")
Back to Journals » The Application of Clinical Genetics » Volume 14
Successful Pregnancy Following Preimplantation Genetic Diagnosis of Adrenoleukodystrophy by Detection of Mutation on the ABCD1 Gene
Authors Trinh The S , Trieu Tien S , Vu Van T, Nguyen Ngoc N , Tran Ngoc Thao M , Tran Van K, Vu Nhat D, Do Nhu B
Received 6 May 2021
Accepted for publication 8 July 2021
Published 14 July 2021 Volume 2021:14 Pages 313—319
DOI https://doi.org/10.2147/TACG.S318884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Son Trinh The,1,* Sang Trieu Tien,2,* Tam Vu Van,3,4 Nhat Nguyen Ngoc,1 My Tran Ngoc Thao,5 Khoa Tran Van,2 Dinh Vu Nhat,6,7 Binh Do Nhu8,9
1Military Institute of Clinical Embryology and Histology, Vietnam Military Medical University, Hanoi, 12108, Vietnam; 2Department of Biology and Genetics, Vietnam Military Medical University, Hanoi, 12108, Vietnam; 3Director Office, Hai Phong Hospital of Obstetrics and Gynecology, Haiphong, 40000, Vietnam; 4Obstetrics and Gynecology Department of Haiphong University of Medicine and Pharmacy, Haiphong, 40000, Vietnam; 5Département de formation Biologie moléculaire et cellulaire, Sorbonne University, Paris, 75006, France; 6Director Office, Military Hospital 103, Hanoi, 12108, Vietnam; 7Department of Trauma and Orthopedic Surgery, Vietnam Military Medical University, Hanoi, 121– 08, Vietnam; 8Division of Military Science, Military Hospital 103, Hanoi, 12108, Vietnam; 9Department of Infectious Disease, Vietnam Military Medical University, Hanoi, 12108, Vietnam
*These authors contributed equally to this work
Correspondence: Dinh Vu Nhat
Military Hospital 103, Hanoi, 12108, Vietnam
Email [email protected]
Background: Adrenoleukodystrophy (ALD) is a rare sex-linked recessive disorder that disrupts adrenal gland function and the white matter of the nervous system. According to recent epidemiological statistics, up to this moment, the disease is the most recorded peroxisomal disorder. ABCD1 is a gene related to ALD, with more than 850 unique mutations have been reported. Early diagnosis of the disease would help to consult families with ALD to plan for interventions to prevent passing along the pathogenic mutations to their children.
Material and Methods: A heterozygous ABCD1 gene mutation related to ALD found in a Vietnamese woman was used to design primers for the polymerase chain reaction (PCR) to amplify the segment spanning the mutation. Then, combining sequencing methods for the PCR products, especially Sanger sequencing and next-generation sequencing (NGS), a protocol was developed to detect mutations on the ABCD1 gene to apply for the DNA samples of in-vitro fertilization (IVF) embryos biopsied at the blastocyst stage to screen for pathogenic alleles.
Results: The established protocol for PGD of ALD detected mutant alleles in 5/8 embryos (62.5%), while the remaining 3 embryos (37.5%) did not carry any mutation. One of the 3 embryos was transferred, and a healthy female baby was born after a full-term pregnancy.
Conclusion: The developed protocol was helpful for the preimplantation genetic diagnosis process to help families with the monogenic disease of ALD but wish to have healthy children.
