")
Back to Journals » International Journal of General Medicine » Volume 14
Survival Analysis of Hepatosplenic T Cell Lymphoma: A Population-Based Study Using SEER
Authors Li Y, Chen K, Zuo C, Zeng R, He Y, Chen X, Xiao L , Zhou H
Received 29 August 2021
Accepted for publication 28 October 2021
Published 17 November 2021 Volume 2021:14 Pages 8399—8411
DOI https://doi.org/10.2147/IJGM.S335464
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Yajun Li,1 Kailin Chen,2 Chaohui Zuo,3 Ruolan Zeng,1 Yizi He,1 Xiaoyan Chen,4 Ling Xiao,5 Hui Zhou1
1Department of Lymphoma and Hematology, Hunan Cancer Hospital and The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, Hunan, 410013, People’s Republic of China; 2Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center of Cancer Medicine, Guangzhou, Guangdong, 510060, People’s Republic of China; 3Department of Gastroduodenal and Pancreatic Surgery, Translation Medicine Research Center of Liver Cancer, Laboratory of Digestive Oncology, Affiliated Cancer Hospital of Xiangya Medical School and Hunan Cancer Hospital, Central South University, Changsha, Hunan, 410013, People’s Republic of China; 4Department of Pathology, Affiliated Cancer Hospital of Xiangya Medical School & Hunan Cancer Hospital, Central South University, Changsha, Hunan, 410013, People’s Republic of China; 5Department of Histology and Embryology, School of Basic Medical Science, Central South University, Changsha, Hunan, 410013, People’s Republic of China
Correspondence: Hui Zhou
Department of Lymphoma and Hematology, Hunan Cancer Hospital and The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, 283 Tongzipo Road, Changsha, Hunan, 410013, People’s Republic of China
Fax +8673189762282
Email [email protected]
Chaohui Zuo
Department of Gastroduodenal and Pancreatic Surgery, Translation Medicine Research Center of Liver Cancer, Laboratory of Digestive Oncology, Affiliated Cancer Hospital of Xiangya Medical School & Hunan Cancer Hospital, Central South University, 283 Tongzipo Road, Changsha, Hunan, 410013, People’s Republic of China
Fax +8673189762142
Email [email protected]
Purpose: Hepatosplenic T cell lymphoma (HSTCL) is a rare tumor that lacks data to guide management decisions. To shed light on the nature and therapy of the entity, we conducted this study.
Patients and Methods: We retrospectively reviewed patients diagnosed with HSTCL between 1975 and 2016 in the Surveillance, Epidemiology, and End Results (SEER) database to analyze the clinical characteristics and survival outcome compared with PTCL-NOS and ALK+ ALCL.
Results: A total of 123 HSTCLs were included in the analysis. Most patients were aged ≤ 60 years (81.3%) and had a male predominance (69.1%). Organs with lymphoma infiltration of HSTCL were more common in the spleen (98.4%). The 1-year, 3-year, and 5-year overall survival (OS) rates in the entire HSTCL cohort were 56.9% (95% CI, 47.5– 66.3%), 37.6% (95% CI, 28.0– 47.2%), and 31.6.0% (95% CI, 22.2– 41.0%), respectively. The overall survival (OS) of HSTCL patients was similar to that of PTCL-NOS patients (P = 0.128) but worse than that of patients with ALK+ ALCL (P < 0.001). The disease-specific survival (DSS) of HSTCL patients was worse than that of PTCL-NOS and ALK+ ALCL patients (P < 0.05). The same tendency was found in the matched data set. Cox regression analyses indicated that the use of chemotherapy combined with topical treatment may improve the survival of patients with HSTCL.
Conclusion: A higher proportion of young patients and a strong male predominance were found in HSTCL. Chemotherapy combined with topical treatment may be an optional regimen. Further studies are needed to intensify efforts in dealing with this rare but unfavorable disease.
Keywords: hepatosplenic T cell lymphoma, survival, SEER, treatment
Introduction
Hepatosplenic T-cell lymphoma (HSTCL) is a rare and aggressive type of extranodal peripheral T-cell lymphoma. It was first described using the designation erythrophagocytic T γ lymphoma in 1981 by Kadin et al, and they reported two adults presenting with hepatosplenomegaly and minimal lymphadenopathy.1 Subsequently, the term hepatosplenic T-cell lymphoma was first used in 1990 in two malignant lymphoma patients with hepatosplenic disease and by the sinusal/sinusoidal tropism and the gamma delta phenotype of the malignant cells.2 The alpha-beta T-cell receptor phenotype is now considered an immunophenotypic variant of the same disease entity in the World Health Organization (WHO) classification.3
HSTCL represents 1.4% of T cell lymphoma diagnosed worldwide.4 A higher incidence rate was reported in the United States population according to the T Cell Project, which accounted for 2% of T cell lymphomas.5 The malignant cells of HSTCL showed the presence of small- to medium-sized mature T cells, which typically involved the sinusoids of the spleen and liver.6 The sinusoids of bone marrow were also reported to be involved in most patients at the time of diagnosis.7 As a consequence, patients commonly present with splenomegaly and/or hepatomegaly. Cytopenias often could be observed. HSTCL mainly affected young adults and had a strong male predominance. It has a rapidly progressive course and no standard therapy for treatment. The efficacy of standard anthracycline-containing chemotherapy may have variable responses with high relapse rates and short median survival in most cases.5,8,9 A more intensive regimen was reported to have a better complete response.8 Moreover, hematopoietic stem cell transplantation was found to be potentially curative.10 Splenectomy may sometimes be a useful therapeutic option.11 The role of radiotherapy still remains unclear. The reported median overall survival (OS) was 6–28.3 months, regardless of the treatment modalities.5,7,12 There are also case reports demonstrating HSCTL patients with hemophagocytic lymphohistiocytosis, which is equally as severe and associated with a rapid clinical course.7,11,13
However, most of these data are obtained from case reports or retrospective studies with very small samples in a single institution.
Due to the limited data and the absence of large clinical studies regarding the clinical course of HSTCL, we identified the clinical characteristics, OS and disease-specific survival (DSS) of patients with HSTCL using the Surveillance, Epidemiology, and End Results (SEER) database.
Patients and Methods
Data were extracted from the SEER 18 (1975–2016) registry database of the US National Cancer Institute using SEER*STAT software version 8.3.8 (https://seer.cancer.gov/, accession number: 15756-Nov2019). Patients with histologically confirmed malignant hepatosplenic T cell lymphoma, peripheral T-cell lymphoma not otherwise specified (PTCL-NOS), and ALK-positive anaplastic large cell lymphoma (ALK+ ALCL), were identified using International Classification of Diseases for Oncology edition 3 histology codes 9716/3, 9702/3 and 9614/3, respectively. In addition, hepatosplenic T cell lymphoma was also identified using organs or systems with lymphoma infiltration and morphology labeling (C22.0 liver and C42.2 spleen).
