")
Back to Journals » Journal of Multidisciplinary Healthcare » Volume 8
Successful management of Barth syndrome: a systematic review highlighting the importance of a flexible and multidisciplinary approach
Authors Reynolds S
Received 6 May 2015
Accepted for publication 28 May 2015
Published 29 July 2015 Volume 2015:8 Pages 345—358
DOI https://doi.org/10.2147/JMDH.S54802
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Stacey Reynolds
Department of Occupational Therapy, Virginia Commonwealth University, Richmond, VA, USA
Abstract: This review describes and summarizes the available evidence related to the treatment and management of Barth syndrome. The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) standards were used to identify articles published between December 2004 and January 2015. The Cochrane Population, Intervention, Control, Outcome, Study Design (PICOS) approach was used to guide the article selection and evaluation process. Of the 128 articles screened, 28 articles matched the systematic review inclusion criteria. The results of this review indicate the need for a flexible and multidisciplinary approach to manage the symptoms most commonly associated with Barth syndrome. It is recommended that a comprehensive care team should include individuals with Barth syndrome, their family members and caregivers, as well as medical, rehabilitative, nutritional, psychological, and educational professionals. The evidence for specific treatments, therapies, and techniques for individuals with Barth syndrome is currently lacking in both quality and quantity.
Keywords: Barth syndrome, rare disorders, rehabilitation, cardiac, systematic review
Introduction
Barth syndrome is a rare X-linked genetic disorder first described by Dr Peter G Barth in 1983.1 It is caused by mutations in the TAZ gene, which is known to encode for the protein tafazzin. Tafazzin plays an important role in the remodeling of cardiolipin, a component of the mitochondrial membrane necessary for maintaining mitochondrial structure as well as for mitochondrial apoptosis and functioning of the electron transport chain.2,3 Heart failure is the most common clinical feature identified at birth and is the leading cause of death in infants with Barth syndrome. The most common cardiac features of Barth syndrome include dilated cardiomyopathy, left ventricular non-compaction, endocardial fibroelastosis, and serious disturbances of heart rhythm such as ventricular fibrillation or tachycardia.4 Sepsis due to neutropenia may also be present at birth and is the second leading cause of infant mortality in Barth syndrome.5 Though survival past infancy is now common in Barth syndrome, cardiovascular and hematological features (ie, neutropenia) in addition to 3-methylglutaconic aciduria often continue throughout the individual’s lifespan.6
Clinical presentation of Barth syndrome also includes neuromuscular features such as skeletal myopathy, hypotonia, delayed motor milestones, exercise intolerance, and abnormal fatigability.7 Growth delay is common in childhood; however, catch-up growth often occurs during late adolescence with achievement of normal adult height in most boys. Often in Barth syndrome, there is the presence of sensory or oral–motor feeding problems, nutritional deficiencies, and episodic or chronic diarrhea.8,9 Specific feeding-related behaviors include difficulty transitioning to solid foods, frequent gagging or difficulty swallowing, and a strong preference for salty or spicy foods while rejecting many other foods eaten by the family.10 There is no definitive cognitive or neurological profile associated with Barth syndrome, though mild learning disabilities, attention deficits, visual spatial deficits, and auditory processing difficulties have been reported.7,9,11
Considering that Barth syndrome is a multisystem disorder, it may be first identified by many different specialists or generalists.7 While there is currently no cure for Barth syndrome, management of clinical features is possible in many cases. Subsequently, individuals with Barth syndrome are likely to interact with a wide range of health care professionals throughout the course of their lifespan in order to address the myriad of symptoms associated with the disorder. The purpose of this review was to identify interventions that are being used to manage symptoms of the disorder and to evaluate the evidence for these interventions related to health and function-related outcomes.
Methods
Prior to initiating a literature search, a protocol using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement was established. Five databases were strategically searched: PubMed, CINAHL, Physiotherapy Evidence Database, Dentistry and Oral Sciences Source, and ProQuest index of Dissertation and Theses. This initial search was followed by secondary selection of articles based on a review of references in relevant articles and articles published online by the Barth Syndrome Foundation. Following PRISMA guidelines, titles of all articles were reviewed for appropriateness, followed by a review of relevant abstracts and appraisal of full-text articles.12 Selection was based on preestablished inclusion and exclusion criteria (the information is described in the paragraph below). The search strategy was initiated using the following question: What treatments, therapies, or techniques are recommended for the management of symptoms seen in males with Barth syndrome? Search terms included Barth AND Syndrome. Only articles published in the last 10 years (December 2004–January 2015) were considered; nonelectronic sources of literature were not considered and only those articles written in English were appraised.
The population was specifically defined as males with a diagnosis of Barth syndrome. Studies using in vitro methods or models (eg, rodent or yeast models) were not considered. To meet the inclusion criteria for intervention, each publication needed to report on the use of a treatment, therapy, or technique to improve the health, well-being, or level of functioning for an individual with Barth syndrome. Interventions focused on end-of-life care were not considered. No comparative intervention or population was required. The studies included were required to assess outcomes relevant to the management of symptoms commonly seen in Barth syndrome including, but not limited to, cardiac function, feeding/eating, nutrition, motor skill development, education, growth, and fatigue. The following standards were used for assessing the levels of evidence and to evaluate the study design and quality of the reviewed articles:
- Level I: systematic reviews, meta-analyses, randomized controlled trials
- Level II: two groups, nonrandomized studies (eg, case– control)
- Level III: one group, nonrandomized (eg, before and after, pretest and posttest)
- Level IV: descriptive studies that include analysis of outcomes (single-subject design, case series)
- Level V: case reports and expert opinion that include narrative literature reviews and consensus statements.13,14
Results
Initial search strategies of the five databases yield 309 articles, with an additional 16 records identified through reference review and supplemental searches (Figure 1). After duplicates were removed (n=19) and titles were screened for relevance, a total of 129 articles remained.2–11,15–133 Within the screened publications, descriptive studies, case reports, narrative reviews, consensus statements, and expert opinion articles were represented. Of the 129 abstracts screened, 86 were excluded with reasons, while 17 appeared to match the Cochrane Population, Intervention, Control, Outcome, Study Design (PICOS) inclusion criteria. An additional 27 articles did not have enough information in the abstract to justify exclusion, received the designation of “maybe”, and were included in the next phase of the review. A comprehensive list of the reviewed abstracts with rationale for exclusion is presented in Table 1. Based on established exclusion criteria, 24 studies did not meet population criteria (28.6%) and 71 studies did not fulfill the intervention criteria (84.5%). Among the abstracts reviewed, there were none that met both population and intervention criteria but were excluded due to irrelevant outcome measures.
