")
Back to Journals » Drug Design, Development and Therapy » Volume 13
SU5416 attenuated lipopolysaccharide-induced acute lung injury in mice by modulating properties of vascular endothelial cells
Authors Huang X, Zhu J, Jiang Y, Xu C, Lv Q, Yu D, Shi K, Ruan Z, Wang Y
Received 26 September 2018
Accepted for publication 12 March 2019
Published 23 May 2019 Volume 2019:13 Pages 1763—1772
DOI https://doi.org/10.2147/DDDT.S188858
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
Xuqing Huang,1,* Junqi Zhu,2,* Yuyue Jiang,1 Changqing Xu,1 Qun Lv,1 Dongwei Yu,1 Kai Shi,1 Zhaoyang Ruan,1 Yan Wang1
1Department of Respiratory Medicine, Affiliated Hospital of Hangzhou Normal University, Hangzhou, Zhejiang, People’s Republic of China; 2Department of Clinical Medicine, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Background and aim: A potent and selective vascular endothelial growth factor receptor (VEGFR) inhibitor SU5416, has been developed for the treatment of solid human tumors. The binding of VEGF to VEGFR plays a crucial role in the pathophysiology of respiratory disorders. However, the impact of SU5416 on lipopolysaccharide (LPS)-induced acute lung injury (ALI) remains unclear. Thus, this study aimed to illuminate the biofunction of SU5416 in the mouse model of ALI.
Methods: Wild-type (WT) and toll-like receptor 4 (TLR4)-deficient (TLR4−/-) C57BL/6 mice were used to establish LPS-induced ALI model. The primary pulmonary microvascular endothelial cell (PMVEC) was extracted for detection of endothelial barrier function.
Results: LPS significantly increased the number of inflammatory cells and inflammatory cytokines in bronchoalveolar lavage fluid (BALF). In addition, LPS increased alveolar epithelial cells injury, inflammation infiltration and vascular permeability of PMVEC in WT and TLR4−/- mice. Western blotting experiment indicated VEGF/VEGFR and TLR4/NF-κB pathways were involved in the progression of LPS-stimulated ALI. Consistent with previous research, dexamethasone treatment appeared to be an effective therapeutic for mice with ALI. Moreover, treatment with SU5416 dramatically attenuated LPS-induced immune responses in mice lung tissues via inhibiting VEGF/VEGFR and TLR4/NF-κB pathways. Finally, SU5416 also decreased vascular permeability of PMVEC in vitro.
Conclusion: SU5416 ameliorated alveolar epithelial cells injury and histopathological changes in mice lung via inhibiting VEGF/VEGFR and TLR4/NF-κB signaling pathways. We also confirmed that SU5416 could restrain vascular permeability in PMVEC through improving the integrity of endothelial cell. These findings suggested that SU5416 may serve as a potential agent for the treatment of patients with ALI.
Keywords: SU5416, lipopolysaccharide, acute lung injury, inflammatory cytokines
Introduction
Acute lung injury (ALI) is a common severe clinical phenomenon which is characterized by expiratory dyspnea, interstitial edema, accumulation of activated inflammatory cells, exuberant migration of activated neutrophils, and diffuse alveolar damage.1,2 Clinically, it appears as acute respiratory distress syndrome (ARDS) caused by various pathologies, including sepsis, trauma, pneumonia, and gram-negative bacterial infection.3,4 ARDS, one of the most serious forms of ALI, is a main cause of death in the patients with sepsis, shock or pneumonia.5
Lipopolysaccharide (LPS) is an endotoxin derived from gram-negative bacteria components, which have been found to lead to ALI via induced inflammatory reaction.6 Accumulating evidence has verified that numerous signal interactions were involved in the progression of LPS-induced ALI.7 Ras homolog family member A (RhoA) regulated cell viability, apoptosis, and ROS activity by activating the downstream Wnt/β-catenin signaling pathway in a cell model of ALI.7 In addition, glycogen synthase kinase-3β (GSK-3β), an important component of Wnt signaling, is reported to be involved in LPS-induced ALI.8 Inactivation of GSK-3β could mitigate LPS-triggered ARDS in mice through restraining the production of proinflammatory cytokines.8 LPS also recruits other membrane receptors to form a complex compound and triggered the downstream signaling cascades such as toll-like receptor 4 (TLR4), cluster of differentiation 14 (CD14), and lymphocyte antigen 96 (MD2).9 TLRs, a class of membrane glycoproteins, could detect a variety of microbial compositions and trigger innate immune responses.10 TLR4, a member of the TLR family, is crucial for the innate immune response through activating intracellular signaling pathway NF-κB.11 Once activated by LPS, TLR4 triggers downstream MyD88/NF-κB signals leading to the production of various proinflammatory cytokines.12
