")
Back to Journals » International Journal of Nanomedicine » Volume 18
Surface-Engineered Monocyte Immunotherapy Combined Graphene Quantum Dots Effective Against Solid Tumor Targets
Authors Xia Q, Tang Y, Li W, Liang T, Zhou Y, Liu J, Liu F
Received 12 January 2023
Accepted for publication 16 April 2023
Published 24 April 2023 Volume 2023:18 Pages 2127—2140
DOI https://doi.org/10.2147/IJN.S404486
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Farooq A. Shiekh
Qing Xia,* Yue Tang,* Wang Li, Tingting Liang, Yue Zhou, Jun Liu, Feila Liu
School of Pharmacy and Bioengineering, Chongqing University of Technology, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Feila Liu, School of Pharmacy and Bioengineering, Chongqing University of Technology, Chongqing, People’s Republic of China, Tel +86-15123002638, Fax +86 2362563190, Email [email protected]
Introduction: The immunosuppressive tumor microenvironment (TME) of solid tumors inhibits most drug delivery system-based nanomaterials from achieving deep penetration in tumor tissue and interferes with T cell activity in terms of differentiation and exhaustion, which is becoming a critical therapy hurdle for solid tumors. Therefore, developing a therapeutic strategy with abilities of rapid establishment of tumor-targeted cells, elimination of immune obstacles, and enhanced active immunization is very important, while is still a big challenge.
Methods: A new strategy was explored to enhance immune therapy via the conjugation of microRNA155 (miR) to the surface of therapeutic monocyte with graphene quantum dots (GQDs).
Results: TME was reversed using surface-engineered monocyte immunotherapy via reprogramming pro-tumoral M2 TAMs into antitumor M1, and thus tumor elimination was dramatically enhanced.
Conclusion: Such a surface-engineered monocyte immunotherapy has been demonstrated to be well tolerated to intravenous administration and bio-compatible, showing the potential to be extended for the solid tumor treatment.
Keywords: solid tumor, surface-engineered monocyte, GQDs, miR155, immunotherapy
Graphical Abstract:
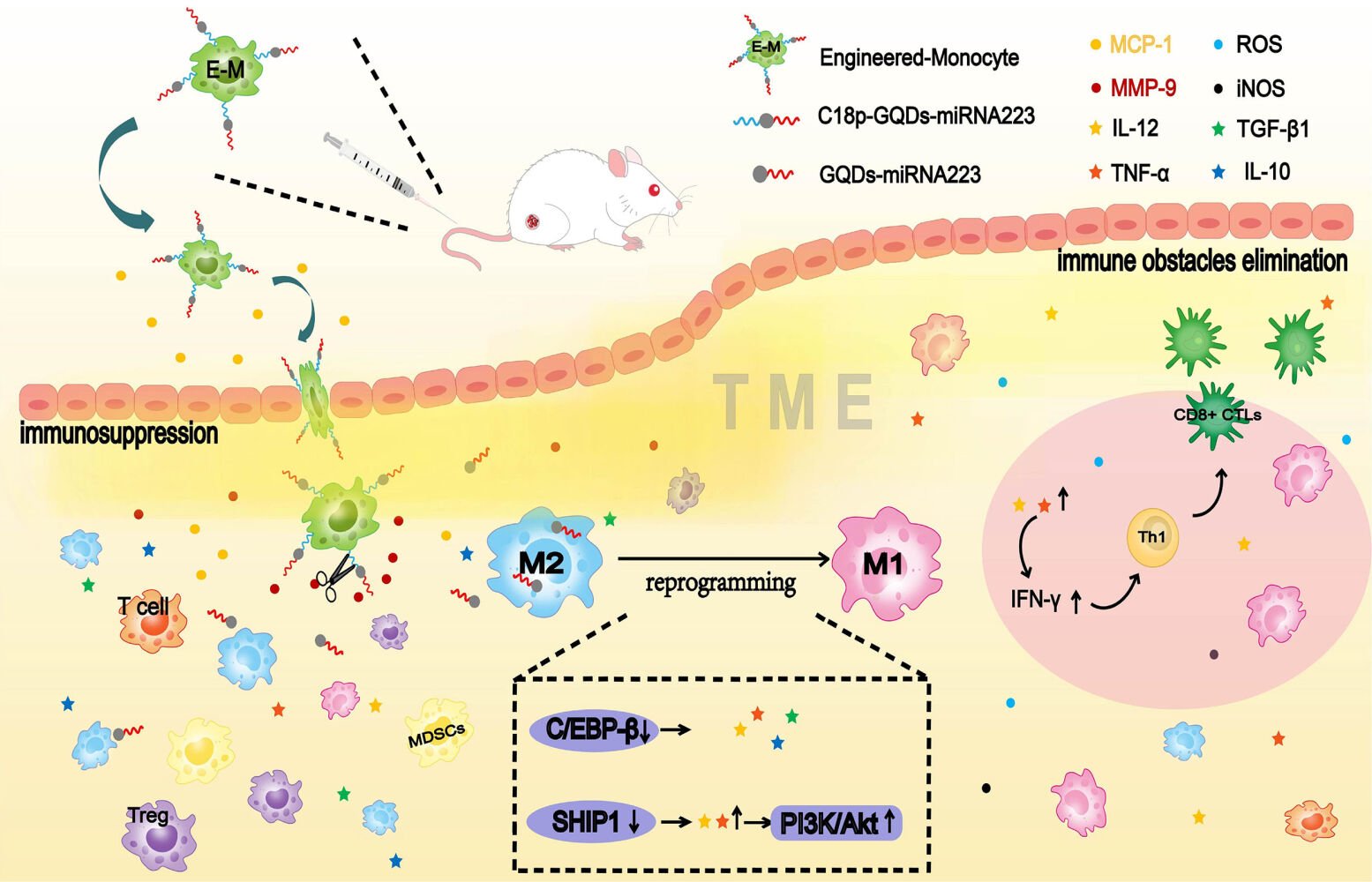
Introduction
The solid tumor can establish a microenvironment including cancer cells, stromal cells and blood vessels. However, in contrast to hematological malignancies that lack the local immune suppression pathways, solid tumors can be strongly infiltrated by different cell types that support tumor growth, angiogenesis, and metastasis and present an immunosuppressed tumor microenvironment (TME).1,2 Regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and M2 tumor-associated macrophages (TAMs) are the most prominent immune suppressor cells in the TME. They facilitate tumor growth and proliferation by producing growth factors, local cytokines, and chemokines in solid tumors, and further impede normal drug-carrying systems from penetrating the interior of tumor tissue.3–5 Even if the drugs are successfully delivered into a solid tumor, their effectiveness is limited through tumor exploitation of counter-regulatory mechanisms.3,5 The existence of an immunosuppressed TME has become a major therapy hurdle for solid tumors.6–9 Therefore, developing an efficient therapy strategy for rapid establishment of tumor-targeted cells, elimination of immune obstacles, and augmentative kinetics of active immunization is critical for solid tumor treatment.
Among the tumor-associated cell species, immune cells are usually applied in cellular immunotherapy tactic due to their exclusive properties.10 Especially monocytes, as a subset of leukocytes, can perform as “living drugs” to overcome transport barrier and penetrate tumor tissue through the vascular endothelium as well as being scarcely interfered with the immunosuppressive TME.11 Inspired by such a natural trafficking activity, monocyte targeted delivery systems (MTDS) with monocytes as carriers or transit medium showed great potential for tumor-targeted accumulation.12–14 Thus, monocytes are considered as an ideal delivery vector based on the collection effect to the tumor lesions. However, augmentation of active immunization in TME impossibly achieves solely depending on the infiltration of monocytes which absent therapeutic function. On the contrary, TAMs in TME partially are derived from circulating monocytes.15
TAMs are a prominent inflammatory cell population primarily polarized to “alternately activated” M2-like macrophages in the TME.16 In contrast to M1, which are highly microbicidal and tumoricidal, M2 subtype TAM plays a crucial role in regulating all aspects of tumor development, namely the growth, survival and metastasis of tumor cells, angiogenesis, and immunosuppression.17 Numerous studies suggest that M2 TAMs in solid tumors is positively associated with the poor prognosis of many human cancers,18,19 thereby reprogramming pro-tumoral M2 TAMs into antitumor M1 macrophages seems as a key point for reversing the immunosuppressed TME and improving the efficacy of cancer therapy.20,21 In the respective of macrophage differentiation and activation, microRNA155 (miR155) significantly attenuates the cytokine production in TAMs via targeting C/EBPβ,22 and re-polarized pro-tumoral M2 TAMs into antitumor M1 macrophages in vitro.23,24 If TAMs-associated miRNAs regulator can be delivered to the inside of the TME with the participation of monocytes, better therapeutic outcomes may be achieved. Nevertheless, loading miRNAs directly onto the surface of monocytes seems impossible because of its ease of degradation. In order to compensate for the limitations of monocytes and miRNAs, an intermediate gene-delivery vector is necessary.