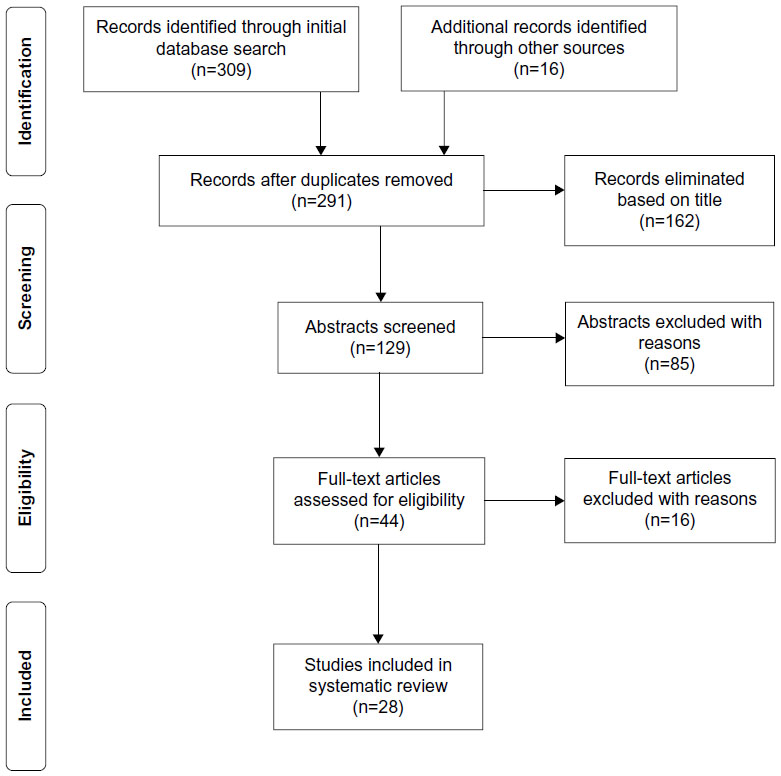 | Figure 1 PRISMA flow diagram. |
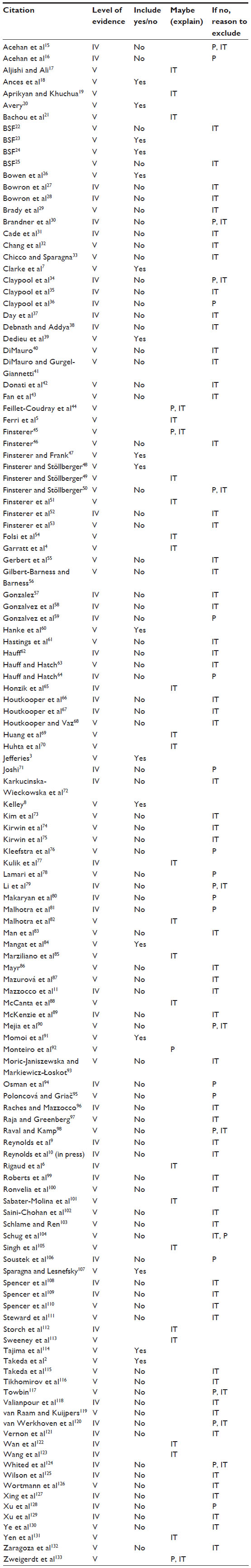 | Table 1 Master citation table of reviewed abstracts |
A total of 44 articles were assessed for eligibility through a full-text review. Each of the 44 articles was read and evaluated based upon the preestablished inclusion/exclusion criteria and level of evidence rating. Sixteen articles did not meet inclusion criteria and were therefore excluded (Table 2). Twenty-eight studies met all inclusion criteria; two articles were ranked at Level IV and 26 were ranked at Level V. Table 3 identifies the characteristics of all the 28 articles meeting inclusion criteria, including all PICOS components.
 | Table 2 Articles excluded after full-text review |
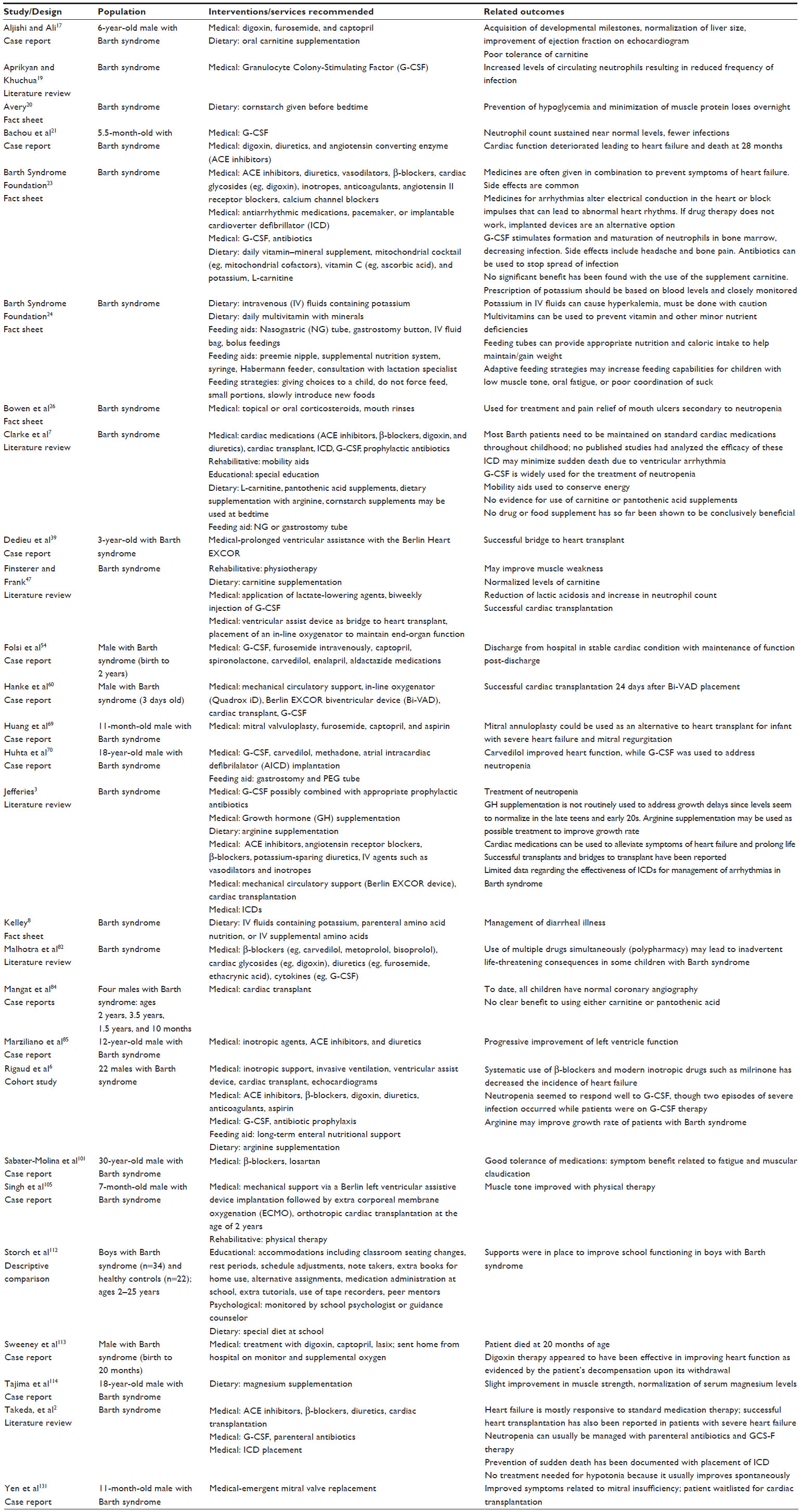 | Table 3 Articles included in systematic review |
Study design
Of the 28 articles selected for this systematic review, 15 were case reports,17,21,39,54,60,69,70,84,85,91,101,105,113,114,131 six were literature reviews,2,3,7,19,47,82 and five were fact sheets.8,20,23,24,26 The selected articles also included one descriptive cohort study6 and one descriptive comparison design.112 Of the 28 articles included in this review, only two used statistical analysis procedures for the analysis of outcomes. Rigaud et al used descriptive and multivariate statistics in a natural history cohort study of 22 individuals with Barth syndrome.6 In this paper, the Kaplan–Meier method was used to estimate survival rates and survival was compared between groups of individuals based on birth year using a log-rank test. Storch et al compared a sample of 34 boys with Barth syndrome with 22 healthy male controls.112 Independent sample t-tests were used to compare the two groups on a variety of psychosocial outcomes. In this study, the alpha level for significance was set at P<0.05; no corrections were made for the possibility of type I error with multiple t-tests.
Population
Across all the 28 reviewed articles, 74 males with the diagnosis of Barth syndrome were enrolled. The sample varied in ethnic origin with representation from American, European, and Asian countries. Case reports described the clinical courses of individuals with Barth syndrome ranging in age from birth to 30 years.
Interventions
Medical interventions
Medical interventions were the most common interventions reported, with 23/28 articles reporting on the use of prescription medications and/or surgical techniques. Medical management of heart failure often used a polypharmacological approach. Cardiac hypertension and congestive heart failure (CHF) were frequently reported to be treated with cardiac glycosides such as digoxin, diuretics such as furosemide, and angiotensin-converting enzyme inhibitors such as captopril; beta-adrenoceptor blockers (β-blockers) such as carvedilol were also reported to be frequently used for the treatment of hypertension, CHF, and/or arrhythmia.2,3,6,7,17,21,23,54,69,70,82,85,91,101,113
In addition to pharmacological therapy to address heart failure, surgical techniques were also identified. Case reports indicated cardiac transplants in six individuals with Barth syndrome,60,84,105 while two additional case reports indicated mitral valve replacement or valvuloplasty.69,131 Three case reports39,60,105 and one cohort study6 identified the use of a ventricular assistance device (eg, the Berlin Heart EXCOR) as a successful bridge to cardiac transplant. An implantable cardioverter defibrillator was utilized in one case to prevent sudden death due to life-threatening ventricular arrhythmias.70