Previous studies have reported TLR4 acted as a vital role in LPS-induced ALI by modulating the release of inflammatory mediator.13,14 TLR-deficient mice exhibited significant resistance to paraquat-induced ALI.15 Both hyaluronan and echinocystic acid ameliorated LPS-stimulated lung inflammation by inhibiting the TLR4/NF-κB signaling pathway,3,16,17 implying targeting TLR4 or its downstream signals is likely to be an effective treatment for ALI. Vascular endothelial growth factor (VEGF) is a sub-family of growth factors that mainly stimulates angiogenesis during the development of an organ. In addition, VEGF overexpression contributes to many diseases including solid cancers, vascular diseases, and ALI.18–20 However, it is ambiguous whether VEGF serves as a protective factor or as a destructive element in the course of ALI. VEGF exerts its biofunction through binding irreversibly to its certain receptor VEGFR-1, VEGFR-2 and VEGFR-3.21 It has been reported that activation of VEGFR-3 protected against endotoxin shock via restraining TLR4-NF-κB signaling.22 TLR4 is required for the protective role of VEGF in primary endothelial cells of the lung. Its ablation repressed VEGF/VEGFR downstream signaling molecules including AKT and ERK, and caused hypersusceptibility to oxidant-induced lung injury in mice.23
SU5416, a potent and selective tyrosine kinase inhibitor, which is commonly used for inhibiting various tumor growth by targeting VEGF/VEGFR and kit signaling pathways.24 In addition, SU5416 could inhibit the vessel permeability during ALI.25 However, the role of SU5416 in LPS-induced ALI remains vague. Thus, this study aimed to illuminate the biofunction of SU5416 in the mice with ALI.
Materials and methods
Animal model and treatment
Wild-type C57BL/6 mice (male, 8–10 weeks, 20–24 g) and genetically engineered TLR4-deficient mice (male, 8–10 weeks, 20–22 g) were obtained from Model Animal Research Center of Nanjing University (Nanjing, China). All mice experiments were approved by the Institutional Animal Care and Use Committee at Affiliated Hospital of Hangzhou Normal University. All animals were raised in a room under controlled light (12 hours/day) and temperature (22±2°C) conditions and housed with free access to food and water. 30 WT and 30 TLR−/- male mice were randomly assigned to five groups (6 mice/group), respectively. Then, the mice were stimulated by intratracheal (posterior pharyngeal instillation) administration of LPS (5 mg/kg, 1 hour; Sigma-Aldrich Co., St Louis, MO, USA) after anesthetization with an intraperitoneal (i.p.) injection of tribromoethanol (0.015 ml/g). isopyknic saline-treated mice served as blank control group. After LPS stimulation, the experimental mice were treated with SU5416 (20 mg/kg BW solution in DMSO, Targetmol, T2064), DXM (DXM, 5 mg/kg BW, Sigma-Aldrich Co., D4902) or DXM + SU5416 by oral administration for 12 hours. DXM-treated mice were served as positive control group. Control group (LPS-induced only)/blank control (saline-treated) were given by gavage with isopyknic saline adding equal concentration of DMSO. Later on, mice were anesthetized by using 1% pentobarbital sodium. Bronchoalveolar lavage fluid (BALF) and lung tissues were collected for the subsequent experiments. National Institutes of Health guide for the care and use of laboratory animals were followed for the animal experiments.
Counting of neutrophil in BALF
After anesthetization, the tracheas of mice were cannulated and lungs were lavaged three times with 1 mL saline at 4°C. Then, 1 mL BALF was collected for the detection of the number of neutrophil with Abbott CD1700 automatic blood cell analyzer (Abbott, Chicago, IL, USA).
