")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Sumoylation in gene regulation and cardiac disease: potential for drug discovery
Authors Beketaev I, Wang J
Received 26 August 2014
Accepted for publication 18 September 2014
Published 6 November 2014 Volume 2014:4 Pages 185—192
DOI https://doi.org/10.2147/AGG.S57218
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr John Martignetti
Ilimbek Beketaev, Jun Wang
Center for Stem Cell Engineering, Department of Basic Research Laboratories, Texas Heart Institute at St Luke’s Episcopal Hospital, Houston, TX, USA
Abstract: Small ubiquitin-related modifier (SUMO) proteins are members of ubiquitin-like super-family proteins that can be covalently conjugated to their targets through multistep enzymatic reactions. Sumoylation has caught much attention due to its versatility, wide involvement in cellular events, and disease association. Sumoylation has been well studied at cellular and molecular levels. A newly emerging role that SUMO conjugation plays is in cardiac pathophysiology. In this review we will update new advances in the study of implications of the sumoylation pathway in the pathogenesis of cardiac diseases, discuss promise of the SUMO pathway as a potential therapeutic target, and conclude with future directions for SUMO research in the heart field.
Keywords: posttranslational modification, SUMO, SENP, heart
Introduction
SUMO proteins and conjugation
Small ubiquitin-related modifier (SUMO), a family of ubiquitin-like proteins, has been studied for over 15 years.1 Sumoylation is a posttranslational modification, in which SUMO proteins are covalently conjugated to target proteins via a series of enzymatic reactions. Conjugatable SUMO proteins (SUMO-1, SUMO-2, and SUMO-3) are synthesized as precursors, and are activated by the cleavage performed by sentrin-/SUMO-specific proteases (SENP1, 2, 3, 5, 6, and 7 in humans) to expose the C-terminal diglycine needed for covalent attachment to target substrates.2–4 SUMO-1 shares ~50% sequence similarity with SUMO-2/3, but the active SUMO-2 and -3 exhibit ~95% homology. The role of SUMO-1 in mouse embryogenesis remains in debate (see “Emerging roles of SUMO in cardiovascular disorders”). However, despite the high similarity at the amino acid sequence level between SUMO-2 and -3, a recent study suggested a differential functional importance of SUMO-2 and -3 in mouse embryonic development. First, SUMO-2 was the most abundantly expressed SUMO isoform during mouse embryogenesis.5 Second, knockout of SUMO-2, but not SUMO-3, caused embryonic lethality.5
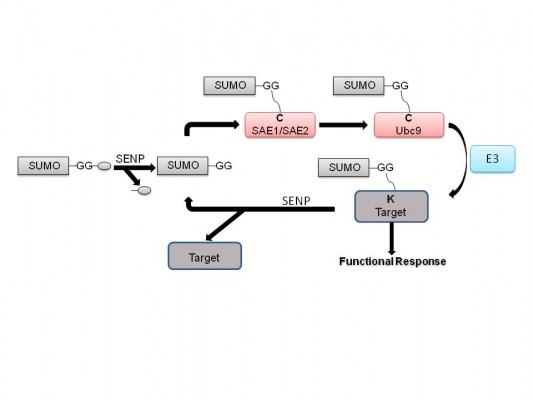
Similarly to ubiquitination, SUMO conjugation to targets occurs through an adenosine triphosphate (ATP)-dependent mode by an enzymatic cascade of activating enzyme (E1), a heterodimeric complex of SAE1/SAE2, conjugating enzyme (E2) Ubc9, and/or an E3-ligase (Figure 1).6,7 When only a single SUMO E1 and SUMO E2 exist in the SUMO conjugation pathway, multiple SUMO E3 ligase enzymes have been defined, including RanBP2/Nup358,8 PIAS family,9 polycomb 2,10 tumor necrosis factor associated protein 7,11 TOPORS,12,13 mitochondrial-anchored protein ligase,14 and Nse2/Mms21.15 SUMO conjugation occurs on the lysine residue(s) that are mainly localized on the consensus sequence ÈKXE/D (È is a bulky hydrophobic amino acid and X is any residue) existent on many SUMO substrates.16,17
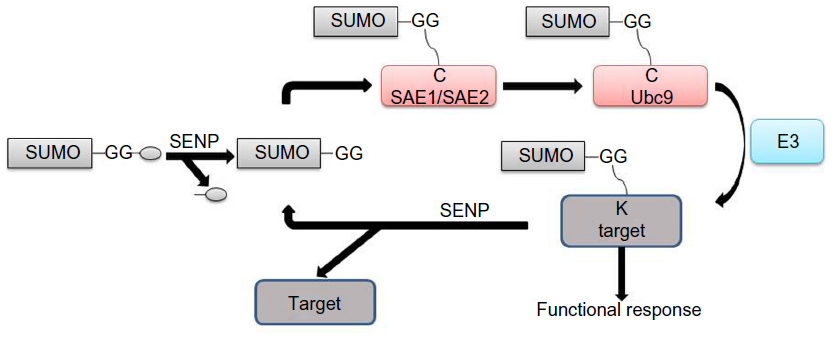 | Figure 1 Reversible sumoylation cycle. |
SUMO deconjugation
SENPs that function for SUMO precursors’ maturation also demodify the conjugated form of SUMO proteins from substrates. The six SENPs identified in humans differ in their isopeptidase activities, subcellular localizations, and SUMO paralogue preferences.2 SENP proteins may have nonredundant roles in mouse embryogenesis, as evidenced by the findings that knockout of either SENP1 or SENP2 caused embryonic lethality.18,19 For more details about structures and functions of SENP proteins, the readers are referred to some previously published comprehensive reviews.3,4,20
SUMO conjugation and cellular activities