Medical management of neutropenia was most frequently accomplished with the use of granulocyte colony-stimulating factor (G-CSF), as reported in four case reports.21,54,60,70 In their cohort study of 22 individuals with Barth syndrome, Rigaud et al indicated that G-CSF was used in six patients, with two individuals receiving long-term G-CSF therapy and the other four using G-CSF “on demand” when an infection occurred.6 In the same study, four individuals also received antibiotic prophylaxis for the treatment of infections secondary to neutropenia.
Dietary interventions
Dietary interventions were identified in 11/28 of the reviewed articles. The use of oral carnitine supplementation (eg, L- carnitine) was identified in one case report,17 one fact sheet,23 and two literature reviews.7,47 Dietary supplementation with argentine was recommended in two literature reviews7,3 and one cohort study.6 Other nutritional supplements recommended as dietary interventions in the Barth population included cornstarch given before bedtime,7,20 parenteral amino acid nutrition or intravenous supplemental amino acids,8,24 magnesium supplementation,114 potassium supplementation,8,23,24 and daily multivitamins.23,24 Storch et al noted that boys with Barth syndrome may also need a special diet at school, suggesting the need for dietary interventions across multiple settings.112
Feeding aids and strategies
Feeding aids were identified in 5/28 of the reviewed articles. Nasogastric and gastronomy tubes were recommended in cases where boys with Barth syndrome could not take adequate amounts of food or drink by mouth and therefore needed nutrients to be placed directly into the stomach.6,7,24,70,91 Other feeding aids recommended by the Barth Syndrome Foundation24 included preemie nipples, Habermann Feeders®, and Supplemental Nutrition Systems™. Feeding strategies for boys with Barth syndrome were also described in cases where the child had strong food and eating preferences (eg, picky eaters or sensory sensitivities). These strategies included giving the child control over food choices or having the option to spit something out, consuming small portions of food throughout the day instead of eating three large meals, and slowly introducing new foods into the child’s diet.24
Rehabilitative
Limited rehabilitative interventions were identified in this review. One case study105 and one literature review47 identified physical therapy (ie, physiotherapy) as a possible intervention to improve muscle weakness and tone in boys with Barth syndrome. Another literature review acknowledged that mobility aids such as wheelchairs may be used to conserve energy.7
Educational
Storch et al reported a significant need for academic accommodations for boys with Barth syndrome as compared to healthy male controls.112 Specific accommodations identified in this study were classroom seating changes, rest periods, schedule adjustments, note takers, extra books for home use, alternative assignments, extra tutorials, use of tape recorders, and peer mentors. In addition, some boys with Barth syndrome may need medication administration at school. As noted by Clarke et al, approximately 33% of boys with Barth syndrome require some form of special education.7
Psychological
In addition to academic and educational supports, Storch et al found that out of the 34 boys – with Barth syndrome – surveyed, 26% were monitored by a school psychologist, while 22% had close contact with a guidance counselor.112 No other psychological interventions were identified in the 28 articles included in this review.