Keywords: adrenoleukodystrophy, ALD, peroxisome disorder, ABCD1 gene mutation, preimplantation genetic diagnosis, PGD
Introduction
ALD is a rare sex-linked hereditary disease with the incidence dropped between 1:20,000 and 1:30,000 male newborns, without significant differences among countries and ethnic groups globally; however, this figure is increasing prior to the widespread application of newborn screening. Mutations in the ABCD1 gene are the pathogenic factors leading to the impaired β-oxidation process in peroxisomes. All patients with ALD carry the mutations; up to date, more than 850 non-recurrent mutations of this disease have been cataloged without correlation to phenotypes.1–3 The gene ABCD1 is located near the end of the X chromosome’s long arm: at Xq28 and has a length of 19.9 kb with 10 exons.4,5 ABCD1 encodes a transmembrane protein made up of 745 amino acids called adrenoleukodystrophy protein, or ALDP. This protein is located across the peroxisome membrane, transporting VLCFA (very long chain fatty acids) from the cytoplasm through the membrane into the organelle to participate in the β-oxidation process.6–8 Mutations in the ABCD1 gene will directly affect the ALD protein, result in a change in the protein’s structure and quantity, making it unable to carry out its transport function, thereby causing the accumulation of VLCFA in the cytoplasm. This accumulation has negative effects on cells, especially on nervous system cells.1,9,10
There are three main phenotypes of ALD that can be listed as follows: cerebral ALD, adrenomyeloneuropathy (AMN), and primary adrenal insufficiency. ALD patients are often asymptomatic at birth; however, clinical symptoms will potentially develop during life, which will eventually cause severe disabilities, especially in males. As the disease progresses, patients might develop primary adrenal insufficiency symptoms and neurologic disease. Hence, ALD is a rapidly progressive disorder that may cause devastating situations for affected patients since an effective treatment such as stem cell transplant can only prevent disease development yet leave them at risk for AMN. In order to find the appropriate therapy, patients must have regular screening to timely detect and diminish the possibility for lethal disease.11–15
ALD is a sex-linked recessive genetic disorder with 95% of reported cases received the ABCD1 pathogenic variant from their parents; only about 5% of reported cases are caused by de novo mutations.1,4,16,17 Heterozygote detection for at-risk females leading to prenatal testing or preimplantation genetic testing for at-risk pregnancies is necessary if the familial pathogenic variant is known.18 In the last decades, preimplantation genetic diagnosis plays a valuable tool to avoid inherited diseases by transferring unaffected in-vitro fertilization (IVF) embryos and having alternative potentiality to prenatal diagnosis. Indeed, PGD first concluded the selection usually embryos for patients at risk of transmitting X-linked recessive disorders.19,20 Furthermore, the widespread application of advances in molecular biology fosters the advent of specific diagnoses for monogenic defects. The fifth report of the European Society for Human Reproduction and Embryology (ESHRE) PGD Consortium recommended over 40 monogenic diseases with PGD indication.21 However, setting up and testing embryonic molecular diagnosis is work-intensive, precise, and costly because the main reason, which is the DNA sample, is limiting. Much effort has been spent to obtain genomic DNA of adequate and quality for genetic analysis. Consequently, whole genome amplification (WGA)20 is a standard solution to solve the existing problem, but its main drawback is the generation of nonspecific amplification artifacts.22,23 As a result, we described a PGD protocol using WGA followed by conventional PCR, then applying Sanger sequencing for detecting the mutation. This work aimed to expand the dependability of PGD for ALD and increase the accuracy and economic effect of the test.
Materials and Methods
Patients Description
A 26-year-old Vietnamese female partner whose family has a background of ALD was enrolled in the study. Her healthy husband was 28 years old and did not present any clinical or genetic alteration of interest. By studying her genetic information with Next-Generation Sequencing (NGS), it was reported that the wife carried a heterozygous mutation c.854G>C (p.Arg285Pro) on exon 1 of ABCD1. According to up-to-date statistics related to ALD (https://adrenoleukodystrophy.info/, accessed April 2021), the mutation c.854G>C has only been reported as pathogenic twice in the past, none of which was found in Vietnam, for that reason, the mutation identified in this female carrier can be considered as the third time recorded in the history of research on ALD and the first report in Vietnam.
All people described in this research were signed written informed consent for the publication of the case details, and the protocol was approved by the Ethical Review Committee of Vietnam Military Medical University (No.1068/2019/VMMU-IRB). This study was also conducted using good clinical practice following the Declaration of Helsinki and its later amendments or comparable ethical standards.