Through targeting many factors, SUMO conjugation is involved in a variety of cellular activities including but not limited to signal transduction, subcellular translocalization, stress regulation, DNA damage and repair.21–24 However, the functional consequence of SUMO conjugation to the target is context-dependent; it may lead to inhibition or activation of the activity of a particular promoter/substrate in a particular pathophysiological setting once conjugated. For instance, serum response factor is sumoylated on lysine 147.25 This SUMO-site mutation enhanced the ability of serum response factor to activate CArG box located in the c-fos promoter,25 but impaired its ability to activate cardiac target gene promoter.26 Given its wide implication in regulation of cellular events, it is conceivable that SUMO conjugation pathway is potentially linked to diseases. Indeed, sumoylation activity is altered in a number of human diseases such as cancer and neurodegenerative diseases.27,28 Recent evidence points to the implication of the SUMO conjugation pathway in cardiac gene regulation as well as in the pathophysiology of cardiovascular disease,29,30 which is the main focus of this updated review.
SUMO-targeted cardiac proteins and roles in cardiac gene regulation
Cardiac transcription factors are a group of proteins that regulates cardiac development/function, and are linked to cardiac disease as well. The work from our lab and others has demonstrated that SUMO targets a multitude of cardiac enriched transcription factors that are important for maintaining normal cardiac development and function (Table 1). For more details, the reader is referred to the previous two reviews written by the author.29,30
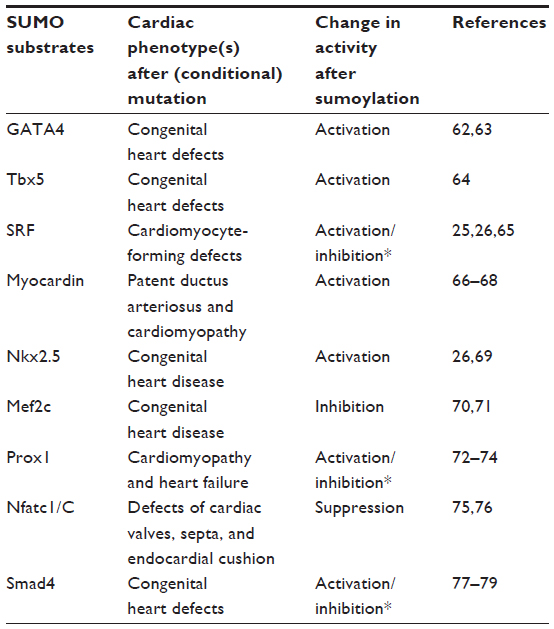 | Table 1 SUMO targets cardiac transcription factors |
Emerging roles of SUMO in cardiovascular disorders
SUMO and murine models of cardiovascular disease
In the animal models, knockout of SUMO-1 may cause cardiac structural defects and premature death,31,32 although the genetic background of mice probably also contributed to the development of this phenotype. This finding indicates the importance of SUMO-1 conjugation in cardiac structural morphogenesis. This importance was further corroborated by a gain-of-function model in which SENP2, a SUMO isopeptidase, was restrictedly activated in cardiomyocytes.33 Overexpressed SENP2 promoted deconjugation of SUMO-1 and caused congenital heart defects (CHDs), which was rescued by simultaneously overexpressed SUMO-1.33 The mice with increased SENP2 expression in cardiomyocytes also developed cardiac hypertrophy and fibrosis with aging, which was probably associated with decreased SUMO-2/3 conjugation, because it was not rescued by improved SUMO-1 conjugation.33 A more recent finding showed that SUMO-1 overexpression was protective against phenylephrine- and pressure-overload-induced cardiac hypertrophy, as well as decreased oxidative stress during cardiac hypertrophy and heart failure.34
Compared with the CHDs caused by activated SENP2, overexpression of SENP5, another SUMO isopeptidase, in cardiomyocytes induced cell death and dilated cardiomyopathy.35 In this gain-of-function murine model, mice with restricted SENP5 overexpression in cardiomyocytes showed decreased conjugation of SUMO-2/3 but not SUMO-1, and exhibited increased cell death that led to cardiomyopathy, fibrosis, and heart failure with aging. Mechanistically, overexpressed SENP5 damaged mitochondrial function, and activated apoptotic factors such as cleaved caspase3 and PARP-1.35 Correspondingly, increased expression of Bcl2, an antiapoptotic factor, rescued cardiac dysfunction triggered by overexpressed SENP5. In the SENP5-linked cardiomyopathic phenotype, at least one mitochondrial SUMO substrate, Drp1, a critical factor for mitochondrial fission, is involved. Desumoylation of Drp1 was observed in the cardiomyocytes with overexpressed SENP5, which showed also enlarged mitochondria, and knockdown of Drp1 by lentiviral-mediated short hairpin RNA reduced activation of apoptotic factors triggered by SENP5.35 These observations are in line with the previous report that SENP5 mainly targeted mitochondria in cultured cells.36,37