Hepatosplenic T cell lymphoma cases were matched 1:1 with PTCL-NOS, ALK+ ALCL cases using case-control matching to compare survival rates. The characteristics were balanced except organs or systems with lymphoma infiltration after application of this case control method.
Patients with only death certificates or autopsy records or without complete survival rate data and follow-up information were excluded from the study. The following variables were extracted from the SEER database: sex, age, race, marital status, Ann Arbor stage, diagnosis year, and treatment. We classified race as white, black, and others. Marital status was divided into married, unmarried, or unknown. Treatment was divided into chemotherapy alone, surgery alone, combination therapy of surgery, radiotherapy and chemotherapy, and combination therapy of radiotherapy and chemotherapy.
Five patients diagnosed with HSTL at Hunan Cancer Hospital were also included in this study. All diagnoses were confirmed by examination of pathologic characteristics and immunohistochemical staining. The detailed clinicopathological information of the enrolled patients was retrospectively extracted from the medical records. The study was approved by the Ethics Committee at Hunan Cancer Hospital. Informed consent was provided by all patients before the study.
Descriptive statistics were used to summarize clinical data. Medians and ranges were used to describe continuous variables. The chi-square test or t-test was used for univariate analysis. Multivariate Cox regression analysis was performed to identify the impact factors. In the Cox regression, the following variables were included: sex, age, race, marital status, Ann Arbor stage and treatment.
Disease-specific survival (DSS) was estimated as the time of diagnosis to the time of death from lymphoma; other deaths were censored at the date of death. OS was determined from the date of diagnosis to the date of either death or the end of follow-up. The survival curves were calculated by the Kaplan–Meier method and compared with the Log rank test. We used a Cox regression model to estimate hazard ratios (HRs) and 95% confidence intervals (95% CIs) for variables. Statistical significance was defined as P < 0.05 (two tailed). Statistical analyses were performed using IBM SPSS Statistics version 23.0 software (IBM Co, New York, Armonk).
Results
Characteristics of Patients with Hepatosplenic T Cell Lymphoma
A total of 123 HSTCLs were included in the study from the SEER database. The baseline characteristics of these patients are shown in Table 1. The median age of all patients at diagnosis was 40 years (range, 6–83), and 81.3% of patients were aged younger than 60 years. Hepatosplenic T cell lymphoma had a male predominance. The majority of patients were diagnosed with Ann Arbor stage III–IV disease. Organs or systems with lymphoma infiltration of HSTCL were more common in the spleen (121/123, 98.4%). In total, only five patients (5/123, 0.4%) underwent surgery of organs or systems with lymphoma infiltration. The majority of patients (102/123, 82.9%) received chemotherapy alone, and (16/123, 13.0%) received radiotherapy alone. Sixteen patients (16/123, 13%) received chemotherapy combined with topical treatment (surgery [n = 5] or radiotherapy [n = 11]).
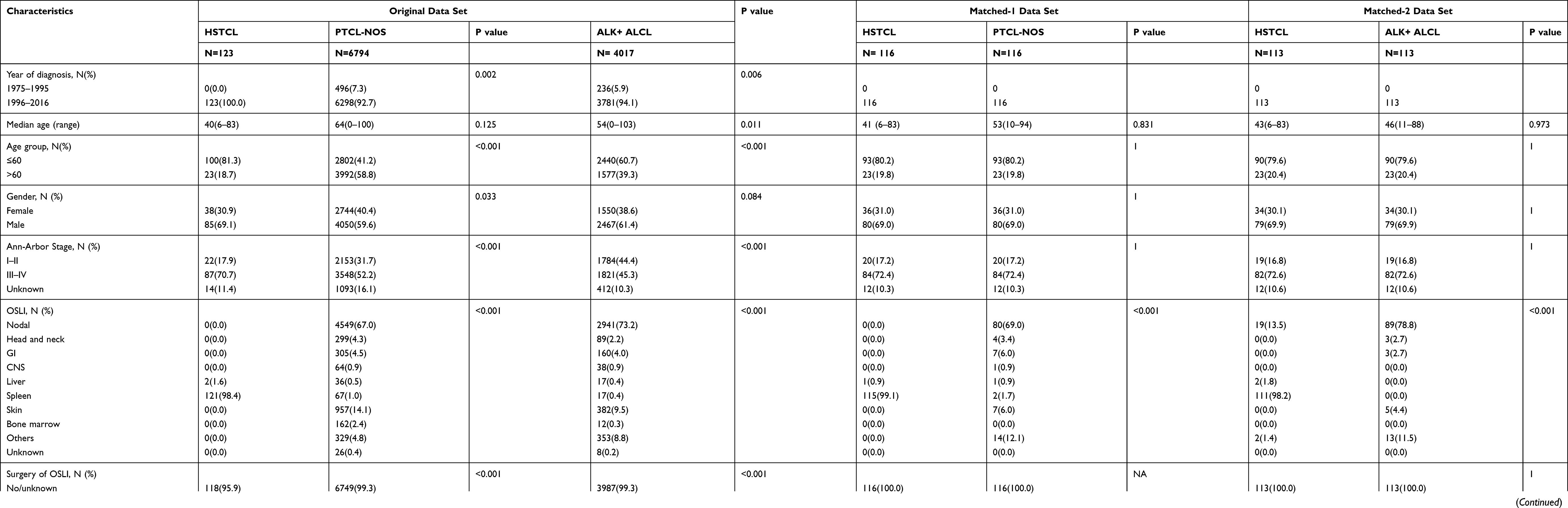 |
Table 1 Baseline Characteristics of Patients with Hepatosplenic T Cell Lymphoma from the SEER database |
Among the five patients from China, the majority were younger and male, and organs with lymphoma infiltration were more common in the spleen, which was consistent with the findings in the SEER database. In addition, we also found that patients were more likely to have B symptoms, bone marrow involvement, cytopenia, abnormal liver transaminase levels, and elevated LDH and alkaline phosphatase (ALP) levels. The clinical characteristics of the five patients with HSTCL are shown in Table 2. Only two patients received anthracycline-based chemotherapy, and in total, the response rate to first-line therapy was not high. One patient had hemophagocytic cells in the bone marrow at diagnosis. The results of the main immunophenotypic studies are summarized in Table 3. TCR expression was determined in one patient, and TCRγ was positive. CD3 was positive in 4/5 cases. All cases tested were positive for CD2 (n = 2), GrB (n =2), and TIA1 (n =4). CD20 (n = 5), Epstein-Barr virus–encoded small RNA (n = 5), CD30 (n = 3), PAX-5 (n = 4), CD79a (n = 2), ALK (n = 3) and TdT (n = 2) were negative in all cases tested. The details of staging, treatment, efficacy, and survival outcome are shown in Table 4. None of the 5 patients received autologous hematopoietic stem cell transplantation.
