")
Back to Journals » OncoTargets and Therapy » Volume 12
Suppressing Hedgehog signaling reverses drug resistance of refractory acute myeloid leukemia
Authors Huang K, Sun Z, Ding B, Jiang X, Wang Z, Zhu Y, Meng F
Received 22 May 2019
Accepted for publication 16 August 2019
Published 11 September 2019 Volume 2019:12 Pages 7477—7488
DOI https://doi.org/10.2147/OTT.S216628
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Kaikai Huang,1–3 Zhiqiang Sun,1 Bingjie Ding,2,4 Xuejie Jiang,2 Zhixiang Wang,2 Yufeng Zhu,2 Fanyi Meng2
1Department of Hematology, Shenzhen Hospital, Southern Medical University, Shenzhen, People’s Republic of China; 2Department of Hematology, Nanfang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 3Department of Hematology, The First Affiliated Hospital of Jinan University, Guangzhou, People’s Republic of China; 4Department of Hematology, Affiliated Tumor Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China
Correspondence: Fanyi Meng
Department of Hematology, Kanghua Hospital, Dongguan 523080, Guangdong, People’s Republic of China
Email [email protected]
Background: Hedgehog (Hh) signaling is involved in the pathogenesis of tumors. By performing gene chip analysis, we predicted that Hh signaling might regulate multiple downstream pathways in acute myeloid leukemia (AML).
Methods: In this study, the potential role of the Hh pathway in refractory AML, and the impact of Hh expression on clinical prognosis were examined. We also investigated the role of the Hh inhibitor NVP-LDE225 in reversing drug resistance of refractory primary AML cells in vitro and the roles of multiple drug-resistant HL60/Adriamycin-resistant cells in vitro and in vivo (in a xenograft mouse model). Finally, we explored the underlying mechanisms.
Results: Hh pathway was highly active in chemotherapy-resistant AML cells; by contrast, activation was less pronounced in chemosensitive cells and non-refractory primary cells. Strong activation of this pathway was associated with higher recurrence rates and poorer relapse-free and overall survival. NVP-LDE225 inhibited MRP1 protein expression, increased intracellular accumulation of Adriamycin, and reversed chemotherapeutic resistance. These effects were likely mediated through inhibition of the IGF-1R/Akt/MRP1 pathway. In the AML xenograft mouse model, NVP-LDE225 plus Adriamycin resulted in marked tumor regression.
Conclusion: These findings suggest that targeting the Hh pathway might be a therapeutic avenue for overcoming MDR resistance and preventing refractory AML.
Keywords: Hedgehog pathway, drug resistance, acute myeloid leukemia
Introduction
Acute myeloid leukemia (AML) is the most common myeloid malignancy in adults. Sixty percent of AML patients achieve complete remission (CR) after standard chemotherapy; unfortunately, most become insensitive to chemotherapeutic drugs, relapse, and eventually die of the disease.1 Previous studies suggest various mechanisms that underlie drug resistance in leukemia; these include genetic mutations, activated/suppressed signaling pathways, the tumor microenvironment, and the presence of highly resistant stem cells.2,3 IGF-1R/PI3K/Akt signaling promotes the growth and survival of human acute leukemia cells.4–6 Previously, we showed that combined treatment of Adriamycin-resistant cells (HL60/ADR) and refractory primary AML cells with the histone deacetylase (HDAC) inhibitor panobinostat plus the proteasome inhibitor bortezomib exerts synergistic effects via inhibition of the PI3K/Akt/MRP1 pathway.7 Gene chip analysis led us to predict that the Hedgehog (Hh) signaling pathway was a major signaling cascade that regulates multiple downstream pathways.
The Hh signaling pathway is complex and functions via two cellular receptors: patched (Ptch) and smoothened (Smo). Under non-ligated conditions, Ptch1 represses Smo, thereby silencing the Hh signaling pathway.4 Once Hh binds to Ptch, Smo is able to transduce the Hh signal, which is mediated via glioma-associated oncogene homologue (Gli) transcription factors.5,6 Dysregulation of Hh signaling, which is an essential pathway for embryonic development, is implicated in a variety of cancers, including human pancreatic carcinoma, colorectal cancer, prostate cancer, small-cell lung cancer, basal cell carcinomas, and hematological malignancies.7–10 For example, the Hh pathway is important for the survival and expansion of Bcr-Abl+ leukemia stem cells.11 In accordance with these findings, others showed that Hh signaling plays an important role in the self-renewal of hematopoietic stem cells and in the maintenance of cancer stem cells in those with leukemia.12 Evidence suggests that Hh might induce chemoresistance in leukemia cells. Previous studies show that Hh signaling induces radio-resistance in esophageal adenocarcinoma cells.13 Min Xu et al found that inhibiting Hh signaling improves the efficacy of chemotherapy in patients with pancreatic ductal adenocarcinoma,14 and others showed that Hh inhibitors restore drug resistance in CD34+ leukemia cells.15
In this study, we evaluated expression of Hh in Adriamycin-sensitive (HL60) and multidrug-resistant (HL60/ADR) cell lines, as well as in non-refractory and refractory primary AML cells. We also analyzed whether expression of Hh pathway components correlates with clinical prognosis. In addition, we investigated the effect of NVP-LDE225, a potent inhibitor of Smo, on the proliferation, apoptosis, and drug resistance of HL60/ADR cells and cells isolated from refractory AML patients, as well as its effect in a mouse model of leukemia. We found that HL60/ADR and refractory primary AML cells showed higher expression of Hh than HL60 or non-refractory primary AML cells. Furthermore, higher expression of Hh was related to higher recurrence rates and poorer relapse-free survival (RFS) and overall survival (OS). The Hh inhibitor NVP-LDE225 reversed resistance to multiple chemotherapy drugs, increased intracellular accumulation of Adriamycin, and induced apoptosis in both HL60/ADR and refractory primary AML cells. In addition, NVP-LDE225 decreased expression of MRP1, Bcl-2, p-IGF-1R, IRS-1, p-Akt, and Gli-1, suggesting that Hh regulates the IGF-1R/Akt/MRP1 pathway, which is consistent with the results obtained from gene chip analysis.