Measurement of inflammatory cytokines
The levels of proinflammatory cytokines (TGF-β, IL-1β, IL-6, and TNF-α) in BALF were performed according to the instruction of ELISA kits (Abcam: ab119558, ab100705, ab178013 and ab181421) (Abcam, Cambridge, MA, USA). The sensitivities of ab119558, ab100705, ab178013 and ab181421 ELSIA kits were 8, 5, 5 and 14 pg/mL, respectively.
Detection of superoxide dismutase (SOD) and nitric oxide (NO)
After centrifugation, the supernatant fluid of BALF was collected for SOD and NO detection. 20 μl and 100 μL supernatant was used to detect the levels of SOD and NO in BALF, respectively. SOD level was analyzed by SOD assay kit (WST-1 method) in accordance to the manufacturer’s directions (Jiancheng Biotechnology, Nanjing, China, A001-3). That was an activity test and the substrate was water soluble tetrazolium salt (WST), which could be easily reduced to purple formazan. Then, the OD value (450 nm) was detected with a microplate reader. Activity of SOD was calculated using the following formula: SOD activity (U/mL)=SOD inhibition rate/50% x (reaction volume/dilution ratio) x dilution ratio of samples before detection. NO level was detected using NO assay kit (microwell plate method) (Jiancheng Biotechnology, Nanjing, China, A013-2) and evaluated by the following formula: NO level (μmol/L)=(samples OD value – blank OD value)/(standard OD value – blank OD value) x standard concentration (20 μm) x dilution ratio.
Histopathology
Four percent paraformaldehyde was injected into the trachea to fix the mice lung tissues. For histomorphological evaluation, the upper left lung lobe was removed and fixed in 3% glutaraldehyde after treatment with LPS for 24 hours. After embedding in paraffin, 4 μm-thick-sections were used to analysis changes of ultrastructure in different groups of mice lung tissues under transmission electron microscope. Additionally, sections were stained with H&E stain. The ALI score was referring to the method mentioned by Mikawa.26 Injury score was evaluated through four indexes: (1) alveolar congestion, (2) haemorrhage, (3) neutrophils infiltration, (4) alveolar septum thickness and hyaline membrane formation. The analysis result of all mice was scored 0–4 with 0 as no injury, 1 as mild injury, 2 as moderate injury, 3 as serious injury, 4 as very severe injury.
Western blot assay
Total proteins were extracted from lung tissues according to the instruction of RIPA lysis buffer kit (Sigma-Aldrich Co.). The concentration of proteins were detected with BCA assay (Pierce Biotechnology; Rockford, IL, USA). Immunoblotting was conducted by using specific primary antibodies: TLR4 (Affinity, AF7017), p-VEGFR (Affinity, AF3279), VEGFR2 (Affinity, AF6281), pho-NF-κB p65 (p-p65) (Abcam, ab86299), NF-κB (Affinity, AF0874), p53 (Affinity, AF0879), and BCl-2 (Affinity, AF6139). β-actin (Boster, BM3873) was served as the internal control. The blots were detected using the ECL reagent (Santa Cruz Biotechnology; Santa Cruz, CA, USA). Blot images were semiquantified in grayscale using Image J (version 2.0; National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry
Slides of lung tissues were blocked with 0.5% H2O2 in methanol for 30 minutes. Then, slides were incubated overnight at 37°C and stained with primary antibody against CD31 (Affinity, AF0077) for 24 hours at 4°C. Then, the sections were stained with second antibody for 20 minutess at room temperature. After chromogenic reaction with DAB agent (Thermo Fisher Scientific, Waltham, MA, USA), the images were photographed with a microscope.
Measurement of ROS
The levels of ROS were analyzed using DCFH-DA fluorescent probe technique in mice lung. Briefly, fresh lung tissue (1 mm3 mass) was digested with trypsin and filtered with a 200-mesh sieve. The cells were collected and resuspended in PBS. After incubation with DCFH-DA or DMSO for 60 minutes, cells were centrifuged at 5000 g. The precipitates were used for detection of fluorescence value at the wavelength of 525 nm.