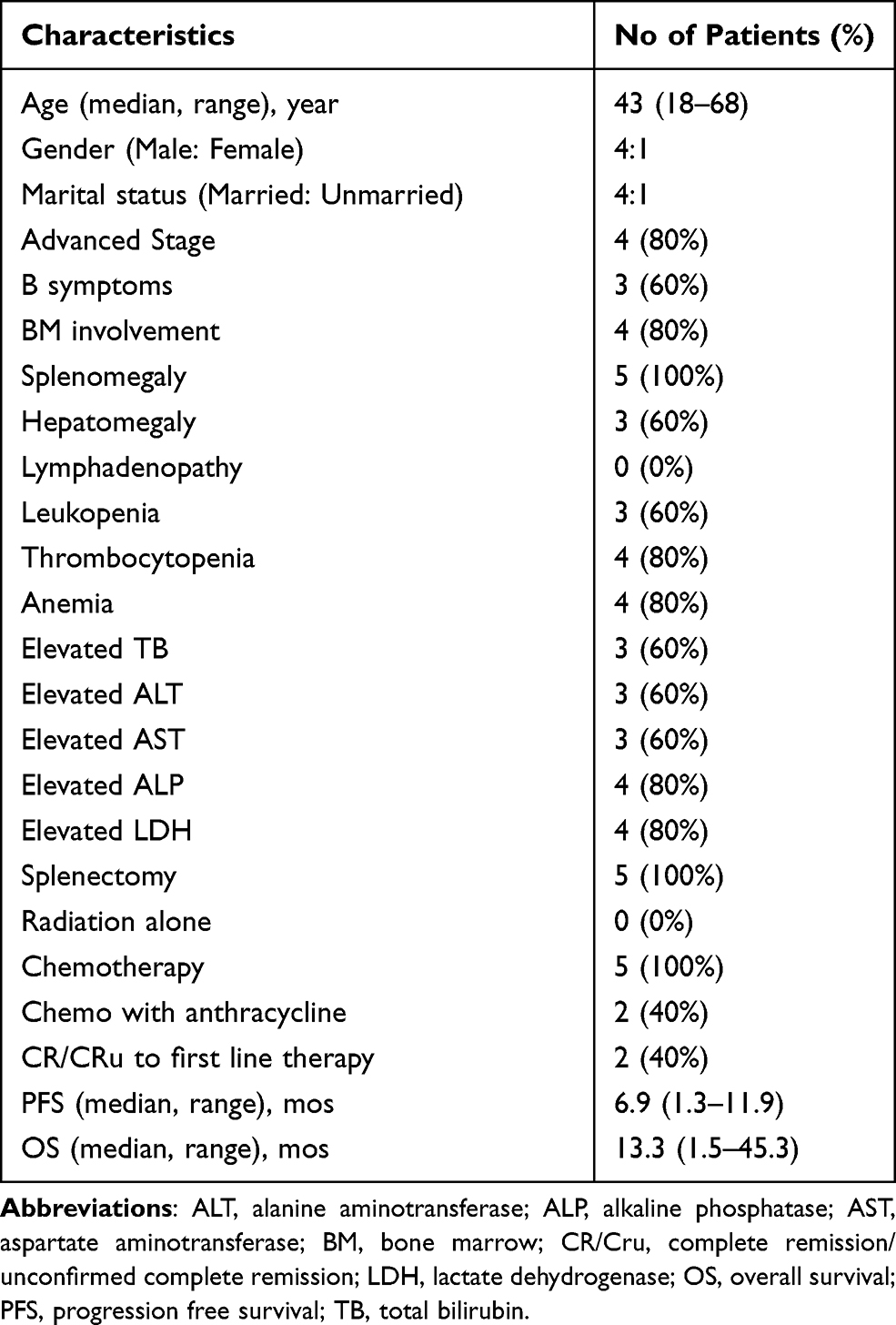 |
Table 2 Clinical Characteristics of Five Chinese Patients with Hepatosplenic T Cell Lymphoma |
 |
Table 3 The Main Immunophenotypic Features of 5 Chinese Cases with Hepatosplenic T Cell Lymphoma |
 |
Table 4 The Staging, Treatment, Outcome, and Survival of 5 Chinese Cases with Hepatosplenic T Cell Lymphoma |
Compared with PTCL-NOS, patients with HSTCL of SEER tended to be younger (81.3% vs 41.2%, P < 0.001), male (69.1% vs 59.6%, P = 0.033), unmarried (56.9% vs 38.6%, P < 0.001) and more common among Blacks (22.7% vs 15.8%, P = 0.005). These can also be observed when compared with patients with ALK+ ALCL, except for sex and marital status. In addition, more patients with HSTCL were diagnosed at an advanced stage than patients with PTCL-NOS (70.7% vs 52.5%, P < 0.001) or ALK+ ALCL (70.7% vs 45.3%, P < 0.001). Presentation in spleen was also more frequent than PTCL-NOS (98.4% vs 1.0%, P < 0.001) or ALK+ ALCL (98.4% vs 0.4%, P < 0.001). The characteristics were well balanced between the patients with HSTCL or with PTCL-NOS except the organs or systems with lymphoma infiltration using the case-control matching method (Table 1, matched-1 dataset). HSTCL cases and ALK+ ALCL cases were also well matched (Table 1, matched-2 dataset).
Overall Survival
After a median follow-up of 9 months (range, 0–131), 76 patients with HSTCL had died; among them, 61/76 (80.3%) had stage III–IV disease at diagnosis. The 1-year, 3-year, and 5-year OS rates in the entire HSTCL cohort were 56.9% (95% CI, 47.5–66.3%), 37.6% (95% CI, 28.0–47.2%), and 31.6.0% (95% CI, 22.2–41.0%), respectively.
The 1-year, 3-year, and 5-year OS rates in all patients with PTCL-NOS were 54.1% (95% CI, 52.9–55.3%), 38.7% (95% CI, 37.5–39.9%), and 32.6% (95% CI, 31.4–33.8%), respectively. The 1-year, 3-year, and 5-year OS rates in all ALK+ ALCL patients were 67.5% (95% CI, 66.1–68.9%), 57.5% (95% CI, 55.9–59.1%), and 52.9% (95% CI, 51.3–54.5%), respectively.
The overall survival of HSTCL patients was similar to that of PTCL-NOS patients (P = 0.128), which was also observed in the matched dataset (P = 0.103). Moreover, the overall survival was better in the ALK+ ALCL cohorts than in the HSTCL cohort (P < 0.001). The same tendency was found in the matched dataset (P =0.001). These results are shown in Figure 1.
