")
Back to Journals » OncoTargets and Therapy » Volume 9
Sunitinib in the treatment of gastrointestinal stromal tumor: patient selection and perspectives
Authors Mulet-Margalef N, Garcia-del-Muro X
Received 1 September 2016
Accepted for publication 26 October 2016
Published 15 December 2016 Volume 2016:9 Pages 7573—7582
DOI https://doi.org/10.2147/OTT.S101385
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Nuria Mulet-Margalef, Xavier Garcia-del-Muro
Sarcoma Multidisciplinary Unit and Medical Oncology Department, Institut Català d’Oncologia Hospitalet, IDIBELL, Barcelona, Spain
Abstract: Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor of the gastrointestinal tract. In advanced setting and after progression to imatinib, the multitargeted receptor tyrosine kinase inhibitor sunitinib has clearly demonstrated a clinical benefit in terms of response rate and progression-free survival with an acceptable toxicity profile. The recommended schedule for sunitinib administration is 50 mg per day 4 weeks ON and 2 weeks OFF; however, potential alternative schedules are also reviewed in the present article. Several biomarkers have been explored to better select candidates for sunitinib therapy, such as the value of early changes in standardized uptake value assessed by positron emission tomography with 18F-fluorodeoxyglucose, circulating biomarkers, clinical biomarkers such as the appearance of arterial hypertension during treatment that correlates with better outcomes, and the GIST genotype. GISTs with KIT mutations at exon 9 and the so-called wild-type GISTs seem to better respond to sunitinib. Nonetheless, further investigation is required to confirm these findings as well as to understand the mechanisms of sunitinib resistance such as the development of new KIT mutations or conformational changes in KIT receptor.
Keywords: sunitinib, GIST, KIT, refractory GIST
GIST: an overview
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor of the gastrointestinal tract. GISTs are more frequently found in stomach (~50%), followed by small intestine (25%), colon/rectum (5%–10%), and esophagus (5%).1 The mean age of presentation of GIST is between 50 and 70 years, although it can be diagnosed at any age.2
Surgical resection followed by adjuvant Imatinib mesylate (Glivec®; Novartis Pharma, Basel, Switzerland) in selected cases is the treatment of choice for GISTs. In metastatic setting (between 15% and 50% of patients develop metastases), GIST represents one of the paradigms of targeted agents era.3 Initially, median survival for patients with advanced disease was around 10–18 months because no effective therapies were available.2 However, after discovering the role of stem cell factor receptor (KIT) gene mutations in the pathogenesis of GIST and the consequent introduction of KIT inhibitor imatinib in GIST treatment dramatically improved the prognosis of these patients.
The KIT, platelet-derived growth factor receptor alpha (PDGFRα) and ABL kinase inhibitor imatinib at doses of 400 mg daily until progression of disease and/or unacceptable toxicity is the standard first-line treatment in patients with unresectable and/or metastatic GIST. Imatinib was approved by Food and Drug Administration (FDA) after the results of a phase II trial in which >60% of patients experimented partial responses (PRs) to imatinib and some of them maintained the benefit for a long period of time.4 Imatinib is supposed to be the first step in advanced GIST treatment, but further investigation is required given that between 10% and 15% of GISTs are primary resistant to imatinib, 50% develop secondary resistance within 2 years of imatinib initiation and ~4% of GIST patients are intolerant to imatinib.5
Sunitinib malate (Sutent®; Pfizer, New York, NY, USA) was approved by regulatory entities after disease progression or intolerance to imatinib in 2006, which is the objective of this review. Recently, another multitargeted receptor tyrosine kinase inhibitor (TKI) regorafenib (Stirvarga; Bayer HealthCare Pharmaceuticals Inc; Montville, NJ, USA) has been approved after failure or intolerance to imatinib and sunitinib. The recommended dose is 160 mg taken orally once per day for the first 21 days of each 28-day cycle. After promising results of a phase II trial,6 the pivotal trial that leads to regorafenib approval was an international, randomized (2:1), placebo-controlled, multicenter phase III trial (the GRID one) that clearly showed improvement in progression-free survival (PFS), but not in overall survival (OS), probably because of the crossover design.7 Other agents for advanced GIST treatment, mostly TKIs, have been investigated unless they are not widespread used either because limited activity in trials or lack of enough data to recommend them. Masatinib, ponatinib, nilotinib, pazopanib, and sorafenib are some examples.
Pharmacologic profile of sunitinib
Mechanism of action
Sunitinib is a multi-targeted TKI with anti-tumor and anti-angiogenic properties. Specifically, sunitinib is an inhibitor of vascular endothelial growth factor receptor (VEGFR) types 1–3, PDGFRα and β, KIT, colony-stimulating factor type 1, glial cell-line-derived neurotrophic factor receptor (RET), and fetal liver tyrosine kinase receptor 3. Table 1 describes the half maximal inhibitory concentration of sunitinib for each tyrosine kinase receptor according to data obtained from several cell lines of solid tumors. As it is shown, sunitinib is a potent inhibitor of VEGFR1 and 3, as well as of PDGFR α/β.8
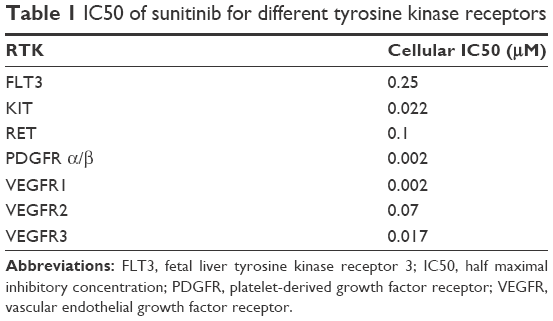 | Table 1 IC50 of sunitinib for different tyrosine kinase receptors |
Pharmacological parameters
Sunitinib is primarily metabolized by cytochrome CYP3A4 and as a result a pharmacologically active metabolite N-desethyl metabolite (SU012662) is formed, which is further metabolized to inactive fraction. SU012662 is considered equipotent to the parent compound regarding the inhibition of VEGFR, PDGFR, and KIT. Both have large volume of distribution, indicating good tissue penetration. Table 2 describes the pharmacokinetic parameters of both sunitinib and SU012662.9
 | Table 2 Pharmacokinetic parameters of sunitinib and SU012662 |
Special situations
Theoretically, the pharmacokinetic profile is not altered by age, body mass index, gender, race, creatinine clearance, previous digestive tube resections, or performance status according to the Eastern Cooperative Oncology Group classification (ECOG). However, consistent data addressing these situations are probably lacking.
