")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 16
Susceptibility of PCSK2 Polymorphism to Hirschsprung Disease in Southern Chinese Children
Authors Wang B , Fang W, Qin D, He Q, Lan C
Received 25 October 2022
Accepted for publication 4 May 2023
Published 15 May 2023 Volume 2023:16 Pages 59—64
DOI https://doi.org/10.2147/CEG.S393340
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Santosh Shenoy
Bingtong Wang,* Wenlin Fang,* Dingjiang Qin,* Qiuming He, Chaoting Lan
Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Clinical Research Center for Child Health, Guangzhou, 510623, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chaoting Lan, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Clinical Research Center for Child Health, Guangzhou, 510623, People’s Republic of China, Email [email protected]
Introduction: Hirschsprung’s disease (HSCR) is a developmental defect of the enteric nervous system (ENS), which is caused by abnormal development of enteric neural crest cells. Its occurrence is caused by genetic factors and environmental factors. It has been reported that single nucleotide polymorphisms (SNPs) of proprotein convertase subtilisin/kexin type 2 (PCSK2) gene are associated with HSCR. However, the correlation of HSCR in southern Chinese population is still unclear.
Methods: We assessed the association of rs16998727 with HSCR susceptibility in southern Chinese children using TaqMan SNP genotyping analysis of 2943 samples, including 1470 HSCR patients and 1473 controls. The association test between rs16998727 and phenotypes was performed using multivariable logistic regression analysis.
Results: We got an unexpected result, PCSK2 SNP rs16998727 was not significantly different from HSCR and its HSCR subtypes: S-HSCR (OR = 1.08, 95% IC: 0.93~1.27, P_adj = 0.3208), L-HSCR (OR = 1.07, 95% IC: 0.84~1.36, P_adj = 0.5958) and TCA (OR = 0.94, 95% IC: 0.61~1.47, P_adj = 0.8001).
Conclusion: In summary, we report that rs16998727 (PCSK2 and OTOR) is not associated with the risk of HSCR in southern Chinese population.
Keywords: Hirschsprung’s disease, HSCR, single nucleotide polymorphism, SNP, proprotein convertase subtilisin/kexin type 2, PCSK2, genetic susceptibility
Introduction
Hirschsprung’s disease (HSCR) is a neonatal congenital disorder of the enteric nervous system (ENS) in which the myenteric and submucosal plexus of the distal intestinal wall are devoid of ganglion cells due to developmental impairment of the ENS during embryonic development.1 The above pathological changes make intestinal peristalsis disorders and tonic contractions in children with HSCR, so that clinical manifestations such as delayed meconium, intractable constipation, abdominal distension and diarrhea happen.2,3 The incidence of HSCR shows gender and racial differences, with a male-to-female ratio of 4/1, and an Asian population incidence of 1/5000.4 There are three subtypes of HSCR, which can be divided into short-segment (S-HSCR), long-segment (L-HSCR), and total colonic aganglionosis (TCA) according to the length of ganglion lesions in the intestine.5 In addition, HSCR can be divided into familial and sporadic, a few inherited in familial, most sporadic.
The occurrence of HSCR is a complex process. Its mechanism has not been fully elucidated, but studies have found that the occurrence of HSCR involves multiple signaling pathways and multiple genes. Many studies have shown that abnormal GDNF-GFRα1-RET and EDN3-EDNRB signaling pathways are high-risk factors for HSCR, which play an important role in the migration, proliferation and differentiation of enteric neural crest cells during ENS development.6–8 Polymorphisms of RET can affect the binding of transcription factors (NXF, ARNT2, SOX10, PHOX2B, and SIM2) to RET, thereby affecting the expression of RET and leading to the occurrence and development of HSCR.9–11 In addition, other mutant genes, like NRG1 and AUTS2 have been reported to be related to the occurrence of HSCR.12 However, only a small fraction of HSCR cases can be explained by these identified variants, the heritability of a large proportion of cases remains to be determined.13 Thus, we believe that the occurrence and development of HSCR may be the interaction of genetic factors and environmental factors. Next, it is necessary to identify unknown genes and other factors of HSCR.