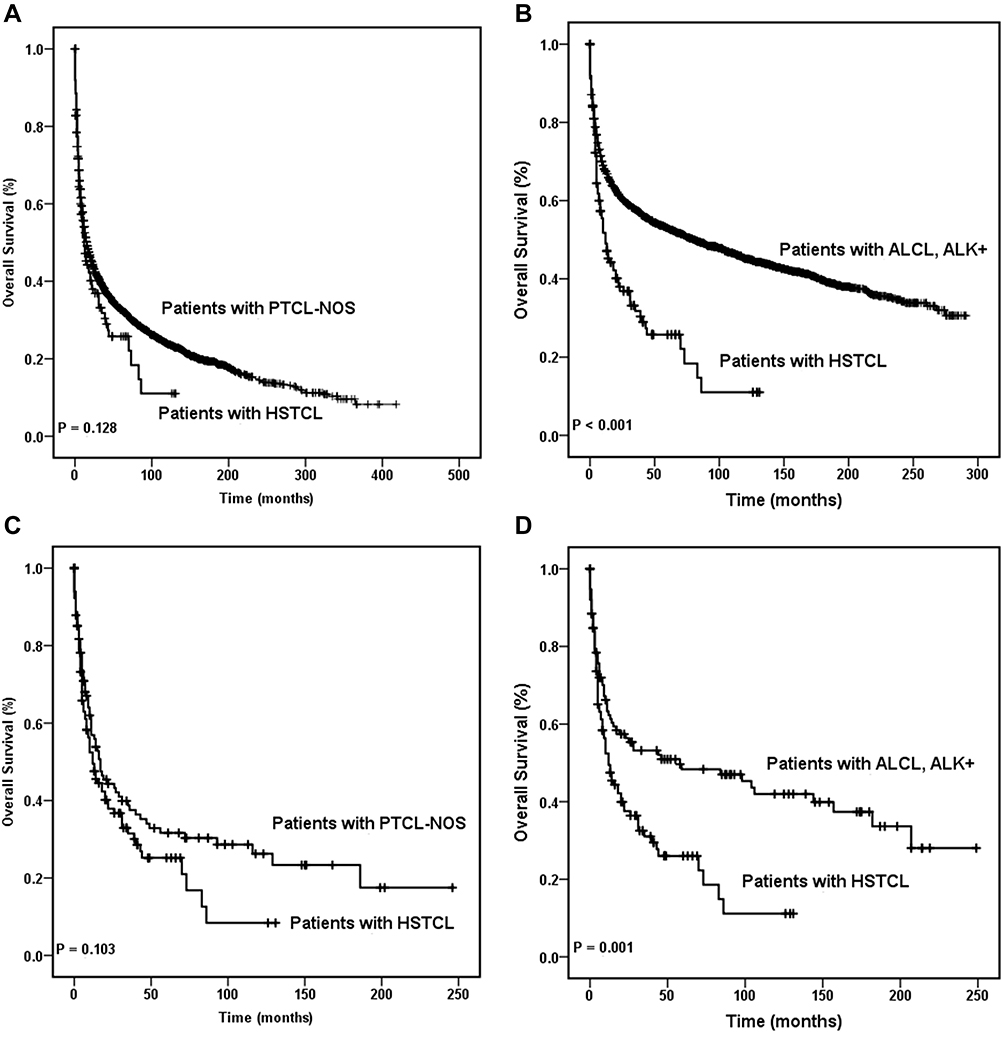 |
Figure 1 Overall survival curves of patients with hepatosplenic T cell lymphoma. OS of patients with HSTCL were compared with that in patients with PTCL-NOS among the entire cohort (A) and the same comparison also made between the entire HSTCL cohort and the entire ALK+ ALCL cohort (B). OS of patients with HSTCL were compared with that of patients with PTCL-NOS among the match cohort (C), and OS of patients with HSTCL were compared with that in patients with ALK+ ALCL among the matched cohort (D). Abbreviations: ALCL, anaplastic large cell lymphoma; ALK, anaplastic lymphoma kinase; HSTCL, hepatosplenic T cell lymphoma; OS, overall survival; PTCL-NOS, peripheral T cell lymphoma-not otherwise specified. |
In the entire HSTCL cohort, age, sex, Ann Arbor stage, race, marital status, and treatment demonstrated no difference in OS. Among patients with primary spleen and liver HSTCL, the Kaplan–Meier curve showed that the treatment regimen affected overall survival (Figures 2A and B). The 1-year OS rates were 75.0% (95% CI, 32.5–100.0%), 63.6% (95% CI, 35.2–92.0%), 51.9% (95% CI, 29.0–74.8%), 49.2% (95% CI, 38.4–60.0%), and 0% for patients who received surgery, radiotherapy and chemotherapy combination therapy, radiotherapy and chemotherapy combination therapy, no or unknown therapy, chemotherapy alone or surgery alone, respectively (P = 0.006). Cox regression analyses among HSTCL indicated that surgery, radiotherapy and chemotherapy combination therapy were significantly associated with better OS (P = 0.001). However, age, sex, Ann Arbor stage, race and marital status were not significantly associated with OS (Table 5).