DNA Extraction from Whole Blood
DNA was extracted from the collected blood samples by following the protocol of the G-spin™ Total DNA Extraction Kit (Lot.No. 105260653; Exp. Oct. 2022). DNA went through a quality check process with a SpectraMax QuickDrop to measure optical density (OD) and A260/A280 index. Hence, the DNA collected from the sample was qualified to be used in the next steps of the research. DNA samples were stored at −20°C.
Blastocyst Embryo Biopsy
There were seven embryos (TR1 – TR7) cultured to the fifth day and one embryo (TR8) cultured to the sixth day of the described couple who had done IVF at the Military Institute of Clinical Embryology and Histology (MICEH). Then, the embryos were biopsied (3–5 cells) and washed with PBS 1X and 1% PVP solution. After that, the washed embryo cells were contained in the 0.2 mL PCR tube. The embryo cells were stored at −20°C.
Whole Genome Amplification for Embryos’ Genome
The DNA from the biopsied embryos was amplified with REPLI-g® Single Cell Kit (Lot.No. 166013027; Exp. Sep. 2021) and diluted by nuclease-free water to reach a concentration of around 20 ng/µL. The concentration and purification were calculated with a SpectraMax QuickDrop. Therefore, the amplified DNA collected from the embryonic cells was qualified to be used in the research. DNA samples were stored at −20°C.
Polymerase Chain Reaction (PCR) Analysis
First, primers were designed to amplify the segment spanning the detected mutation c.854G>C. Afterward, a PCR was performed by using the designed primers and DNA collected from the mother – the mutated allele carrier and DNA amplified from biopsied embryonic cells. PCR products were then electrophoresed on 2% agarose gel on multiSUB Choice, Wide Midi Horizontal Electrophoresis System (Cleaver Scientific, SKU: MSCHOICE10) to check for the appropriate desired products. The detailed information on the primers and PCR reaction was presented in Supplementary 1.
Sanger Sequencing and the Next-Generation Sequencing Analysis
The amplified PCR products showing the accurate bands on electrophoresis results would be sequenced by Sanger sequencing to scan for the c.854G>C mutation. Next, any embryos that did not have any mutated allele would be screened for chromosomal abnormalities with Next-Generation Sequencing system. The flowchart of the study protocol was shown in Figure 1.
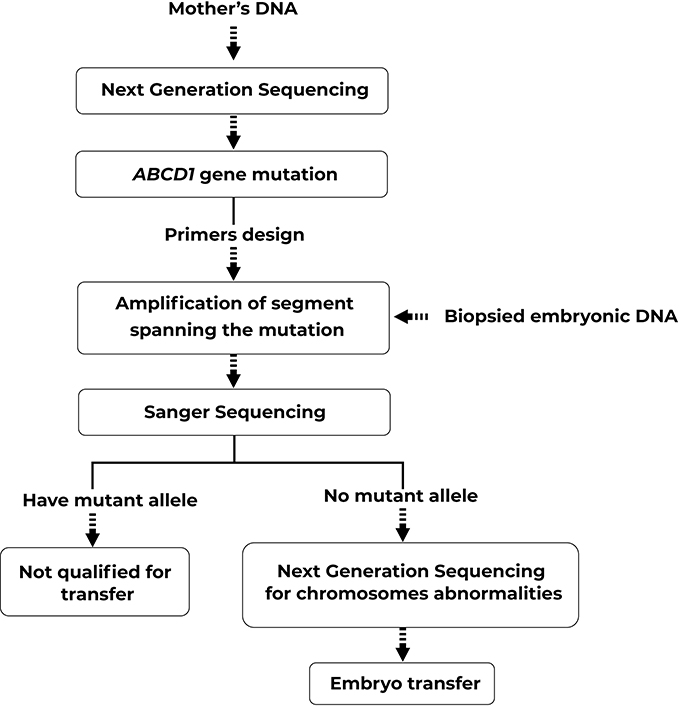 |
Figure 1 Flowchart of PGD protocol for ALD. |
Results
Preimplantation Genetic Diagnosis Program for ALD
The PCR reaction using the described component and the thermal cycle was performed in triplicate. After that, the PCR products were electrophoresed on 2% agarose gel and observed under UV light, and the results obtained were consistent in all reactions. By annotating the gel electrophoresis (Supplementary 2), it could be stated that the PCR reaction successfully amplified the desired gene segment of all embryos in comparison with the positive control that was the heterozygous woman’s DNA. The product bands appeared bright and clear at the position corresponding to the standard scale size of about 200 bp, proving that the amplified segment had the size of 199 bp, consistent with the initial expectation. From here, the PCR products would be purified and sequenced via the Sanger method to detect the c.854G>C mutation in these cells.