Patients and methods
AML patients and cells
The patient subject study was approved by the Institutional Ethics Committee of the First Affiliated Hospital Of Jinan University. And was performed in accordance with the Declaration of Helsinki and the guidelines of the Ethics Committee of Jinan University. Written informed consent was also provided by all participants. Sixteen refractory and fifteen non-rsefractory AML patients recruited from the Department of Hematology in the First Affiliated Hospital Of Jinan University and Nanfang Hospital and were enrolled in the study. All patients provided informed consent to participate in the study. Refractory patients were defined as persons who either failed to achieve CR following at least two induction cycles, or persons in whom disease recurred within 12 months of CR (<5% marrow blasts) and who were resistant to the reinduction protocol.2 Bone marrow mononuclear cells (BMMCs) were isolated on Lymphocyte Separation Medium (TBD, China) and suspended in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS; GIBCO, USA). HL60 and HL60/ADR cells (Chinese Academy of Sciences, Shanghai, China) were cultured in RPMI-1640 medium (Hyclone, USA) supplemented with 10% heat-inactivated FBS (GIBCO) in a humidified atmosphere of 5% CO2 at 37 °C. HL60/ADR cells were maintained in 1.0 µg/mL Adriamycin (Doxorubicin) (Sigma, USA) to ensure drug resistance. HL60/ADR displayed 79-fold higher resistance to Adriamycin than the parental line.3 Cells (1×105) cells/mL were incubated with different chemotherapy agents as described below. All experiments were repeated at least three times.
Reagents
Panobinostat was provided by Novartis Pharmaceuticals (Basel, Switzerland) and bortezomib was purchased from Millennium Pharmaceuticals (Cambridge, MA, USA). NVP-LDE225 was obtained from Selleck BioSciences Corporation (Shanghai, China) and chemotherapeutic drugs were purchased from Nanfang Hospital. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and anti-GAPDH and anti-β-actin antibodies were purchased from Sigma (St. Louis, MO, USA). An Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining kit was purchased from NanJing KeyGen Biotechnology (NanJing, China). Primary antibodies specific for phosphorylated IGF-1R (p-IGF-1R), IGF-1R, IRS-1, phosphorylated AKT (p-AKT), AKT, and Bcl-2 were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies specific for Sonic hedgehog (Shh), Ptch, Smo, and Gli-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) or Abcam (USA). An antibody specific for MRP1 and secondary horseradish peroxidase-conjugated goat anti-mouse IgG and goat anti-rabbit IgG antibodies were obtained from Abcam (USA).
RNA extraction
HL60/ADR cells were seeded in 75 cm2 plates at a concentration of 1×107 cells/plate and then treated for 48 h with panobinostat and bortezomib (final concentrations of 21 nM and 12 nM, respectively), or with a combination of both. RNA was extracted using Trizol (Invitrogen, USA), according to the manufacturer’s recommended protocol. The quantity and purity of RNA were determined by measuring A260 and A280 in a NanoDrop ND-1000 spectrometer (NanoDrop). The integrity of the RNA was determined by 1.5% agarose gel electrophoresis.
Gene expression microarray
Total RNA was harvested from different groups of cells as described above. First-strand cDNA was transcribed with a primer containing a T7 oligo (dT) sequence. Next, multiple copies of biotin-modified aRNA were synthesized using the double-stranded cDNA as templates. The labeled aRNA was fragmented prior to hybridization with GeneChip® 3ʹ expression arrays. In the Affymetrix Genechip system, hybridization is performed using the Human Genome U133 Plus 2.0 Array. Differences in the gene expression profiles of treated and control HL60/ADR cells were then analyzed. A difference in gene expression was defined as a -fold change ≥2. After removing negative-flagged probes and data normalization using R’s “limma” package, differentially expressed genes were selected based on the fold-changes for functional enrichment using R programming (www.cran.r-project.org) and the NextBio platform (www.nextbio.com). Gene set enrichment analysis was performed using an in-house web tool, in which the main data sources are provided by MSigDB (Broad Institute), Reactome, BioCarta, and the Gene Ontology Consortium, in addition to gene sets collected from literature mining.
Assessment of cell viability
Cell lines or primary AML cells were treated with chemotherapeutic drugs, either alone or in combination with NVP-LDE225. Antiproliferative effects were measured in an MTT assay. Briefly, 20 µL MTT solution (5 mg/mL) in 200 µL medium plus drug was added to each well for the last 4 h of a 48 h culture. Absorbance was measured in a spectrophotometer (Thermo Scientific Evolution 600, China) at 540 nm. Cell viability was calculated and expressed as a percentage.
Apoptosis analysis
After 48 h of culture, 1×104 cells were collected and washed twice (each for 5 min) with cold PBS (pH =6.9). Cells were then stained for 30 min in the dark with 5 µL Annexin V-FITC and 5 µL PI prior to flow cytometry analysis (Becton Dickinson, CA, USA) of apoptosis.
Intracellular adriamycin accumulation assay
After pretreatment for 48 h with NVP-LDE225, HL60/ADR cells were incubated for 1 h at 37 °C with Adriamycin (0.5 µg/mL), washed twice with cold PBS, and subjected to flow cytometry analysis. Intracellular mean fluorescence intensity (MFI) associated with Adriamycin was measured at excitation/emission wavelengths of 488/575 nm, and intracellular accumulation of Adriamycin was calculated.
Western blot analysis
Western blot analysis was carried out as described previously.3 Briefly, HL60/ADR and primary AML cells were cultured for 48 h with different concentrations of NVP-LDE225. Cells were harvested and lysed in 200 µL cold lysis buffer (0.5 M Tris-HCl, pH 6.8, 2 mM EDTA, 10% glycerol, 2% SDS, 5% β-mercaptoethanol) containing 0.5 mM dithiothreitol (DTT) and 0.1 mM phenylmethylsulfonyl fluoride (PMSF). The supernatant was collected after centrifugation (12,000 rpm, 20 min), and protein concentration was determined using a BCA protein determination kit (Pierce Chemical, Rockford, IL, USA). Cell extracts were subjected to sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked with 5% skimmed milk or 5% BSA in Tris-buffered saline containing 0.1% Tween 20 and then incubated overnight at 4 °C with appropriate primary antibodies, followed by a horseradish peroxidase-linked secondary antibody. Reactions were visualized by enhanced chemiluminescence (Millipore, USA). Antibodies specific for β-actin or GAPDH were used to ensure equivalent loading.
AML xenograft mouse model
Xenograft tumor experiment was performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Ethics Review Committee of Nanfang Hospital, and was conducted in accordance with the Decalration of Helsiki. were established in female BALB/c nu/nu mice (4–6 weeks old) purchased from the Animal Resources Centre (Guangdong, China). Approximately 1×107 HL60/ADR cells in 200 µL RPMI medium were injected subcutaneously into the right flank of each mouse. When tumors reached 100–300 mm3 in volume, mice were divided into cohorts (n=6) and treated with: (a) vehicle alone, (b) NVP-LDE225 (80 mg/kg), (c) ADM (3 mg/kg), and (d) ADM (3 mg/kg) + NVP-LDE225 (80 mg/kg). Vehicle and LDE225 were administered by gavage once daily and ADM was administered i.p. q3d (three injections). In the combination therapy group, mice received a daily oral administration of NVP-LDE225 on days 1–10. Tumor size was measured every 2 days using a caliper and the tumor volume (V) was calculated as follows: V=1/2× a × b2, where a and b are the longest and the shortest diameters of the tumor (in mm). The rate of tumor inhibition was expressed in terms of relative tumor volume, calculated as follows: (1-Vt/V0) ×100%, where Vt and V0 are the average tumor volumes in the treatment and control groups, respectively. All tumors were excised and weighed. Mice were euthanized when the set endpoint of the experiment was reached.