Isolation of pulmonary microvascular endothelial cell
After anesthetization with 10% phenobarbital, mice lungs were collected and cut into pieces. Then, pieces of lung tissue were mixed with DMEM and filtered with a 200-mesh sieve. Cells (5 x 105 cells/well) were cultured with endothelial cell medium in the dish covered with 0.1% gelatin at 37°C in a 5% CO2 humidified atmosphere. Cell morphology was observed using microscope and cell purity was verified utilizing immunofluorescence staining with factor VIII (endothelial cells marker). Then, the isolated cells were treated with DMSO, LPS, LPS+SU5416, LPS+DXM or LPS+SU5416+DXM for 24 hours, respectively.
Endothelial barrier function detection
Endothelial barrier function was evaluated through detecting endothelial cell integrity and vascular permeability. For endothelial cell integrity assay, cells were incubated with fluorescein diacetate (FDA) (100 μg/mL) and propidium iodide (PI) (60 μg/mL) for 10 minutes at room temperature in darkness and photographed with a fluorescence microscope. For vascular permeability detection, pulmonary microvascular endothelial cells (PMVECs) were grown in transwell at a density of 105/cm2. After 2–3 days culture, 125 mg/L FITC-LDL was added into upper chamber, while 600 µL DMEM was added into the substrate of transwell. OD values of upper chamber and substrate were measured by using microplate reader. And vascular permeability was calculated by the following formula: Pa (%)=(Vr × Cr)/(Vd × Cd)×100%. Cr: FITC-LDL concentration in substrate; Cd: FITC-LDL concentration in upper chamber; Vr: medium volume in substrate; Vd: medium volume in upper chamber.
Immunofluorescence
PMVECs were seeded on cover glasses for 24 hours, then fixed with 3% paraformaldehyde for 30 minutes at 4°C. After permeabilizing with 0.1% Triton-100 for 15 minutess, cells were incubated with factor VIII at room temperature for 1 hour followed by incubation with a secondary antibody for another 1 hour. The immuno-labeled cells were evaluated with Carl Zeiss LSM5 EXITER laser scanning confocal microscope (Carl Zeiss, Jena, Germany).
Statistical analyses
All experiments were repeated at least three times. Statistical analysis was processed using SPSS software with a two-tailed Student's t-test and one-way ANOVA. Values are presented as the mean ± SEM. Statistically significant differences were defined at P<0.05.
Results
SU5416 suppresses immune response in mice with ALI
Excessive activation and penetration of neutrophils is the common pathological process in LPS-induced ALI.2 Activated neutrophils subsequently enhanced the release of proinflammatory cytokines, which can further aggravate lung injury.27 To determine the biofunction of SU5416 in LPS-induced ALI, we collected BALF from the mice. In comparison with saline group, LPS significantly increased neutrophil cell numbers in BALF of WT and TLR4−/- mice, while neutrophil cells were significantly diminished in TLR4−/- mice (Figure 1A, P<0.01). As a positive control, dexamethasone (DXM) observably inhibited cell population of neutrophil in BALF isolated from two genotype mice (P<0.01). In addition, SU5416 exhibited the similar inhibitory effect on the population of neutrophil cell (P<0.01). Furthermore, co-treatment with SU5416 and DXM significantly alleviated LPS-induced ALI (P<0.01) (Figure 1A). The levels of proinflammatory cytokines (TGF-β, IL-1β, IL-6, and TNF-α) in BALF showed the same trend with the level of neutrophil cells in mice. Moreover, SU5416 and/or DXM significantly reversed LPS-induced proinflammatory factors in BALF (Figure 1B–E).