Outcomes
The most frequent outcomes reported in the 28 reviewed articles were related to the core features of Barth syndrome, which are cardiac complications (eg, CHF, hypertension, and arrhythmia) and neutropenia. Specific outcomes related to heart function included improvement of ejection fraction on echocardiogram,17 progressive improvement of left ventricular function,85 improvement in cardiomegaly with reduced pulmonary edema,91 and decreased incidence of heart failure6 with the use of cardiac medications. However, since most articles reported on the use of multiple medications simultaneously, the individual contributions of each medication or medication type are indistinguishable from the overall effects of the medication regime. In contrast, neutropenia was primarily treated using G-CSF, and therefore outcomes such as increased neutrophil count and reduced infection can be more specifically linked with this medical treatment.6,21 Interestingly, the simultaneous use of multiple drugs (polypharmacy) was questioned in one review, indicating that coadministration of contraindicated medications may prove to be life threatening in some children with Barth syndrome.82
Objective outcomes related to dietary interventions or use of feeding aids were limited in this systematic review. While one literature review suggested that carnitine deficiency in Barth syndrome may be ameliorated by means of substitution,47 other articles suggested no significant benefit with the use of carnitine supplements.7,17,23 Similarly, one published review7 and one case report84 suggested that there is no evidence for the use of pantothenic acid supplements in this population. Magnesium supplementation was utilized in one case report of an 18-year-old male with Barth syndrome.114 In this report, magnesium supplementation led to a slight improvement in muscle strength and normalization of serum magnesium levels. Specific outcomes data related to caloric intake, weight gain, and/or growth were not reported in the other reviewed articles which utilized dietary interventions, feeding aids, or feeding strategies.
Rehabilitative outcomes were also limited in this review. Singh et al reported on a 7-month-old African-American male with Barth syndrome whose muscle tone improved with physical therapy; however, the frequency and duration of physical therapy were not reported, nor were any specific measures of muscle strength or tone.105 In line with this outcome, Finsterer and Frank indicated in their 2013 review that physiotherapy may help to improve muscle weakness in boys with Barth syndrome.47 Interestingly, Takeda et al argued in their review of the literature that no treatment is needed for hypotonia because it usually resolves spontaneously.2
No outcomes related to educational or psychological interventions were reported.
Discussion
Medical management of Barth syndrome
The interventions and outcomes identified in this review reinforce that treatment of medical complications associated with Barth syndrome is of primary importance in this population. However, the types of articles available for this systematic review represent reports that are not based on scientific analysis of clinical outcomes. Of the 28 reviewed articles, 15 were case reports. While case reports have a role in medical literature, particularly for detecting novelties and describing unique cases, findings from case reports often lack generalizability and are reliant upon the author’s memory of events or availability of information in medical records.134
Though lacking direct evidence, the accumulation of descriptive data from case reports and expert clinical opinion suggest that cardiac medications (eg, β-blockers, angiotensin-converting enzyme inhibitors) can be used to alleviate symptoms of heart failure and prolong life in boys with Barth syndrome. In addition, several sources reported increased levels of circulating neutrophils following G-CSF treatment, suggesting that G-CSF, possibly combined with appropriate prophylactic antibiotics, is the best available treatment for neutropenia in this population.19,21,70 While neutropenia appears to respond well to G-CSF, Rigaud et al noted that 2 of the 22 Barth patients in their cohort study had episodes of severe infection while on G-CSF therapy.6 It is also important to note that some medications, either in isolation or coadministered with other medications, may cause symptoms to worsen, and therefore starting or stopping of any new medications or increasing the dosage of the existing ones should be closely monitored in boys with Barth syndrome.23
While several successful surgeries, including cardiac transplants, were reported in the 28 reviewed articles,60,84,105 none of the articles provided data on how many boys with Barth syndrome failed to survive the surgery or what was the long-term survival rates post discharge. Since journals often prefer publication of cases with positive outcomes, there is a risk in overstating or generalizing the surgical outcomes gleaned in this review.134
Despite the lack of specific outcomes in this review, it is clear that medical management of Barth syndrome is essential for growth and survival. Medical team members involved in the care of boys with Barth syndrome may include specialists in cardiology, hematology, metabolism, endocrinology, neurology, and genetics, as well as developmental pediatricians and primary care physicians.
Feeding and dietary interventions for Barth syndrome
The literature supporting nonmedical interventions for Barth syndrome is much scarcer. While it has been identified that feeding issues are stressful for families of boys with Barth syndrome, very little research has been directed toward interventions that support feeding behaviors or nutritional concerns.9,10 Further, there does not appear to be consensus about which nutritional supplements are effective in this population or what process should go into the selection of a supplemental regime.
Given the complexity of feeding and nutritional issues in boys with Barth syndrome, multiple disciplines may be involved in the management of these clinical features. Dietitians and nutritional specialists will likely work closely with medical doctors, including metabolic specialists, to determine what and how much food/liquid the child should be consuming. Decisions about the placement of nasogastric tubes or other long-term enteral nutritional support will likely be made by a team of individuals familiar with the specific child and familiar with the complexities of Barth syndrome. Throughout the lifespan of a boy with Barth syndrome, the needs of the child and the family will likely require consultation or intervention by a variety of other health care professionals. During infancy, lactation consultants, pediatric nurses, and occupational therapists may work with families in order to facilitate good positioning for breastfeeding and recommend appropriate feeding aids and strategies to assist the child with oral sucking and self-regulation during the feeding process. When the child begins transitioning to solid foods, rehabilitative professionals (ie, physical therapists, occupational therapists, and speech and language pathologists) may be consulted for issues related to proper positioning, coordination of chewing and swallowing, or sensory sensitivities leading to picky eating patterns. As the child grows, nutritional concerns related to growth will likely become important and ongoing case management with the medical team may be necessary. During the school years, special dietary or feeding needs may need to be documented on the child’s individualized education plan and consultation between the school team and medical team may be required. Since eating is a big part of normal socialization patterns, practitioners should be mindful of the need for children, adolescents, and adults with Barth syndrome to participate in these types of activities as part of their overall psychosocial health.
Interventions to support participation and well-being
This systematic review identified a relative gap in the literature in regard to the interventions that support the participation and well-being of boys with Barth syndrome and their families. Physical therapy was identified as one rehabilitative service that could potentially help enhance muscle strength and tone; however, it was unclear from the review whether these improvements in strength and tone led to increased play with peers, greater participation due to lack of fatigue, or an improvement in self-concept or self-esteem. Research using mouse models of Barth syndrome are beginning to provide insights into the physiological benefits of exercise and endurance training;135 however, translation of these findings into practice are nonexistent. As basic science research begins to work its way from bench to bedside, it will be important for researchers and clinicians alike to focus on the functional and participation benefits of exercise in addition to changes in muscle physiology.