Graphene quantum dots (GQDs) have already been adopted as an intermediate gene-delivery vectors in our previous research.25,26 As a species of graphene, GQDs possesses excellent gene loading capability and stable interactions with various molecules through π–π stacking. Moreover, its smaller size allows facile cellular uptake,27 improves biocompatibility,28 and minimizes cytotoxicity.28 Considering all the advantages, GQDs is expected to be appropriate intermediate gene-delivery vector for the protection, transfection and release of miR155.
In this study, we demonstrate a new strategy based on GQDs-mediated surface engineered monocyte therapy system consisting of monocytes and miR155 for the solid tumor treatment. In the unique architecture of engineered monocyte, disulfide linkage was formed between GQDs and miR155 (GQDs-S-S-miR155) and then grafted on the monocyte surface via a well-designed metalloprotease responsive peptide, as the natural trafficking of monocytes reached to the interior of tumor and further effected on TAMs. The successful construction of engineered monocyte therapy system achieves multifunction including tumor targeting, immune obstacles elimination, and active immunization enhancement, which will provide a brand-new universal strategy for cellular immunotherapy on solid tumor treatment.
Materials and Methods
Cell Culture and Animals
Primary monocytes were isolated from peripheral blood of BALB/c mice and differentiated using standard protocols. 4T1 mammary breast cancer cells and RAW264.7 cells were obtained from cell bank of American Type Culture Collection (ATCC, Shanghai agency). Primary monocytes, 4T1 and RAW264.7 cells were grown in complete DMEM containing 10% fetal calf serum (FCS) (Gibco), 2×10−3 M glutamine (Hyclone), 100 U mL−1 penicillin and 100 µg mL−1 streptomycin (Hyclone). Female BALB/c mice (4–5-week-old, 18–24g) were purchased from Chongqing Tengxin Biotechnology Co., Ltd. (Chongqing). All the animal procedures were carried out under the guideline approved by the Institutional Animal Care and Use Committee (IACUC) of Chongqing University of Technology, Chongqing, China (No.202321).
Preparation and Characterization of GQDs-S-S-miR155
Sulfosuccinimidyl 6-(3′-[2-pyridyldithio]-propionamido) hexanoate (2.5 mg, sulfo-LC-SPDP, Sigma) was added to a solution of amino-functionalized GQDs (GQDs-NH2 purchased from Nanjing XFNANO Materials Tech Co., Ltd.) in 5X PBS (pH = 7.44). The mixture was reacted at room temperature with stirring gently for 1 h, after which the conjugate was centrifuged twice through centrifugal filters with a molecular weight cutoff of 100 kDa (Millipore) for 5 min to remove excess sulfo-LC-SPDP crosslinkers. The thiolated miR (8 µL) was added to the solution of sulfhydrylated GQDs with avoiding light and stored overnight at 4°C. SH-miR155 (Genepharma, Shanghai, China) was double stranded with sense sequence 5′-SH-UUAAUGCUAAUUGUGA UAGGGGU-3′ and antisense sequence 5′-CCCUAUCACAA UUAGCAUUAAUU-3′ (FAM fluorescent labeling). Conjugated GQDs-S-S-miR155 (GQDs-miR155) was then ready for in vitro experiments. The morphology of GQDs-miR155 was photographed by a transmission electron microscope (TEM, Tecnai, USA). A UV spectrophotometer (ShIMADZU, Japan), as well as a fluorescence spectrophotometer F-7000 (HITACHI, Japan), were used for qualitative analysis of disulfide bonds.
Preparation and Characterization of Surface Engineered-Monocyte
The construction of surface engineered-monocyte was achieved by a short peptide: C18-GVFHQTVSR (abbreviated C18p, ChinaPeptide Co., Ltd, Shanghai, China). The conjugation between the GQDs-NH2 and C18p was achieved by coupling with 1-(3-(dimethylamino) propyl)-3-ethylcarbodiimide and N-hydroxysuccinimide (EDC/NHS, Sigma). To remove residual EDC and NHS, the solution was dialyzed (MWCO, 2000) for 48 h (dialysate was replaced every 12 h). Then, 2.5 mg of sulfo-LC-SPDP was added to the product of the conjugation of GQDs-NH2 and C18p (pH ≈ 7.4) and the mixture was allowed to react at room temperature for 1 h. After removing excess sulfo-LC-SPDP, 20 μL of miR155 was added in the dark, and the mixture was stored in a 4°C refrigerator overnight. The prepared C18p -GQD-miR was co-incubated with the monocytes (Dio labeled) for 12 h to afford the immunocyte surface-engineered therapy system. A 90 Plus PALS laser particle sizer (Brookhaven, USA) was used for recording the C18p-GQDs-miR155 particle size and a laser scanning confocal microscope (LSCM, Nikon, Japan) was used for image information acquirement. The characteristic bands of C18p-GQD-miR were detected by FT-IR spectroscopy (Bruker, USA).
More detailed methods are provided in the Supporting Information.
Results
Preparation of GQDs-S-S-miR155 and miR155 Release
The cross-linking process between GQDs and miR155 is shown in Figure 1a. According to our previously reported protocol,29 disulfide-containing linkage can be formed between GQDs and miR via utilizing Sulfo-SPDP-LC. A typical high-resolution transmission electron microscopy (HRTEM) image shows GQDs-NH2 is approximately 10 nm in size (Figures 1b and S1) and GQDs-S-S-miR155 increase to around 30 nm (Figure S2). UV-vis spectrum reveals the existence of disulfide-containing linkage in GQDs-miR155 (Figure 1c). To further verify the successful formation of disulfide-containing linkage between GQDs and miR155, the extracellular cleavage of disulfide linkage by DTT was conducted. It can be seen from the fluorescence spectrometry results in Figure 1d that the DTT-treated GQD-miR155 solution exhibited significantly fluorescence decay (red curve). The above results indicated that a cleavable disulfide bond30 was indeed formed between GQDs and miR155.