One of the main concerns when oral antineoplastic agents are administered is the drug absorption when patients have previously underwent resections of digestive tube. This issue is of special relevance in GIST patients, and it was assessed in a retrospective analysis of 305 individuals. The conclusion was that gastrectomy does not imply variations in sunitinib or SU012662 exposure, but it does if a small bowel resection has also been done in addition to gastrectomy (lower blood levels of sunitinib and SU012662), nonetheless without meaningful clinical relevance.10
Related with renal impairment, sunitinib seems to be safe and effective in patients with renal insufficiency, even in end-stage, although these studies have been done in renal carcinoma population and not in GIST ones.11 For mild or moderate hepatic alteration, no special precautions are needed for sunitinib dose, but increases or decreases in plasma drug concentration can occur if CYP3A4 inhibitors or inducers, respectively, are concomitantly taken.12 Regarding body weight, it is important to consider that it has effects on clearance and distribution volume of sunitinib and SU12662. Although no dose adjustments are recommended by regulatory entities, individual analysis demonstrates that sunitinib plasma levels in patients with severe obesity can be below the required for drug activity at conventional sunitinib doses.13 This issue is of special interest given that steady-state area under the curve of total drug (pharmacologically active at levels between 50 and 100 ng/mL) is correlated with patient outcome in terms of time to progression (TTP) and OS.14 If fasting state can influence in sunitinib, pharmacokinetics has to be evaluated in a phase I trial with healthy volunteers, and it has been concluded that sunitinib can be taken either with or without food.15
Development of sunitinib in GIST: from bench to bedside
Preclinical data
Molecular mechanisms by sunitinib that exerts its antitumor function are not clearly elucidated, partly because available preclinical data are scarce. Preclinical studies with GIST cell lines suggest that SU11248 induces growth arrest and apoptosis of GIST cells. In addition, GIST cells exposition to SU11248 inhibits c-KIT autophosphorylation and the phosphorylation of AKT and ERK, key components of PI3K-Akt-mTOR and MAPK pathways, respectively, involved in cell survival and proliferation. This fact provides a rational for combining sunitinib with other target therapies directed to the mentioned pathways.16
Early trials
An open-label, single-arm, dose-escalation phase I/II trial in Western population enrolled 97 patients with metastatic GIST who have progressed to imatinib or they were intolerant to it.17 Several doses and schedules were tested in different cohorts in order to evaluate treatment safety: schedule 2/2 (2 weeks ON sunitinib, 2 weeks OFF) at doses of 25, 50, or 75 mg/day, and schedules 4/2 and 2/1 starting at 50 mg/day. The dose of 50 mg/day was defined as maximum tolerated dose because two of four patients treated at 75 mg/day 2/2 experienced dose-limiting toxicities during the first cycle (fatigue, nausea, and vomiting). Pharmacokinetic analysis revealed that steady state was achieved by days 7–10 and 7–21 for sunitinib and SU12662, respectively. In order to maximize sunitinib exposure, the schedule 4/2 was selected for further development. Promising sunitinib activity was observed in this trial since 54% of patients benefited from the treatment. More concisely, 7 patients presented PR with a median time of 8.3 months to achieve it and 45 patients experienced long-lasting stable disease (SD) for a minimum of 6 months. Median PFS was 7.8 months (95% confidence interval [CI], 5.1–10.4 months), and median OS was 19 months (95% CI, 12.9–21.5 months). Approximately 60 participants of this trial had a baseline positron emission tomography with 18F-fluorodeoxyglucose (FDG-PET) and another on day 7 of cycle 1. Even if it will be detailed later, early metabolic responses correlated with better clinical outcomes.
In addition, sunitinib activity was also demonstrated in a preclinical setting because approximately half of the patients included had pre- and post-sunitinib biopsies. After 1 week of sunitinib treatment, levels of phospho-KIT in tumor samples as well as the expression of proteins involved in cell proliferation (cyclin A and AKT) in a percentage of patients were reduced. Mentioned early changes related to lower cell proliferation could correlate with better clinical outcomes, but it is a hypothesis to be further demonstrated.
Another phase I/II nonrandomized, open-label, and dose-escalating study aimed to evaluate the safety and preliminary efficacy of sunitinib in Asiatic population.18 About 12 patients were enrolled in part I and doses of 25, 50, and 75 mg/day of sunitinib on schedule 4/2 were tested; 50 mg/day on schedule 4/2 until progression disease (PD) and/or unacceptable toxicity was defined as recommended phase II dose and after that several dose-limiting toxicities were observed in the cohort of 75 mg/day on schedule 4/2. A total of 36 patients were included in part II of the study and received the previously defined dose. According to Response Evaluation Criteria in Solid Tumors (RECIST), 11% of patients experiment a PR and, globally, the disease control rate was ~61%. Median TTP was 28.3 weeks. Regarding safety, all patients included experienced at least one adverse treatment-related event, but 84% of them were grade 1/2 and generally manageable and reversible.