Statistical analysis
The statistical significance of differences between groups was determined using a two-sided Student’s t-test or an independent-sample t-test. Kaplan–Meier survival curves and log-rank tests were used to estimate RFS and OS. The statistical significance of differences among tumor volumes was assessed by one-way analysis of variance (ANOVA). p<0.05 was considered significant. All data were obtained from three independent experiments.
Results
Panobinostat and bortezomib induce gene expression changes in HL60/ADR cells
Compared with the control group, HL60/ADR cells treated with panobinostat expressed 460 upregulated genes and 150 downregulated genes. After treatment with bortezomib, 221 genes were upregulated and 65 genes were downregulated. Finally, combined treatment led to upregulation of 1,301 genes and downregulation of 1,446 genes (Supplementary data). These genes are closely associated with EZH2 (p<0.05) as well as the proteasome (p<0.05). Many gene sets associated with histone methylation or the proteasome, both of which regulate downstream pathways such as the E2F transcription factor network, the C-MYC transcription factor network, TNF-mediated apoptosis, HSC progenitors, and the NF-kB, JAK-STAT, ERK/AKT, WNT, and PI3K/AKT pathways, were also enriched (Supplementary data). In addition, bioinformatics analyses using the literature mining tool FABLE (www.fable.chop.edu) revealed that the molecular interactions regulated the genes via a network (Figure 1). These data strongly suggest a non-random association among these genes. Within the network, the Hh pathway (a pathway associated with HSC progenitors and self-renewal) was considered to be the upstream molecular switch.
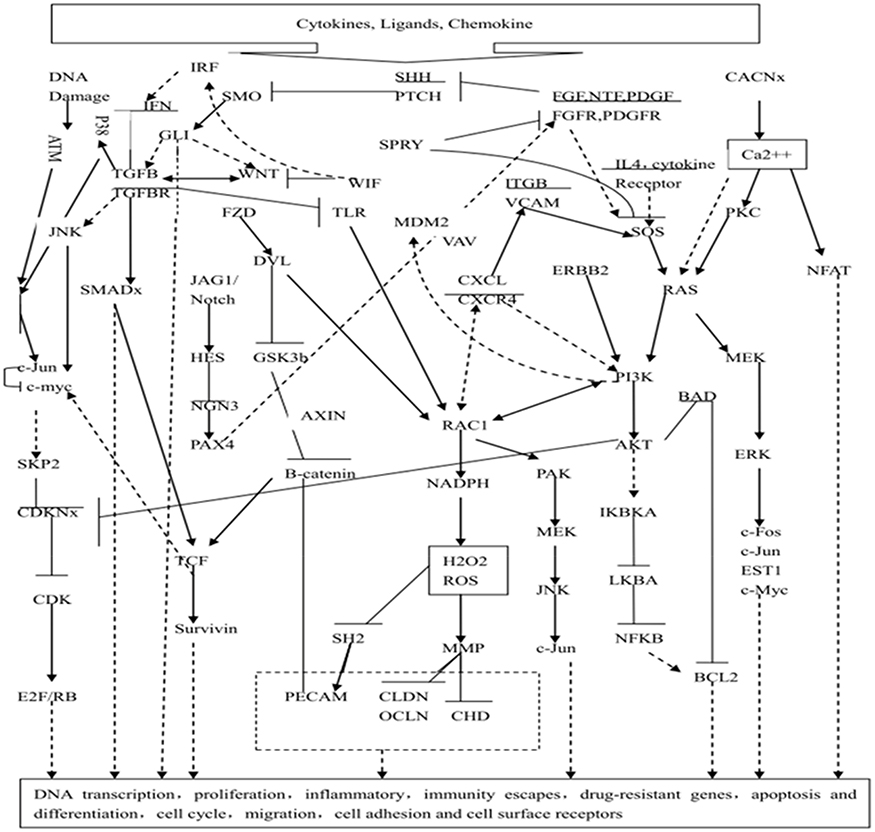 |
Figure 1 A hypothetical model of gene expression regulation by panobinostat and bortezomib. We propose a model in which hedgehog is the upstream molecular switch, thereby turning on/off downstream targets/pathways. Dash line: inhibition; solid line: activation. |
High expression of Hh is associated with refractory primary AML cells and a poor clinical prognosis
To explore the potential role of the Hh pathway in refractory AML, we examined expression of the Gli-1 protein, which is a surrogate marker of Hh signaling pathway activity.16 BMMC fractions from 31 diagnosed AML patients (16 refractory and 15 non-refractory patients; median age, 43 years (range, 16–62 years)) were examined. As shown in Figure 2A, 13/16 refractory patients expressed Gli-1 (81.3%); by contrast, only 4/15 non-refractory cases expressed Gli-1 (26.7%, p=0.002) (Figure 2B). The primary samples were divided into a Gli-1high group and a Gli-1low group. There were no differences between these groups in terms of gender distribution, age, WBC counts, bone marrow blast cell counts, and FAB subtypes (including treatment regimen and therapy after CR) (p>0.05). However, there were significant differences between the groups with respect to relapse rates, RFS, and OS. High expression of Gli was associated with rapid leukemia relapse or with several relapses (p=0.031). The RFS and OS of the Gli-1high group were significantly shorter than those of the Gli-1low group (p=0.009 and p=0.026). Survival curves are shown in Figure 2C and D.