Proprotein convertase subtilisin/kexin type 2 (PCSK2), which is mainly expressed in neurons and endocrine cells, belongs to the family of serine proteases that are responsible for the hydrolysis of precursor proteins to produce a variety of biologically active molecules such as hormones, neuropeptides, growth factors, cytokines and transcription factors.14–17 The production of most biologically active molecules depends on proprotein convertase subtilisin/kexins (PCSKs), especially the proper processing of most neuropeptide and endocrine precursors by PCSK2.18 It is worth noting that the regulation of gastrointestinal motility is controlled by a variety of biologically active peptides in central and intestinal neurons and peripheral endocrine cells.19 Otoraplin (OTOR), recognized as a novel cochlear gene, has expression in the cochlear nerve. It has a predicted secretory signal peptide sequence and harbors a high degree of cross-species conservation. The rs16998727 SNP was validated in a study from the HSCR group (n = 181) and control group (n = 346) from the Hong Kong Chinese.20 Considering that this result was not revalidated, we conducted a correlation study with a case group (n = 1470) and a control group (n = 1473) in a southern Chinese population to repeat the association of PCSK2 polymorphisms with HSCR.
Materials and Methods
Study Subjects
The subjects of this study were from Guangzhou Women’s and Children’s Medical Center. This research project was approved by the hospital’s Institutional Review Committee (Ethical Approval Number: 2018052406) and obtained written informed consent from each subject’s guardian. The study included 1470 HSCR patients and 1473 controls, and specific clinical data can be found in previous studies, as shown in Table 1.21 All cases were diagnosed as HSCR by postoperative biopsies after pull-through surgery showing lack of submucosal and intermuscular ganglia, and three subgroups were divided according to the length of enteric ganglionopathy, including S-HSCR (n = 1033), L-HSCR (n = 294), and TCA (n = 82). The control group had healthy populations without a history of nervous system diseases.