A meta-analysis including GIST and renal cell carcinoma patients treated with sunitinib underlined the importance of maintaining sunitinib dosage and schedule. The achievement of the steady state of sunitinib and SU12662 correlated with better response rate, TTP, and OS, however, with the increased incidence of some adverse events (AEs) also.14
Pivotal and expanded access trials
After phase I/II trial, sunitinib efficacy was further demonstrated in a phase III trial.19 This one was multicenter, randomized, double-blind, and placebo-controlled in patients who had presented imatinib resistance or intolerance. A total of 302 patients were randomly assigned 2:1 to receive sunitinib at doses established in phase I (n: 207) or placebo (n: 105). However, the trial was early unblinded due to the results of planned interim analysis that clearly favored sunitinib in terms of TTP. Median TTP in sunitinib arm was 27.3 weeks (95% CI 16.0–32.1) versus 6.4 weeks in placebo ones (95% CI 4.4–10.0; hazard ratio [HR] 0.33; 95% CI 0.23–0.47; P<0.0001). After these results, all patients treated with placebo were allowed to receive open-label sunitinib. OS data were more difficult to analyze because of the crossover. According to Kaplan–Meier method, OS did not reveal statistically significant differences between sunitinib and placebo (73.9 weeks versus 64.9 weeks; 95% CI 45.7–96.0; P=0.161). Nonetheless, a posterior long-term OS analysis was performed using another statistical method that accounts for the bias introduced by the crossover from placebo to sunitinib, the rank-preserving structural failure time (RPSFT). RPSFT method identified clear differences in median OS favoring sunitinib group (73.9 weeks; 95% CI 61.3–85.7 versus 35.7 weeks; 95% CI 25.7–49.8; P<0.001).20
An expanded-access study for patients ineligible for previous trials or for whom sunitinib was unavailable enrolled 1,126 patients who received sunitinib 50 mg/day 4/2 until PD and/or unacceptable toxicity.21 The results were better than pivotal trial, thus median TTP was 41.0 weeks (95% CI 36–47) and OS was ~75.0 weeks (95% CI 68–84). Subgroup analyses were performed and age ≤65 years, prior imatinib dose (400 mg daily or lower), and Eastern Cooperative Oncology Group performance status of one or less favored longer OS. Regarding elderly population (≥65 years) who are usually under-represented in clinical trials, a retrospective French series of 71 patients showed similar benefit of sunitinib than in younger patients. However, dose reductions or interruptions were more frequent and often the comorbidities implied a higher risk of AE.22
Although the sound trials of sunitinib in GIST have mostly been performed in Western countries, retrospective series in both Taiwanese and Chinese population as well as a small prospective study in Chinese patients suggest similar efficacy and toxicity profile compared with Western population.23–25
Sunitinib safety
In general terms, sunitinib AEs are mild and reversible, but their optimal management is crucial in order to maintain patients on sunitinib therapy. In the pivotal phase III trial, the most common AEs were fatigue (47%), diarrhea (43%), nausea (36%), anorexia (28%), and dysgeusia (25%). However, most common grade 3–4 AEs were fatigue (10%), arterial hypertension (AH, 7%), and hand–foot syndrome (6%). Regarding hematological AEs, they were mainly grade 1–2 and consisted of hemoglobin, neutrophils, and platelet reductions. As a consequence of AEs, in 36% of patients dose interruptions were required and in 28% of patients dose reductions were required.19 The AE profile in the expanded-access study was very close to the observed one in the phase III.
As mentioned in the section “Patient selection for sunitinib therapy,” appearance of AH during sunitinib therapy has been postulated as a positive predictive biomarker of sunitinib response. However, it is also important to consider if AH induces higher risk of cardiovascular AE not only in GIST patients but also in renal cell carcinoma patients. This issue was evaluated in a retrospective series, and cardiovascular events seemed to be slightly more frequent in those patients with AH (5% of grade 3 cardiac events in patients with AH versus 3% in patients without AH). Nevertheless, mortality related with cardiac events in patients receiving sunitinib is very low.26–28
In addition, sunitinib can induce reductions in left ventricular ejection function (LVEF), being ≥20% from basal level in ~2% of cases. Worsening a pre-existing AH and developing AH during sunitinib therapy are risk factors for LVEF impairment. In these patients and in those with other cardiac comorbidities, close monitoring of LVEF should be performed, despite cardiac failure related with sunitinib tends to be reversible after stopping the drug.29 The previous described findings come from analysis including renal and GIST patients. In the expanded use program limited to GIST patients, grade 3 or 4 heart failure, myocardial infarction or pulmonary edema were <1%.30
No predictive biomarkers of sunitinib-related AH development have been approved, although VEGF polymorphisms were evaluated as a putative biomarkers and the presence of VEGF-A rs699947 correlated with AH appearance during treatment in patients with renal cell cancer.31
Hypothyroidism is considered an AE clearly related to sunitinib, although it was not taken into consideration since the beginning. A prospective and an observational study with 42 patients treated with sunitinib revealed abnormal serum thyroid-stimulating hormone in as much as 62% of participants. The biological explanation for this AE is that suntinib also inhibits ret proto-oncogene, and it binds to proteins that assemble with thyroid hormone receptor (for example, retinoid X receptor alpha and retinoic acid receptors beta and gamma). Hypothyroidism can be considered as off-target AE, and regular control of thyroid profile since the beginning of the therapy is mandatory. Its management is usually easy with hormone replacement that allows to continue sunitinib therapy without developing hypothyroidism complications like fatigue and other potentially life-threatening as myxedema coma.32–34
Alternative schedules of sunitinib
Alternative schemes of sunitinib have been investigated in order to improve the safety profile and tolerance. Sunitinib 37.5 mg once daily until PD and/or unacceptable toxicity were evaluated in an open-label, multicenter, phase II trial in which patients were randomized in a ratio of 1:1 in order to receive the mentioned dose in the morning or in the evening.35 The results of this trial in terms of both efficacy and toxicity overlapped with the phase III patients, with a median PFS of 34 weeks (95% CI, 24–49) and a median OS of 107 weeks (95% CI, 72 to not calculable). Consequently, sunitinib 37.5 mg once daily could be considered as an alternative dosing strategy, although it has not been directly compared with standard scheme. Regarding the optimal condition in sunitinib intake, no major differences were found between morning and evening dosing. In both the cases, no drug accumulation was observed across cycles and effective drug concentration was achieved.