Sanger Sequencing and NGS Results
Sanger sequencing was carried out using only one primer to amplify the forward strand so that the interpretation of the results would be more homogeneous. The obtained electropherograms were analyzed using BioEdit software (Supplementary 3) and the annotated results are shown in Table 1.
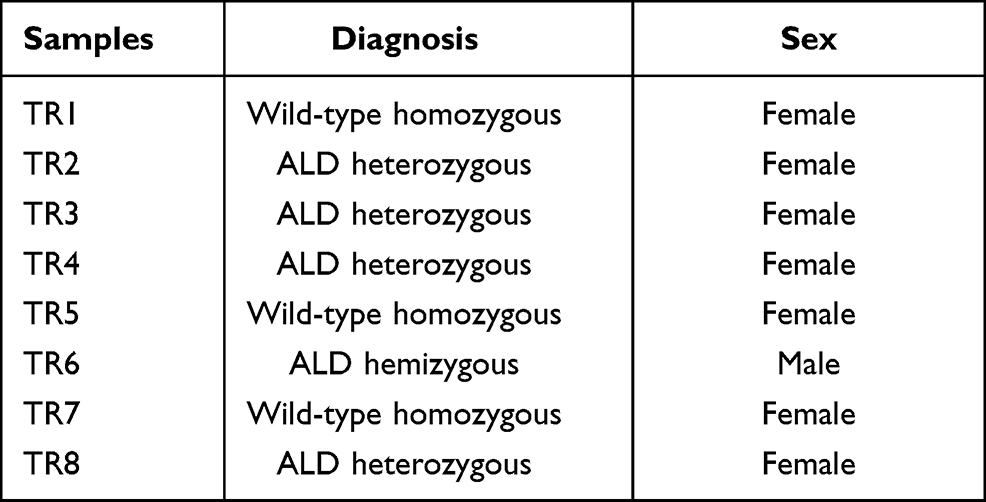 |
Table 1 Genotypes of Eight in vitro Fertilization Embryos by Annotating Sanger Sequencing Results |
Sanger sequencing results showed 3 embryos (37.5%) that did not carry any pathogenic mutant alleles. In comparison, 4 embryos (50%) had the allele with the pathogenic mutation in the heterozygous form, which had the same mutation peak as the mother or the positive control, and 1 embryo (12.5%) carried the pathogenic allele in the hemizygote form. The father was known to carry no ALD mutation proves that embryos carrying pathogenic alleles during meiosis to form gametes have received the mutated alleles from the mother. These embryos will be assessed as unsuitable for use in embryo transfer, and those that do not carry the mutant allele will continue to be subjected to preimplantation genetic screening tests for evaluating the quality of embryos before transferring. For DNA samples of TR1, TR5, and TR7 embryos with normal results which do not carry any pathogenic mutant allele will be sequenced according to the next-generation sequencing method to screen for chromosomal abnormalities. NGS screening results for these 3 embryos without ALD are shown in Supplementary 4.
Both TR1 and TR7 samples had abnormalities in the chromosomes; in detail, the TR1 sample had abnormalities in Chromosome 11 when TR7 has abnormalities in Chromosome 2, 4, 6, 8, 11, 17, 18, whereas the sample TR5 had none of these. Therefore, we concluded that the TR5 embryo’s quality was acceptable and should be transferred into the mother’s uterus.