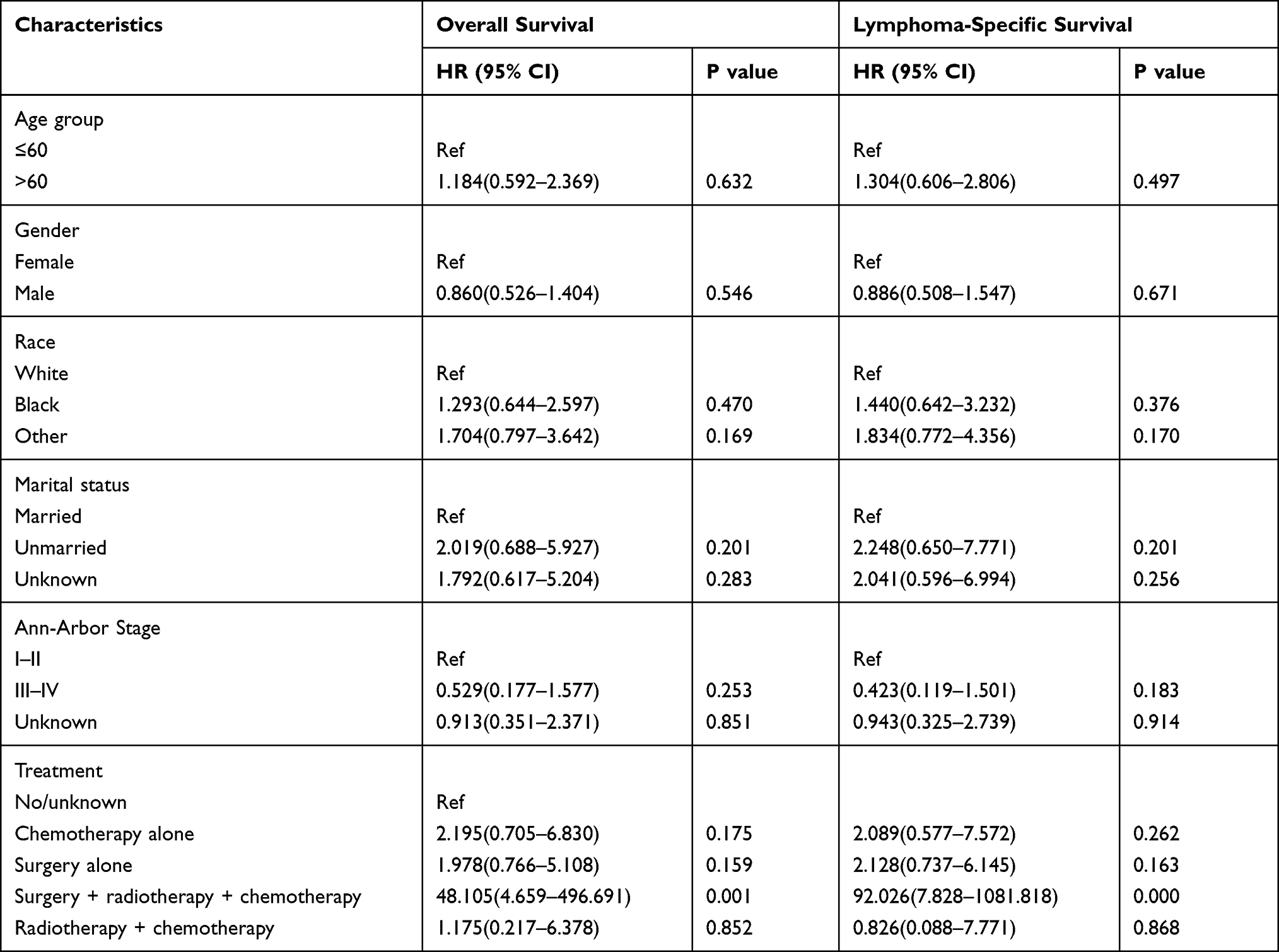 |
Table 5 Cox Regression Analysis for Survival of Hepatosplenic T Cell Lymphoma Patients |
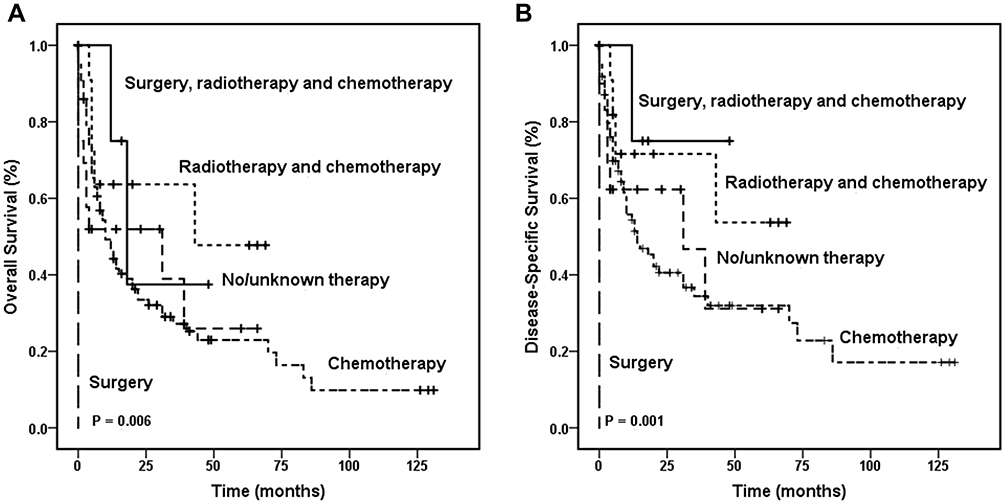 |
Figure 2 Kaplan–Meier survival curves according to the different treatment. Overall survival curves (A) and disease-specific survival curves (B) were made in the patients with primary spleen and liver HSTCL, respectively. Abbreviation: HSTCL, hepatosplenic T cell lymphoma. |
Disease-Specific Survival
In total, 66/83 (79.5%) patients with HSTCL died of causes related to HSTCL, whereas 17/83 (20.5%) died of causes not related to lymphoma. The 5-year DSS rates in all patients with HSTCL, PTCL-NOS and ALK+ ALCL were 46.4% (95% CI, 35.4–57.4%), 49.8% (95% CI, 48.4–51.2%), and 66.3% (95% CI, 64.7–67.9%), respectively.
A significant difference in DSS was detected between patients with HSTCL and those who were diagnosed with PTCL-NOS (Figure 3A, P = 0.002) and between patients with HSTCL and patients with ALK+ ALCL (Figure 3B, P < 0.001) among the entire cohort. Concordantly, this tendency of DSS was also found in the matched dataset. Patients with PTCL-NOS showed better DSS than those who were diagnosed with HSTCL (Figure 3C, P = 0.033), and patients with ALK+ ALCL showed better DSS than those who were diagnosed with HSTCL (Figure 3D, P = 0.016). Kaplan–Meier curves showed that the treatment regimen also affected DSS (P = 0.001). Cox regression analyses demonstrated that patients who received surgery, radiotherapy and chemotherapy combination therapy had better DSS than those who received other treatments (P < 0.001). Age, sex, Ann Arbor stage, race, and marital status were not significantly associated with DSS.