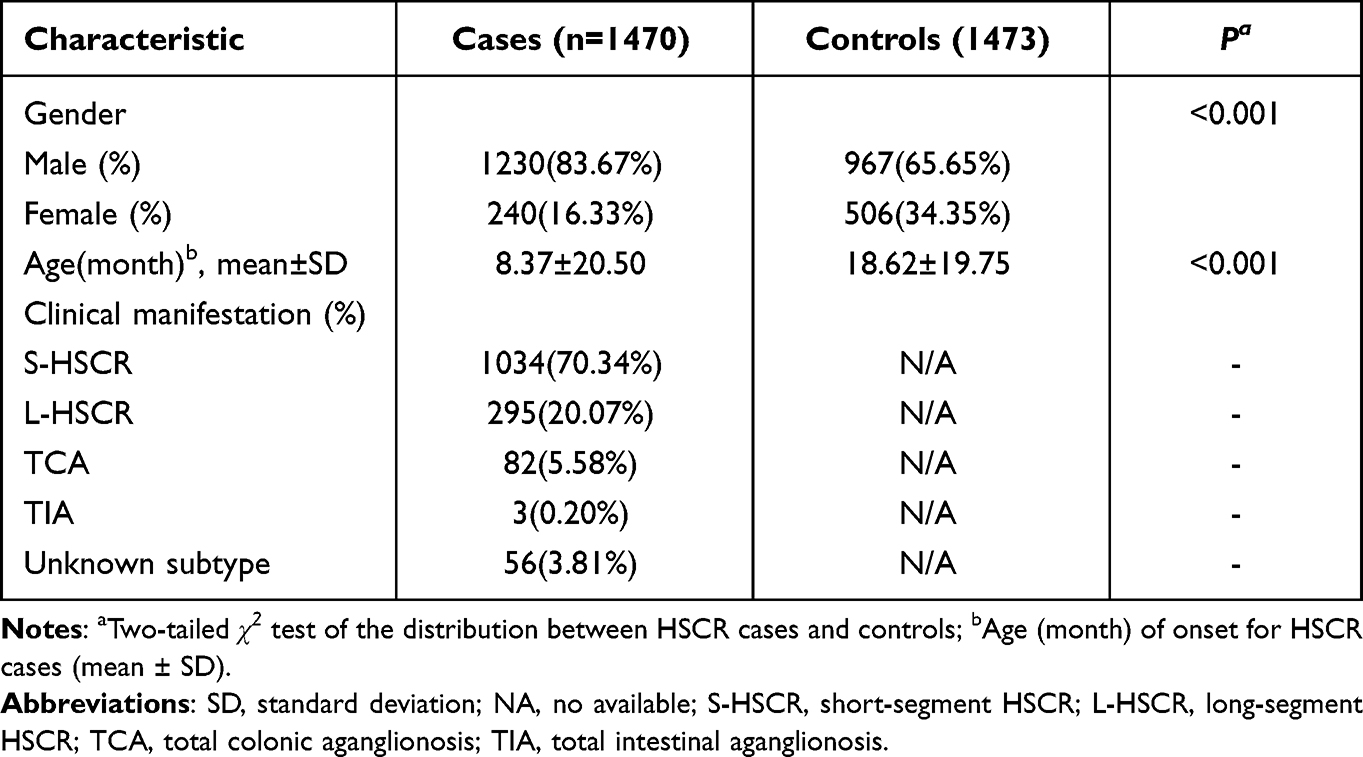 |
Table 1 Clinical Characteristics of the Study Subjects |
SNP Selection and Genotyping
The selection criteria for rs16998727 (PCSK2 and OTOR) were the same as described in our previous study.22 Briefly, candidate SNPs to be validated are selected to satisfy minor allele frequency, Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium criteria. TIANamp Blood Genomic DNA Kit (Tiangen Biotechnology Co., Ltd.) was used to extract genomic DNA from peripheral blood, then genotyping of PCSK2 and OTOR polymorphisms was performed using the TaqMan real-time PCR blind method. Each sample was repeated three times. Moreover, we randomly selected 10% of individuals and obtained two DNA samples per person to evaluate the consistency of the genotyping.
Statistical Analysis
We evaluated risk associations between PCSK2 and OTOR polymorphisms with HSCR and HSCR subtypes by odds ratios (ORs) and 95% confidence intervals (CIs) of multivariable logistic regression analysis. P and P_adj denote the significance of the effect association without and with age and sex adjustment, respectively. Differences in age and gender between groups were compared using a two-tailed chi-square test. Hardy-Weinberg equilibrium test was used to assess genotyping quality, and P > 0.05 was considered to indicate satisfactory goodness of fit. PLINK 1.9 software was used for statistical analysis and testing of additive, recessive and dominant genetic models.
Results
eQTL Analysis
To explore the potential functions of SNPs, we used the Genotype Tissue Expression (GTEx) dataset to evaluate the association of the rs16998727 with PCSK2 and OTOR gene expression. According to Figure 1, PCSK2 rs16998727 was significantly different at the splicing quantitative trait locus (eQTLs) in colon tissue (P= 1.3e−7) and nerve (P = 5.4e−6), but no OTOR association was observed.