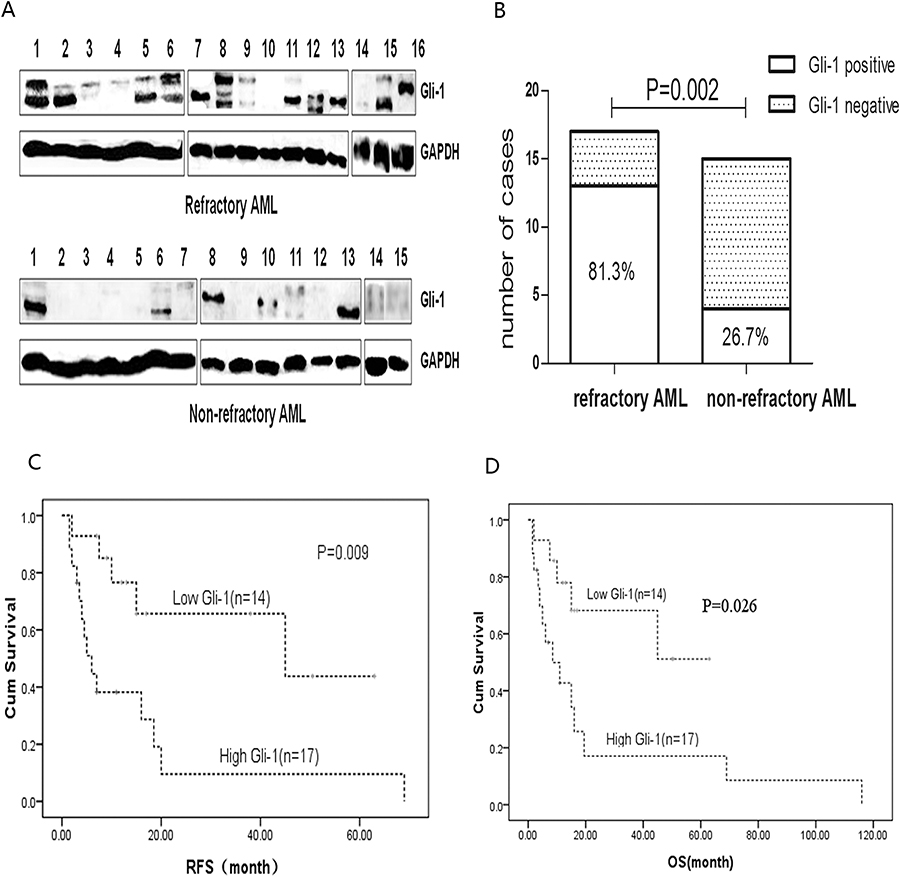 |
Figure 2 Protein expression of Hh pathway components in primary cells. (A) Protein expression of Gli-1 was detected by Western blotting of primary bone marrow mononuclear cells from 16 refractory and 15 non-refractory AML patients. (B) Significantly more samples from refractory than from non-refractory patients highly expressed Gli-1. Expression of Gli-1 was higher in refractory cells than in non-refractory cells (P=0.002). (C, D) Kaplan-Meier analysis of RFS and OS of patients. |
Expression of Hh pathway components is higher in refractory AML cell lines, and pharmacological inhibition of Hh signaling reverses drug resistance
So far, the data suggest that the Hh signaling pathway is more active in primary refractory AML cells and is associated with a poor prognosis. However, it is not clear whether high expression of Hh contributes to the drug resistance of AML cells, or to what extent the chemoresistant phenotype depends on this pathway. Figure 3A and B show RT-PCR and Western blot data. Expression of components of the Hh signaling pathway by the multidrug-resistant AML cell line HL60/ADR was higher than that by parental HL-60 cells; this was true at both the mRNA and protein level. Pharmacological inhibition of the Hh pathway in HL60 and HL60/ADR cells using NVP-LDE225 revealed that the inhibitor did not affect survival of HL60 and HL60/ADR cells when used at concentrations up to 10 µM for 48 h (data not shown). However, combination treatment with the chemotherapeutic agent LDE225 (10 µM) led to a marked increase in the sensitivity of HL60/ADR cells to Adriamycin, daunorubicin, homoharringtonine (HHT), and cytarabine (Figure 3C and Table 1). However, this effect was not observed in HL60 cells (Figure 3D).
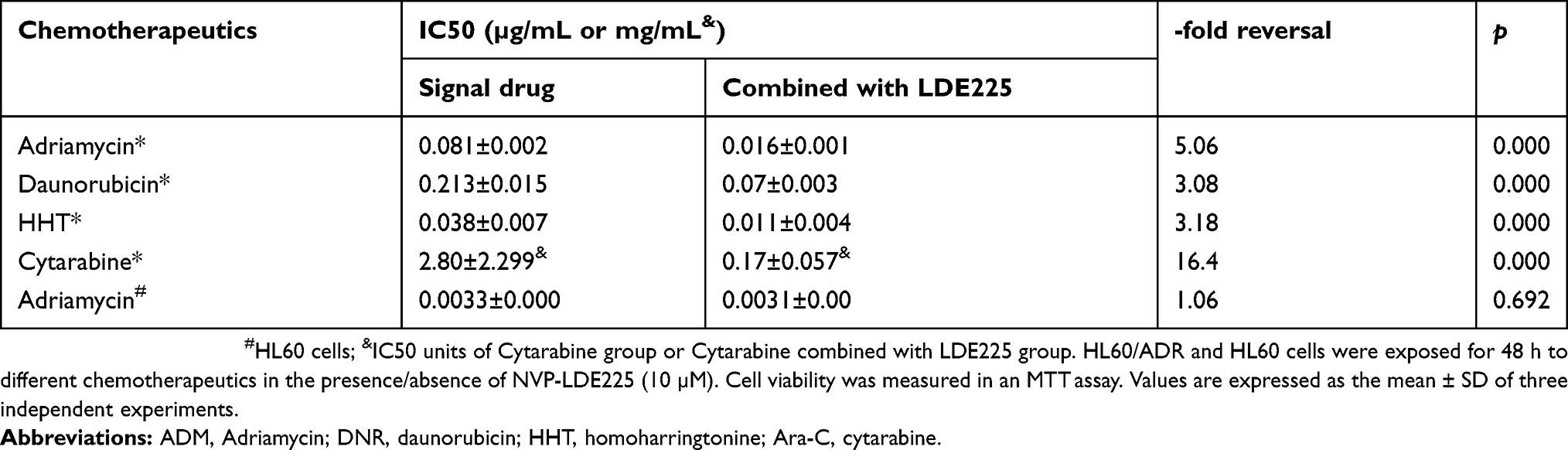 |
Table 1 NVP-LDE225 increases the cytotoxicity of chemotherapeutic agents in vitro |
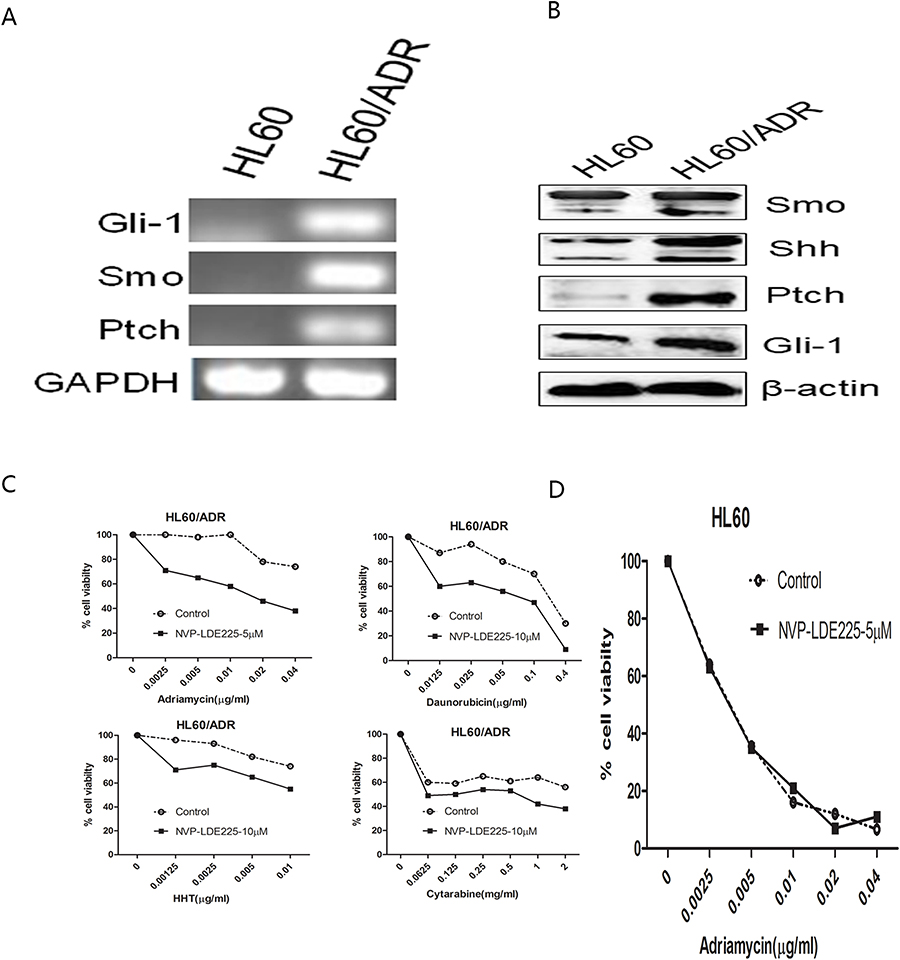 |
Figure 3 Expression of Hh signaling components is higher in HL60/ADR cells than in HL60 cells, and inhibition of Hh signaling reverses drug resistance of HL60/ADR cells. (A) Agarose gel electrophoresis demonstrated that Hh signaling molecules were highly expressed in HL60/ADR cells. (B) Protein expression of Hh signaling molecules was higher in HL60/ADR cells than in HL60 cells. (C) HL60/ADR cells were treated with increasing concentrations of Adriamycin, Daunorubicin, Cytarabine, or Homoharringtonine in the presence of 10 µM NVP-LDE225 for 48 h, and then cell viability was determined. (D) HL60 cells were treated with NVP-LDE225 (5µM) in combination with different concentrations of Adriamycin for 48 h. |