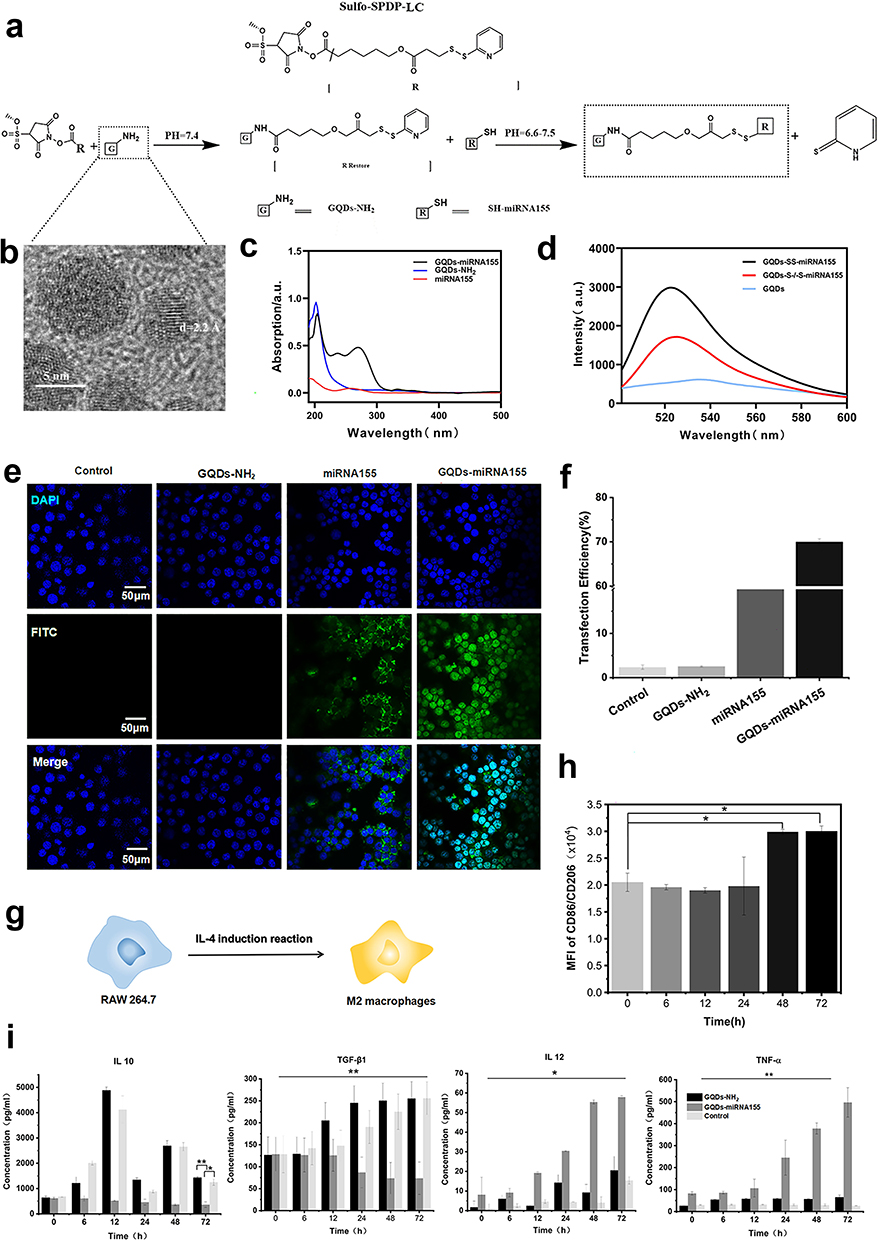 |
Figure 1 Preparation, characterization and cellular regulation of GQDs-miR155. (a) Schematic diagram of the crosslinking process between GQDs and miR155. (b) HRTEM image of the GQDs-NH2. (c) UV-vis spectrum characterization of prepared GQDs-miR155. (d) Fluorescence spectra of GQDs (blue) and FAM-labeled GQDs-miRNA before (black) and after DTT treatment and filtration (red), respectively. (e) Immunofluorescent staining images of GQDs-miR155 transfected into macrophages for determination of the transfection efficiency. (f) The calculated transfection efficiency results. (g) Scheme depicting the polarization of macrophages using IL-4 stimuli. (h) Intracellular NR fluorescence intensity determined by flow cytometry before and after miR155 regulation during 72h. (i) ELISA detection of macrophage cytokine production. Images are representative of n = 3 independent experiments. The data in the figures represent the averages ± SD. Significant differences: *p < 0.05, **p < 0.01. |
Intracellular delivery of gene molecules by GQDs are devised to release biological cargos from nano carrier via biologically triggered disulfide bond cleavage.31 Disulfide-containing conjugates (GQDs-S-S-miR) can not only respond to the redox microenvironment in the presence of high concentrations of glutathione/reactive oxygen species at tumor sites,32 but also be cleaved by thiol-reducing enzymes (γ-interferon-inducible lysosomal thiol reductase, GILT)25 aided by the acidic conditions in lysosomes upon endocytosis.33 The presence of a cleavable disulfide linkage in the GQDs-S-S-miR conjugates, representing such a simple functionality installed on GQDs can facilitate the subsequent miR155 transfection and release in cells. Subsequently, the gene delivery efficiency of GQDs for miR155 transfection using RAW264.7 cells was tested.
Gene-Delivery Efficiency of GQDs in vitro
A total of 10 independent groups were transfected. Confocal microscopy images revealed fluorescence colocalization of miR155 (green) in the cell nucleus, giving rise to green fluorescent spots within the nucleus (Figure 1e). The transfection efficiency of the GQDs-miR155 can reach ≈ 70% within 6 h calculated from flow cytometry analysis (Figures 1f and S3). From the perspective of RNA delivery and transfection, GQDs, as one kind of a biocompatible nanomaterial, has been developed as safe substitute to viral vector.34 In addition, as a nonviral vector, GQDs with 70% transfection efficiency is a guarantee for the effective uptake of miR by TAMs in tumors. Herein, we evaluated the effect of cellular uptake of miR on TAMs phenotype as following.
GQDs-S-S-miR155 Reprogrammed TAMs into Antitumor M1 Macrophages in vitro
TAMs closely resemble “alternative” (M2) macrophages, which produce high amounts of interleukin (IL)-10 but not IL-12, express scavenger receptors, and exhibit anti-inflammatory. In contrast, M1 macrophages produce large amounts of pro-inflammatory cytokines, express high levels of major histocompatibility complex molecules, and are potent killers of pathogens and tumor cells.35 Thus, reprograming TAMs toward the anti-tumor M1-like phenotype is a promising strategy for reversing the TME. To determine whether miR155 transfection could reprogram TAMs into M1 macrophage, RAW264.7 were pre-polarized to the M2-type with interleukin-4 (IL-4) (Figure 1g) and monitored the in situ phenotype change using flow cytometry (Figure S4). We observed that M2 protein marker CD206 was fully activated in 48 h, suggesting that the tumor-related M2 macrophage model was successfully established. To determine whether GQDs-miR155 transfection could reprogram M2 TAMs into M1, the mean fluorescence intensity (MFI) of CD86+ and CD206+ macrophages within 72 h using flow cytometric analysis was calculated (Figures 1h, S5 and S6). CD86 is an activation marker of macrophages, providing stimulatory signals for T cell activation.36 Obviously, the up regulation CD86 may induce the immune activation of TAMs. As expected, GQDs-miR155 significantly elevated CD86 and inhibited CD206 on TAMs as a function of time, suggesting the reprogram of M2 TAMs into anti-tumor M1 macrophages.
Moreover, we measured the serum level of macrophage-produced cytokines including IL-10, TGF-β1, IL-12 and TNF-α in a 72 h period to further verify the successful re-polarization. As shown in Figure 1i, the levels of the pro-inflammatory cytokine tumor necrosis factor alpha (TNF-α), Interleukin-12 (IL-12) in the GQDs-miR155 treated group significantly increased as a function of time, whereas the levels of the anti-inflammatory cytokines interleukin-10 (IL-10) and TGF-β1 both decreased. It is worth noting that the independent GQDs treated group presented an up-regulated anti-inflammatory cytokines, suggesting the down regulation of miR155 could be a key mechanism promoting TAM polarization to immunosuppressive M2 phenotype.37 Previous studies have already demonstrated that miR-155-regulated inflammatory cytokine production in TAMs or proliferation, survival and cell activation in hematopoietic cells via targeting C/EBP-β, a critical negative regulator of macrophage programming and activation.38 Overexpression of miR155 can indirectly upregulate PI3K/Akt pathway by the decrease in SHIP1 concentration, inducing M1 phenotype polarization and stimulating the release of pro-inflammatory cytokines.39 Furthermore, IL-12 is required for the differentiation and activation of tumor-specific cytotoxic T cells (CTLs).40 Hence, the up-regulation of IL-12 and TNF-α confirmed the immune activated potential of TAMs after GQDs-miR155 treatment, which is essential for active immunization enhancement.
The Architecture of Surface-Engineered Monocyte Therapy System
Targeted delivery of miR155 in vivo only depending on GQDs may lead to nonspecific delivery and accumulation in non-lesion organs, such as the spleen or the liver, because most nanoparticles-based delivery system have a insufficient long blood half-life and cannot realize deep penetration in tumor tissue.41 Herein, we constructed a surface engineered therapy system based on monocytes. The detail linking process of C18p-GQDs-miR155 is shown in Figure 2. The designed C18p consists of two parts: a short peptide (GVFHQTVSRK) with a hydrophobic terminal that could be directly digested by matrix metalloproteinases (MMP) in the tumor environment,18 and a straight-chain alkene consisting of 18 carbon atoms that could extend the hydrophobic terminal of the short peptide.