Sunitinib 50 mg/daily in a schedule of 2 weeks ON/1 week OFF has been investigated in metastatic renal cell carcinoma. The RESTORE trial accrued 76 patients, and they were randomized to sunitinib 4 weeks ON/2 weeks OFF schedule or to the 2 weeks ON/1 week OFF regimen. The results of this trial demonstrated better toxicity profile and better compliance with the 2/1 schedule.36 A retrospective analysis with 249 patients concluded with similar results.37 Even though this scheme has not been evaluated in GIST patients, it could be considered in some patients with poor tolerance to the conventional schedule.38
Response evaluation of sunitinib
One of the main concerns regarding GIST treatment is to define the best method to evaluate treatment response. Target therapies induce several radiologic changes not only in size lesions but also in lesion structure (that is, variations in density) and in the pattern of contrast enhancement of intratumoral nodules. RECIST are exclusively based on changes in size lesions, and as a result the RECIST assessment could mislead the treatment response, for example, GIST lesions enlarge because of necrosis or cystic degeneration, but not because of true tumor progression.
Choi criteria combine both biologic response and tumor volume response, and they were described in order to improve the sensitivity and specificity of the RECIST in the aforementioned setting.39 No prospective trials have compared RECIST and Choi criteria, but Choi criteria seem to be less robust than in first-line setting with imatinib. Prospective–retrospective series with small number of patients have failed in demonstrating superiority of Choi criteria in terms of better prediction of patient outcome depending on sunitinib response and even have recommended the RECIST.40–42
Surgery after sunitinib treatment
Unless treatment with sunitinib in metastatic GIST patients should be considered as palliative, a potentially radical surgery could be occasionally planned in the clinical practice if the response has been good enough. Nonetheless, the scientific evidence supporting this surgical management is very scarce. Two retrospective series with a very limited number of patients (10 and 50) suggest that post-sunitinib surgery is feasible, but the patients should be selected carefully because no clear improvement in terms of survival has been suggested. In addition, in the largest series, the surgery was frequently incomplete (not clearly related with the magnitude of the previous sunitinib response) and significant complications occurred in >50% of patients.43,44
Sunitinib rechallenge
The current approved treatment in advanced GIST after sunitinib failure is regorafenib based on data obtained in the pivotal phase III trial (the GRID trial).45 However, systemic treatment options are limited and the rechallenge with imatinib and/or sunitinib emerges as a possibility in the daily practice. No trials have been designed in order to assess the efficacy of sunitinib rechallenge. The limited evidence comes from the report of 2 cases in whom prolonged clinical and radiological benefit was achieved after the rechallenge. Both the cases previously presented PR to sunitinib that lasted for >2 years and the primary tumor was located in stomach.46 In a post hoc analysis of the expanded use program, those patients who had progressed to sunitinib and did not discontinue the therapy presented better outcomes than those who discontinued, suggesting a potential benefit of sunitinib beyond progression.30
Sunitinib or imatinib escalation treatment after first-line imatinib failure
At present, the best choice in second line is still unclear, being both imatinib dose escalation and directly switching to sunitinib widely used options. A formal prospective comparison of both the strategies as a first approach in patients with GIST that progress after a first-line therapy with imatinb at 400 mg. A large retrospective analysis from the MetaGIST group proved that a dose escalation to 800 mg/day at the time of progression on first-line imatinib 400 mg/day was associated with new disease stabilization in ~30% of cases.47 Unfortunately, these stabilizations are often of limited duration. Although dose escalation is widely recommended within clinical guidance documents, the current evidence for its effectiveness in GIST after progression at the standard imatinib dose is based only on the analysis of several sets of observational data.
A retrospective series of 123 patients with KIT exon 11 mutated GIST who progressed after first-line imatinib 400 mg showed a TTP of 10 months (95% CI 9.7–10.9) in the group of patients treated with sunitinib in comparison with 5 months in those receiving high doses of imatinib (95% CI 3.6–6.7) (P=0.012). No difference was found in OS (P=0.883). In imatinib arm, KIT exon 11 deletions were associated with a shorter TTP (7 vs 17 months; P=0.02), with a trend in OS (54 vs 71 months; P=0.063). No difference was found in patients treated with sunitinib (P=0.370).48 These results suggest that some GIST patients harboring KIT exon 11 deletions may acquire secondary mutations more resistant to escalated dose of imatinib than to sunitinib. On the basis of these results, sunitinib as second-line treatment may be mostly considered in patients with KIT exon 11 deletion after imatinib being failure. However, validation in prospective studies on larger series of patients would be of great interest.