NVP-LDE225 increases apoptosis of HL60/ADR cells and intracellular accumulation of adriamycin
To determine whether NVP-LDE225 alone induces apoptosis, HL60/ADR cells were treated with the drug (0, 5 µM, 10 µM, or 20 µM) for 48 h. Apoptosis was determined by flow cytometry, including early apoptotic (Annexin V-FITC positive, PI negative) and late apoptotic/necrotic (Annexin V-FITC and PI positive) cells (Figure 4A). Significant apoptotic cell death was observed at only 20 µM (p<0.0001). Next, cells were treated for 48 h with NVP-LDE225 and incubated for 1 h with Adriamycin. Intracellular accumulation of Adriamycin was measured by flow cytometry. As shown in Figure 4B, the MFI associated with intracellular Adriamycin was significantly higher in cells exposed to LDE225 (5 µM or 10 µM) than in untreated HL60/ADR cells (p<0.0001). Importantly, at these doses, NVP-LDE225 did not induce marked cell death.
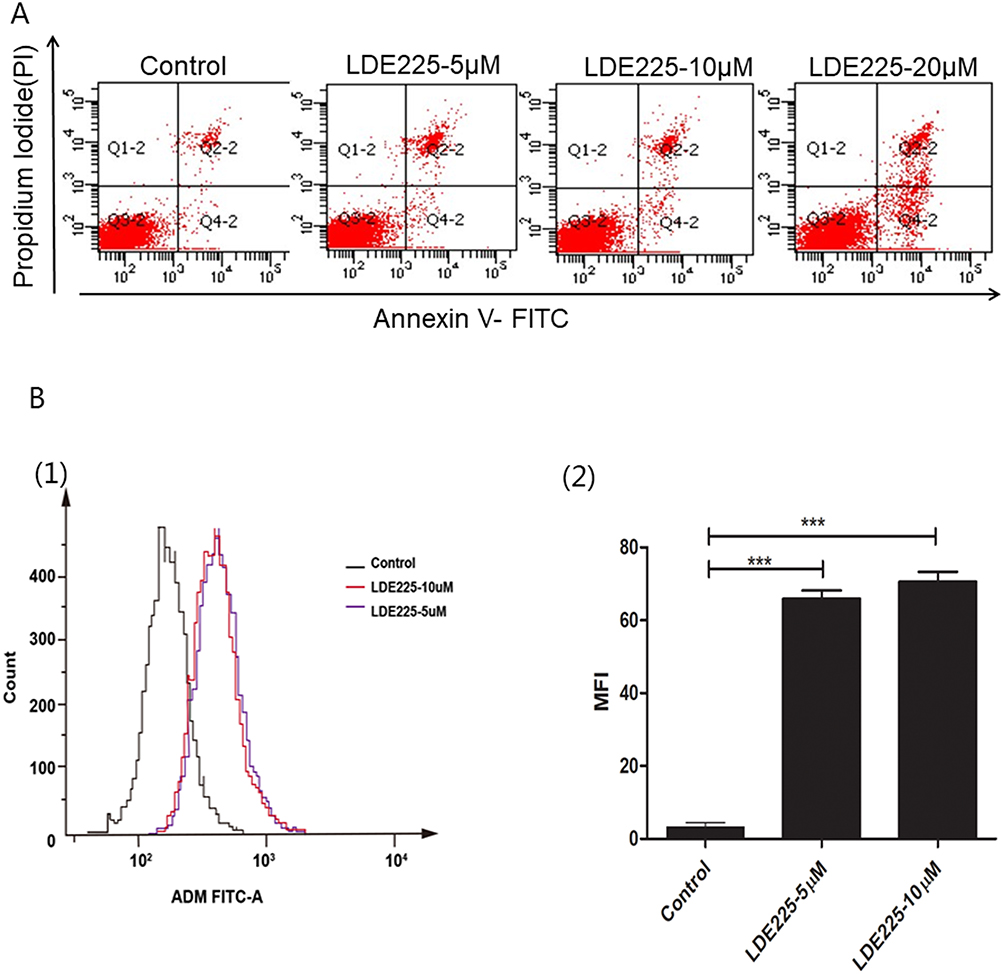 |
Figure 4 Inhibition of Hh signaling increases apoptosis of HL60/ADR cells and intracellular accumulation of Adriamycin. (A) After exposure to different doses of NVP-LDE225 for 48 h, HL60/ADR cells were stained with Annexin V-FITC/PI and subjected to flow cytometry to detect apoptotic cells. The percentage of apoptotic cells was significantly higher in the 20 µM-treated group than in the low-dose-treated group. (B) Effects of NVP-LDE225 on ADR accumulation in HL60/ADR cells. MFI was significantly increased in the LDE225-treated groups. Means ± SD of three experiments are presented. Results marked with an asterisk were statistically significant (***P<0.0001). |
NVP-LDE225 reverses resistance to chemotherapeutics by inhibiting the IGF-1R/Akt/MRP-1 pathway
Previously, we showed that bortezomib plus panobinostat inhibits PI3K/Akt/MRP1 signaling to reverse drug resistance.3 Here, gene chip analysis revealed that Hh signaling might act upstream of the IGF-1R/Akt/MRP1 signaling pathway. To investigate whether the Hh inhibitor NVP-LDE225 also inhibited IGF-1R/Akt/MRP1, we treated HL60/ADR cells with NVP-LDE225 and examined expression of signaling molecules by Western blotting. As shown in Figure 5, compared with the control, treatment with different concentrations of LDE225 could decrease expression of p-IGF-1R, p-Akt, and Gli-1 (a marker of Hh pathway activation).