An earlier study showed a globally increased SUMO-1 conjugation in response to hypoxic insult in heart, resulting in elevated levels of SUMO-1-attached HIF1α.38 However, how sumoylation affects HIF1α’s stability and function remains in debate. Although several lines of evidence indicate that SUMO conjugation to HIF1α increased its stability and activity,39,40 HIF1α activity and stability may also be decreased by SUMO conjugation.18,41 Very intriguingly, a more recent study suggested that SUMO E3 ligases CBX4 and PIASy promoted HIF1α sumoylation on different lysine residues.42 As a result, while CBX4-enhanced sumoylation stabilized HIF1α and increased its activity, PIASy-stimulated sumoylation did the opposite.42 Thus, it seems likely that different sumoylation sites of HIF1α may mediate different functional outcomes once they are SUMO-conjugated.
Another study has provided evidence that extracellular signal-regulated kinase 5 (Erk5), an important mediator of ischemic/reperfused injury and inhibitor of apoptosis, is inhibited by sumoylation.43 Sumoylation of Erk5 is elevated in myocardial infarction, in the H2O2-induced inflammation, and in the diabetic aortas of mice.43,44 An increase in the level of SUMO-1-conjugated Erk5 appears to promote the inflammation, therefore worsening the injury. Consistent with this, SENP2 was shown to protect endothelial dysfunction at least partially via desumoylation of Erk5.45 Given that a multitude of SUMO substrates are involved in regulating ischemic/reperfused heart injury, the net functional outcome of the increased SUMO-1 conjugation in this particular pathophysiological setting in vivo merits further examination.
SERCA2a, an ATPase and a critical handler of Ca2+ homeostasis during excitation–contraction coupling, is a SUMO substrate.46 The levels of both free and SUMO-1-conjugated SERCA2a were significantly decreased in transverse-aortic-constriction-induced failing hearts of murine model.46 The delivery of SUMO-1 gene by cardiotropic recombinant adeno-associated viruses serotype 9 into failing hearts greatly improved heart function, and was accompanied by increased SERCA2a levels in hearts.46 Mechanistically, SUMO-1 increased SERCA2a stability and its ATP binding affinity. Thus, SERCA2a is one of those factors that mediate SUMO-1 function in cardiac pathophysiology. Additionally, SUMO-1 also abolished the H2O2-induced negative impacts on SERCA2a function.34
SUMO and human cardiovascular diseases
The evidence that the SUMO conjugation pathway is implicated in human cardiovascular disorders has been emerging recently. First, human CHD-linked mutations of some transcription factors present deficiency in SUMO conjugation. For instance, several naturally occurring missense mutations of Nkx2.5, particularly those in homeodomain, impaired its sumoylation.47 Moreover, expression of sumoylation-deficient Nkx2.5 mutant (K51R, conversion of lysine 51 to arginine) in mice with the background of Nkx2.5 haploinsufficiency caused CHDs,47 further supporting the deficient Nkx2.5 sumoylation underlying Nkx2.5-linked CHDs. Sumoylation of ZIC3 is another example. ZIC3 is an X-linked zinc finger transcription factor that is causally linked to human heterotaxy including CHDs.48 The nucleocytoplasmic shuttling of ZIC3, which is governed by nuclear export/import signals, is an important mechanism that mediates the activity of ZIC3. A number of naturally occurring missense mutations that are etiologically associated with CHDs negatively affect ZIC3 nuclear occupancy,49,50 consequently leading to its decreased activity. Interestingly, our work identified ZIC3 as a novel SUMO substrate on lysine 248. Mutation of lysine 248 to arginine reduced its nuclear localization, although it is not located in any of those nuclear export/import signals identified.51 Coincidently, a number of human missense mutations that exhibit diminished nuclear occupancy also showed decreased sumoylation, while those which have normal subcellular distribution also showed normal levels of sumoylation compared with wild type ZIC3.51 Moreover, recovery of SUMO-1 conjugation to these sumoylation-defective ZIC3 mutants by PIAS1, a SUMO E3 ligase, promoted their nuclear occupancy.51 These findings clearly demonstrate that sumoylation deficiency underpins the ZIC3-linked human congenital defects.