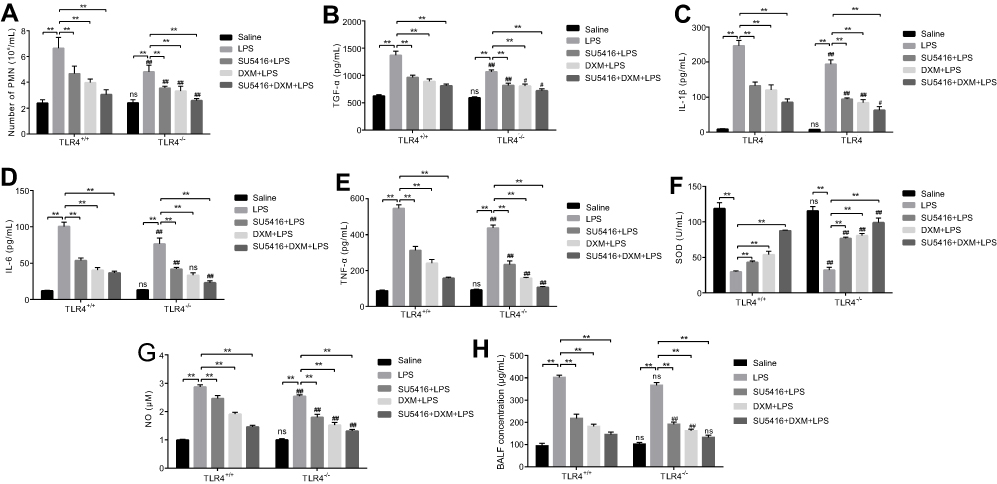 | Figure 1 Effects of SU5416 on the levels of inflammatory factors in mice treated with LPS. (A) Numbers of neutrophil cells in BALF. Levels of proinflammatory cytokines TGF-β (B), IL-1β (C), IL-6 (D) and TNF-α (E) of BALF in each group was detected with ELISA kits, respectively. Activity of SOD (F) and NO level (G) in BALF was measured with WST-1 method and microwell plate method, respectively. (H) The concentration of BALF was detected with BCA assay. *P<0.01; **P<0.05; ***P<0.01 vs TLR4+/+. Abbreviations: BALF, bronchoalveolar lavage fluid; LPS, lipopolysaccharide; DXM, dexamethasone ; TLR4, toll-like receptor 4. |
It is known that levels of SOD and NO are the indicators for lung injury. Compared to saline group, LPS restrained SOD in BALF, which was obviously revered by SU5416 and/or DXM treatment (Figure 1F). Moreover, SU5416 and/or DXM treatment dramatically remitted LPS-triggered the production of NO in BALF (Figure 1G). Similarly, the concentration of BALF in each group exhibited the similar trend (Figure 1H). These data suggested SU5416 could significantly reverse LPS-induced ALI in mice, and exert an even better protective effect in TLR4 knockout mice.
SU5416 attenuates LPS-mediated lung injury in mice
As we know, accumulation of inflammatory cytokines could damage adjacent cells and result in severe lung injury. Transmission electron microscope examinations showed LPS severely damaged the integrity of the alveolar epithelial cells, for instance, cell nucleus and mitochondria swelling, mitochondria number reduction, and osmiophilic lamellar bodies’ vacuolation. (Figure 2A); and TLR4−/- mice moderately resistant to LPS-mediated lung injury (Figure 2A, second row). Similar to DXM, SU5416 or SU5416 plus DXM significantly reversed LPS-induced structure disturbance of alveolar epithelial cells in both WT and TLR4−/- mice (Figure 2A). Next, H&E staining was performed to assess LPS-induced histopathological changes in each group. As indicated in Figure 2B, numerous neutrophil infiltration, lung tissue injury, and alveolar wall thickening were observed in LPS-stimulated WT and TLR4−/- mice. However, thinning alveolar wall, normal alveolar structure, and no obvious inflammatory infiltration were observed in SU5416 or DXM-treated groups. In addition, co-treatment of SU5416 with DXM showed better protective effect on LPS-treated ALI mice than DXM alone treatment (Figure 2B). Moreover, TLR4 deletion not only mitigated LPS-triggered histopathological disturbance of alveolar epithelial cells, but also presented superimposed protective effect on LPS-induced ALI after treatment with SU5416 and/or DXM. Total ALI score analysis based on H&E staining showed the same tendency with histopathological analysis (Figure 2C). Furthermore, compared to saline-treated mice, SU5416 and/or DXM repressed the production of ROS in lung tissues (WT and TLR4−/- mice) (Figure 2D). All these results demonstrated that SU5416 acted as a protective agent against LPS-induced ALI in mice.