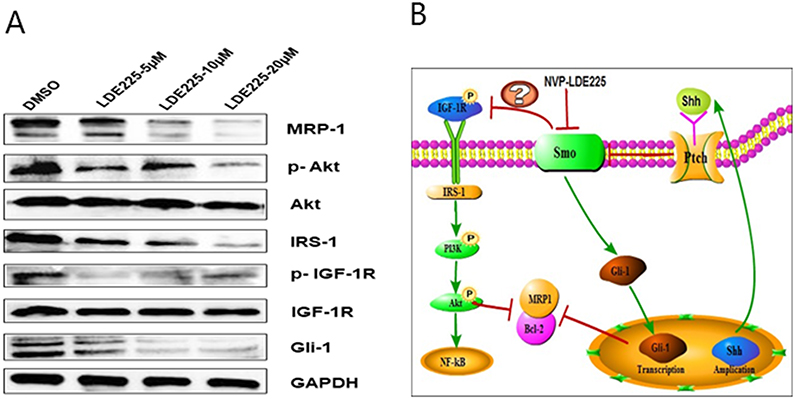 |
Figure 5 Protein expression changes in HL60/ADR cells treated with different doses of NVP-LDE225 for 48 h. (A) Cellular protein expression was determined by Western blotting. Expression of Gli-1, p-IGF-1R, p-Akt, and MRP1 was decreased, whereas expression of IGF-1R and Akt did not change. (B) Proposed model to explain the relationship between the Shh/Gli-1 and IGF-1R/Akt/MRP1 pathways in refractory AML. |
Effects of the Hh inhibitor NVP-LDE225 on cells from refractory AML patients
Next, we examined the biological effects of NVP-LDE225 on cells isolated from refractory or relapsed AML patients. Representative results from a single relapse patient are shown in Figure 6A (six patients were tested in all). Cell viability in the presence of 10 µM NVP-LDE225 plus different doses of Adriamycin was significantly lower than that in the presence of Adriamycin alone. In addition, NVP-LDE225 itself induced apoptosis and increased uptake of Adriamycin (Figure 6B and C). Finally, NVP-LDE225 reduced expression of MRP1 in a dose-dependent manner (Figure 6D).
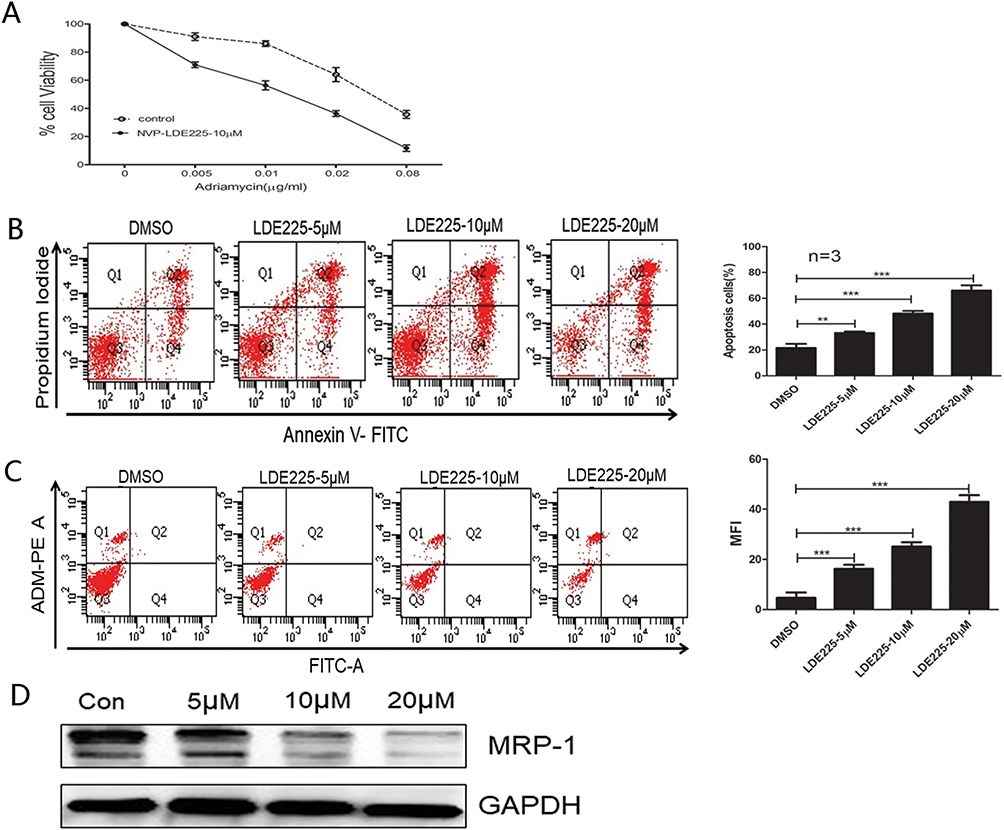 |
Figure 6 Refractory primary cells were treated with NVP-LDE225 for 48 h. (A) MTT cytotoxicity assay of primary cells exposed to Adriamycin in combination with 10µM NVP-LDE225. (B) Primary cells were exposed to different concentrations of NVP-LDE225 for 48 h and then analyzed by the Annexin V/PI assay. Means ± SD of three experiments are presented. Results marked with an asterisk were statistically significant (**P<0.005; ***P<0.0001). (C) After primary cells were treated with LDE225 for 48 h, accumulation of Adriamycin in the medium (right upper quadrant) with 0.5 µg/mL Adriamycin was measured and analyzed by flow cytometry. Means ± SD of three experiments are presented. Results marked with an asterisk were statistically significant (***P<0.0001). (D) Primary cells were treated with different doses of NVP-LDE225 for 48 h. MRP1 was detected by Western blotting. |
NVP-LDE225 plus adriamycin reduces the volume of tumors in mice
Finally, we used a mouse subcutaneous xenograft model (bearing HL60/ADR tumors) to determine whether the combination of ADM plus NVP-LDE225 reverses ADM resistance in vivo. ADM (3 mg/kg on days 1, 4, and 7) plus LDE225 (80 mg/kg on days 1–10) inhibited tumor growth to a significantly (p<0.05) greater extent than either drug alone or the control. Although ADM and LDE225 alone inhibited tumor growth in mice when compared with the control, the differences were not significant (Figure 7). Daily oral administration of NVP-LDE225 (80 mg/kg) and intraperitoneal injection of ADM (3 mg/kg) showed only modest antitumor efficacy (6.1% and 16.1% tumor growth inhibition, respectively, compared with untreated control mice). Combined treatment with ADM plus LDE225 inhibited tumor growth by 75.8% compared with the control, suggesting that combination therapy with a Smo inhibitor improves responses to ADM.