A subset of familial cardiomyopathy is also etiologically associated to the SUMO conjugation pathway. Mutations of lamin A, a nuclear structural protein, are associated with inherited dilated cardiomyopathy.52 Interestingly, SUMO targets lamin A on the lysine residue 201 in the consensus sequence MKEE, and the mutation of glutamic acid 203 to either glycine (E203G)53 or lysine (E203K),54 which are causally linked to familial dilated cardiomyopathy and conduction disease, negatively influences the lamin A’s sumoylation, consequently altering its nuclear distribution,55 which resembles the molecular phenotype exhibited by the sumoylation-resistant mutant E201R.55 These data support the argument that a defective SUMO conjugation of its substrate lamin A is directly implicated in the initiation/development of dilated cardiomyopathy.
Deficient SUMO conjugation may be involved in another cardiac conduction disease, the progressive familial cardiac conduction block I. This conduction disease exhibits autosomal-dominant inheritance, and the gene TRPM4, which encodes a Ca2+-activated nonselective cation channel, is associated with its pathogenesis.56,57 TRPM4 is a SUMO substrate,58 although the SUMO attachment site or sites remain elusive. A missense mutation in TRPM4, E7K (mutation of glutamic acid 7 to lysine), displays a resistance to the desumoylation by SENP1, subsequently protecting it from proteasomal degradation and leading to a gain of function.58
In human failing hearts, SUMO-1 conjugation was decreased and so was its conjugation to SERCA2a,46 indicating the implication of both in human heart failure. In the large animal model, SUMO-1 gene transfer greatly improved cardiac function in porcine models with ischemic-induced heart failure.59 SERCA2a as a SUMO substrate is partially involved in mediating SUMO-1 gene transfer-achieved cardiac protection; the protection obtained by the high levels of SUMO-1 expression in this large animal heart failure model may be independent of SERCA2a,59 indicating the involvement of other substrates/mechanisms.
More recently, our work has revealed that SENP5 was elevated in the human failing hearts at both transcription and protein levels.35 In the gain-of-function mouse model, overexpressed SENP5 in cardiomyocytes recapitulated the pathogenesis of dilated cardiomyopathy and heart failure of humans.35 Our finding demonstrates for the first time that SENP5, a desumoylation enzyme, is implicated in cardiomyopathy and heart failure of both human and murine models, therefore presenting it as a potential therapeutic target for cardiomyopathy and heart failure.
The ubiquitin proteasome pathway (UPP) is the major mechanism for degradation of cytosolic and nuclear proteins. Dysfunctional UPP may cause a number of human diseases, including cardiomyopathy.60 Ubc9, the sole SUMO E2, was found to be required for normal function of UPP.61 Overexpression of Ubc9 in cultured cardiomyocytes potentiated the activity of UPP, whereas depletion of Ubc9 inhibited the UPP function, leading to increased mis-folded protein aggregations. Thus, Ubc9 is an important factor for UPP to execute its essential function to eliminate aggregated proteins. It will be interesting to pursue if any SUMO targets are involved in mediating UPP function by Ubc9.
The main SUMO conjugation pathway components and substrates that are discussed in this section are summarized in Table 2.