Nevertheless, two additional retrospective studies did not show differences in benefit between imatinib dose escalation and switching to sunitinib in second-line therapy of GIST (Hsu et al, 2014; Hislop, 2012).49,50 In these studies, however, mutational status was not analyzed. In the absence of prospective comparative trials, imatinib escalation and switch to sunitinib are both acceptable options after imatinib failure. Physicians can choose either treatment considering the specific kinase mutations or distinct adverse events associated with the two treatments.
Mechanisms of sunitinib resistance
The mechanisms of sunitinib resistance are still under investigation, but some hypotheses have been established in cell line studies. Long-term exposure of sunitinib could induce methylation of the promoter of phosphatase and tensin homolog deleted on chromosome ten (PTEN) gene, which encodes the negative regulator of PI3K. The epigenetic silencing of PTEN could elicit a constitutive activation of the PI3K-AKT-mTOR pathway, one of the sunitinib targets as it has been previously mentioned.51
Sunitinib mainly blocks the autoactivation of KIT because it binds to the adenosine triphosphate (ATP)-binding pocket of the unactivated conformation of KIT. A shift in the equilibrium of activated/inactivated form of KIT toward the active conformation in which sunitinib is less effective has been postulated as a potential mechanism of sunitinib resistance.52
Similar to imatinib resistance, preclinical studies suggest that long exposure to sunitinib elicits the appearance of new mutations in the KIT activation loop conferring this resistance to sunitinib (mainly in exon 17, for example, D816V, D816F, and T670I).53,54
Patient selection for sunitinib therapy
No predictive biomarkers of sunitinib response have been validated, although several putative clinical and molecular parameters have been studied in order to be useful in the selection of appropriate candidates for this therapy and avoiding exposure to it in patients in whom the treatment will be ineffective. Some of them would be pre-treatment predictive markers of response, whereas other would be early markers of resistance or benefit when the therapy has been started.
Arterial hypertension and other clinical biomarkers
AH can be considered an on-target AE due to the inhibition of VEGFR caused by sunitinib. Activation of VEGFR-2 by vascular endothelial growth factor (VEGF) induces hypotension and vasodilatory effects in vascular endothelial cells. As a consequence, the inhibition of VEGFR-2 and VEGF interaction by sunitinib increases the peripheral vascular resistance. The molecular mechanisms of this effect include apoptosis of endothelial cells, reduction of nitric oxide levels (a vasodilator) and activation of a constrictor pathway endothelin-1.26
In renal cell carcinoma, AH experienced during sunitinib treatment has been related to better patient outcome.55,56 Whether AH related to sunitinib in GIST patients can be used as biomarker has also been assessed. In a prospective series of 137 patients, the AH appearance of any grade was a strong predictor of better PFS (HR 0.20, P=0.000) and OS (HR 0.2056, P=0.000).57 Another retrospective series of >1,000 patients included in the phase I/II and III and in the expanded use program also correlated AH of any grade with overall response (16% versus 3%, P=0.004), PFS (34 weeks versus 16 weeks, 95% CI, P<0.0001) and OS (87 weeks versus 53 weeks, 95% CI, P=0.0003).26
Regarding other potential clinic biomarkers, a subgroup analysis of the phase III trial revealed the benefit of sunitinib in terms of TTP regardless of age (<65 or contrary), ECOG (0 or 1) and previous imatinib exposure (>6 months or the contrary).19
Imatinib intolerance
Although imatinib toxicity profile is in general acceptable, ~4% of patients are intolerant to imatinib and discontinuation is required. The major evidence of the efficacy of sunitinib in this setting comes from the phase III trial in which 13 patients enrolled were considered as imatinib intolerants. Nine of 13 patients were randomized to sunitinib, and 8 of them showed clinical benefit to sunitinib (4 PRs were observed) and only 1 patient experienced PD. These results emerge the possibility of better efficacy of sunitinib in first-line scenario, but further studies are required because number of patients evaluated was very few.19
Imaging biomarkers
FDG-PET could be a potential and feasible tool for early sunitinib response assessment. A small study with 23 patients evaluated the maximal standardized uptake value (SUV) before and after the first 4 weeks of sunitinib therapy. Changes in SUV value were clearly related to PFS, specifically, basal SUV reductions of >25% were correlated with a PFS of 29 weeks, in contrast to those patients whose SUV did not reduce at least a 25% in whom PFS was ~4 weeks.58 In addition, it seems that early SUV reductions of at least 25% in FDG-PET performed earlier at day 7 of cycle 1 of sunitinib correlate with treatment responses months earlier than could be appreciated with CT scan. These data come from the phase I trial in Western population, in which ~85%–89% of patients with clinical benefit of sunitinib (PR or SD) showed early metabolic response.17
Nonetheless, it is important to consider that GIST lesions can be negative in FDG-PET due to the small diameter (between 1 and 5 cm) or to the histological tumor changes (necrosis, myxoid degeneration, or scarring).59 However, this approach would require further validation in order to be used to routinely modify patient management.