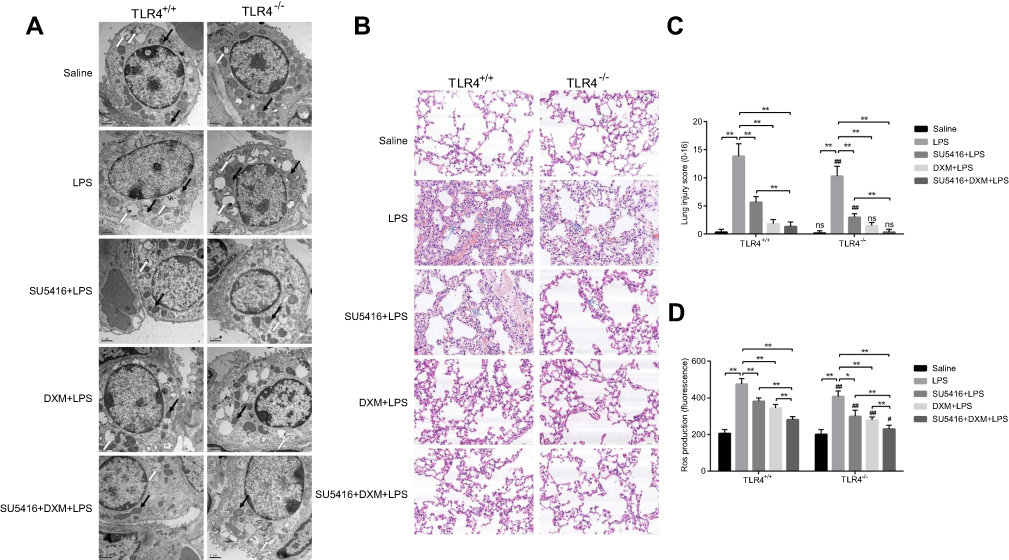 | Figure 2 Impacts of SU5416 on LPS-induced ALI. (A) Mice pulmonary ultrastructure was observed with transmission electron microscope. White and black arrows indicate osmiophilic multilamellar body and mitochondria, respectively. (B) Histopathologic staining images of mice lungs. Total injury score (C) and ROS production (D) in all groups of mice lungs. *P<0.01; **P<0.05; ***P<0.01 vs TLR4+/+.Abbreviations: TLR4, toll-like receptor 4; LPS, lipopolysaccharide; DXM, dexamethasone; ALI, acute lung injury |
SU5416 reverses LPS-induced ALI partly via modulating TLR4/NF-κB signaling in mice
To analyze the role of SU5416 on LPS-induced alterations of alveolar endothelial cells, vascular endothelial cell marker CD31 was detected using immumohistochemical staining. The results indicated that CD31 was significantly reduced after LPS treatment in the two genotype mice (Figure 3A, second row). However, SU5416 and/or DXM dramatically restored the reduction of CD31 expression mediated by LPS, suggesting SU5416 could rescue LPS-induced dysfunction of pulmonary endothelial barrier (Figure 3A, third to fifth rows). To explore the molecule mechanisms underlying the protective effect of SU5416 on LPS-launched ALI, Western blots were performed. As shown in Figure 3B–D, both p-VEGFR2 and VEGFR2 expressions were inhibited by SU5416 in WT and TLR4−/- mice.
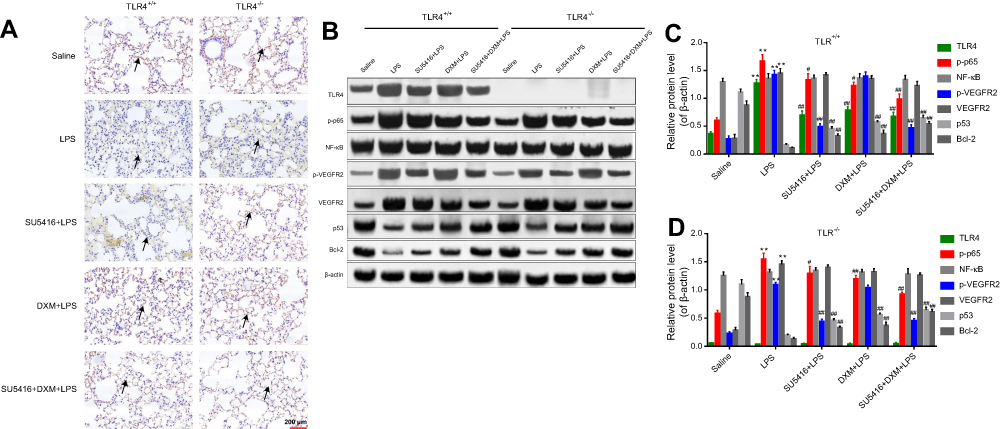 | Figure 3 TLR4/NF-κB signaling was involved in the progression of LPS-stimulated ALI. (A) Immunohistochemical staining of CD31 in LPS-stimulated WT and TLR4−/- mice after treatment with DXM and/or SU5416+DXM. (B) Mice were treated with DXM and/or SU5416 for 12 hours, and the expressions of TLR4, p-p65, NF-κB, p-VEGFR2, VEGF2R, p53, Bcl-2, and β-actin in lung tissues were detected with Western blot. (C, D). Relative expressions of TLR4, p-p65, NF-κB, p-VEGFR2, VEGF2R, p53 and Bcl-2. *P<0.01 vs saline; **P<0.05; ***P<0.01 vs LPS. Abbreviations: TLR4, toll-like receptor 4; LPS, lipopolysaccharide; DXM, ; ALI, acute lung injury; VEGFR, vascular endothelial growth factor receptor; DXM, dexamethasone. |