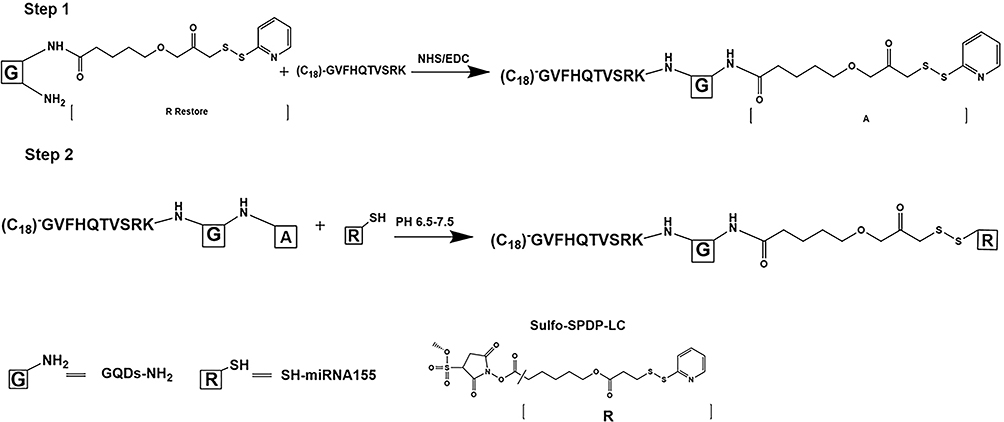 |
Figure 2 Schematic diagram of the crosslinking process of C18p-GQDs-miR155. |
TEM image in Figure 3a shows that the C18p-GQDs-miR155 combination was nanometer-sized (~200 nm) spherical particle, significantly larger than individual GQDs-NH2 or GQDs-miR155. The successful cross-linking of C18p and GQDs-miR155 was confirmed by the FT-IR spectra (Figure 3b). DTT cleavage experiment results further showed the miR155 concentration in the collected filtrate was 7-fold higher than the untreated group (Figure 3c), indicating that C18p-GQDs-miR155 was successfully synthesized and a cleavable disulfide linkage was formed in this combination. In order to prove the special disulfide-containing linkage formation in C18p-GQDs-miR155 can effectively protect miR in the RNase exposed environment and facilitate subsequent in vivo delivery, we tested and determined the remaining miR155 concentration in different combinations. From Figure 3d, the combination of GQDs-miR155 containing ingredient obviously retains more miR content in comparison with the solitary miR group or C18p, GQDs and miR-mixed group. We speculated that special spatial structure via the stacking of GQD layers could effectively protect partial miR from enzymatic degradation when the RNA was crosslinked with the GQDs via disulfide linkages, which provided an experimental basis for the safe delivery of miRNAs in vivo. In other word, GQDs facilitate miR uptake and cytosolic releasing as well as avoid miR from degradation during delivery.
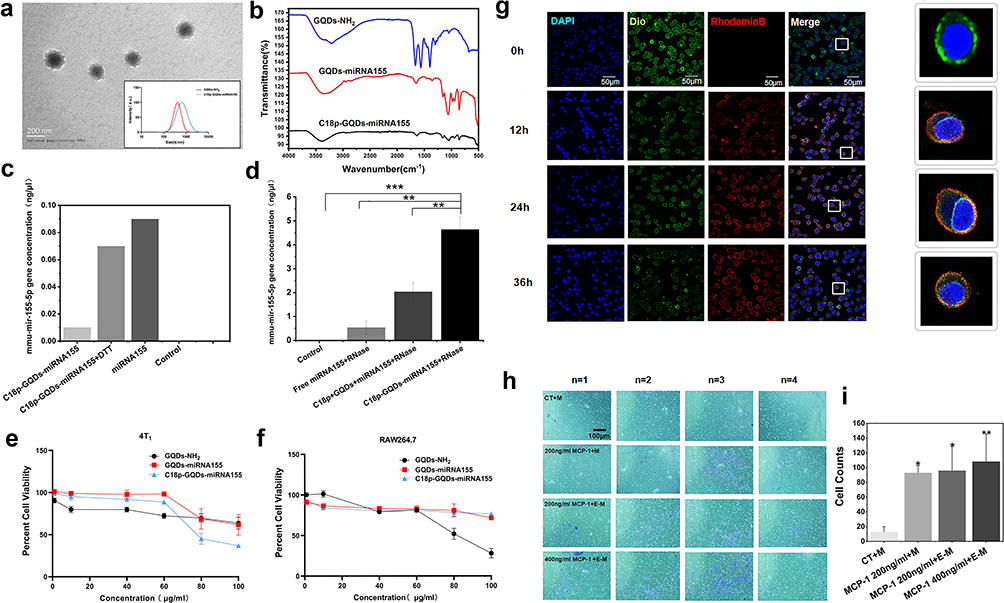 |
Figure 3 Preparation, characterization and performance of surface engineered monocyte therapy system. (a) TEM image of C18p-GQDs-miR155. The inset shows the size distribution of GQDs-NH2 and C18p-GQDs-miR155. (b) FT-IR spectra of the GQDs-NH2, GQDs-miR155 and C18p-GQDs-miR155. (c) Comparison of miR155 concentrations in C18p-GQDs-miR155 before and after DTT treatment. (d) MiR155 concentration levels after 6 h in the presence of RNase as detected by Ultra-Micro spectrophotometer in the control, free miR155+RNase (1mL miR155 was mixed with 2μL RNase), C18p+GQDs+miR155+RNase (2μL RNase was added into the 1mL mixed C18p, GQDs and miR155 solution) and C18p-GQDs-miR155 (1mL C18p-GQDs were mixed with 2μL RNase for half an hour and then mixed with 10 µL of miR155) group. Evaluation of the effect of GQDs-NH2, GQDs-miR155 and C18p-GQDs-miR155 at different concentrations on cell viability in 4T1 cell (e) and RAW246.7 cell (f). (g) Confocal fluorescence images of the C18p-GQDs-miR155 surface-engineered monocyte at different time from 0 to 36h. Red fluorescence indicates rhodamine B bound to the peptide, green fluorescence indicates cell membrane labeling Dio, and blue fluorescence indicates DAPI. (h) The images of Transwell migration of cells in each group: CT, 200 ng/mL MCP-1+M, 200 ng/mL MCP-1+E-M and 400 ng/mL MCP-1+E-M, respectively. M indicates monocyte, E-M indicates C18p-GQDs-miR155 engineered monocyte. Transwell chamber assay showing that MCP-1 (200 ng/mL) increases the migration of normal monocyte and surface engineered-monocyte cells after 24 h incubation, respectively. (i) Cell counts of Transwell migration in each group. Images are representative of n ≥ 3 independent experiments, unless otherwise noted. The data in the figures represent the averages ± SD. Significant differences: *p < 0.05, **p < 0.01, ***p < 0.001. |
Following the morphology and structural analysis of C18p-GQDs-miR155, we investigated the optimal experimental concentration toward 4T1 and RAW264.7 to determine that C18p-GQDs-miR155 inside living cells had no ill effect on cell viability. The results obtained through Cell Counting Kit-8 (CCK8) showed that the C18p-GQDs-miR155 started to reduce cell viability at a concentration of 60 µg mL−1 for both 4T1 (Figure 3e) and RAW264.7 (Figure 3f). Herein, we adopted 40 µg mL−1 as the maximum working concentration for the following studies.
The hydrophobic terminal of C18p was confined to the hydrophobic region after passing through the cell membrane, and GQDs-miR155 were grafted onto the surface of the monocyte. As shown in Figure 3g, after incubating the C18p (rhodamine B – labeled)-GQDs-miR155 system with the primary monocytes (Dio-labeled membrane) for 12 h ~ 36 h, fluorescence superposition was observed on the cell membrane. This result not only confirmed the successful attachment of C18p-GQDs-miR155 onto the surface of monocyte, but also suggested no significant endocytosis by monocyte. In other words, surface-engineered monocyte therapy system (M-C18p-G-miR155) was successfully constructed.
Innate Trafficking Behavior of Surface-Engineered Monocyte Therapy System
It is not clear that whether C18p-GQDs-miR155-engineered monocyte could maintain the natural trafficking behavior of tumor recruitment as normal monocyte did, thus we tested it using in vitro transwell chamber assays. The migration results from Figure 3h and i showed that the number of migrated cells per field in the MCP-1 (200 ng/mL) treated cells were significantly higher than those in the control group, indicating that MCP-1 can promote the migration of both monocytes and engineered monocytes and high concentrations of MCP-1 (400 ng/mL) even favors migration of engineered monocytes.
As one of the most prominent tumor-homing leukocytes,11 monocytes are constantly recruited to the tumor through the mediation of monocyte chemoattractant protein-1 (MCP-1) and specifically target tumor tissue through binding to the vascular cell adhesion molecule-1 (VCAM-1) of various cancer cells.42,43 Moreover, surface-engineered monocytes still maintain the natural trafficking behavior of tumor recruitment, which is essential for rapid establishment of tumor-targeted cells and elimination of immune barrier.
In vivo Anticancer Effect of Surface-Engineered Monocyte
Solid tumors are known to secrete immuno-factors, including chemokine, cytokine, and growth factors, preventing effector T cells from infiltrating into the tumor bed.44,45 Active immunization restricts in established tumors, factors release in the TME (such as IL-10, IL-4, and IL-13)46–48 polarize M1 macrophages to M2 macrophages,49 therefore exhibits immunosuppressive activity. Reprogrammed TAMs have been reported to inhibit tumor progression and metastasis through direct cytotoxic effect.50,51 Therefore, we need to evaluate cytotoxic effect of re-educated TAMs against tumor cell in vivo. Here, we adopted the 4T1 subcutaneous tumor as a model and surface-engineered monocyte as an anticancer therapy system to evaluate the strategy of M2 macrophage precision regulation.