Mutational status
Refractory GIST is a heterogeneous disease composed of a mixture of clones; each of them harbors different mutations mainly in KIT or PDGFRA. Despite every lesion in a given patient has the primary GIST mutation (except of wild-type GIST), secondary mutations can appear under treatment pressure and confer resistance to therapies. The percentage of secondary mutations in GIST with primary mutations is estimated to range between 44% and 90%, depending on the sensitivity of the method used to determine them. In addition, the development of several secondary mutations at the same time seems to be a common event.60 After imatinib exposure, secondary mutations are more commonly found in GIST with primary KIT exon 11 mutations than in GIST with primary KIT exon 9 mutations and not found in GIST wild-type. Secondary mutations after imatinib treatment are usually located at exons 13 (for example, V654A mutation) and 14 (for example, T607I mutation), both encode the ATP-binding pocket, or in exon 17 (encodes kinase activation loop).61
The potential role of primary and secondary mutations as predictor factors of sunitinib response has been investigated. A retrospective analysis using samples from patients who are included in a phase I/II sunitinib trial (17) concluded that patients with KIT exon 9 mutations clearly benefited more of sunitinib than those patients who harbor KIT exon 11 mutations in terms of objective response rate (37% versus 5%; P=0.002), PFS (19.4 months versus 5.1 months; P=0.0005), and OS (26.9 months versus 12.3 months; P=0.012).61 These results have also been reported in a series of 137 patients in whose tumors carried a KIT exon 9 mutations or were wild-type and presented clearly better 1-year PFS compared with those whose tumors carried a KIT exon 11 or PDGFRA mutations (68% and 57% versus 34% and 15%, respectively).57 KITAY502-3ins mutations at exon 9 is the most sensitive to sunitinib.53
Regarding secondary mutations, in vitro studies with GIST cell lines suggest that sunitinib is highly active against kinase activity of KIT containing secondary mutations at ATP-binding pocket (exons 13 and 14), in contrast to GIST cell lines harboring imatinib resistant mutations at activation loop (exons 17, for example, D820Y, D820E and NK822K, and exon 18).61,62 These findings correlate with better PFS and OS of patients treated with sunitinib with exon 13 and 14 mutations, compared with patients with exon 17 and 18 mutations, although these results should be further validated.
The 10%–15% of GIST patients defined as “wild-type” (WT, no mutations in KIT neither in PDGFRA) are of special interest, since the vast majority do not respond to imatinib. In these cases, the deficiency of succinate dehydrogenase (due to either inactivating mutations or through epigenetic mechanisms)63 and sporadic mutations in the MAPK pathway have a major a role in tumor development.64 Among pediatric population, GIST WT is the most frequently found, sporadically or as a part of congenital syndromes such as Carneid triad or Neurofibromatosis type I. In this subset of patients, sunitinib shows promising substantial antitumor activity and acceptable tolerability.65 In addition, preclinical data suggest higher antitumor efficacy of sunitinib compared with imatinib.66
Circulating biomarkers
Circulating plasma levels of the extracellular domain of soluble KIT (sKIT) have been evaluated as potential biomarker in patients enrolled in the phase III trial of sunitinib versus placebo. sKIT level reduction from the baseline was a significant predictor of time to tumor progression.67 However, this finding deserves further evaluation because sKIT changes could also be related to changes in KIT synthesis or metabolism, or even changes that sunitinib could elicit in the sKIT in non-neoplastic cells.68
The plasma levels of different isoforms of soluble vascular endothelial growth factor receptor (sVEGFR) have been correlated with the outcome during sunitinib therapy. sVEGFR3 showed to have the greatest predictive potential for OS.69
New perspectives
Sunitinib has demonstrated efficacy against advanced GIST after imatinib progression and an acceptable toxicity profile. Currently, it is considered the standard second-line treatment in metastatic GIST after progression or intolerance to imatinib. However, there are still several open questions that would help to better select the candidates for this treatment. Further investigation that could correlate GIST genotype with sunitinib response is necessary probably because mutational status seems to be the most promising potential biomarker. Therefore, determination of GIST genotype to help the optimal selection of therapy should be considered mandatory. Moreover, the repetition of the mutational analysis with new diagnostic tool such as liquid biopsy, if feasible, after disease progression, may be an option in the future that allows identification of secondary mutations or other alterations, choosing the best therapeutic options in accordance with their sensitivity to TKIs.
Furthermore, the deep knowledge of mechanism of sunitinib resistance represents an unmet need. Apart from developing new therapies, the study of molecular events that occur at sunitinib progression could facilitate potentially effective drug combinations.
Disclosure
Xavier Garcia-del-Muro reports advisory role for Pfizer and Bayer. Nuria Mulet-Margalef reports no conflicts of interest in this work.
References
Rubin BP. Gastrointestinal stromal tumours: an update. Histopathology. 2006;48(1):83–96. | ||
DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231(1):51–58. | ||
Nilsson B, Bümming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era – a population-based study in western Sweden. Cancer. 2005;103(4):821–829. | ||