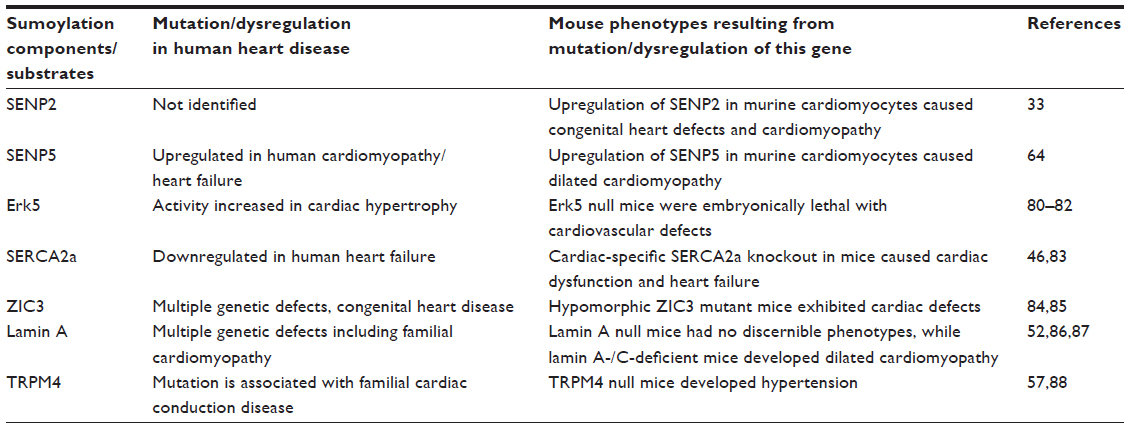 | Table 2 Summary of the role of the major SUMO pathway components and substrates discussed in this review that regulate cardiac function and disease |
SUMO as a therapeutic target in cardiac disease
Increasing evidence has pointed to the important role the SUMO conjugation pathway plays in the pathogenesis of cardiovascular diseases. Thus, the sumoylation pathway may represent a new potential therapeutic target for a number of cardiac diseases including cardiomyopathy and heart failure. Given the existence of multiple sumoylation machinery with less specificity and widely distributed targets in various signaling pathways and gene regulatory networks, the guiding principles to develop the small molecules for therapeutic purpose via altering sumoylation activity should be high selectivity and specificity. For instance, increased SUMO-1 conjugation appears to provide a general protection of heart against insults such as oxidative stress and ischemia/reperfusion; however, whether and how enhanced conjugation of SUMO-2/3 affects cardiac function and disease progression remains unclear. Thus, a small molecule that increases pan-SUMO conjugation should be cautiously considered as a practical treatment method. Instead, a chemical that favorably improves conjugation of SUMO-1 but not SUMO-2/3 should be more applicable in clinic. Also, the agents that specifically repress the activity of SENP5 instead of all SENPs are desired, because recent evidence shows that only SENP5 was involved in human failing heart.35 Another challenge will be how to preferably increase/decrease SUMO attachment to a particular target in vivo. The additional challenge also lies in the fact of the ubiquitous expression pattern of SUMO conjugation machinery; the molecules that are to be developed should only exert impacts on sumoylation activity specifically in heart as desired but not in other tissues/organs to treat cardiac-specific diseases and reduce nondesired effects.
Conclusion and prospects
Sumoylation is a versatile and fascinating posttranslational modification that underlies a wide spectrum of cellular functions. Recently, much attention has been paid to sumoylation in the heart field due to its importance in cardiac pathology/pathophysiology. Indeed, recent studies from our lab and others have implicated SUMO conjugation in human cardiac diseases. As mentioned above, dysregulated levels of SENP5 and SUMO-1 conjugation are present in the human failing hearts, and overexpression of SENP5 in mouse cardiomyocytes recapitulates the development of human cardiomyopathy and heart failure. Still, many questions remain open. For instance, increased SUMO-1 did not reduce cardiac hypertrophy and improve cardiac function of SENP2 transgenic mice.33 Does that suggest that SUMO-2/3 is also involved in cardiac muscle disorders? Also, do any SUMO E3 ligases such as PIAS family members play a role in the pathogenesis of human cardiac diseases? Additionally, in a particular cardiovascular disease setting, does SUMO conjugation to only one substrate, or to a group of substrates that work in the same functional network or pathway, make a contribution to the disease development? Furthermore, it remains largely unexplored how the expression of the SUMO conjugation pathway components such as SENP5 or SUMO-1 is regulated in heart, although SENP5 was found to positively mediate SUMO-2/3 transcription.35
In conclusion, to fully understand the molecular mechanisms by which the SUMO conjugation pathway contributes to cardiovascular diseases, it is necessary to have more systemic studies by employing various genetically modified murine models along with stress stimulation. Given that this field is currently catching more attention, we anticipate that in the foreseeable future the studies will advance our knowledge about the importance of sumoylation in heart disease initiation and/or progression, and will pave the way to develop highly specific small molecules for therapeutic purpose to ameliorate heart afflictions by targeting the SUMO conjugation pathway.