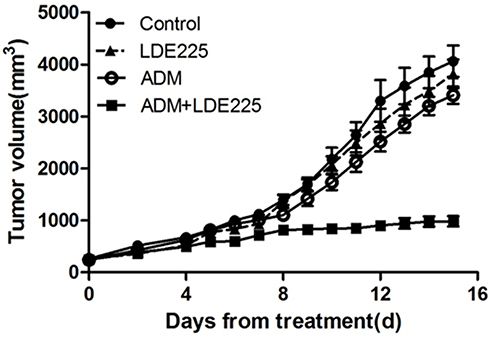 |
Figure 7 A mouse model with subcutaneous HL60/ADR xenografts was used to evaluate the in vivo antitumor efficacy of NVP-LDE225 in combination with Adriamycin (ADM). Mice were treated with vehicle, NVP-LDE225, ADM, or NVP-LDE225 plus ADM as detailed in the Methods section. Antitumor activity was higher in mice treated with NVP-LDE225 plus ADM than in mice treated with the vehicle control, ADM alone, and LDE225 alone (P=0.003, P=0.006, and P=0.045, respectively). |
Discussion
Multidrug resistance (MDR) is characterized mainly by simultaneous resistance to a variety of antitumor drugs with different structures and molecular targets.2,3 Intrinsic and acquired MDR against chemotherapy agents remains one of the major problems when treating leukemia.13,16–18 The mechanism underlying MDR is complex and includes reduced drug accumulation in target cells, increased DNA repair, and blockade of signaling pathways. Activation of the Hh pathway plays a crucial role in the proliferation, survival, and drug resistance of leukemic cells and CML stem cells.19
In this study, we showed that Hh pathway signaling components are overexpressed in refractory primary AML cells and drug-resistant AML cells. Also, we demonstrated that expression of Hh pathway components correlates with a poor clinical prognosis. Moreover, targeting the Hh pathway sensitized drug-resistant AML cells to chemotherapeutic agents induced tumor cell apoptosis and increased intracellular accumulation of Adriamycin. Also, we demonstrated that the Hh pathway might regulate the IGR-1R/Akt/MRP1 pathway in AML cells. Participation of Hh signaling in AML cell survival and chemotherapy resistance makes it an attractive target, especially in patients who relapse and succumb to chemoresistant disease. All of the findings presented herein are consistent with those of a previous study by Hiba Ahamd Zahreddine et al20. They also found that overexpression of Gli-1 contributes to drug resistance and clinical relapse in some AML patients and that inhibiting Gli-1 restores sensitivity to clinically relevant doses of Ara-C. In addition, Hh mutations are associated with resistance to induction chemotherapy in patients with T-cell acute lymphoblastic leukemia.21
HL-60/ADR is a multidrug-resistant AML cell line that overexpresses MRP1, which is a drug efflux pump responsible for drug resistance and a poor prognosis in AML.22,23 Also, the Bcl-2 protein regulates cell apoptosis via mitochondria-intrinsic pathways and may thus be a therapeutic target for leukemia.24 We demonstrated that a combination of panobinostat and bortezomib shows synergistic cytotoxic effects against HL60/ADR cells and chemoresistant primary AML cells by down-regulating the PI3K/Akt/MRP1 pathway and Bcl-2 expression. Here, gene chip analysis, cluster analysis, and signal transduction pathway analysis revealed that bortezomib affects the ubiquitin proteasome system, NF-kB, P53, reactive oxygen species, and mitochondria; these findings are in agreement with those in patients with leukemia and multiple myeloma.25,26 In addition, panobinostat acts on the FOS, JNK, P53, and TGFβ/TGFβR pathways,27–29 all of which have been investigated in the context of other tumors. In this study, we analyzed gene networks and found that the Hh pathway acts upstream of all dysregulated pathway networks; therefore, it may regulate other signaling pathways such as PI3K/Akt. Next, we analyzed the differential expression of Hh pathway components in refractory and non-refractory AML cells, investigated the ensuing biological effects, and explored the relationship between Hh and the PI3K/Akt pathway. We found that Hh may regulate the IGF-1R/PI3K/Akt pathway to reverse drug resistance.
Hh signaling plays a role in solid tumors and hematological diseases, including acute or chronic myeloid leukemia, multiple myeloma, lymphoma, and others. However, crosstalk between Hedgehog and other signaling pathways, especially PI3K/Akt, is rarely reported. Francisco Vega et al reported that Shh and Gli-1 proteins are upregulated in ALK+ ALCL cell lines and tumor samples, and that inhibition of Shh/Gli1 signaling pathways reduces cell viability, colony formation, and induction of cycle arrest. They also showed synergism between PI3K/Akt and Hh signaling, and that activation of Akt contributes to activation of the Hh signaling pathway.18 Others found that Hedgehog signaling regulates Stat3 activation but does not induce phosphorylation of Akt or p44/42-MAPK in CD34+ CML cells.30 Jillian/Brechbiel summarized the crosstalk between Hh and other pathways such as RAS/RAF/MEK/ERK, PI3K/AKT/mTOR, EGFR, and Notch.31 The results reported herein indicate that a combination of an Hh inhibitor plus another therapeutic agent would be a better choice for treating leukemia. Figure 5B shows a schematic diagram summarizing the proposed relationship between Hh/Gli-1 and PI3K/Akt in refractory AML. These findings are in agreement with the previous findings.