The established 4T1 subcutaneous model mice were injected with phosphate-buffered saline (PBS, pH 7.2–7.4, control group), monocyte, GQDs-miR155, and M-C18p-G-miR155 (50 nM, 100 μL) on days 1, 3, 5, 7, and 9 and sacrificed on day 12 (Figure 4a). Based on the previous in vitro research (Figure 3), 40 µg mL−1 of C18p-GQDs-miR155 showed no cytotoxicity on cells, however, biosafety is always a major concern of nano material-involved gene therapy. In this study, blood urea nitrogen (BUN)/creatinine (CRE) and serum alanine aminotransferase (ALT)/aspartate aminotransferase (AST) were measured after finishing the entire cell therapy on the model animal, which are critical biomarkers of renal and liver damage, respectively.52–55 The results showed that serum BUN/CRE (Figure 4b and c) and serum ALT/AST (Figure 4d and e) were not significantly different among all groups, indicating no significant renal or liver toxicity of monocyte/GQDs-miR155/M-C18p-G-miR155 in comparison with control group. The potential toxicity of monocyte/GQDs-miRNA155/M-C18p-G-miR155 complexes was further investigated in vital organs using H&E staining. From the Figure 4f, after five consecutive treatments, mouse organs including heart, liver, spleen, lung and kidney did not show histopathological abnormalities, lesions or degenerations in all four groups, confirming that both the GQDs-miR155 and M-C18p-G-miR155 therapy system were biocompatible.
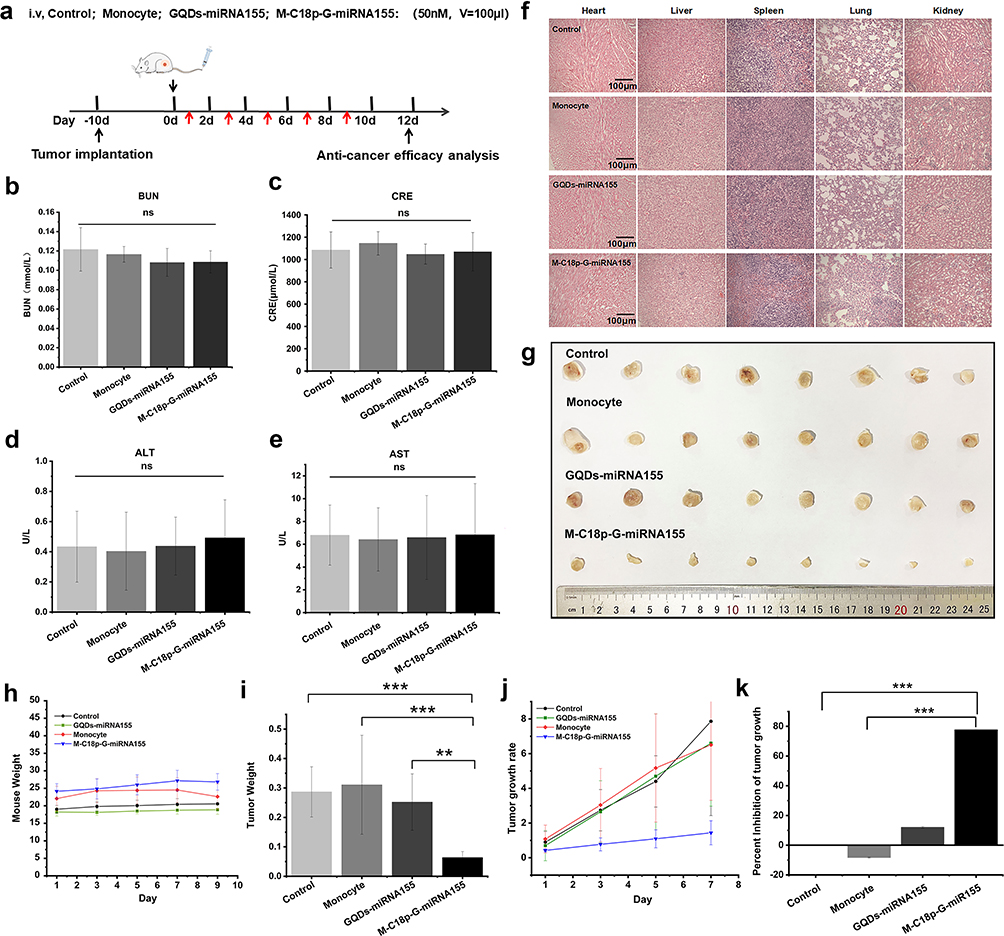 |
Figure 4 In vivo anticancer therapy efficacy analysis. (a) Scheme depicting the BALB/c mice bearing 4T1 subcutaneous tumor receiving the treatment of control/monocyte/GQDs-miRNA155/M-C18p-G-miR155 at day 1, 3, 5, 7, and 9. Histological/biochemical analysis was performed on day 12. Biosafety of monocyte/GQDs-miR155/M-C18p-G-miR155 in vivo. Serum levels of (b) BUN and (c) CRE (renal function) at 24 h after finishing the last treatment. Serum levels of (d) ALT and (e) AST (liver function) at 24 h after finishing the last treatment. Data are presented as mean ± SE. The differences of all the groups were determined using one-way ANOVA. (f) H&E staining of mouse organs (hearts, livers, spleens, lungs, and kidneys) after finishing the entire treatments. (g) Photographs of 4T1 subcutaneous tumors after finishing the entire treatments. (h) Weight change of 4T1 tumor-bearing mice injected with different treatments. (i) 4T1 subcutaneous tumor weight of each mouse in groups with the treatment of control, monocyte, GQDs-miRNA155 and M-C18p-G-miR155, respectively. The calculated tumor growth rate (j) and tumor inhibition rate (k) of each mouse in groups with different treatments. Images are representative of n = 8 independent experiments. Significant differences: **p < 0.01, ***p < 0.001. |
Figure 4g showed the photographs of 4T1 subcutaneous tumors after finishing all the treatments in the different groups. It can be seen that M-C18p-G-miR155 strongly inhibited tumor progression and presented a greater potency for eliminating the tumor. Weight change of 4T1 tumor-bearing mice with different treatment options were not obvious (Figure 4h), whereas tumor weight (Figure 4i) showed significant differences, consistent with the results in Figure 4g. The calculated tumor growth rate (Figure 4j) and tumor inhibition rate (Figure 4k) all showed the tumor inhibition effect of M-C18p-G-miR155. M-C18p-G-miR155 were recruited into the interior of tumor lesions and digested by matrix metalloproteinase, such as MMP-9.56 The isolated GQDs-miR155 which released by matrix metalloproteinase efficiently delivered miR155 into the TAMs by macrophages endocytosis. Macrophages are professional phagocytes that can internalize large particles such as debris, apoptotic cells, and pathogens to maintain homeostasis in the human body.57,58
Subsequently, cleavage of the disulfide linker in GQDs-miR155 in the lysosomes caused the miRNA cargos to be released into the cytosol under the protection of the GQDs. Nanomaterials/miR effectively facilitated miR uptake by macrophages rather than tumor cells had been confirmed.59,60 Finally, miR155 coordinated the immune function of the body and inhibited tumors. Moreover, paraffin sections from the collected tumors were stained via Ki67 cell proliferation kit, which also confirmed the above speculations. From Figure 5a and d, M-C18p-G-miR155 treated group showed the significant tumor proliferation inhibition.