Finally, this review did not identify any currently researched interventions to address the psychosocial needs of boys with Barth syndrome and their families. As noted by Storch et al, youth with Barth syndrome tend to have more internalizing and externalizing symptoms, social problems, and loneliness compared to healthy children; in addition, they rate their overall quality of life much lower than that of healthy controls and other children with cardiac problems.112 Parents of boys with Barth syndrome also report higher stress levels compared to parents of health controls. Psychologists, licensed clinical social workers, occupational therapists, and other mental health professionals may be able to help children and families with these issues through a variety of therapeutic interventions. There is a current need to examine the usefulness and applicability of these types of services in the Barth population.
Conclusion
This systematic review highlights the paucity of objective, high-level evidence supporting interventions for males with Barth syndrome. While case reports and literature reviews on this rare disorder are important contributions to the literature, future studies should aim to include systematic and objective analyses of outcomes using research designs with methodological rigor. This level of research is needed across multiple disciplines in order to develop best practices for the management of the symptoms most commonly associated with the Barth syndrome. Clinically, it is recommended that a comprehensive care team be established, which includes individuals with Barth syndrome, their family members and caregivers, as well as medical, rehabilitative, nutritional, psychological, and educational professionals. While medical management of symptoms that preserves life is of primary importance, services that enhance the child’s and family’s quality of life and participation should also be included as part of the individual’s care plan.
Acknowledgment
Stacey Reynolds would like to thank Hope Caracci, OTD, OTR/L, Division of Pediatric Rehabilitation, Children’s Hospital of The Kings Daughter, for her assistance in preparing this paper.
Disclosure
The author reports no conflicts of interest in this work.
References
Barth PG, Scholte HR, Berden JA, et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci. 1983;62(1–3):327–355. | |
Takeda A, Sudo A, Yamada M, et al. Eponym: Barth syndrome. Eur J Pediatr. 2011;170(11):1365–1367. | |
Jefferies JL. Barth syndrome. Am J Med Genet C Semin Med Genet. 2013;163C(3):198–205. | |
Garratt V, Riddiford D, Steward C, Tsai-Goodman B, Newbury-Ecob R. What is Barth syndrome? Midwives. 2011;14(4):32–33. | |
Ferri L, Donati MA, Funghini S, et al. New clinical and molecular insights on Barth syndrome. J Rare Disord. 2013;8:27. | |
Rigaud C, Lebre, AS, Touraine R, et al. Natural history of Barth syndrome: a national cohort study of 22 patients. J Rare Disord. 2013;8:70. | |
Clarke SL, Bowron A, Gonzalez IL, et al. Barth syndrome. J Rare Disord. 2013;8(23):1–17. | |
Kelley RI. Management of Diarrheal Illness in Patients With Barth Syndrome. 2008. Available from: https://www.barthsyndrome.org/CMFiles/Resources/DiarrheaBarthFINAL2008.pdf. Accessed January 18, 2015. | |
Reynolds S, Kreider CM, Bendixen R. A mixed-methods investigation of sensory response patterns in Barth syndrome: a clinical phenotype? Am J Med Genet A. 2012;158A(7):1647–1653. | |
Reynolds S, Kreider CM, Meeley LE, Bendixen R. Feeding history and chemical taste sensitivity in boys with Barth syndrome. J Rare Disord. In press 2015;3(1). | |
Mazzocco MM, Henry AE, Kelley RI. Barth syndrome is associated with a cognitive phenotype. J Dev Behav Pediatr. 2007;28(1):22–30. | |
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol. 2009;62:1006–1012. | |
American Occupational Therapy Association (AOTA). Guidelines to Critically Appraised Paper Worksheet. 2014. Available from: http://www.aota.org/-/media/Corporate/Files/Practice/EvidenceExchange/CAP%20Guidelines%20for%20Evidence%20Exchange.pdf. Accessed August 4, 2014. | |
Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn’t. Clin Orthop Relat Res. 2007;455:3–5. | |
Acehan D, Xu Y, Stokes DL, Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Investig. 2007;87(1):40–48. | |
Acehan D, Vaz F, Houtkooper RH, et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem. 2011;286(2):899–908. | |
Aljishi E, Ali F. Barth syndrome: an X-linked cardiomyopathy with a novel mutation. Indian J Pediatr. 2010;77(2):1432–1433. | |
Ances BM, Sullivan J, Weigele JB, et al. Stroke associated with Barth syndrome. J Child Neurol. 2006;21(9):805–807. | |
Apikyan AA, Khuchua Z. Advances in the understanding of Barth syndrome. Br J Haematol. 2013;161(3):330–338. | |
Avery R. Cornstarch Fact Sheet. 2006. Available from: https://www.barthsyndrome.org/CMFiles/Resources/Cornstarch.pdf. Accessed January 18, 2015. | |
Bachou T, Giannakopoulos A, Trapali C, Vazeau A, Kattamis A. A novel mutation in the G4.5 (TAZ) gene in a Greek patient with Barth syndrome. Blood Cells Mol Disord. 2009;42(3):262–264. | |
Barth Syndrome Foundation. Metabolism of Barth syndrome. 2005. Available from: https://www.barthsyndrome.org/CMFiles/Resources/MetabolismBarthSyndromeFINAL.pdf. Accessed January 18, 2015. | |
Barth Syndrome Foundation. Medications Used in Barth syndrome. 2005. Available from: https://www.barthsyndrome.org/CMFiles/Resources/MedicationsFactSheetfinaledit82105.pdf. Accessed January 18, 2015. | |
Barth Syndrome Foundation. Nutrition Facts in Barth syndrome. 2006. Available from: https://www.barthsyndrome.org/CMFiles/Resources/NutritionFactSheetFINALRevisedMay06.pdf. Accessed January 18, 2015. | |
Barth Syndrome Foundation. Neurological Manifestations in Barth Syndrome. 2006. Available from: https://www.barthsyndrome.org/CMFiles/Resources/NeurologicalManifestationsBarthedited.pdf. Accessed January 18, 2015. | |