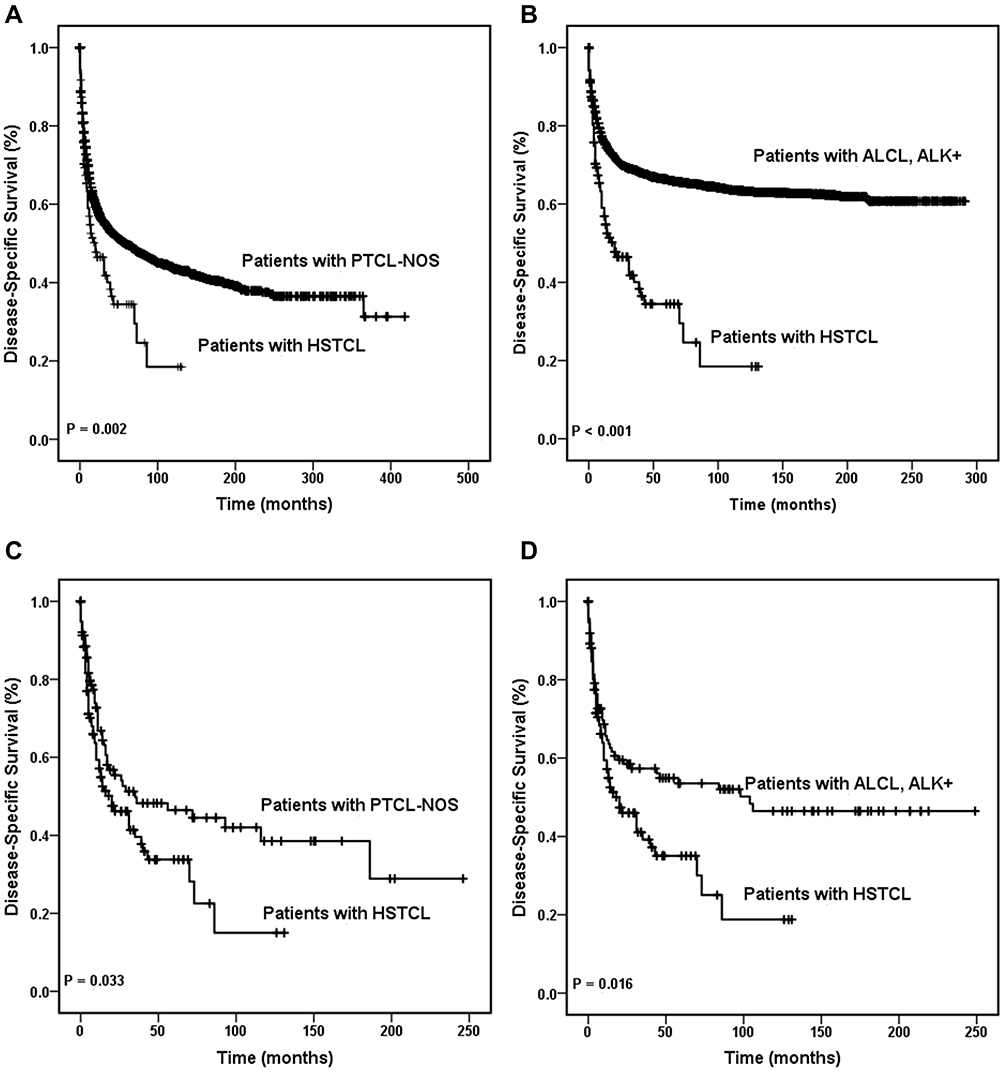 |
Figure 3 Disease-specific survival curves of patients with hepatosplenic T cell lymphoma. DSS of patients with HSTCL were compared with that in patients with PTCL-NOS among the entire cohort (A) and the same comparison also made between the entire HSTCL cohort and the entire ALK+ ALCL cohort (B). DSS of patients with HSTCL were compared with that of patients with PTCL-NOS among the match cohort (C), and DSS of patients with HSTCL were compared with that in patients with ALK+ ALCL among the matched cohort (D). Abbreviations: ALCL, anaplastic large cell lymphoma; ALK, anaplastic lymphoma kinase; HSTCL, hepatosplenic T cell lymphoma; DSS, disease-specific survival; PTCL-NOS, peripheral T cell lymphoma-not otherwise specified. |
Discussion
Since the name hepatosplenic T-cell lymphoma was used in 1990, few case series of HSTCL have been reported.5,7–10,12,14 The largest reported cohort of 31 patients was from a prospective trial T Cell Project.5 However, to the best of our knowledge, this is the first population-based study of HSTCL patients using the SEER database.
The etiology of the majority of patients with HSTCL is unclear. Most of HSTCL were de novo. Approximately 20% of HSTCL was found in immunocompromised patients, including patients with solid organ transplant, chronic steroid use, some types of autoimmune disorders, and primary hematologic malignancies.15 In addition, HSTCL was reported to be associated with inflammatory bowel disease such as Crohn’s disease, which is thought to have contributed to 10% of HSTCL patients.15 In our study, only one patient had allergic rhinitis among the five patients with a medical history from the medical records at Hunan Cancer Hospital. Several studies have also suggested that some drugs, such as tumor necrosis factor alpha (TNF alpha) inhibitors, may increase the risk of HSTCL,7,16 however, it was controversial.16–18 These results indicated that the impairment of the immune system and chronic antigenic stimulation may play a role in the genesis of HSTCL. HSTCL is not associated with Epstein-Barr virus (EBV), and EBER (EBV-encoded small RNA) is usually negative,11 which is consistent with our findings.
Our study showed that patients with HSTCL in both the SEER and Hunan cohorts had a higher proportion of young patients and a strong male predominance, which is in concordance with prior studies.2,7,9 The proportion of spleen invasion in HSTCL is higher than that in ALK+ ALCL and PTCL-NOS. Primary liver involvement is rare. In most reported cases, bone marrow involvement was common. Among the five patients from China, 80% had bone marrow involvement, and this ratio was also very high. It is worth noting that one patient aged 18 was diagnosed with hemophagocytic syndrome at baseline but recovered after the first cycle of chemotherapy. The study by showed that hemophagocytic syndrome is uncommon in patients with HSTCL.7 However, it may be a major cause of morbidity in HSTL.
As the pathologic data of HSTL from SEER are unavailable, it is impossible to analyze the correlation between the pathological parameters and prognosis in the SEER cohort. The main immunophenotypic features of 5 Chinese patients with HSTCL are in concordance with those of patients from prior studies.5,7,11 It is worth noting that all but one patient had low expression of Ki-67 (<70%). The expression of Ki-67 has been widely used as an index to evaluate the proliferative activity of lymphoma. High Ki-67 expression is considered to be a poor prognostic factor in many types of lymphoma, such as mantle cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, and peripheral T-cell lymphomas.19–24 However, its prognostic value for HSTCL remains unclear. Among the 5 Chinese cases, the survival time of Patient 3 with a high expression of Ki-67 (80%) was only 1.3 months, which was clearly shorter than the survival time of the other 4 patients (31.6 months, 45.3 months, 12.5 months, 13.3 months, respectively). This suggests that Ki-67 may be a very effective prognostic indicator for HSTCT, which needs to be confirmed by more studies with larger sample sizes. Another notable pathological parameter is CD56, which is positive in the majority of HSTCL cases.11 All but one patient had negative expression of CD56 (Patient 1) in our Hunan cohort, which is in concordance with prior studies.8,11 The prognostic value of CD56 expression for lymphoma is still contradictory and inconclusive. CD56 expression is considered an indicator of poor prognosis in patients with acute myeloid leukemia, anaplastic large cell lymphoma and diffuse large B-cell lymphoma.25–27 However, Wang et al did show that CD56-negative extranodal NK/T cell lymphoma had significantly inferior survival outcomes.28 The CD56-negative patient (Patient 1) in our study was alive for 31.6 months until the last follow-up. It appears that the correlation between CD56 expression and outcomes in patients with HSCTL should be clarified by investigating a larger number of cases in the future.