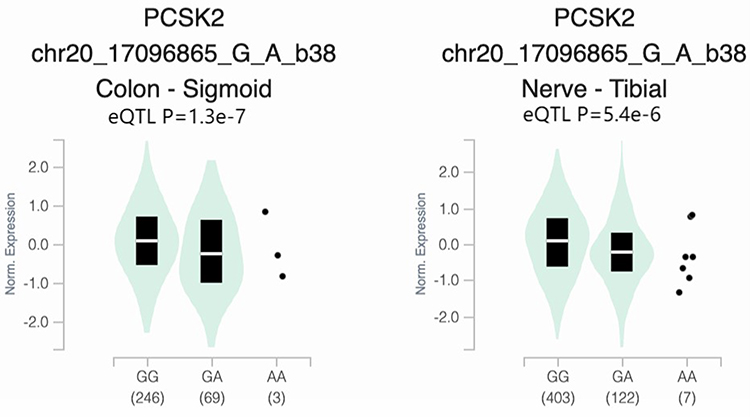 |
Figure 1 Based on data from the GTEx Portal database (https://www.gtexportal.org/home/), boxplots show that the rs16998727 (chr20_170968565_G_A_b38) PCSK2 genotype is associated with tibial nerve and sigmoid colon tissue expression. |
Association of rs16998727 (PCSK2 and OTOR) with HSCR Susceptibility
We calculated the association between the genotype frequency of rs16998727 and HSCR and are summarized in Table 2. The genotype of this SNP in the control group was in HWE (P>0.05). Additionally, logistic regression was performed on four different genetic models (allelic, genotypic, dominant and recessive) to predict the pattern of effects of rs16998727 PCSK2 and OTOR. The results in Table 2 showed that the rs16998727 (PCSK2 and OTOR) was not significantly associated with HSCR (P>0.05).
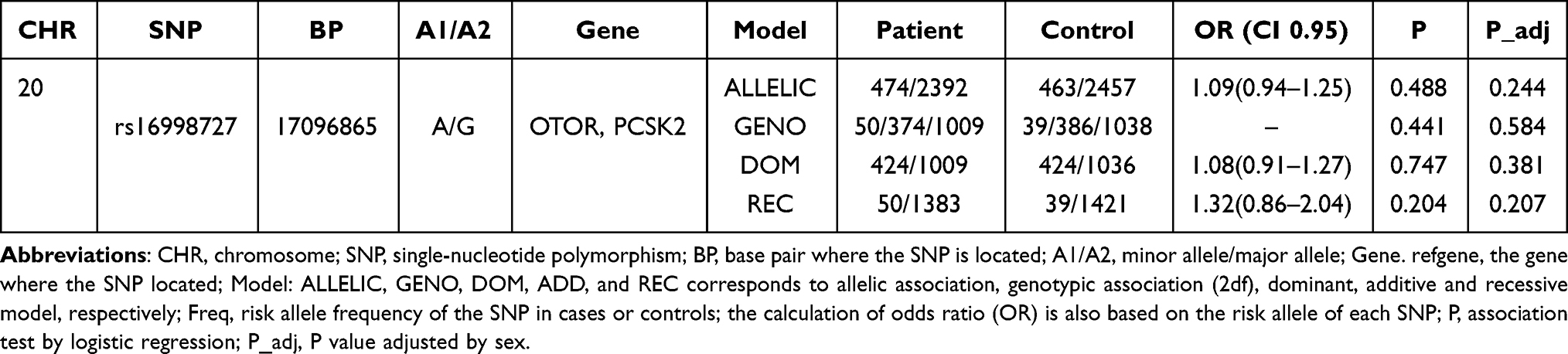 |
Table 2 Replication Results for rs16998727 A>G in a Southern Chinese Population of 1470 Cases and 1473 Controls |
Stratification Analysis
Considering that HSCR is divided into different subtypes, SNP may play an important role in HSCR subtypes. Therefore, we want to analyze further whether there is an association between rs16998727 (PCSK2 and OTOR) and HSCR subtypes (S-HSCR, L-HSCR and TCA). The results in Table 3 show that there is no significant difference between rs16998727 and S-HSCR (OR = 1.08, 95%IC = 0.93–1.27, P_adj = 0.3208), L-HSCR (OR = 1.07, 95%IC = 0.84–1.36, P_adj = 0.5958) and TCA (OR = 0.94, 95%IC = 0.61–1.47, P_adj = 0.8001).