Bowen S, Kelley RI, Mann S. Oral Hygiene For Children With Barth Syndrome. 2007. Available from: https://www.barthsyndrome.org/CMFiles/Resources/OralHygieneBarthSyndromeFINAL.pdf. Accessed January 18, 2015. | |
Bowron A, Frost R, Powers VE, Thomas PH, Heales SJ, Steward CG. Diagnosis of Barth syndrome using a novel LC-MS/MS method for leukocyte cardiolipin analysis. J Inherit Metab Dis. 2013;36(5):741–746. | |
Bowron A, Honeychurch J, Williams M, et al. Barth syndrome without tetralinoleoyl cardiolipin deficiency: a possible ameliorated phenotype. J Inherit Metab Dis. 2014;38(2):279–286. | |
Brady AN, Shehata BM, Fernhoff PM. X-linked fetal cardiomyopathy caused by a novel mutation in the TAZ gene. Prenat Diagn. 2006;25(5):462–465. | |
Brandner K, Mick DU, Frazier AE, Taylor RD, Meisinger C, Rehling P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth syndrome. Mol Biol Cell. 2005;16(11):5202–5214. | |
Cade WT, Spencer CT, Reeds DN, et al. Substrate metabolism during basal and hyperinsulinemic conditions in adolescents and young-adults with Barth syndrome. J Inherit Metab Dis. 2013;36(1):91–101. | |
Chang B, Momoi N, Shan L, et al. Gonadal mosaicism of a TAZ (G4.5) mutation in a Japanese family with Barth syndrome and left ventricular noncompaction. Mol Genet Metab. 2010;200(2):198–203. | |
Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007;292(1): C33–C44. | |
Claypool SM, Boontheung P, McCaffery JM, Loo JA, Koehler CM. The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome. Mol Biol Cell. 2008;19(12):5143–5155. | |
Claypool SM, McCaffery JM, Koehler CM. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J Cell Biol. 2006;174(3):379–390. | |
Claypool SM, Whited K, Srijumnong S, Han X, Koehler CM. Barth syndrome mutations that cause tafazzin complex lability. J Cell Biol. 2011;192(3):447–462. | |
Day JA, Spencer CT, Byrne B. Grip strength in a population of boys with Barth syndrome. Pediatr Phys Ther. 2006;18(1):88–89. | |
Debnath S, Addya S. Mis-sense mutations in tafazzin (TAZ) that escort to mild clinical symptoms of Barth Syndrome is owed to the minimal inhibitory effect of the mutations on the enzyme function: in-silico evidence. Interdiscip Sci. 2014;7(1):21–25. | |
Dedieu N, Giardini A, Steward CG, et al. Successful mechanical circulatory support for 251 days in a child with intermittent severe neutropenia due to Barth syndrome. Pediatr Transplant. 2013;17(2):E46–E49. | |
DiMauro S. Mitochondrial myopathies. Curr Opin Rheumatol. 2006;18(6):636–641. | |
DiMauro S, Gurgel-Giannetti J. The expanding phenotype of mitochondrial myopathy. Curr Opin Neurol. 2005;18(5):538–542. | |
Donati MA, Malvagia S, Pasquini E, et al. Barth syndrome presenting with acute metabolic decompensation in the neonatal period. J Inherit Metab Disord. 2006;29(5):684. | |
Fan Y, Steller J, Gonzalez IL, et al. A novel exonic splicing mutation in the TAZ (G4.5) gene in a case with atypical Barth syndrome. J Inherit Metab Disord. 2013;11:99–106. | |
Feillet-Coudray C, Fouret G, Casas F, Coudray C. Impact of high dietary lipid intake and related metabolic disorders on the abundance and acyl composition of the unique mitochondrial phospholipid, cardiolipin. J Bioenerg Biomembr. 2014;46(5):447–457. | |
Finsterer J. Hematological manifestations of primary mitochondrial disorders. Acta Haematol. 2007;118(2):88–98. | |
Finsterer J. Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr Cardiol. 2009; 30(5):659–681. | |
Finsterer J, Frank M. Haematological features in Barth syndrome. Curr Opin Hematol. 2013;20(1):36–40. | |
Finsterer J, Stöllberger C. Atrial fibrillation/flutter in myopathies. Int J Cardiol. 2008a;128(3):304–310. | |
Finsterer J, Stöllberger C. Primary myopathies and the heart. Scand Cardiovasc J. 2008;42(1):9–24. | |
Finsterer J, Stöllberger C. Unclassified cardiomyopathies in neuromuscular disorders. Wien Med Wochenschr. 2013;163(21–22):505–513. | |
Finsterer J, Stöllberger C, Blazek G. Neuromuscular implications in left ventricular hypertrabeculation/noncompaction. Int J Cardiol. 2006; 110(3):288–300. | |
Finsterer J, Stöllberger C, Blazek G. Prevalence of Barth syndrome in adult left ventricular hypertrabeculation/noncompaction. Scand Cardiovasc J. 2008;42(2):157–160. | |
Finsterer J, Stöllberger C, Fazio G. Neuromuscular disorders in left ventricular hypertrabeculation/noncompaction. Curr Pharm Des. 2010; 16(26):2895–2904. | |
Folsi V, Miglietti N, Lombardi A, et al. Cardiomyopathy in a male patient with neutropenia and growth delay. Ital J Pediatr. 2014;40:45. | |
Gerbert N, Joshi AS, Kutik S, et al. Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr Biol. 2009;19(24):2133–2139. | |
Gilbert-Barness E, Barness LA. Pathogenesis of cardiac conduction disorders in children genetic and histopathologic aspects. Am J Med Genet A. 2006;140(19):1993–2006. | |
Gonzalez IL. Barth syndrome: TAZ gene mutations, mRNAs, and evolution. Am J Med Genet A. 2005;134(4):409–414. | |
Gonzalvez F, D’Aurelio M, Boutant M, et al. Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim Biophys Acta. 2013;1832(8):1194–1206. | |
Gonzalvez F, Schug ZT, Houtkooper RH, et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183(4):681–696. | |
Hanke SP, Gardner AB, Lombardi JP, et al. Left ventricular noncompaction cardiomyopathy in Barth syndrome: an example of an undulating cardiac phenotype necessitating mechanical circulatory support as a bridge to transplantation. Pediatr Cardiol. 2012;33(8):1430–1434. | |
Hastings R, Steward C, Tsai-Goodman B, Newbury-Ecob R. Dysmorphology of Barth syndrome. Clin Dysmorphol. 2009;18(4):185–187. | |
Hauff KD. Hypocholesterolemia in Barth syndrome (NR52998). ProQuest Dissertations and Theses. Canada: University of Manitoba. 2009;335. Available from: http://search.proquest.com/docview/305061005?accountid=14780. (305061005). Accessed February 15, 2015. | |