The majority of patients with HSTCL in SEER received chemotherapy. Unfortunately, treatment of HSTCL is not successful using chemotherapy alone. The median follow-up of patients who received chemotherapy was 9 (0–131) months. The 1-year and 3-year overall survival rates of HSTCL patients who received chemotherapy were 52.9% and 31.2%, respectively, which were not significantly different from those of patients who did not receive chemotherapy. The combination of surgery, chemotherapy and radiotherapy seemed to be better. Currently, no standard therapy is recommended. In SEER patients, the regimens of chemotherapy could be split into with or without anthracycline. Chemotherapy regimens without anthracycline were mainly based on pegaspase and gemcitabine. The chemotherapy regimen with anthracycline was CHOP-like in most cases. The median survival of patients who received anthracycline-based chemotherapy seemed to be longer than that of patients without anthracycline-based chemotherapy. However, there was no difference in survival between these two types of regimens (median OS: 13.2 months vs 10.3 months, P = 0.474). The efficacy of anthracycline-containing chemotherapy was also previously reported to be disappointing.5,8,9 Patients who received these chemotherapy regimens survived for less than 9 months.29 More intensive regimens, such as HyperCIDDDoxil (fractionated cyclophosphamide, liposomal doxorubicin, vincristine and dexamethasone) alternating with methotrexate and high-dose cytarabine, tended to perform better. The median OS was 11 months.8 Several studies have suggested that both autologous (autoSCT) and allogeneic (allo-SCT) transplants may be a potentially curative treatment option. A systematic review of 44 patients who received allo-SCT demonstrated that the estimated 3-year OS for transplants was 56%.10 Moreover, allo-SCT tended to have a better result than autoSCT.11,30 However, the nonrelapse mortality rate of allo-SCT was as high as 68%.10 The data mentioned above are limited to small retrospective studies and case series, and further multicenter studies with larger samples are needed to quantify this benefit.
It is worth noting that the 5-year OS rate is 31.6% for HSTCL from SEER, which is much better than that in a previously published study (5-year OS for nontransplanted patients <10%; Pro et al, Blood 2020).11 The possible reason for the difference in the 5-year OS rate between the SEER cohort and patients who of Pro et al are as follows: a) the sample size of the study from Pro was small (only 18 patients); however, the SEER cohort included 123 cases. b) In the study of Pro et al, up to 95% of patients with stage 3 or stage 4 disease, while only 70.7% of the patients in the SEER cohort were staged advanced. c) The enrollment time of patients in the study of Pro et al was from 1990 to 2002, while the enrollment time of patients in the SEER database was from 1996 to 2016. This makes patients from SEER more likely to use new drugs (such as brentuximab vedotin, romidepsin, pralatrexate, etc.). d) The final reason is that some patients in the SEER database may undergo hematopoietic stem cell transplantation, but we cannot obtain the specific proportion of transplanted patients because SEER does not provide publicly available data.
The prognosis of patients with HSTCL is usually poor. We found that patients with HSTCL had similar overall survival compared with PTCL-NOS patients but significantly worse overall survival than ALK+ ALCL patients. T-cell lymphoma is a group of heterogeneous malignancies. Advances in gene expression profiling, next-generation sequencing and other genomics studies provide the chance to have a better understanding of the pathogenetic mechanisms underlying the diverse biology of HSTCL, which helps the differential diagnosis from other types of lymphoma and suggests potential targets for therapy, prognosis factors and further improvement of diagnostic criteria.
The source of PTCL cells is associated with prognosis. The vast majority of PTCLs arise from alpha-beta T cells, while cases of HSTCL are more frequently composed of gamma delta T-cells and rare of the alpha-beta phenotype. Gamma–delta T cells are important to mucosal immunity.15 Previous studies have demonstrated that the alpha-beta phenotype showed a higher incidence in women.12,31 Patients with HSTCL expressing the alpha-beta T-cell receptor had a poorer prognosis.7 One of the five patients from the Hunan cohort detected TCR expression, which suggested that TCRγ was positive. He received chemotherapy without anthracycline, and the survival time was 13.3 months. We found that chemotherapy combined with topical treatment (surgery and radiotherapy) was significantly associated with better OS among patients with HSTCL.
There are some limitations in our study. First, the SEER database does not provide detailed information about chemotherapy regimens, making it impossible to conduct subgroup analysis to compare the efficacy differences of different chemotherapy regimens. Second, the time span is relatively large, which may increase the heterogeneity of the treatment of patients. Third, the lack of biomarker information of HSTCL in the SEER database makes prognostic analysis related to tumor markers impossible. Finally, selection bias was inevitable in this retrospective study.
Conclusion
In conclusion, this is the first population-based study of patients with HSTCL using the SEER database. We demonstrated a higher proportion of young patients with HSTCL and a strong male predominance. The organs or systems with lymphoma infiltration of HSTCL were more common in spleen than ALK+ ALCL and PTCL-NOS. Patients with HSTCL had similar overall survival compared with PTCL-NOS cases, but significantly worse than ALK+ ALCL. In addition, we identified that chemotherapy combined with topical treatment (surgery and radiotherapy) was an independent impact factor among patients with HSTCL. Broader multi-institutional collaborations are needed to intensify efforts in dealing with this rare but unfavorable disease.
Ethical Disclosure
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations.
Acknowledgments
The authors thanks to the SEER for allowing us to obtain the original data.
Funding
This work is supported by Hunan Provincial Natural Science Foundation of China (No. 2021JJ30425), Changsha Municipal Natural Science Foundation (No. kq2014206), the grants from Natural Science Foundation of Hunan Province National Health Commission (No. 20201659), and the grants from the Hunan Provincial Science and Technology Department (No. 2016JJ3083, No. 2017SK2130).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Kadin ME, Kamoun M, Lamberg J. Erythrophagocytic T gamma lymphoma: a clinicopathologic entity resembling malignant histiocytosis. N Engl J Med. 1981;304(11):648–653. doi:10.1056/NEJM198103123041106
2. Farcet JP, Gaulard P, Marolleau JP, et al. Hepatosplenic T-cell lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma delta. Blood. 1990;75(11):2213–2219. doi:10.1182/blood.V75.11.2213.2213
3. Jaffe ESHN, Stein H, Vardiman J. World Health Organization Classification of Tumours/Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2001.
4. Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–4130.