Discussion
As in many X-linked recessive disorders, it can be assumed that females remain asymptomatic carriers; they often have an association with their extended family having reported ALD patients. However, Engelen et al suggested that heterozygous women with ALD did show milder symptoms of neurologic diseases compared to men at a later stage of their lives, especially in their 40s and 60s.24 Consequently, a genetic diagnosis of ALD is necessary to detect heterozygous women who could be offered prenatal or preimplantation diagnosis for pregnancies in the future.2,25 Prenatal diagnosis of ALD was first performed in 1982 when Moser et al carried out amniocentesis in heterozygous women for the VLCFA level determination.26 Later on, Imamura et al documented the mutation by genome analysis. Hence, prenatal diagnosis was a crucial measure for families with ALD history by analyzing the fetal DNA from amniotic fluid or placenta source.27,28 Nevertheless, amniocentesis is an invasive measure that may have a drawback of insufficient fetal DNA level collected for diagnosis and adversely affects the mother’s health.
Because of solving the matters mentioned above, the preimplantation diagnosis was developed with molecular biology advances. In the beginning, PGD was mainly the selection of female embryos for patients having a background of X-linked recessive diseases.19 However, half of the discarded male embryos which are not transferred are unaffected by the disease. In this way, not only does the pregnancy outcome decrease, but it also brings up many ethical problems. Otherwise, fifty percent of selected female embryos carried the mutant gene that would transmit the mutated allele to the next generation, so the inheritance of the disease to the offspring is not permanently stopped. There are prior reports of sex selection using different techniques to identify affected Xq28 loci. They had the same procedure and outcome: to use multiple displacement amplification technologies for adequate input DNA materials and, in the end, to determine the sex of the embryos. Only one prior study reported by Miriam Iglesias et al in 2008 that made use of PGD to prevent ALD indicated a successful pregnancy.29–32 Up to date, molecular biology becoming more and more essential with many advances has led to the fact that, once the mutation is identified, the couples would undergo specific genetic diagnosis instead of simple sex selection. Hence, our study with the successful live birth after a full-term pregnancy is more suitable with the modern trend and avoids ethical concerns by not choosing the sex but applying sequencing technology to determine the pathogenic gene.33–35
Setting up the new PGD protocol for monogenic diseases is always time-consuming and demanding much of the labor’s effort to improve the technique. Firstly, multiple biopsy methods are now available with their advantages. Among them, 5–10 trophectoderm (TE) cells biopsied at the blastocyst stage provide more genetic material as well as make the diagnosis more reliable and have fewer errors. Even though the chosen biopsy method generates the highest DNA yields, WGA is still necessary to increase the amount of template DNA available up to 106 times for diagnostic reactions, then template DNA is further amplified in a targeted fashion using conventional PCR.36–38 PCR primers must be explicitly designed to amplify the mutation and optimize other PCR conditions to diminish the amplification failure and ADO rate.
The Sanger sequencing method was first developed in 1977. Though spreading current NGS circulation, improvements in the Sanger sequencing methodology, commercialization, and automation have enabled it to remain the most suitable sequencing method for many current applications. Sanger sequencing technology remains vastly advantageous for applications where high throughput is not required.39 Our PGD procedure was carried out on the embryos of one family and owing to its rapidity and efficiency. However, the embryonic aneuploidy prevalence was high even in young patients with a monogenic disorder background. The majority of the patient population (53.2%) had at least one blastocyst with unaffected single gene defect were aneuploid, approximately 26.5% of normal or non-pathogenic blastocysts diagnosed based on monogenic disease PGD were aneuploid which would have been transferred, resulting in negative impacts in pregnancy outcome unless PGS had performed.40 Conversely, concurrent screening demonstrably aided in embryo selection, as evidenced by the significant improvement in single embryo transfer rates, so 24-chromosome aneuploidies screening became more and more popular to apply in embryos biopsied for PGD to reduce the risk of miscarriage in the gestational period.41–43 Indeed, PGD combined with PGS allowed opting for an embryo that did not carry the mutated allele and euploid embryos to transfer into the mother’s uterus then, fortunately, achieved pregnancy with a baby girl. Later on, her peripheral blood sample was collected and applied to the developed protocol to confirm the accuracy. The baby girl was a wild-type homozygote and completely free of ALD.