Hauff KD, Hatch GM. Cardiolipin metabolism and Barth syndrome. Prog Lipid Res. 2006;45(2):91–101. | |
Hauff KD, Hatch GM. Reduction in cholesterol synthesis in response to serum starvation in lymphoblasts of a patient with Barth syndrome. Biochem Cell Biol. 2010;88(4):595–602. | |
Honzik T, Tesarova M, Magner M, et al. Neonatal onset of mitochondrial disorders in 129 patients: clinical and laboratory characteristics and a new approach to diagnosis. J Inherit Metab Disord. 2012;35(5):749–759. | |
Houtkooper RH, Rodenburg RJ, Thiels C, et al. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal Biochem. 2009;387(2):230–237. | |
Houtkooper RH, Turkenburg M, Poll-The BT, et al. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim Biophys Acta. 2009; 1788(10):2003–2014. | |
Houtkooper RH, Vaz FM. Cardiolipin, the heart of mitochondrial metabolism. Cell Mol Life Sci. 2008;65(16):2493–2506. | |
Huang SC, Wu ET, Chiu SN, Hwu WL, Wu MH, Wang SS. Mitral annuloplasty in an infant with Barth syndrome and severe mitral insufficiency: first case report and determination of annular diameter. J Thorac Cardiovasc Surg. 2008;136(4):1095–1097. | |
Huhta JC, Pomerance HH, Barnsss EG. Clinicopathologic conference: Barth syndrome. Fetal Pediatr Pathol. 2005;24(4–5):239–254. | |
Joshi AS. Identification of cellular functions of cardiolipin as physiological modifiers of Barth syndrome (354426). ProQuest Dissertations and Theses. Detroit, MI: Wayne State University. 2012;154. Available from: http://search.proquest.com/docview/1225792947?accountid=14780. (122579247). | |
Karkucinska-Wieckowska A, Trubicka J, Werner B, et al. Left ventricular noncompaction (LVNC) and low mitochondrial membrane potential are specific for Barth syndrome. J Inherit Metab Disord. 2013;36(6):929–937. | |
Kim GB, Kwon BS, Bae EJ, Noh CI, Seong MW, Park SS. A novel mutation of the TAZ gene in Barth syndrome: acute exacerbation after contrast-dye injection. J Korean Med Sci. 2013;28(5):784–787. | |
Kirwin SM, Manolakos A, Barnett SS, Gonzalez IL. Tafazzin splice variants and mutations in Barth syndrome. Mol Genet Metab. 2014; 111(1):26–32. | |
Kirwin SM, Vinette KM, Schwartz SB, Funanage VL, Gonzalez IL. Multiple transmissions of Barth syndrome through an oocyte donor with a de novo TAZ mutation. Fertil Steril. 2007;87(4):976. e5–e7. | |
Kleefstra T, Wortmann SB, Rodenburg RJ, et al. Mitochondrial dysfunction and organic aciduria in five patients carrying mutations in the Ras-MAPK pathway. Eur J Hum Genet. 2011;19(2):138–144. | |
Kulik W, van Lenthe H, Stet FS, et al. Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin Chem. 2008;54(2):371–378. | |
Lamari F, Mochel F, Sedel F, Saudubray JM. Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: toward a new category of inherited metabolic diseases. J Inherit Metab Disord. 2013;36(3):411–425. | |
Li G. The role of mitochondrial anionic phospholipids in signaling pathways and essential functions (3259009). ProQuest Dissertations and Theses. Detroit, MI: Wayne State University. 2007;157. Available from: http://search.proquest.com/docview/3048022665?accountid=14780. (304802665). | |
Makaryan V, Kulik W, Vaz FM, et al. The cellular and molecular mechanisms for neutropenia in Barth syndrome. Eur J Haematol. 2012; 88(3):195–209. | |
Malhotra A, Edelman-Novemsky I, Xu Y, et al. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc Natl Acad Sci U S A. 2009;106(7):2337–2341. | |
Malhotra A, Kahlon P, Donoho T, Doyle IC. Pharmacogenomic considerations in the treatment of the pediatric cardiomyopathy called Barth syndrome. Recent Pat Biotechnol. 2014;8(2):136–143. | |
Man E, Lafferty KA, Funke BH, et al. NGS identifies TAZ mutation in a family with X-linked dilated cardiomyopathy. BMJ Case Report. 2013. | |
Mangat J, Lunnon-Wood T, Rees P, Elliott M, Burch M. Successful cardiac transplantation in Barth syndrome – single-centre experience of four patients. Pediatr Transplant. 2007;11(3):327–331. | |
Marziliano N, Mannarino S, Nespoli L, et al. Barth syndrome associated with compound hemizygosity and heterozygosity of the TAZ and LDB3 genes. Am J Med Genet A. 2007;143A(9):907–915. | |
Mayr JA. Lipid metabolism in mitochondrial membranes. J Inherit Metab Disord. 2015;38(1):137–144. | |
Mazurová S, Tesařová M, Magner M, et al. Novel mutations in the TAZ gene in patients with Barth syndrome. Prague Med Rep. 2013;114(3):139–153. | |
McCanta AC, Chang AC, Weiner K. Cardiomyopathy in a child with neutropenia and motor delay. Curr Opin Pediatr. 2008;20(5):605–607. | |
McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J Mol Biol. 2006;361(3):462–469. | |
Mejia EM, Cole LK, Hatch GM. Cardiolipin metabolism and the role it plays in heart failure and mitochondrial supercomplex formation. Cardiovasc Hematol Disord Drug Targets. 2014;14(2):98–106. | |
Momoi N, Chang B, Takeda I, Aoyagi Y, Endo K, Ichida F. Differing clinical courses and outcomes in two siblings with Barth syndrome and left ventricular noncompaction. Eur J Pediatr. 2012;171(3):515–520. | |
Monteiro JP, Oliveira PJ, Jurado AS. Mitochondrial membrane lipid remodeling in pathophysiology: a new target for diet and therapeutic interventions. Prog Lipid Res. 2013;52(4):513–528. | |
Moric-Janiszewska E, Markiewicz-Łoskot G. Genetic heterogeneity of left-ventricular noncompaction cardiomyopathy. Clin Cardiol. 2008;31(5):201–204. | |
Osman C, Haag M, Wieland FT, Brügger B, Langer T. A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase GEP4. EMBO J. 2010;29(12):1976–1987. | |
Poloncová K, Griač P. Phospholipid transport and remodeling in health and disease. Gen Physiol Biophys. 2011;30 Spec No:S25–S35. | |
Raches D, Mazzocco MM. Emergence and nature of mathematical difficulties in young children with Barth syndrome. J Dev Behav Pediatr. 2012;33(4):328–335. | |
Raja V, Greenberg ML. The functions of cardiolipin metabolism-potential modifiers of the Barth syndrome phenotype. Chem Phys Lipids. 2014;179:49–56. | |