Acknowledgments
The authors apologize to the colleagues whose work was not cited due to page limitation. The work from the author’s lab was supported by the grants from the Texas Higher Education Coordinating Board (THECB), the American Heart Association, and the National Institutes of Health (to JW).
Disclosure
The authors report no conflicts of interest in this review.
References
Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88:97–107. | |
Hay RT. Sumo-specific proteases: A twist in the tail. Trends Cell Biol. 2007;17:370–376. | |
Drag M, Salvesen GS. DeSUMOylating enzymes – SENPs. IUBMB Life. 2008;60:734–742. | |
Hickey CM, Wilson NR, Hochstrasser M. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol. 2012;13:755–766. | |
Wang L, Wansleeben C, Zhao S, Miao P, Paschen W, Yang W. SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep. 2014;15:878–885. | |
Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11:861–871. | |
Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–956. | |
Pichler A, Gast A, Seeler JS, Dejean A, Melchior F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell. 2002;108:109–120. | |
Rytinki MM, Kaikkonen S, Pehkonen P, Jääskeläinen T, Palvimo JJ. Pias proteins: pleiotropic interactors associated with SUMO. Cell Mol Life Sci. 2009;66:3029–3041. | |
Kagey MH, Melhuish TA, Wotton D. The polycomb protein Pc2 is a SUMO E3. Cell. 2003;113:127–137. | |
Morita Y, Kanei-Ishii C, Nomura T, Ishii S. TRAF7 sequesters c-Myb to the cytoplasm by stimulating its sumoylation. Mol Biol Cell. 2005;16:5433–5444. | |
Weger S, Hammer E, Heilbronn R. Topors acts as a SUMO-1 E3 ligase for p53 in vitro and in vivo. FEBS Lett. 2005;579:5007–5012. | |
Pungaliya P, Kulkarni D, Park HJ, et al. TOPORS functions as a SUMO-1 E3 ligase for chromatin-modifying proteins. J Proteome Res. 2007;6:3918–3923. | |
Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. | |
Andrews EA, Palecek J, Sergeant J, Taylor E, Lehmann AR, Watts FZ. Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol Cell Biol. 2005;25:185–196. | |
Rodriguez MS, Dargemont C, Hay RT. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J Biol Chem. 2001;276:12654–12659. | |
Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108:345–356. | |
Cheng J, Kang X, Zhang S, Yeh ET. Sumo-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–595. | |
Kang X, Qi Y, Zuo Y, et al. SUMO-specific protease 2 is essential for suppression of polycomb group protein-mediated gene silencing during embryonic development. Mol Cell. 2010;38:191–201. | |
Mukhopadhyay D, Dasso M. Modification in reverse: The SUMO proteases. Trends Biochem Sci. 2007;32:286–295. | |
Ulrich HD. Two-way communications between ubiquitin-like modifiers and DNA. Nat Struct Mol Biol. 2014;21:317–324. | |
Feligioni M, Nisticò R. SUMO: a (oxidative) stressed protein. Neuromolecular Med. 2013;15:707–719. | |
Andreou AM, Tavernarakis N. SUMOylation and cell signalling. Biotechnol J. 2009;4:1740–1752. | |
Wang YE, Pernet O, Lee B. Regulation of the nucleocytoplasmic trafficking of viral and cellular proteins by ubiquitin and small ubiquitin-related modifiers. Biol Cell. 2012;104:121–138. | |
Matsuzaki K, Minami T, Tojo M, et al. Serum response factor is modulated by the SUMO-1 conjugation system. Biochem Biophys Res Commun. 2003;306:32–38. | |
Wang J, Zhang H, Iyer D, Feng XH, Schwartz RJ. Regulation of cardiac specific nkx2.5 gene activity by small ubiquitin-like modifier. J Biol Chem. 2008;283(34):23235–23243. | |
Rodriguez JA. Interplay between nuclear transport and ubiquitin/SUMO modifications in the regulation of cancer-related proteins. Semin Cancer Biol. 2014;27:11–19. | |
Krumova P, Weishaupt JH. Sumoylation in neurodegenerative diseases. Cell Mol Life Sci. 2013;70:2123–2138. | |
Wang J, Schwartz RJ. Sumoylation and regulation of cardiac gene expression. Circ Res. 2010;107:19–29. | |
Wang J. Cardiac function and disease: emerging role of small ubiquitin-related modifier. Wiley Interdiscip Rev Syst Biol Med. 2011;3:446–457. | |
Wang J, Chen L, Wen S, et al. Defective sumoylation pathway directs congenital heart disease. Birth Defects Res A Clin Mol Teratol. 2011;91:468–476. | |