A study has reported that DXM could attenuate LPS-mediated ALI through modulating NF-κB activation and TLR4/NF-κB signaling.1 In the present study, we observed that TLR4 ablation moderately inhibited LPS-induced upregulation of VEGF/VEGFR and NF-κB pathway, and decreased downstream molecules p53 and Bcl-2 (Figure 3B–D). This data indicated that TLR4 plays an important role in the process of LPS-induced ALI. In addition, SU5416 and/or DXM suppressed VEGF/VEGFR and TLR4/NF-κB signaling pathways and upregulated the expressions of p53 and Bcl-2 in LPS-treated WT and TLR4−/- mice (Figure 3B). Furthermore, SU5416 and DXM exhibited better protective effect in TLR4−/- mice than WT mice implying that the anti-ALI functions of SU5416 and DXM were potentially independent of TLR4.
SU5416 regulates endothelial barrier dysfunction in PMVECs
We further performed in vitro experiments to illustrate the role of SU5416 in LPS-stimulated inflammatory responses of PMVECs. The morphology of PMVECs isolated from WT and TLR4−/- mice were in good condition (Figure 4A). In addition, immunofluorescence staining indicated the purity of PMVECs was 98% and cells were suitable for subsequent experiments (Figure 4B).
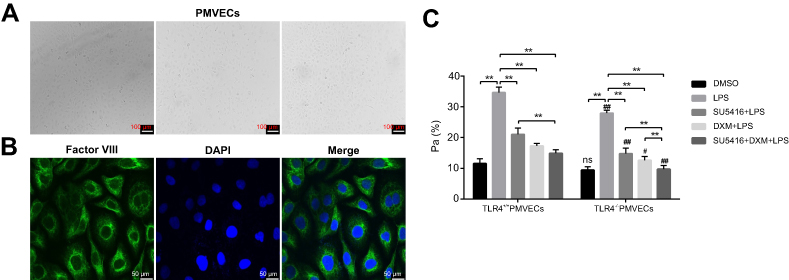 | Figure 4 Effects of SU5416 on vascular permeability in LPS-treated mice. (A) Morphology images of PMVECs isolated from LPS-treated WT and TLR4-/- mice. (B) Immunofluorescence staining of factor VIII in PMVECs after treatment with DXM and/or SU5416. (C) Analysis result of endothelial cell permeability in PMVECs after treatment with DXM and/or SU5416. *P<0.01; **P<0.05; ***P<0.01 vs TLR4+/+. Abbreviations: PMVECs, pulmonary microvascular endothelial cells; TLR4, toll-like receptor 4; LPS, lipopolysaccharide; DXM, dexamethasone. |
Next, PMVECs from WT and TLR4−/- mice were treated with DMSO, LPS, LPS+SU5416, LPS+DXM, or LPS+SU5416+DXM, respectively. As shown in Figure 4C, LPS obviously enhanced the membrane permeability of endothelial cells, which was prominently reversed by SU5416 or DXM. Additionally, SU5416 plus DXM exerted a more strongly protective effect on the membrane permeability. Consistent with the in vivo experiments, TLR4 deletion could further improve the endothelial barrier dysfunction of PMVECs (Figure 4C).