Raval KK, Kamp TJ. Cardiomyopathy, mitochondria and Barth syndrome: iPSCs reveal a connection. Nat Med. 2014;20(6):585–586. | |
Roberts AE, Nixon C, Steward CG, et al. The Barth Syndrome Registry: Distinguishing disease characteristics and growth data from a longitudinal study. Am J Med Genet A. 2012;158A(11):2726–2732. | |
Ronvelia D, Greenwood J, Platt J, Hakim S, Zaragoza MV. Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great uncle. Mol Genet Metab. 2012; 107(3):428–432. | |
Sabater-Molina M, Guillén-Navarro E, Garcia-Molina E, Ballesta-Martinez MJ, Escudero F, Ruiz-Espejo F. Barth syndrome in adulthood: a clinical case. Rev Esp Cardiol (Engl Ed). 2013;66(1):68–70. | |
Saini-Chohan HK, Mitchell RW, Vaz FM, Zelinski T, Hatch GM. Delineating the role of alterations in lipid metabolism to the pathogenesis of inherited skeletal and cardiac muscle disorders: Thematic Review Series: Genetics of human lipid disease. J Lipid Res. 2012; 53(1):4–27. | |
Schlame M, Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006;580(23):5450–5455. | |
Schug ZT, Frezza C, Galbraith LC, Gottlieb E. The music of lipids: how lipid composition orchestrates cellular behavior. Acta Oncol. 2012;51(3):301–310. | |
Singh HR, Yang Z, Siddiqui S, et al. A novel Alu-mediated Xq28 microdeletion ablates TAZ and partially deletes DNL1L in a patient with Barth syndrome. Am J Med Genet A. 2009;149A(5):1082–1085. | |
Soustek MS, Falk DJ, Mah CS, et al. Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum Gene Ther. 2011;22(7):865–871. | |
Sparagna GC, Lesnefsky EJ. Cardiolipin remodeling in the heart. J Cardiovasc Pharmacol. 2009;53(4):290–301. | |
Spencer CT, Bryant RM, Day J, et al. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. 2006;118(2):e337–e346. | |
Spencer CT, Byrne BJ, Bryant RM, et al. Impaired cardiac reserve and severely diminished skeletal muscle O2 utilization mediate exercise intolerance in Barth syndrome. Am J Physiol Heart Circ Physiol. 2011; 301(5):H2122–H2129. | |
Spencer CT, Byrne BJ, Gewitz MH, et al. Ventricular arrhythmia in the X-linked cardiomyopathy Barth syndrome. Pediatr Cardiol. 2005; 26(5):632–637. | |
Steward CG, Newbury-Ecob RA, Hastings R, et al. Barth syndrome: an X-linked cause of fetal cardiomyopathy and stillbirth. Prenat Diagn. 2010;30(10):970–976. | |
Storch EA, Keeley M, Merlo LJ, et al. Psychosocial functioning in youth with Barth syndrome. Child Health Care. 2009;38(2):137–156. | |
Sweeney RT, Davis GJ, Noonan JA. Cardiomyopathy of unknown etiology: Barth syndrome unrecognized. Congenit Heart Dis. 2009; 3(6):433–438. | |
Tajima T, Satoh K, Okuhara K, Tsubaki J, Fujieda K. Hypomagnesemia in a patient with Barth syndrome. J Pediatr Endocrinol Metab. 2005;18(5):523. | |
Takeda A, Sudo A, Yamada M, et al. Barth syndrome diagnosed in the subclinical stage of heart failure based on the presence of lipid storage myopathy and isolated noncompaction of the ventricular myocardium. Eur J Pediatr. 2011;170(11):1481–1484. | |
Tikhomirov E, Aeryanova N, Basargina E, Degtyareva T. Gene symbol: TAZ. disease: Barth syndrome. Hum Genet. 2008;124(3):315–316. | |
Towbin JA. Left ventricular noncompaction: a new form of heart failure. Heart Fail Clin. 2010;62(6):459–462. | |
Valianpour F, Mitsakos V, Schlemmer D, et al. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J Lipid Res. 2005;46(6):1182–1195. | |
van Raam BJ, Kuijpers TW. Mitochondrial defects lie at the basis of neutropenia in Barth syndrome. Curr Opin Hematol. 2009;16(1):14–19. | |
van Werkhoven MA, Thorburn DR, Gedeon AK, Pitt JJ. Monolysocardiolipin in cultured fibroblasts is a sensitive and specific marker for Barth syndrome. J Lipid Res. 2006;47(10):2346–2351. | |
Vernon HJ, Sandlers Y, McClellan R, Kelley RI. Clinical laboratory studies in Barth syndrome. Mol Genet Metab. 2014;112(2):143–147. | |
Wan C, Yu HH, Lu MY, et al. Clinical manifestations and outcomes of pediatric chronic neutropenia. J Formos Med Assoc. 2012;111(4):220–227. | |
Wang G, McCain ML, Yang L, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20(6):616–623. | |
Whited K, Baile MG, Currier P, Claypool SM. Seven functional classes of Barth syndrome mutation. Hum Mole Genet. 2013;22(3):483–492. | |
Wilson LD, Al-Majid S, Rakovski CS, Md CD. Higher IL-6 and IL6:IGF ratio in patients with Barth syndrome. J Inflamm (Lond). 2012;9(1):25. | |
Wortmann SB, Espeel M, Almeida L, et al. Inborn errors of metabolism in the biosynthesis and remodeling of phospholipids. J Inherit Metab Disord. 2015;38(1):99–110. | |
Xing Y, Ichida F, Matsuoka T, et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. 2006;88(1):71–77. | |
Xu Y, Malhotra A, Ren M, Schlame M. J Biol Chem. 2006;281(51):39217–39224. | |
Xu Y, Sutachan JJ, Plesken H, Kelley RI, Schlame M. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab Invest. 2005;85(6):823–830. | |
Ye C, Shen Z, Greenberg ML. Cardiolipin remodeling: a regulatory hub for modulating cardiolipin metabolism and function. J Bioenerg Biomembr. Epub November 29, 2014. | |
Yen TY, Hwu WL, Chien YH, et al. Acute metabolic decompensation and sudden death in Barth syndrome: report of a family and a literature review. Eur J Pediatr. 2008;167(8):941–944. | |
Zaragoza MV, Arbustini E, Narula J. Noncompaction of the left ventricle: primary cardiomyopathy with an elusive genetic condition. Curr Opin Pediatr. 2007;19(6):619–627. | |
Zweigerdt R, Gruh I, Martin U. Your heart on a chip: iPSC-based modeling of Barth-syndrome-associated cardiomyopathy. Cell Stem Cell. 2014;15(1):9–11. | |
Nissen T, Wynn R. The clinical case report: a review of its merits and limitations. BMC Res Notes. 2014;7(264):1–7. | |
Soustek MS, Baligand C, Falk DJ, Walter GA, Lewin AS, Byrne BJ. Endurance training ameliorates complex 3 deficiency in a mouse model of Barth syndrome. J Inherit Metab Dis. Epub April 10, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.