1467:15–21. doi:10.1007/978-1-4939-4023-3_2
8. Demmer L, Primack W, Loik V, Brown R, Therville N, Mcelreavey K. Frasier Syndrome. J Am Soc Nephrol. 1999;10(10):2215–2218. doi:10.1681/ASN.V10102215
9. Sun S, Xu L, Bi Y, et al. Early diagnosis of WT1 nephropathy and follow up in a Chinese multicenter cohort. Eur J Med Genet. 2020;63(11):104047. doi:10.1016/j.ejmg.2020.104047
10. Ruf RG, Schultheiss M, Lichtenberger A, Karle SM. Prevalence of WT1 mutations in a large cohort of patients with steroid-resistant and steroid-sensitive nephrotic syndrome. Kidney Int. 2004;66(2):564–570. doi:10.1111/j.1523-1755.2004.00775.x
11. Melo KFS, Martin RM, Costa EMF, et al. An unusual phenotype of Frasier syndrome due to IVS9 +4C>T mutation in the WT1 gene: predominantly male ambiguous genitalia and absence of gonadal dysgenesis. J Clin Endocrinol Metab. 2002;87(6):2500–2505. doi:10.1210/jcem.87.6.8521
12. Liu EK, Suson KD. Syndromic Wilms tumor: a review of predisposing conditions, surveillance and treatment. Transl Androl Urol. 2020;9(5):2370–2381. doi:10.21037/tau.2020.03.27
13. Yang YH, Zhao F, Feng DN, et al. Wilms’ tumor suppressor gene mutations in girls with sporadic isolated steroid-resistant nephrotic syndrome. Genet Mol Res. 2013;12(4):6184–6191. doi:10.4238/2013.December.4.5
14. Avni EF, Vandenhoute K, Devriendt A, et al. Update on congenital nephrotic syndromes and the contribution of US. Pediatr Radiol. 2011;41(1):76–81. doi:10.1007/s00247-010-1793-5
15. Chiba Y, Inoue CN. Once-daily low-dose cyclosporine a treatment with angiotensin blockade for long-term remission of nephropathy in Frasier syndrome. Tohoku J Exp Med. 2019;247(1):35–40. doi:10.1620/tjem.247.35
16. Koziell A, Grundy R. Frasier and Denys-Drash syndromes: different disorders or part of a spectrum? Arch Dis Child. 1999;81:365–369. doi:10.1136/adc.81.4.365
17. Tasic V, Gucev Z, Polenakovic M. Steroid resistant nephrotic syndrome-genetic consideration. Pril. 2015;36(3):5–12. doi:10.1515/prilozi-2015-0073
18. Hashimoto H, Zhang X, Zheng Y, Wilson GG, Cheng X. Denys-Drash syndrome associated WT1 glutamine 369 mutants have altered sequence-preferences and altered responses to epigenetic modifications. Nucleic Acids Res. 2016;44(21):10165–10176. doi:10.1093/nar/gkw766
19. Matsuoka D, Noda S, Kamiya M, et al. Immune-complex glomerulonephritis with a membranoproliferative pattern in Frasier syndrome: a case report and review of the literature. BMC Nephrol. 2020;21(1). doi:10.1186/s12882-020-02007-0
20. Lipsk BS, Lipsk BS, Ranchin B, et al. Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int. 2014;85(5):1169–1178. doi:10.1038/ki.2013.519
21. Trautmann A, Bodria M, Ozaltin F, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the podoNet registry cohort. Clin J Am Soc Nephrol. 2015;10(4):592–600. doi:10.2215/CJN.06260614
22. Park E, Lee C, Kim NKD, et al. Genetic study in Korean pediatric patients with steroid-resistant nephrotic syndrome or focal segmental glomerulosclerosis. J Clin Med. 2020;9(6):2013. doi:10.3390/jcm9062013
23. Chan AOK, But WM, Lee CY, et al. Aetiological bases of 46, XY disorders of sex development in the Hong Kong Chinese population. Hong Kong Med J. 2015;21(6):499–510. doi:10.12809/hkmj144402
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.