Taken together, the results of this study suggest differential expression of Hh pathway components in AML, which affect the proliferation, apoptosis, and drug resistance of cancer cells. Also, the results from an AML xenograft mouse model reveal that combined treatment with NVP-LDE225 and ADM results in marked tumor regression. These findings provide evidence that the Hh/IGF-1R/Akt/MRP1 pathway plays a role in refractory AML. Inhibiting this pathway may overcome drug resistance and improve therapeutic outcomes for patients with refractory AML.
Acknowledgments
This work was supported by grants from the Science Foundation of Guangdong Province (no. S2011010003807), the National High Technology Research and Development Program of China (863) (no. 2012AA02A505), and the Natural Science Foundation of China (no. 81073099).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106(4):1154–1163. doi:10.1182/blood-2005-01-0178
2. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. doi:10.1038/nrc706
3. Tsuruo T, Naito M, Tomida A, et al. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. 2003;94(1):15–21. doi:10.1111/j.1349-7006.2003.tb01345.x
4. Doepfner KT, Spertini O, Arcaro A. Autocrine insulin-like growth factor-I signaling promotes growth and survival of human acute myeloid leukemia cells via the phosphoinositide 3-Kinase/Akt pathway. Leukemia. 2007;21(9):1921–1930. doi:10.1038/sj.leu.2404813
5. Tamburini J, Chapuis N, Lacombe C, Bouscary D. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood. 2008;111(1):379–382. doi:10.1182/blood-2007-03-080796
6. Chapuis N, Tamburini J, Cornillet-Lefebvre P, Lacombe C, Bouscary D. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica. 2010;95(3):415–423. doi:10.3324/haematol.2009.010785
7. Jiang X-J, Huang -K-K, Yang M, Meng F-Y. Synergistic effect of panobinostat and bortezomib on chemoresistant acute myelogenous leukemia cells via AKT and NF-kB pathways. Cancer Lett. 2012;326(2):135–142. doi:10.1016/j.canlet.2012.07.030
8. Karhadkar SS, Bova GS, Abdallah N, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707–712. doi:10.1038/nature02962
9. Vestergaard J, Pedersen MW, Pedersen N. Hedgehog signaling in small-cell lung cancer: frequentin vivo but a rare event in vitro. Lung Cancer. 2006;52(3):281–290. doi:10.1016/j.lungcan.2005.12.014
10. Oro AE, Higgins KM, Hu Z. Basal cell carcinomas in mice overexpressing Sonic Hedgehog. Science. 1997;276(5313):817–821. doi:10.1126/science.276.5313.817
11. Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14(3):238–249. doi:10.1016/j.ccr.2008.08.003
12. Hofmann I, Stover EH, Cullen DE, Mao J, Morgan KJ, Lee BH. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell. 2009;4(6):559–567. doi:10.1016/j.stem.2009.03.016
13. Sims-Mourtada J, Izzo JG, Apisarnthanarax S, Wu TT, Malhotra U, Luthra R. Hedgehog: an attribute to tumor regrowth after chemoradiotherapy and a target to improve radiation response. Clin Cancer Res. 2006;12(21):6565–6572. doi:10.1158/1078-0432.CCR-06-0176
14. Xu M, Li L, Liu Z, Zhang Y. ABCB2(TAP1) as the downstream target of SHH signaling enhances pancreatic ductal adenocarcinoma drug resistance. Cancer Lett. 2013;333(2):152–158. doi:10.1016/j.canlet.2013.01.002
15. Kobune M, Takimoto R, Murase K, Kato J. Drug resistance is dram-atically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci. 2009;100(5):948–955. doi:10.1111/j.1349-7006.2009.01111.x
16. O’Hare T, Corbin AS, Druker BJ. Targeted CML therapy: controlling drug resistance, seeking cure. Curr Opin Genet Dev. 2006;16(1):92–99. doi:10.1016/j.gde2005.11.002
17. Diehl KM, Keller ET, Ignatoski KM. Why should we still care about oncogenes? Mol Cancer Ther. 2007;6(2):418–427. doi:10.1158/1535-7163.MCT-06-0603
18. Lowenberg B. On the road to new drugs in acute myeloid leukemia. JCO. 2007;25(1):1–2. doi:10.1200/JCO.2006.08.6462
19. Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–779. doi:10.1038/nature07737
20. Zahreddine HA, Clujkovic-Kralijacic B, Assouline S, Miller WH
21. Burns MA, Liao ZW, Yamagata N, Sallan SE, Gutierrez A. Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia. 2018;32(10):2126–2137. doi:10.1038/s41375-018-0097-x
22. Bonhoure E, Pchejetski D, Aouali N, et al. Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting sphingosine kinase-1. Leukemia. 2006;20(1):95–102. doi:10.1038/sjleu.2404023
23. Mahadevan D, List AF. Targeting the multidrug resistance-1 transporter in AML: molecular regulation and therapeutic strategies. Blood. 2004;104(7):1940–1951. doi:10.1182/blood-2003-07-2490
24. Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than Bcl-2-dependent counterparts. J Cell Biol. 2009;187(3):429–442. doi:10.1083/jcb.200904049
25. Yin L, Kufe T, Avigan D, Kufe D. Targeting MUC1-C is synergistic with bortezomib in downregulating TIGAR and inducing ROS-mediated myeloma cell death. Blood. 2014;123(19):2997–3006. doi:10.1182/blood-2013-11-539395
26. Uttenveiler-Joseph S, Bouyssie D, Calligaris D, Lutz PG, Monsarrat B, Burlet-Schilltz O. Quantitative proteomic analysis to decipher the differential apoptotic response of bortezomib-treated APL cells before and after retinoic acid differentiation reveals involvement of protein toxicity mechanisms. Proteomics. 2013;13(1):37–47. doi:10.1002/pmic.201200233
27. Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32(3):157–165. doi:10.1016/j.ctrv.2005.12.006
28. Kelly WK. Marks PA.Drug insight:histone deacetylase inhibitors development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol. 2005;2(3):150–157. doi:10.1038/ncponc0106
29. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6(1):38–51. doi:10.1038/nrc1779
30. Sengupta A, Banerjee D, Chandra S, et al. Deregulation and cross talk among Sonic hedgehog, Wnt, Hox and Notch signaling in chronic myeloid leukemia progression. Leukemia. 2007;21(5):945–950. doi:10.1038/sj.leu.2404657
31. Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treatment Reviews. 2014;40(6):750–759. doi:10.1016/j.ctrv.2014.02.003
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.