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. | ||
Van Glabbeke M, Verweij J, Casali PG, et al. Initial and late resistance to imatinib in advanced gastrointestinal stromal tumors are predicted by different prognostic factors: A European Organisation for Research and Treatment of Cancer-Italian Sarcoma Group-Australasian Gastrointestinal Trials Group. J Clin Oncol. 2005;23(24):5795–5804. | ||
George S, Wang Q, Heinrich MC, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012;30(19):2401–2407. | ||
Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302. | ||
Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337. | ||
Izzedine H, Buhaescu I, Rixe O, Deray G. Sunitinib malate. Cancer Chemother Pharmacol. 2007;60(3):357–364. | ||
de Wit D, van Erp NP, Khosravan R, et al. Effect of gastrointestinal resection on sunitinib exposure in patients with GIST. BMC Cancer. 2014;14(1):575. | ||
Czarnecka AM, Kawecki M, Lian F, Korniluk J, Szczylik C. Feasibility, efficacy and safety of tyrosine kinase inhibitor treatment in hemodialyzed patients with renal cell cancer: 10 years of experience. Future Oncol. 2015;11(16):2267–2282. | ||
Bello CL, Garrett M, Sherman L, Smeraglia J, Ryan B, Toh M. Pharmacokinetics of sunitinib malate in subjects with hepatic impairment. Cancer Chemother Pharmacol. 2010;66(4):699–707. | ||
Desar IM, Burger DM, Van Hoesel QG, Beijnen JH, Van Herpen CM, Van der Graaf WT. Pharmacokinetics of sunitinib in an obese patient with a GIST. Ann Oncol. 2009;20(3):599–600. | ||
Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol. 2010;66(2):357–371. | ||
Bello CL, Sherman L, Zhou J, et al. Effect of food on the pharmacokinetics of sunitinib malate (SU11248), a multi-targeted receptor tyrosine kinase inhibitor: results from a phase I study in healthy subjects. Anticancer Drugs. 2006;17(3):353–358. | ||
Ikezoe T, Yang Y, Nishioka C, et al. Effect of SU11248 on gastrointestinal stromal tumor-T1 cells: enhancement of growth inhibition via inhibition of 3-kinase/Akt/mammalian target of rapamycin signaling. Cancer Sci. 2006;97(9):945–951. | ||
Demetri GD, Heinrich MC, Fletcher JA, et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res. 2009;15(18):5902–5909. | ||
Shirao K, Nishida T, Doi T, et al. Phase I/II study of sunitinib malate in Japanese patients with gastrointestinal stromal tumor after failure of prior treatment with imatinib mesylate. Invest New Drugs. 2010;28(6):866–875. | ||
Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–1338. | ||
Demetri GD, Huang X, Garrett CR, et al. Novel statistical analysis of long-term survival to account for crossover in a phase III trial of sunitinib (SU) vs. placebo (PL) in advanced GIST after imatinib (IM) failure. J Clin Oncol. 2008;26(15S):559s. | ||
Reichardt P, Kang Y, Ruka W, et al. Detailed analysis of survival and safety with sunitinib (SU) in a worldwide treatment-use trial of patients with advanced GIST. J Clin Oncol. 2008;26(15S):565s. | ||
Duffaud F, Ray-Coquard I, Marchal F, et al. Clinical experience with sunitinib (SU) in patients over age 65 with metastatic gastrointestinal stromal tumors (GIST): a retrospective study from the French Sarcoma Group (FSG). J Clin Oncol. 2013;31 (suppl; abstr 10546). | ||
Chen Y-Y, Yeh C-N, Cheng C-T, et al. Sunitinib for Taiwanese patients with gastrointestinal stromal tumor after imatinib treatment failure or intolerance. World J Gastroenterol. 2011;17(16):2113–2119. | ||
Li J, Gao J, Hong J, Shen L. Efficacy and safety of sunitinib in Chinese patients with imatinib-resistant or -intolerant gastrointestinal stromal tumors. Future Oncol. 2012;8(5):617–624. | ||
Shen L, Qin S, Sun Y, et al. Sunitinib in Chinese patients with advanced gastrointestinal stromal tumor (GIST). J Clin Oncol. 2015;33 (suppl; abstr e15208). | ||
George S, Reichardt P, Lechner T, Li S, Cohen DP, Demetri GD. Hypertension as a potential biomarker of efficacy in patients with gastrointestinal stromal tumor treated with sunitinib. Ann Oncol. 2012;23(12):3180–3187. | ||
Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370(9604):2011–2019. | ||
Di Lorenzo G, Autorino R, Bruni G, et al. Cardiovascular toxicity following sunitinib therapy in metastatic renal cell carcinoma: a multicenter analysis. Ann Oncol. 2009;20(9):1535–1542. | ||
Galizia D, Ortega C, Palesandro E, et al. Hypertension monitoring as a tool to predict congestive heart failure (CHF) during sunitinib (SU) therapy in GIST and renal cell carcinoma (RCC). J Clin Oncol. 2011;29 (suppl, abtr10061). | ||
Reichardt P, Kang Y-K, Rutkowski P, et al. Clinical outcomes of patients with advanced gastrointestinal stromal tumors: safety and efficacy in a worldwide treatment-use trial of sunitinib. Cancer. 2015;121(9):1405–1413. | ||
Rodriguez de Antona C, Grande E, Leandro-Garcia LJ, et al. Evaluation of CYP3A5, VEGF-a, and VEGFR2 polymorphisms as markers of sunitinib toxicity. J Clin Oncol. 2012;30 (suppl; abstr 10546). | ||
Wolter P, Stefan C, Decallonne B, et al. The clinical implications of sunitinib-induced hypothyroidism: a prospective evaluation. Br J Cancer. 2008;99(3):448–454. | ||
Desai J, Yassa L, Marqusee E, et al. Hypothyroidism after sunitinib treatment for patients with gastrointestinal stromal tumors. Ann Intern Med. 2006;145(9):660–664. | ||
Shu M, Zai X, Zhang B, Wang R, Lin Z. Hypothyroidism side effect in patients treated with sunitinib or sorafenib: clinical and structural analyses. PLoS One. 2016;11(1):e0147048. | ||
George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45(11):1959–1968. | ||
Lee JL, Kim MK, Park I, et al. Randomized phase II trial of Sunitinib four weeks on and two weeks off versus Two weeks on and One week off in metastatic clear-cell type REnal cell carcinoma: RESTORE trial. Ann Oncol. 2015;26(11):2300–2305. | ||