Conclusion
The established protocol was helpful for the preimplantation genetic diagnosis process to help families with the monogenic disease of ALD but wish to have healthy children. This process not only has the potential to identify embryos with pathogenic mutations but also prevent their inheritance among generations at an early stage.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request. If you have concerns about sharing the data, please contact [email protected].
Ethical Statements
All people described in this research were signed written informed consent for the publication of the case details, and the protocol was approved by the Ethical Review Committee of Vietnam Military Medical University (No.1068/2019/VMMU-IRB). This study was conducted following the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent for Publication
I understand that The Application of Clinical Genetics journal may be available in both print and on the internet, and will be available to a broader audience through marketing channels and other third parties. Therefore, anyone can read material published in the journal. I understand that readers may include not only medical professionals and scholarly researchers but also journalists and general members of the public. If you have any queries, please contact [email protected].
Acknowledgments
We thank all the staff in the Department of Biology and Genetics, Military institute of clinical Embryology and Histology, and the Director board of Military Hospital 103 for supporting this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Disclosure
The authors declared no potential conflicts of interest for this work nor the research, authorship, and publication of this article.
References
1. Wiesinger C, Eichler FS, Berger J. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis. Appl Clin Genet. 2015;8:109–121. doi:10.2147/TACG.S49590
2. Kemp S, Pujol A, Waterham HR, et al. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum Mutat. 2001;18(6):499–515. doi:10.1002/humu.1227
3. Lee S, Clinard K, Young SP, et al. Evaluation of X-linked adrenoleukodystrophy newborn screening in North Carolina. JAMA Netw Open. 2020;3(1):e1920356. doi:10.1001/jamanetworkopen.2019.20356
4. Mosser J, Douar AM, Sarde CO, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361(6414):726–730. doi:10.1038/361726a0
5. Migeon BR, Moser HW, Moser AB, Axelman J, Sillence D, Norum RA. Adrenoleukodystrophy: evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci U S A. 1981;78(8):5066–5070. doi:10.1073/pnas.78.8.5066
6. van Roermund CWT, Visser WF, Ijlst L, et al. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008;22(12):4201–4208. doi:10.1096/fj.08-110866
7. Wiesinger C, Kunze M, Regelsberger G, Forss-Petter S, Berger J. Impaired very long-chain acyl-CoA β-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J Biol Chem. 2013;288(26):19269–19279. doi:10.1074/jbc.M112.445445
8. Kawaguchi K, Mukai E, Watanabe S, et al. Acyl-CoA thioesterase activity of peroxisomal ABC protein ABCD1 is required for the transport of very long-chain acyl-CoA into peroxisomes. Sci Rep. 2021;11(1):2192. doi:10.1038/s41598-021-81949-3
9. Galea E, Launay N, Portero-Otin M, et al. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: a paradigm for multifactorial neurodegenerative diseases? Biochim Biophys Acta. 2012;1822(9):1475–1488. doi:10.1016/j.bbadis.2012.02.005