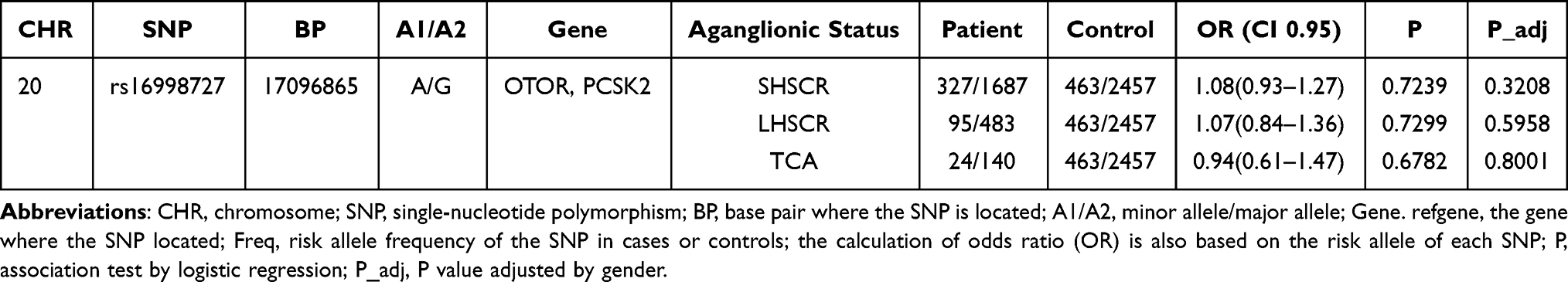 |
Table 3 Stratification Analysis for the Association Between OTOR and PCSK2 rs16998727 A>G and Hirschsprung Disease Susceptibility (by Subtype) |
Discussion
HSCR is a neonatal ENS disease with congenital loss of enteric ganglion cells. Currently, the only treatment option for HSCR is to resect the ganglionic bowel and reconstruct the normally innervated bowel.23 However, postoperative complications such as enterocolitis and intractable constipation often occur in children with HSCR.24 It is generally known that HSCR is related to genetic factors, but only a few mutant genes of HSCR have been found so far, and it is still necessary to continue to identify the unknown mutant genes of HSCR.
The rs16998727 SNP was found to be associated with HSCR in a study from Hong Kong Chinese.20 This SNP has not been replicated, so we replicated in a cohort of HSCR (n = 1470) in southern China. The closest genes to this SNP site are PCSK2 and OTOR. The above studies in Hong Kong population in China only show that PCSK2 rs16998727 is associated with HSCR. Additionally, the GTEx database searched for the association of potential functions of rs16998727 (PCSK2 and OTOR) genes and found that the eQTLs of PCSK2 rs16998727 were significantly different in colon tissue and nerves. Therefore, we next focused on the relationship between PCSK2 rs16998727 and HSCR.