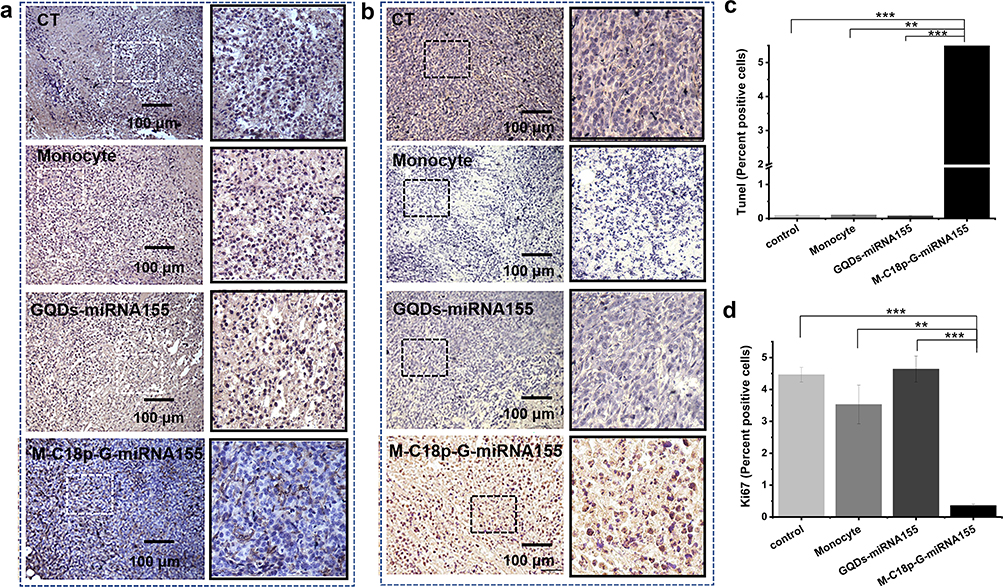 |
Figure 5 Immunohistochemistry analysis of tumor after treatment. Ki67 (a) and TUNEL (b) staining of tumor sections, respectively. (c) Detection of apoptotic cells in tumor sections. (d) Detection of proliferative cells in tumor sections. Data are shown as means ± SD (n ≥ 3). Significant differences: **p < 0.01, ***p < 0.001. |
To further determine the M-C18p-G-miR155-targeting specificity of treatment, we used a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay to measure the apoptosis of 4T1 cells in the collected tumors (Figure 5b and c). As shown in Figure 5c, compared to the mice treated with GQDs-miR155 and individual monocytes, M-C18p-G-miR155 induced the most significant rate of apoptotic cells. This observation indicated that the M-C18p-G-miR155 therapy system could facilitate the apoptosis of tumor cells in vivo. The potent anti-tumor effect of M-C18p-G-miR155 could be attributed to its ability to elevate miR155 in tumor TAMs. However, whether M-C18p-G-miR155 can reprogram M2 TAMs into M1 in the tumor environment requires further validation.
Surface-Engineered Monocyte Reprogrammed M2 TAMs into Antitumor M1 Macrophages in vivo
To determine whether M-C18p-G-miR155 could reprogram TAMs into M1 macrophage in vivo, we monitored the in-situ change in the macrophage phenotype using immunofluorescent staining, as shown in Figure 6a and b. Consistent with the results of in vitro experiments in Figure 1j, the M1 protein marker CD86 (green fluorescence) was activated, whereas the M2 protein marker CD206 (red fluorescence) was strongly inhibited. In addition, the number of CD86+ macrophages were significantly higher than that of CD206+ macrophages (Figure 6c and d), indicating the ability of M-C18p-G-miR155 reprogramming M2 TAMs into M1 macrophages in vivo. Previously reported results indicated that M1 TAMs secrete proinflammatory cytokines, such as TNF-α, IL-12, and reactive oxygen species (ROS) to induce tumor cell apoptosis and enhance tumor immunotherapy,61 which has also been demonstrated by us via in vitro macrophage cytokine production before and after regulation by miR155 in the Figure 1k. In addition to direct cytotoxic effect against tumor through releasing a variety of cytotoxic soluble factors, activated TAMs have been reported that could consequently activate and recruit multiple effector cells (eg, CD4+/CD8+ T cells and NK cells) for tumor inhibition.62–64 Therefore, the increased CD86+ cells and higher CD86+/CD206+ ratio (Figure 6e) significantly affected tumor progression. In Figure 1k, we have discussed that GQDs-miR155 significantly inhibited production of anti-inflammatory cytokines TGF-β, IL-10, and promoted IL-12 secretion. IL-12, as one of the cytotoxic soluble factors and typical marker of activated TAMs, can facilitate CD8+ and CD4+ T cell activation.65 Moreover, activated TAMs can also present tumor antigen to CD4 and CD8 T cells, triggering anti-tumor adaptive immune responses. Herein, M-C18p-G-miR155 could not only facilitate TAM-targeted miR delivery, but also played a vital role in reversing immunosuppressive tumor microenvironment through reprogramming M2 TAMs into M1 subtype indicated by infiltration deduction of TAMs, finally promoted CD4+ T cells and CD8+ T cells activation and proliferation.
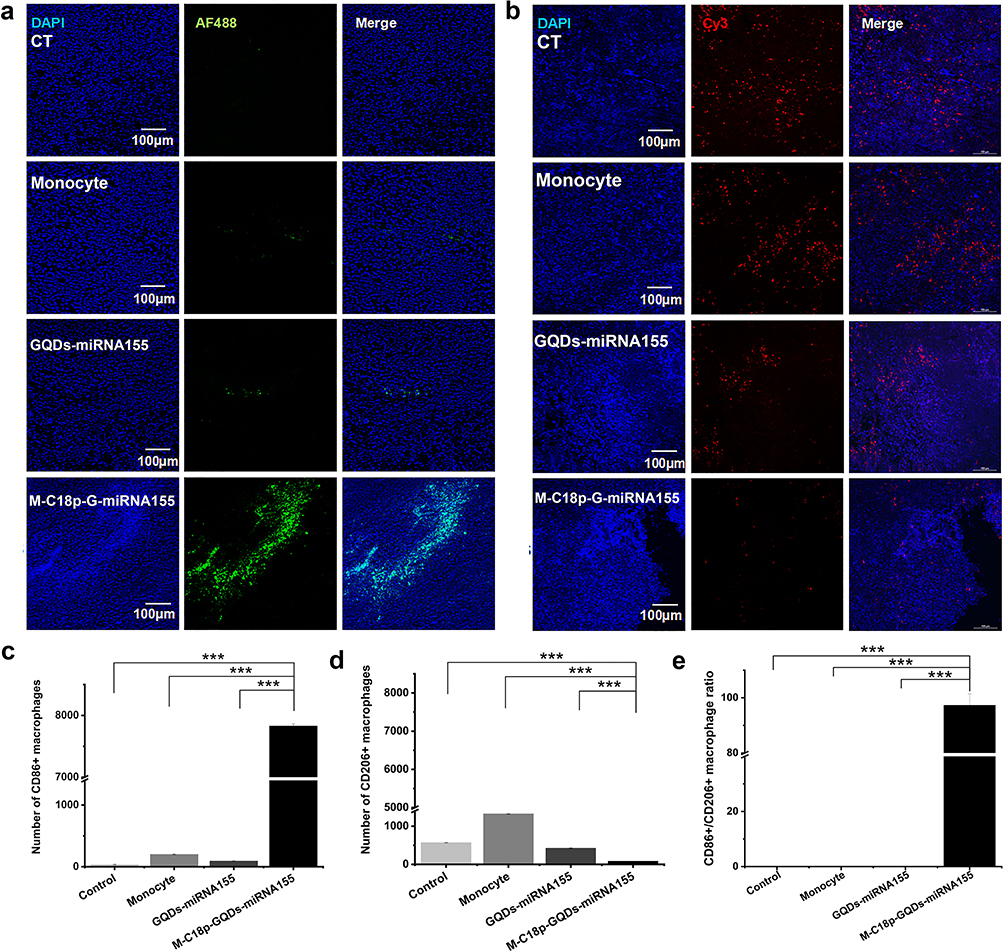 |
Figure 6 Biochemical analysis of tumor after treatment. Immunostaining of (a) CD86+ (AF488) and (b) CD206+ (Cy3) macrophages in tumor sections after treatment. (c) CD86+ cells in tumor sections, (d) CD206+ cells in tumor sections and (e) the calculated CD86+/CD206+ ratio. Data are shown as means ± SD (n ≥ 3). Significant differences: ***p < 0.001. |
Discussion
Cell therapies are in common clinical practice for certain indications, such as islet cell transplants, and are also being aggressively developed in other areas of medicine, such as T-cell therapy of tumor.66 Take T-cell therapy for tumor treatment as an example, chimeric antigen receptor (CAR) T cells are genetically engineered T cells to express a receptor for the recognition of the particular surface marker that has given rise to advances in the treatment of blood disorders. However, their efficacy in solid tumor treatment has not yet been supported. Antigen heterogeneity of solid tumors,67 a stumbling block of penetrating tumor tissue through the vascular endothelium68 and the immunosuppressive TME4 all restrict the influence of T cell-based cellular therapies.
Monocyte-based cell therapies may overcome the above limitations, given the constant trafficking of mononuclear phagocytes into tumors.69 In previous study, Allavena et al proved that monocytes replenished with drug-loaded nanoparticles and intravenously injected into tumor-bearing mice were able to reach the tumor site with superior efficiency than free nanoparticles.70 Hence, using monocytes to bring small molecules, especially gene drugs to the tumor site and consequently activate an immune response is becoming a new strategy for solid tumor treatment.