10. Raymond GV, Seidman R, Monteith TS, et al. Head trauma can initiate the onset of adreno-leukodystrophy. J Neurol Sci. 2010;290(1–2):70–74. doi:10.1016/j.jns.2009.11.005
11. Engelen M, Kemp S, De visser M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. 2012;7:51. doi:10.1186/1750-1172-7-51
12. Zhu J, Eichler F, Biffi A, Duncan CN, Williams DA, Majzoub JA. The changing face of adrenoleukodystrophy. Endocr Rev. 2020;41(4):577–593. doi:10.1210/endrev/bnaa013
13. Turk BR, Theda C, Fatemi A, Moser AB. X‐linked adrenoleukodystrophy: pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int J Dev Neurosci. 2020;80(1):52–72. doi:10.1002/jdn.10003
14. Mallack EJ, Turk BR, Yan H, et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: meta-analysis and consensus guidelines. J Inherit Metab Dis. 2021;44(3):728–739. doi:10.1002/jimd.12356
15. Regelmann MO, Kamboj MK, Miller BS, et al. Adrenoleukodystrophy: guidance for adrenal surveillance in males identified by newborn screen. J Clin Endocrinol Metab. 2018;103(11):4324–4331. doi:10.1210/jc.2018-00920
16. Bezman L, Moser AB, Raymond GV, et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49(4):512–517. doi:10.1002/ana.101
17. Wang Y, Busin R, Reeves C, et al. X-linked adrenoleukodystrophy: ABCD1 de novo mutations and mosaicism. Mol Genet Metab. 2011;104(1–2):160–166. doi:10.1016/j.ymgme.2011.05.016
18. Raymond GV, Moser AB, Fatemi A, et al. X-linked adrenoleukodystrophy. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews®. Seattle: University of Washington; 1993.
19. Handyside AH, Kontogianni EH, Hardy K, Winston RM. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature. 1990;344(6268):768–770. doi:10.1038/344768a0
20. Imudia AN, Plosker S. The past, present, and future of preimplantation genetic testing. Clin Lab Med. 2016;36(2):385–399. doi:10.1016/j.cll.2016.01.012. PMID: 27235919.
21. Harper JC, Boelaert K, Geraedts J, et al. ESHRE PGD Consortium data collection V: cycles from January to December 2002 with pregnancy follow-up to October 2003. Hum Reprod Oxf Engl. 2006;21(1):3–21. doi:10.1093/humrep/dei292
22. Cheung VG, Nelson SF. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA. Proc Natl Acad Sci U S A. 1996;93(25):14676–14679. doi:10.1073/pnas.93.25.14676
23. Czyz ZT, Kirsch S, Polzer B. Principles of whole-genome amplification. Methods Mol Biol Clifton NJ. 2015;1347:1–14. doi:10.1007/978-1-4939-2990-0_1
24. Engelen M, Barbier M, Dijkstra IM, et al. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain. 2014;137(Pt 3):693–706. doi:10.1093/brain/awt361. PMID: 24480483.
25. Semmler A, Köhler W, Jung HH, Weller M, Linnebank M. Therapy of X-linked adrenoleukodystrophy. Expert Rev Neurother. 2008;8(9):1367–1379. doi:10.1586/14737175.8.9.1367
26. Moser HW, Moser AB, Powers JM, et al. The prenatal diagnosis of adrenoleukodystrophy. Demonstration of increased hexacosanoic acid levels in cultured amniocytes and fetal adrenal gland. Pediatr Res. 1982;16(3):172–175. doi:10.1203/00006450-198203000-00002
27. Ke L, Wang Z, Huang H, et al. [Prenatal molecular diagnosis of four fetuses at high risk for X-linked adrenoleukodystrophy]. Zhonghua Fu Chan Ke Za Zhi. 2008;43(1):25–28. Chinese.