5. Foss FM, Horwitz SM, Civallero M, et al. Incidence and outcomes of rare T cell lymphomas from the T cell project: hepatosplenic, enteropathy associated and peripheral gamma delta T cell lymphomas. Am J Hematol. 2020;95(2):151–155. doi:10.1002/ajh.25674
6. Yabe M, Medeiros LJ, Wang SA, et al. Distinguishing between hepatosplenic T-cell lymphoma and γδ T-cell large granular lymphocytic leukemia: a clinicopathologic, immunophenotypic, and molecular analysis. Am J Surg Pathol. 2017;41(1):82–93. doi:10.1097/PAS.0000000000000743
7. Yabe M, Medeiros LJ, Tang G, et al. Prognostic factors of hepatosplenic T-cell lymphoma: clinicopathologic study of 28 cases. Am J Surg Pathol. 2016;40(5):676–688. doi:10.1097/PAS.0000000000000614
8. Falchook GS, Vega F, Dang NH, et al. Hepatosplenic gamma-delta T-cell lymphoma: clinicopathological features and treatment. Ann Oncol. 2009;20(6):1080–1085. doi:10.1093/annonc/mdn751
9. Belhadj K, Reyes F, Farcet JP, et al. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood. 2003;102(13):4261–4269. doi:10.1182/blood-2003-05-1675
10. Rashidi A, Cashen AF. Outcomes of allogeneic stem cell transplantation in hepatosplenic T-cell lymphoma. Blood Cancer J. 2015;5(6):e318. doi:10.1038/bcj.2015.43
11. Pro B, Allen PB, Behdad A. Hepatosplenic T-cell lymphoma: a rare but challenging entity. Blood. 2020;136(18):2018–2026. doi:10.1182/blood.2019004118
12. Macon WR, Levy NB, Kurtin PJ, et al. Hepatosplenic alphabeta T-cell lymphomas: a report of 14 cases and comparison with hepatosplenic gammadelta T-cell lymphomas. Am J Surg Pathol. 2001;25(3):285–296. doi:10.1097/00000478-200103000-00002
13. Paes VR, de Lima PP, Siqueira SAC. Hemophagocytic lymphohistiocytosis associated with hepatosplenic T-cell lymphoma: case report. Autops Case Rep. 2014;4(4):19–24. doi:10.4322/acr.2014.035
14. Voss MH, Lunning MA, Maragulia JC, et al. Intensive induction chemotherapy followed by early high-dose therapy and hematopoietic stem cell transplantation results in improved outcome for patients with hepatosplenic T-cell lymphoma: a single institution experience. Clin Lymphoma Myeloma Leuk. 2013;13(1):8–14. doi:10.1016/j.clml.2012.09.002
15. Thai A, Prindiville T. Hepatosplenic T-cell lymphoma and inflammatory bowel disease. J Crohns Colitis. 2010;4(5):511–522. doi:10.1016/j.crohns.2010.05.006
16. Yabe M, Medeiros LJ, Daneshbod Y, et al. Hepatosplenic T-cell lymphoma arising in patients with immunodysregulatory disorders: a study of 7 patients who did not receive tumor necrosis factor-α inhibitor therapy and literature review. Ann Diagn Pathol. 2017;26:16–22. doi:10.1016/j.anndiagpath.2016.10.005
17. Montgomery M, van Santen MM, Biemond BJ, Diamond RH, Pals ST. Hepatosplenic T-cell lymphoma: a Population-Based Study assessing incidence and association with immune-mediated disease. Gastroenterol Hepatol. 2015;11(3):160–163.
18. Subramaniam K, Yeung D, Grimpen F, et al. Hepatosplenic T-cell lymphoma, immunosuppressive agents and biologicals: what are the risks? Intern Med J. 2014;44(3):287–290. doi:10.1111/imj.12363
19. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: results from randomized trials of the European mantle cell lymphoma network. J Clin Oncol. 2016;34(12):1386–1394. doi:10.1200/JCO.2015.63.8387
20. Huang X, Sun Q, Fu H, Zhou X, Guan X, Wang J. Both c-Myc and Ki-67 expression are predictive markers in patients with extranodal NK/T-cell lymphoma, nasal type: a retrospective study in China. Pathol Res Pract. 2014;210(6):351–356. doi:10.1016/j.prp.2014.02.004
21. Koh YW, Hwang HS, Park CS, Yoon DH, Suh C, Huh J. Prognostic effect of Ki-67 expression in rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone-treated diffuse large B-cell lymphoma is limited to non-germinal center B-cell-like subtype in late-elderly patients. Leuk Lymphoma. 2015;56(9):2630–2636. doi:10.3109/10428194.2015.1004169
22. Wang C, Wang Y, Cao J, et al. Elevated expression of HMGB1 is prognostic of poor survival in patients with relapsed/refractory T/NK-CL. Ann Hematol. 2021;100(9):2293–2302. doi:10.1007/s00277-021-04473-4
23. Xu P, Yu D, Wang L, Shen Y, Shen Z, Zhao W. Analysis of prognostic factors and comparison of prognostic scores in peripheral T cell lymphoma, not otherwise specified: a single-institution study of 105 Chinese patients. Ann Hematol. 2015;94(2):239–247. doi:10.1007/s00277-014-2188-8
24. Xue T, Yu BH, Yan WH, et al. Prognostic significance of histologic grade and Ki-67 proliferation index in follicular lymphoma. Hematol Oncol. 2020;38(5):665–672. doi:10.1002/hon.2778
25. Liu Y, Shen J, Awal Issah M, Liu T, Zhou H, Fu H. CD56-positive diffuse large B-cell lymphoma/leukemia with BCL6/MYC double-hit and multiple gene mutations: an indicator of poor prognosis? J Int Med Res. 2020;48(5):300060520918087.
26. Sekiguchi Y, Shimada A, Imai H, et al. CD56+ angioimmunoblastic T-cell lymphoma with evans syndrome: a case report and review of the literature. J Clin Exp Hematop. 2013;53(1):37–47. doi:10.3960/jslrt.53.37
27. Sun Y, Wan J, Song Q, et al. Prognostic significance of CD56 antigen expression in patients with de novo non-M3 acute myeloid leukemia. Biomed Res Int. 2021;2021:1929357. doi:10.1155/2021/1929357
28. Wang L, Wang Z, Xia ZJ, Lu Y, Huang HQ, Zhang YJ. CD56-negative extranodal NK/T cell lymphoma should be regarded as a distinct subtype with poor prognosis. Tumour Biol. 2015;36(10):7717–7723. doi:10.1007/s13277-015-3485-0
29. Saito T, Matsuno Y, Tanosaki R, Watanabe T, Kobayashi Y, Tobinai K. Gamma delta T-cell neoplasms: a clinicopathological study of 11 cases. Ann Oncol. 2002;13(11):1792–1798. doi:10.1093/annonc/mdf293
30. Tanase A, Schmitz N, Stein H, et al. Allogeneic and autologous stem cell transplantation for hepatosplenic T-cell lymphoma: a retrospective study of the EBMT lymphoma working party. Leukemia. 2015;29(3):686–688. doi:10.1038/leu.2014.280
31. Lai R, Larratt LM, Etches W, et al. Hepatosplenic T-cell lymphoma of alphabeta lineage in a 16-year-old boy presenting with hemolytic anemia and thrombocytopenia. Am J Surg Pathol. 2000;24(3):459–463. doi:10.1097/00000478-200003000-00016
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.