Under the influence of the tumor TME, TAMs play an essential role in tumor occurrence, metastasis, and invasion. The presence of a large number of infiltrating macrophages in tumor tissues differentiating into the M2 type is associated with poor prognosis, therefore, several strategies have been proposed for reducing the M2 TAMs. Reprogramming of M2 TAMs into M1 TAMs in our study is one of the strategies. Sufficient evidences have reported the ability of miR155 in polarizing macrophages to the M1 phenotype23,24 and elevated miR155 in TAM might be a potential strategy to repolarize TAM and overcome immunosuppression in TME, which would consequently inhibit tumor progression. However, we must overcome several biological barriers in order to achieve successful TAM-targeted miR therapy, including 1) to specifically target TAM in the TME; 2) to facilitate miR uptake and cytosolic releasing; 3) to avoid miR degradation or capture by other cells during delivery. Based on the above considerations, we successfully designed the monocyte surface nanoengineered miR155 therapy system. In this study, we confirmed the GQD layers could effectively protect partial miR155 from enzymatic degradation when the RNA was crosslinked with the GQDs via disulfide linkages (Figure 3d), which provided an experimental basis for the safe delivery of miRNAs in vivo. Additionally, GQDs are only engulfed by macrophages other than endothelial cells or smooth muscle cells,25 which is beneficial to specifically target TAM in the TME. Although GQDs-mediated cell transfection can effectively regulate TAMs, their accumulation in tissues or organs is inevitable due to the non-degradability. Cellular immunotherapy is the future trend of precise treatment for solid tumors, therefore, using non-toxic, degradable and functional biomaterials to completely replace GQDs will be our research objective.
Finally, inspired by CAR-T therapy, chimeric antigen receptor monocytes (CAR-M) armed with receptors recognizing carcinoembryonic antigen are expected to be developed for the treatment of solid tumors in the future.
Conclusion
As an advanced solid tumor treatment concept in its infancy, a strategy to enhance therapeutic cell therapy using engineering means to integrate nanomaterials and genetic molecules is emerging. Unlike traditional nanoparticles that target accumulation at the tumor site based on passive targeting, or active targeting via special cell membrane camouflage, the surface engineered monocyte has an advantage of natural trafficking activity toward tumor targeting. Additionally, such a cell therapy system can protect gene molecules from degradation as well as facilitate them uptake by TAMs through GQDs intermediation. Particularly, different from the chimeric antigen receptor (CAR)-T therapy in solid tumors, the surface-engineered monocyte therapy system takes little account of which based on the impediments posed by TME in association with tumor heterogeneity. Moreover, similar with CAR-T cell, this strategy will offer new opportunities to modify endogenous immunocytes and reinfuse, which may have a potential to be translated into clinically utilizing monocyte harvested from humans in the future.
Acknowledgments
This work was supported by Scientific Research Initiation Fund of Chongqing University of Technology (No. 2019ZD120).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16(7):447–462. doi:10.1038/nrc.2016.54
2. Vitale I, Manic G, Coussens LM, et al. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36–50. doi:10.1016/j.cmet.2019.06.001
3. McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov. 2013;12(3):217–228. doi:10.1038/nrd3870
4. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med. 2016;94(5):509–522. doi:10.1007/s00109-015-1376-x
5. Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov. 2020;19(1):39–56. doi:10.1038/s41573-019-0044-1
6. Tian H, Zhang M, Jin G, et al. Cu-MOF chemodynamic nanoplatform via modulating glutathione and H(2)O(2) in tumor microenvironment for amplified cancer therapy. J Colloid Interface Sci. 2021;587:358–366. doi:10.1016/j.jcis.2020.12.028
7. Li Q, Zhao Z, Qin X, et al. A checkpoint-regulatable immune niche created by injectable hydrogel for tumor therapy. J Adv Function Mater. 2021;31(37):2104630 doi:10.1002/adfm.202104630 doi:.
8. Zhao Z, Li Q, Qin X, et al. An injectable hydrogel reshaping adenosinergic axis for cancer therapy. Adv Funct Mater. 2022;24:32 doi:10.1002/adfm.202200801.
9. Ren X, Wang N, Zhou Y, et al. An injectable hydrogel using an immunomodulating gelator for amplified tumor immunotherapy by blocking the arginase pathway. Acta biomaterialia. 2021;124:179–190. doi:10.1016/j.actbio.2021.01.041
10. Stephan M, Moon JJ, Um SH, et al. Abstract SSY01-05: therapeutic cell engineering using surface-conjugated synthetic nanoparticles. Cancer Res. 2010;70(8 Supplement):SSY01–SSY05. doi:10.1158/1538-7445.AM10-SSY01-05
11. Xia Y, Rao L, Yao H, et al. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater. 2020;32(40):e2002054. doi:10.1002/adma.202002054
12. Choi J, Kim HY, Ju EJ, et al. Use of macrophages to deliver therapeutic and imaging contrast agents to tumors. Biomaterials. 2012;33(16):4195–4203. doi:10.1016/j.biomaterials.2012.02.022
13. Ding X, Sun X, Cai H, et al. Engineering macrophages via nanotechnology and genetic manipulation for cancer therapy. Front Oncol. 2021;11:786913. doi:10.3389/fonc.2021.786913
14. Zheng L, Hu X, Wu H, et al. In vivo monocyte/macrophage-hitchhiked intratumoral accumulation of nanomedicines for enhanced tumor therapy. J Am Chem Soc. 2020;142(1):382–391. doi:10.1021/jacs.9b11046
15. Ge Z, Ding S. The crosstalk between Tumor-Associated Macrophages (TAMs) and tumor cells and the corresponding targeted therapy. Front Oncol. 2020;10:590941. doi:10.3389/fonc.2020.590941
16. Zhang Y, Choksi S, Chen K, et al. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013;23(7):898–914. doi:10.1038/cr.2013.75
17. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi:10.1038/nri3073
18. Yeung OW, Lo CM, Ling CC, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. 2015;62(3):607–616. doi:10.1016/j.jhep.2014.10.029
19. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv Drug Deliv Rev. 2017;114:206–221. doi:10.1016/j.addr.2017.04.010
20. Gunassekaran GR, Poongkavithai Vadevoo SM, Baek MC, et al. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomaterials. 2021;278:121137. doi:10.1016/j.biomaterials.2021.121137
21. Ramesh A, Kumar S, Nandi D, et al. CSF1R- and SHP2-inhibitor-loaded nanoparticles enhance cytotoxic activity and phagocytosis in tumor-associated macrophages. Adv Mater. 2019;31(51):e1904364. doi:10.1002/adma.201904364
22. Johansson J, Berg T, Kurzejamska E, et al. MiR-155-mediated loss of C/EBPbeta shifts the TGF-beta response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene. 2013;32(50):5614–5624. doi:10.1038/onc.2013.322
23. O’Connell RM, Taganov KD, Boldin MP, et al. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104(5):1604–1609. doi:10.1073/pnas.0610731104
24. Essandoh K, Li Y, Huo J, et al. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock. 2016;46(2):122–131. doi:10.1097/SHK.0000000000000604
25. Liu F, Ding N, Huo D, et al. Surface-engineered monocyte inhibits atherosclerotic plaque destabilization via graphene quantum dot-mediated MicroRNA delivery. Adv Healthc Mater. 2019;8(15):e1900386. doi:10.1002/adhm.201900386
26. Li N, Liu Y, Cai J. LncRNA MIR155HG regulates M1/M2 macrophage polarization in chronic obstructive pulmonary disease. Biomed Pharmacother. 2019;117:109015. doi:10.1016/j.biopha.2019.109015
27. Wang XY, Lei R, Huang HD, et al. The permeability and transport mechanism of graphene quantum dots (GQDs) across the biological barrier. Nanoscale. 2015;7(5):2034–2041. doi:10.1039/C4NR04136D