Bracarda S, Iacovelli R, Boni L, et al. Sunitinib administered on 2/1 schedule in patients with metastatic renal cell carcinoma: the RAINBOW analysis. Ann Oncol. 2015;26(10):2107–2113. | ||
Khosravan R, Motzer RJ, Fumagalli E, Rini BI. Population Pharmacokinetic/pharmacodynamic modeling of sunitinib by dosing schedule in patients with advanced renal cell carcinoma or gastrointestinal stromal tumor. Clin Pharmacokinet. 2016;55(10):1251–1269. | ||
Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25(13):1753–1759. | ||
Dudeck O, Zeile M, Reichardt P, Pink D. Comparison of RECIST and Choi criteria for computed tomographic response evaluation in patients with advanced gastrointestinal stromal tumor treated with sunitinib. Ann Oncol. 2011;22(8):1828–1833. | ||
Schramm N, Englhart E, Schlemmer M, et al. Tumor response and clinical outcome in metastatic gastrointestinal stromal tumors under sunitinib therapy: comparison of RECIST, Choi and volumetric criteria. Eur J Radiol. 2013;82(6):951–958. | ||
Shinagare AB, Barysauskas CM, Braschi-Amirfarzan M, O’Neill AC, Catalano PJ, George SRN. Comparison of performance of various tumor response criteria in assessment of sunitinib activity in advanced gastrointestinal stromal tumors. Clin Imaging. 2016;40(5):880–884. | ||
Tielen R, Verhoef C, van Coevorden F, et al. Surgery after treatment with imatinib and/or sunitinib in patients with metastasized gastrointestinal stromal tumors: is it worthwhile? World J Surg Oncol. 2012;10:111. | ||
Raut CP, Wang Q, Manola J, et al. Cytoreductive surgery in patients with metastatic gastrointestinal stromal tumor treated with sunitinib malate. Ann Surg Oncol. 2010;17(2):407–415. | ||
Demetri GD, Reichardt P, Kang Y-K, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. | ||
Bracci R, Maccaroni E, Cascinu S. Transient sunitinib resistance in gastrointestinal stromal tumors. N Engl J Med. 2013;368(21):2042–2043. | ||
Zalcberg JR, Verweij J, Casali PG, et al. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer. 2005;41(12):1751–1757. | ||
Vincenzi B, Nannini M, Fumagalli E, et al. Imatinib dose escalation versus sunitinib as a second line treatment in KIT exon 11 mutated GIST: a retrospective analysis. Oncotarget. Epub 2015 Sep 24. | ||
Hsu C-C, Wu C-E, Chen J-S, et al. Imatinib escalation or sunitinib treatment after first-line imatinib in metastatic gastrointestinal stromal tumor patients. Anticancer Res. 2014;34(9):5029–5036. | ||
Hislop J, Mowatt G, Sharma P, et al. Systematic review of escalated imatinib doses compared with sunitinib or best supportive care, for the treatment of people with unresectable/metastatic gastrointestinal stromal tumours whose disease has progressed on the standard imatinib dose. J Gastrointest Cancer. 2012;43(2):168–176. | ||
Yang J, Ikezoe T, Nishioka C, et al. Long-term exposure of gastrointestinal stromal tumor cells to sunitinib induces epigenetic silencing of the PTEN gene. Int J Cancer. 2012;130(4):959–966. | ||
Gajiwala KS, Wu JC, Christensen J, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci U S A. 2009;106(5):1542–1547. | ||
Guo T, Hajdu M, Agaram NP, et al. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res. 2009;15(22):6862–6870. | ||
Nishida T, Takahashi T, Nishitani A, et al. Sunitinib-resistant gastrointestinal stromal tumors harbor cis-mutations in the activation loop of the KIT gene. Int J Clin Oncol. 2009;14(2):143–149. | ||
Rini BI, Cohen DP, Lu DR, et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst. 2011;103(9):763–773. | ||
Lévy BI. Blood pressure as a potential biomarker of the efficacy angiogenesis inhibitor. Ann Oncol. 2009;20(2):200–203. | ||
Rutkowski P, Bylina E, Klimczak A, et al. The outcome and predictive factors of sunitinib therapy in advanced gastrointestinal stromal tumors (GIST) after imatinib failure – one institution study. BMC Cancer. 2012;12(1):107. | ||
Prior JO, Montemurro M, Orcurto M-V, et al. Early prediction of response to sunitinib after imatinib failure by 18F-fluorodeoxyglucose positron emission tomography in patients with gastrointestinal stromal tumor. J Clin Oncol. 2009;27(3):439–445. | ||
Choi H, Charnsangavej C, de Castro Faria S, et al. CT evaluation of the response of gastrointestinal stromal tumors after imatinib mesylate treatment: a quantitative analysis correlated with FDG PET findings. AJR Am J Roentgenol. 2004;183(6):1619–1628. | ||
Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216(1):64–74. | ||
Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. | ||
Judson IR. Prognosis, imatinib dose, and benefit of sunitinib in GIST: knowing the genotype. J Clin Oncol. 2008;26(33):5322–5325. | ||
Boikos SA, Pappo AS, Killian JK, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors: a report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016;2(7):922–928. | ||
Huss S, Elges S, Trautmann M, et al. Classification of KIT/PDGFRA wild-type gastrointestinal stromal tumors: implications for therapy. Expert Rev Anticancer Ther. 2015;15:623–628. | ||
Janeway KA, Albritton KH, Van Den Abbeele AD, et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer. 2009;52(7):767–771. | ||
Agaram NP, Laquaglia MP, Ustun B, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14(10):3204–3215. | ||
Deprimo SE, Huang X, Blackstein ME, et al. Circulating levels of soluble KIT serve as a biomarker for clinical outcome in gastrointestinal stromal tumor patients receiving sunitinib following imatinib failure. Clin Cancer Res. 2009;15(18):5869–5877. | ||
Blanke CD. Biomarkers in GIST: partly ready for prime-time use. Clin Cancer Res. 2009;15(18):5603–5605. | ||
Hansson EK, Amantea MA, Westwood P, et al. PKPD modeling of VEGF, sVEGFR-2, sVEGFR-3, and sKIT as predictors of tumor dynamics and overall survival following sunitinib treatment in GIST. CPT Pharmacometrics Syst Pharmacol. 2013;2:e84. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.