PCSK enzymes are a family of nine related serine proteases, of which PCSK2 is abundant in endocrine and neuronal cells. There is a report of low birth weight and mild hypoglycemia in pcsk2-KO mice, in which part of this phenotype may be due to altered gastrointestinal physiology (GI) peptide response systems, primarily by altered bioactive peptide molecules produced from PCSK2 precursors in the gastrointestinal tract, delaying gastrointestinal motility.19 In addition, a study on the genome-wide association screening of sporadic amyotrophic lateral sclerosis (ALS) among American veterans showed that PCSK2 rs6080539 was significantly different from the occurrence of ALS.25 Another study found that PCSK2 was strongly positive in most pheochromocytomas and paraganglioma in midgut neuroendocrine tumors (NETs).17 Studies have shown that bioactive peptide molecules catalyzed by PCSK2 are closely related to gastrointestinal motility and neural development. The regulation of intestinal peristalsis in HSCR is controlled by a variety of bioactive peptides in central and intestinal neurons and peripheral endocrine cells.19
The results of this study were surprising, because we were not able to reproduce the same results as the original study, no correlation was observed between PCSK2 rs16998727 HSCR and in the southern population (Table 2). Besides, a variant may only have a slight impact on a specific type of HSCR. Therefore, we speculate that this may partially explain the ineffective effects of PCSK2 rs16998727. Each specific type of HSCR may have a specific genetic background. For further verification, we conducted a stratified analysis of all HSCR subtypes. The results showed that PCSK2 rs16998727 and all specific subtypes were not significant (Table 3). The above results are contrary to our hypothesis, the reason why the results were not replicated consistently may be due to the limitations of this study. Firstly, only one SNP was selected in this study. The influence of other PCSK2 SNPs has not been explored, and the functional variants responsible for the gene effects remain undiscovered. Secondly, HSCR is a disease that combines genetic factors, environmental factors and other factors, so it is possible that the effect of PCSK2 rs16998727 on HSCR is masked by other factors. Lastly, our sample size is relatively large, but our study population is limited to Han people in southern China. Further studies should focus on the relationship between genotypes-phenotypes in multiple regions and ethnicities. In conclusion, this study demonstrated no correlation between rs16998727 and HSCR.
Ethics Statement
This study was approved by the Institutional Review Board of Guangzhou Women and Children’s Medical Center (Ethical Approval Number: 2018052406). All procedures performed in this study involving human participants were by the Declaration of Helsinki (as revised in 2013).
Acknowledgments
We thank Lifeng Lu and Tuqun Hu for their help in collecting samples and medical histories.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by grants from the Medical Science and Technology Research Foundation of Guangdong Province of China (Grant NO. A2020076), Guangzhou’s science and technology innovation and development special project (Grant NO. 202102080511) and National Natural Science Foundation of China (Grant NO. 82070528).
Disclosure
The authors state that the study is published without any conflict of interest.
References
1. Butler Tjaden NE, Trainor PA. The developmental etiology and pathogenesis of Hirschsprung disease. Transl Res. 2013;162(1):1–15. doi:10.1016/j.trsl.2013.03.001
2. Heuckeroth RO. Hirschsprung disease - integrating basic science and clinical medicine to improve outcomes. Nat Rev Gastroenterol Hepatol. 2018;15(3):152–167. doi:10.1038/nrgastro.2017.149
3. Singh SJ, Croaker GD, Manglick P, et al. Hirschsprung’s disease: the Australian Paediatric Surveillance Unit’s experience. Pediatr Surg Int. 2003;19(4):247–250. doi:10.1007/s00383-002-0842-z
4. Spouge D, Baird PA. Hirschsprung disease in a large birth cohort. Teratology. 1985;32(2):171–177. doi:10.1002/tera.1420320204
5. Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45(1):1–14. doi:10.1136/jmg.2007.053959