Alkuraya FS, Saadi I, Lund JJ, Turbe-Doan A, Morton CC, Maas RL. SUMO1 haploinsufficiency leads to cleft lip and palate. Science. 2006;313:1751. | |
Kim EY, Chen L, Ma Y, et al. Enhanced desumoylation in murine hearts by overexpressed SENP2 leads to congenital heart defects and cardiac dysfunction. J Mol Cell Cardiol. 2012;52:638–649. | |
Lee A, Jeong D, Mitsuyama S, et al. The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid Redox Signal. Epub June 3, 2014. | |
Kim EY, Zhang Y, Beketaev I, et al. SENP5, a SUMO isopeptidase, induces apoptosis and cardiomyopathy. J Mol Cell Cardiol. Epub August 12, 2014. | |
Zunino R, Schauss A, Rippstein P, Andrade-Navarro M, McBride HM. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J Cell Sci. 2007;120:1178–1188. | |
Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem. 2009;284:17783–17795. | |
Shao R, Zhang FP, Tian F, et al. Increase of SUMO-1 expression in response to hypoxia: direct interaction with HIF-1alpha in adult mouse brain and heart in vivo. FEBS Lett. 2004;569:293–300. | |
Bae SH, Jeong JW, Park JA, et al. Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem Biophys Res Commun. 2004;324:394–400. | |
Carbia-Nagashima A, Gerez J, Perez-Castro C, et al. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell. 2007;131:309–323. | |
Berta MA, Mazure N, Hattab M, Pouysségur J, Brahimi-Horn MC. SUMOylation of hypoxia-inducible factor-1alpha reduces its transcriptional activity. Biochem Biophys Res Commun. 2007;360:646–652. | |
Li J, Xu Y, Long XD, et al. Cbx4 governs Hif-1α to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell. 2014;25:118–131. | |
Shishido T, Woo CH, Ding B, et al. Effects of MEK5/ERK5 association on small ubiquitin-related modification of ERK5: Implications for diabetic ventricular dysfunction after myocardial infarction. Circ Res. 2008;102:1416–1425. | |
Woo CH, Shishido T, McClain C, et al. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res. 2008;102:538–545. | |
Heo KS, Chang E, Le NT, et al. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced sumoylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res. 2013;112:911–923. | |
Kho C, Lee A, Jeong D, et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. | |
Kim EY, Chen L, Ma Y, et al. Expression of sumoylation deficient Nkx2.5 mutant in Nkx2.5 haploinsufficient mice leads to congenital heart defects. PLoS One. 2011;6:e20803. | |
Cowan J, Tariq M, Ware SM. Genetic and functional analyses of ZIC3 variants in congenital heart disease. Hum Mutat. 2014;35:66–75. | |
Bedard JE, Purnell JD, Ware SM. Nuclear import and export signals are essential for proper cellular trafficking and function of ZIC3. Hum Mol Genet. 2007;16:187–198. | |
Hatayama M, Tomizawa T, Sakai-Kato K, et al. Functional and structural basis of the nuclear localization signal in the ZIC3 zinc finger domain. Hum Mol Genet. 2008;17:3459–3473. | |
Chen L, Ma Y, Qian L, Wang J. Sumoylation regulates nuclear localization and function of zinc finger transcription factor ZIC3. Biochim Biophys Acta. 2013;1833:2725–2733. | |
van Tintelen JP, van Spaendonck-Zwarts KY, van den Berg MP. Lamin A/C-related cardiac disease and pregnancy. Eur J Heart Fail. 2010;12:532–534. | |
Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. | |
Jakobs PM, Hanson EL, Crispell KA, et al. Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J Card Fail. 2001;7:249–256. | |
Zhang YQ, Sarge KD. Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial cardiomyopathies. J Cell Biol. 2008;182:35–39. | |
Brink PA, Ferreira A, Moolman JC, Weymar HW, van der Merwe PL, Corfield VA. Gene for progressive familial heart block type I maps to chromosome 19q13. Circulation. 1995;91:1633–1640. | |
Kruse M, Schulze-Bahr E, Corfield V, et al. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest. 2009;119:2737–2744. | |
Liu H, El Zein L, Kruse M, et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ Cardiovasc Genet. 2010;3:374–385. | |
Tilemann L, Lee A, Ishikawa K, et al. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci Transl Med. 2013;5:211ra159. | |
Schlossarek S, Frey N, Carrier L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J Mol Cell Cardiol. 2014;71:25–31. | |