28. Nurunnabi M, Khatun Z, Huh K, et al. In vivo biodistribution and toxicology of carboxylated graphene quantum dots. ACS nano. 2013;7(8):6858–6867. doi:10.1021/nn402043c
29. Wei K, Liu F, Yang J, et al. Engineered-macrophage induced endothelialization and neutralization via graphene quantum dot-mediated MicroRNA delivery to construct small-diameter tissue-engineered vascular grafts. J Biomed Nanotechnol. 2019;15(7):1492–1505. doi:10.1166/jbn.2019.2787
30. de la Rica R, Aili D, Stevens MM. Enzyme-responsive nanoparticles for drug release and diagnostics. Adv Drug Deliv Rev. 2012;64(11):967–978. doi:10.1016/j.addr.2012.01.002
31. Arunachalam B, Phan UT, Geuze HJ, et al. Enzymatic reduction of disulfide bonds in lysosomes: characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT). Proc Natl Acad Sci U S A. 2000;97(2):745–750. doi:10.1073/pnas.97.2.745
32. Fu S, Rempson CM, Puche V, et al. Construction of disulfide containing redox-responsive polymeric nanomedicine. Methods. 2022;199:67–79. doi:10.1016/j.ymeth.2021.12.011
33. Rausch MP, Hastings KT. Diverse cellular and organismal functions of the lysosomal thiol reductase GILT. Mol Immunol. 2015;68(2 Pt A):124–128. doi:10.1016/j.molimm.2015.06.008
34. Chung S, Revia RA, Zhang M. Graphene quantum dots and their applications in bioimaging, biosensing, and therapy. Adv Mater. 2021;33(22):e1904362. doi:10.1002/adma.201904362
35. Saccani A, Schioppa T, Porta C, et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66(23):11432–11440. doi:10.1158/0008-5472.CAN-06-1867
36. Duluc D, Corvaisier M, Blanchard S, et al. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int J Cancer. 2009;125(2):367–373. doi:10.1002/ijc.24401
37. Teng C, Lin C, Huang F, et al. Intracellular codelivery of anti-inflammatory drug and anti-miR 155 to treat inflammatory disease. Acta Pharm Sin B. 2020;10(8):1521–1533. doi:10.1016/j.apsb.2020.06.005
38. He M, Xu Z, Ding T, et al. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell Mol Immunol. 2009;6(5):343–352. doi:10.1038/cmi.2009.45
39. Rothchild AC, Sissons JR, Shafiani S, et al. MiR-155-regulated molecular network orchestrates cell fate in the innate and adaptive immune response to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2016;113(41):E6172–E6181. doi:10.1073/pnas.1608255113
40. Kerkar SP, Muranski P, Kaiser A, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70(17):6725–6734. doi:10.1158/0008-5472.CAN-10-0735
41. Lee H, Lytton-Jean AK, Chen Y, et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat Nanotechnol. 2012;7(6):389–393. doi:10.1038/nnano.2012.73
42. Deshmane SL, Kremlev S, Amini S, et al. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29(6):313–326. doi:10.1089/jir.2008.0027
43. Franklin RA, Liao W, Sarkar A, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–925. doi:10.1126/science.1252510
44. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80. doi:10.1126/science.aaa6204
45. Sawant DV, Yano H, Chikina M, et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. 2019;20(6):724–735. doi:10.1038/s41590-019-0346-9
46. Yao M, Ventura P, Jiang Y, et al. Astrocytic trans-differentiation completes a multicellular paracrine feedback loop required for medulloblastoma tumor growth. Cell. 2020;180(3):502–520.e519. doi:10.1016/j.cell.2019.12.024
47. Gaydosik AM, Queen DS, Trager MH, et al. Genome-wide transcriptome analysis of the STAT6-regulated genes in advanced-stage cutaneous T-cell lymphoma. Blood. 2020;136(15):1748–1759. doi:10.1182/blood.2019004725
48. Dey P, Li J, Zhang J, et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov. 2020;10(4):608–625. doi:10.1158/2159-8290.CD-19-0297
49. Goswami KK, Bose A, Baral R. Macrophages in tumor: an inflammatory perspective. Clin Immunol. 2021;232:108875. doi:10.1016/j.clim.2021.108875
50. Dey P, Kimmelman AC, DePinho RA. Metabolic codependencies in the tumor microenvironment. Cancer Discov. 2021;11(5):1067–1081 doi:10.1158/2159-8290.CD-20-1211.
51. Di Pilato M, Kfuri-Rubens R, Pruessmann JN, et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell. 2021;184(17):4512–4530 e4522. doi:10.1016/j.cell.2021.07.015
52. Parlakpinar H, Ozer MK, Sahna E, et al. Amikacin-induced acute renal injury in rats: protective role of melatonin. J Pineal Res. 2003;35(2):85–90. doi:10.1034/j.1600-079X.2003.00059.x
53. Romagnani P, Rinkevich Y, Dekel B. The use of lineage tracing to study kidney injury and regeneration. Nat Rev Nephrol. 2015;11(7):420–431. doi:10.1038/nrneph.2015.67
54. Bakhautdin B, Das D, Mandal P, et al. Protective role of HO-1 and carbon monoxide in ethanol-induced hepatocyte cell death and liver injury in mice. J Hepatol. 2014;61(5):1029–1037. doi:10.1016/j.jhep.2014.06.007
55. Chiew AL, James LP, Isbister GK, et al. Early Acetaminophen-protein adducts predict hepatotoxicity following overdose (ATOM-5). J Hepatol. 2020;72(3):450–462. doi:10.1016/j.jhep.2019.10.030
56. van Rijt SH, Bolukbas DA, Argyo C, et al. Protease-mediated release of chemotherapeutics from mesoporous silica nanoparticles to ex vivo human and mouse lung tumors. ACS Nano. 2015;9(3):2377–2389. doi:10.1021/nn5070343
57. Bakalar MH, Joffe AM, Schmid EM, et al. Size-dependent segregation controls macrophage phagocytosis of antibody-opsonized targets. Cell. 2018;174(1):131–142 e113. doi:10.1016/j.cell.2018.05.059
58. Jaumouille V, Cartagena-Rivera AX, Waterman CM. Coupling of beta(2) integrins to actin by a mechanosensitive molecular clutch drives complement receptor-mediated phagocytosis. Nat Cell Biol. 2019;21(11):1357–1369. doi:10.1038/s41556-019-0414-2
59. Frank AC, Ebersberger S, Fink AF, et al. Apoptotic tumor cell-derived microRNA-375 uses CD36 to alter the tumor-associated macrophage phenotype. Nat Commun. 2019;10(1):1135. doi:10.1038/s41467-019-08989-2
60. Liu L, Yi H, He H, et al. Tumor associated macrophage-targeted microRNA delivery with dual-responsive polypeptide nanovectors for anti-cancer therapy. Biomaterials. 2017;134:166–179. doi:10.1016/j.biomaterials.2017.04.043
61. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi:10.1016/j.cell.2010.03.014
62. Kersten K, Hu KH, Combes AJ, et al. Spatiotemporal co-dependency between macrophages and exhausted CD8(+) T cells in cancer. Cancer Cell. 2022;40(6):624–638 e629. doi:10.1016/j.ccell.2022.05.004
63. Hong S, Zhang Z, Liu H, et al. B cells are the dominant antigen-presenting cells that activate naive CD4(+) T cells upon immunization with a virus-derived nanoparticle antigen. Immunity. 2018;49(4):695–708 e694. doi:10.1016/j.immuni.2018.08.012
64. Chiossone L, Dumas PY, Vienne M, et al. Author correction: natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18(11):726. doi:10.1038/s41577-018-0077-4
65. Etxeberria I, Bolanos E, Quetglas JI, et al. Intratumor adoptive transfer of IL-12 mRNA transiently engineered antitumor CD8(+) T cells. Cancer Cell. 2019;36(6):613–629 e617. doi:10.1016/j.ccell.2019.10.006
66. Chen K, Wang S, Qi D, et al. Clinical investigations of CAR-T cell therapy for solid tumors. Front Immunol. 2022;13:896685. doi:10.3389/fimmu.2022.896685
67. Kirtane K, Elmariah H, Chung CH, et al. Adoptive cellular therapy in solid tumor malignancies: review of the literature and challenges ahead. J Immunother Cancer. 2021;9(7):e002723. doi:10.1136/jitc-2021-002723
68. Nikitenko LL. Vascular endothelium in cancer. Cell Tissue Res. 2009;335(1):223–240. doi:10.1007/s00441-008-0707-4
69. Wofford KL, Singh BS, Cullen DK, et al. Biomaterial-mediated reprogramming of monocytes via microparticle phagocytosis for sustained modulation of macrophage phenotype. Acta Biomater. 2020;101:237–248. doi:10.1016/j.actbio.2019.11.021
70. Allavena P, Palmioli A, Avigni R, et al. PLGA based nanoparticles for the monocyte-mediated anti-tumor drug delivery system. J Biomed Nanotechnol. 2020;16(2):212–223. doi:10.1166/jbn.2020.2881
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.