6. Obermayr F, Hotta R, Enomoto H, Young HM. Development and developmental disorders of the enteric nervous system. Nat Rev Gastroenterol Hepatol. 2013;10(1):43–57. doi:10.1038/nrgastro.2012.234
7. Barlow A, de Graaff E, Pachnis V. Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron. 2003;40(5):905–916. doi:10.1016/S0896-6273(03)00730-X
8. Gianino S, Grider JR, Cresswell J, Enomoto H, Heuckeroth RO. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development. 2003;130(10):2187–2198. doi:10.1242/dev.00433
9. Sribudiani Y, Metzger M, Osinga J, et al. Variants in RET associated with Hirschsprung’s disease affect binding of transcription factors and gene expression. Gastroenterology. 2011;140(2):572–582 e572. doi:10.1053/j.gastro.2010.10.044
10. Di Zanni E, Adamo A, Belligni E, et al. Common PHOX2B poly-alanine contractions impair RET gene transcription, predisposing to Hirschsprung disease. Biochim Biophys Acta Mol Basis Dis. 2017;1863(7):1770–1777. doi:10.1016/j.bbadis.2017.04.017
11. Leon TY, Ngan ES, Poon HC, et al. Transcriptional regulation of RET by Nkx2-1, Phox2b, Sox10, and Pax3. J Pediatr Surg. 2009;44(10):1904–1912. doi:10.1016/j.jpedsurg.2008.11.055
12. Zhang Y, Xie X, Zeng J, et al. Association of NRG1 and AUTS2 genetic polymorphisms with Hirschsprung disease in a South Chinese population. J Cell Mol Med. 2018;22(4):2190–2199. doi:10.1111/jcmm.13498
13. Alves MM, Sribudiani Y, Brouwer RW, et al. Contribution of rare and common variants determine complex diseases-Hirschsprung disease as a model. Dev Biol. 2013;382(1):320–329. doi:10.1016/j.ydbio.2013.05.019
14. Seidah NG, Chretien M. Proprotein and prohormone convertases: a family of subtilases generating diverse bioactive polypeptides. Brain Res. 1999;848(1–2):45–62. doi:10.1016/S0006-8993(99)01909-5
15. Seidah NG, Mayer G, Zaid A, et al. The activation and physiological functions of the proprotein convertases. Int J Biochem Cell Biol. 2008;40(6–7):1111–1125. doi:10.1016/j.biocel.2008.01.030
16. Benjannet S, Rondeau N, Paquet L, et al. Comparative biosynthesis, covalent post-translational modifications and efficiency of prosegment cleavage of the prohormone convertases PC1 and PC2: glycosylation, sulphation and identification of the intracellular site of prosegment cleavage of PC1 and PC2. Biochem J. 1993;294(Pt 3)):735–743. doi:10.1042/bj2940735
17. Remes SM, Leijon H, Vesterinen T, et al. PCSK2 expression in neuroendocrine tumors points to a midgut, pulmonary, or pheochromocytoma-paraganglioma origin. APMIS. 2020;128(11):563–572. doi:10.1111/apm.13071
18. Seidah NG, Benjannet S, Hamelin J, et al. The subtilisin/kexin family of precursor convertases. Emphasis on PC1, PC2/7B2, POMC and the novel enzyme SKI-1. Ann N Y Acad Sci. 1999;885:57–74. doi:10.1111/j.1749-6632.1999.tb08665.x
19. Gagnon J, Mayne J, Chen A, et al. PCSK2-null mice exhibit delayed intestinal motility, reduced refeeding response and altered plasma levels of several regulatory peptides. Life Sci. 2011;88(5–6):212–217. doi:10.1016/j.lfs.2010.11.010
20. Tang S. Genetic dissection of Hirschsprung disease Unpublished Doctoral dissertation. The University of Hong Kong, China; 2009:67–76.
21. Liu Y, Lan C, Li B, et al. Associations of CYP2B6 genetic polymorphisms with Hirschsprung’s disease in a southern Chinese population. J Clin Lab Anal. 2021;35(12):e24074. doi:10.1002/jcla.24074
22. Xie X, He Q, Huang L, et al. Associations of SLC6A20 genetic polymorphisms with Hirschsprung’s disease in a Southern Chinese population. Biosci Rep. 2019;39:8. doi:10.1042/BSR20182290
23. Menezes M, Corbally M, Puri P. Long-term results of bowel function after treatment for Hirschsprung’s disease: a 29-year review. Pediatr Surg Int. 2006;22(12):987–990. doi:10.1007/s00383-006-1783-8
24. Baillie CT, Kenny SE, Rintala RJ, Booth JM, Lloyd DA. Long-term outcome and colonic motility after the Duhamel procedure for Hirschsprung’s disease. J Pediatr Surg. 1999;34(2):325–329. doi:10.1016/S0022-3468(99)90201-4
25. Kwee LC, Liu Y, Haynes C, et al. A high-density genome-wide association screen of sporadic ALS in US veterans. PLoS One. 2012;7(3):e32768. doi:10.1371/journal.pone.0032768
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.