28. Imamura A, Suzuki Y, Song XQ, et al. Prenatal diagnosis of adrenoleukodystrophy by means of mutation analysis. Prenat Diagn. 1996;16(3):259–261. doi:10.1002/(SICI)1097-0223(199603)16:3<259::AID-PD840>3.0.CO;2-E
29. Gigarel N, Frydman N, Burlet P, et al. Single cell co-amplification of polymorphic markers for the indirect preimplantation genetic diagnosis of hemophilia A, X-linked adrenoleukodystrophy, X-linked hydrocephalus and incontinentia pigmenti loci on Xq28. Hum Genet. 2004;114(3):298–305. doi:10.1007/s00439-003-1063-9
30. Lledó B, Bernabeu R, Ten J, Galán FM, Cioffi L. Preimplantation genetic diagnosis of X-linked adrenoleukodystrophy with gender determination using multiple displacement amplification. Fertil Steril. 2007;88(5):1327–1333. doi:10.1016/j.fertnstert.2007.01.034
31. Iglesias M, Ceballos P, Giménez C, García-Nebreda MI, Domínguez R, García-Enguídanos A. Pregnancy outcome after preimplantation genetic diagnosis in an affected couple with X-linked adrenoleukodystrophy. Fertil Steril. 2008;90(5):
32. Borgulova I, Putzova M, Soldatova I, et al. Preimplantation genetic diagnosis of X-linked diseases examined by indirect linkage analysis. Bratisl Lek Listy. 2015;116(9):542–546. doi:10.4149/bll_2015_103. PMID: 26435019.
33. Sermon K. Current concepts in preimplantation genetic diagnosis (PGD): a molecular biologist’s view. Hum Reprod Update. 2002;8(1):11–20. doi:10.1093/humupd/8.1.11
34. Harper JC, Wilton L, Traeger-Synodinos J, et al. The ESHRE PGD Consortium: 10 years of data collection. Hum Reprod Update. 2012;18(3):234–247. doi:10.1093/humupd/dmr052
35. Basille C, Frydman R, El Aly A, et al. Preimplantation genetic diagnosis: state of the art. Eur J Obstet Gynecol Reprod Biol. 2009;145(1):9–13. doi:10.1016/j.ejogrb.2009.04.004
36. Sciorio R, Tramontano L, Catt J. Preimplantation genetic diagnosis (PGD) and genetic testing for aneuploidy (PGT-A): status and future challenges. Gynecol Endocrinol. 2020;36(1):6–11. doi:10.1080/09513590.2019.1641194
37. Capalbo A, Bono S, Spizzichino L, et al. Sequential comprehensive chromosome analysis on polar bodies, blastomeres and trophoblast: insights into female meiotic errors and chromosomal segregation in the preimplantation window of embryo development. Hum Reprod Oxf Engl. 2013;28(2):509–518. doi:10.1093/humrep/des394
38. Haapaniemi Kouru K, Malmgren H, Nordenskjöld M, Fridström M, Csemiczky G, Blennow E. One-cell biopsy significantly improves the outcome of preimplantation genetic diagnosis (PGD) treatment: retrospective analysis of 569 PGD cycles at the Stockholm PGD centre. Hum Reprod Oxf Engl. 2012;27(9):2843–2849. doi:10.1093/humrep/des235
39. Slatko BE, Gardner AF, Ausubel FM. Overview of next-generation sequencing technologies. Curr Protoc Mol Biol. 2018;122(1):e59. doi:10.1002/cpmb.59
40. Li G, Niu W, Jin H, et al. Importance of embryo aneuploidy screening in preimplantation genetic diagnosis for monogenic diseases using the karyomap gene chip. Sci Rep. 2018;8(1):3139. doi:10.1038/s41598-018-21094-6
41. Forman EJ, Hong KH, Franasiak JM, Scott RT. Obstetrical and neonatal outcomes from the BEST Trial: single embryo transfer with aneuploidy screening improves outcomes after in vitro fertilization without compromising delivery rates. Am J Obstet Gynecol. 2014;210(2):
42. Giménez C, Sarasa J, Arjona C, et al. Karyomapping allows preimplantation genetic diagnosis of a de-novo deletion undetectable using conventional PGD technology. Reprod Biomed Online. 2015;31(6):770–775. doi:10.1016/j.rbmo.2015.08.017
43. Goldman KN, Nazem T, Berkeley A, Palter S, Grifo JA. Preimplantation Genetic Diagnosis (PGD) for monogenic disorders: the value of concurrent aneuploidy screening. J Genet Couns. 2016;25(6):1327–1337. doi:10.1007/s10897-016-9975-4
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.