Gupta MK, Gulick J, Liu R, Wang X, Molkentin JD, Robbins J. Sumo E2 ligase UBC9 is required for efficient protein quality control in cardiomyocytes. Circ Res. Epub August 5, 2014. | |
Wang J, Feng XH, Schwartz RJ. SUMO-1 modification activated GATA4-dependent cardiogenic gene activity. J Biol Chem. 2004; 279:49091–49098. | |
Belaguli NS, Zhang M, Garcia AH, Berger DH. PIAS1 is a GATA4 SUMO ligase that regulates GATA4-dependent intestinal promoters independent of SUMO ligase activity and GATA4 sumoylation. PLoS One. 2012;7:e35717. | |
Beketaev I, Kim EY, Zhang Y, Yu W, Qian L, Wang J. Potentiation of Tbx5-mediated transactivation by SUMO conjugation and protein inhibitor of activated STAT 1 (PIAS1). Int J Biochem Cell Biol. 2014;50:82–92. | |
Niu Z, Iyer D, Conway SJ, et al. Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci U S A. 2008;105:17824–17829. | |
Huang J, Cheng L, Li J, et al. Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J Clin Invest. 2008;118:515–525. | |
Wang J, Li A, Wang Z, Feng X, Olson EN, Schwartz RJ. Myocardin sumoylation transactivates cardiogenic genes in pluripotent 10T1/2 fibroblasts. Mol Cell Biol. 2007;27:622–632. | |
Huang J, Min Lu M, Cheng L, et al. Myocardin is required for cardiomyocyte survival and maintenance of heart function. Proc Natl Acad Sci U S A. 2009;106:18734–18739. | |
Costa MW, Lee S, Furtado MB, et al. Complex SUMO-1 regulation of cardiac transcription factor Nkx2-5. PLoS One. 2011;6:e24812. | |
Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–1407. | |
Grégoire S, Yang XJ. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol Cell Biol. 2005;25:2273–2287. | |
Pan MR, Chang TM, Chang HC, Su JL, Wang HW, Hung WC. Sumoylation of Prox1 controls its ability to induce VEGFR3 expression and lymphatic phenotypes in endothelial cells. J Cell Sci. 2009;122:3358–3364. | |
Shan SF, Wang LF, Zhai JW, et al. Modulation of transcriptional corepressor activity of prospero-related homeobox protein (Prox1) by SUMO modification. FEBS Lett. 2008;582:3723–3728. | |
Risebro CA, Searles RG, Melville AA, et al. Prox1 maintains muscle structure and growth in the developing heart. Development. 2009;136:495–505. | |
Nayak A, Glöckner-Pagel J, Vaeth M, et al. Sumoylation of the transcription factor NFATc1 leads to its subnuclear relocalization and interleukin-2 repression by histone deacetylase. J Biol Chem. 2009;284:10935–10946. | |
de la Pompa JL, Timmerman LA, Takimoto H, et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. | |
Moskowitz IP, Wang J, Peterson MA, et al. Transcription factor genes Smad4 and Gata4 cooperatively regulate cardiac valve development. [corrected]. Proc Natl Acad Sci U S A. 2011;108:4006–4011. | |
Lin X, Liang M, Liang YY, Brunicardi FC, Melchior F, Feng XH. Activation of transforming growth factor-beta signaling by SUMO-1 modification of tumor suppressor Smad4/DPC4. J Biol Chem. 2003;278:18714–18719. | |
Long J, Wang G, He D, Liu F. Repression of Smad4 transcriptional activity by SUMO modification. Biochem J. 2004;379:23–29. | |
Regan CP, Li W, Boucher DM, Spatz S, Su MS, Kuida K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc Natl Acad Sci U S A. 2002;99:9248–9253. | |
Yan L, Carr J, Ashby PR, Murry-Tait V, Thompson C, Arthur JS. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev Biol. 2003;3:11. | |
Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007;116:1413–1423. | |
Li L, Louch WE, Niederer SA, et al. Sodium accumulation in SERCA knockout-induced heart failure. Biophys J. 2012;102:2039–2048. | |
Herman GE, El-Hodiri HM. The role of ZIC3 in vertebrate development. Cytogenet Genome Res. 2002;99:229–235. | |
Haaning AM, Quinn ME, Ware SM. Heterotaxy-spectrum heart defects in Zic3 hypomorphic mice. Pediatr Res. 2013;74:494–502. | |
Nikolova V, Leimena C, McMahon AC, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113:357–369. | |
Fong LG, Ng JK, Lammerding J, et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest. 2006;116:743–752. | |
Mathar I, Vennekens R, Meissner M, et al. Increased catecholamine secretion contributes to hypertension in TRPM4-deficient mice. J Clin Invest. 2010;120:3267–3279. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.