Endothelial cell integrity was also a key determinant for the endothelial barrier. By using FDA-PI assay, we evaluated damage degree of endothelial cell in all groups. As showed in Figure 5A and B, LPS severely damaged endothelial cell integrity and promoted PMVECs apoptosis. Moreover, the endothelial cell integrity was modestly improved in the TLR4−/- group compared with WT mice (Figure 5A and B, second row). SU5416 or DXM also exhibited protective effects on endothelial cell integrity. Similarly, SU5416 plus DXM exerted a better protective effect than used alone (Figure 5A and B). Furthermore, knockout of TLR4 showed less invasive to endothelial cells in each group. Our findings suggested SU5416 exhibited a similar protective effect as DXM on endothelial cell integrity and vascular permeability in PMVECs.
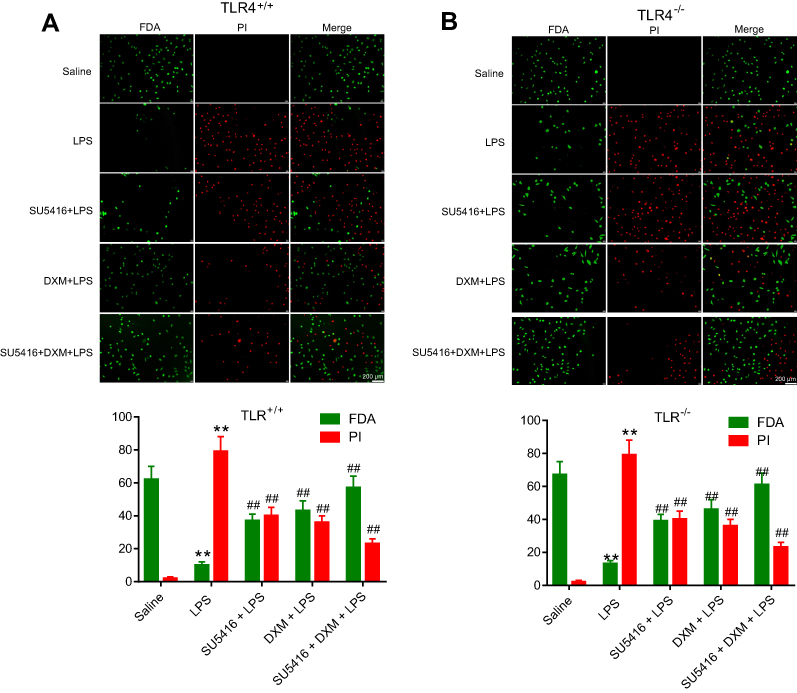 | Figure 5 Effects of SU5416 on the endothelial cell integrity in mice treated with LPS. Measurement of endothelial cell integrity using FDA-PI staining method in WT (A) and TLR4−/- (B) mice after treatment with DMSO, LPS, SU5416, DXM, or SU5416+DXM. Green indicates positive endothelial cell. Red indicates endothelial cell nucleus. *P<0.01 vs saline; **P<0.01 vs LPS. Abbreviations: FDA, fluorescein diacetate; TLR4, toll-like receptor 4; PI, propidium iodide; LPS, lipopolysaccharide; DXM, dexamethasone. |
Discussion
SU5416 is a potent and selective inhibitor of Flk1/KDR (VEGFR-2). Previous study indicated that SU5416 combined with chronic hypoxia could produce a model of severe pulmonary arterial hypertension.28 In addition, the combined utilization of radiation and SU5416 observably repressed the survival in human umbilical vein endothelial cells (HUVECs) via induction of apoptosis, implying that SU5416 was potentially served as an adjuvant for cancer treatment with radiotherapy.29 In addition to tumor inhibition effective, SU5416 mitigated bleomycin-induced pulmonary fibrosis via targeting VEGF/VEGFR (Flk-1) signaling in mice.30 In this study, we indicated that SU5416 could attenuate LPS-induced ALI through modulating the VEGF/VEGFR and NF-κB pathways, which suggested SU5416 might be used for the treatment of patients with inflammation-mediated ALI.
LPS can induce transcription and production of inflammatory cytokines such as IL-6, TNF-α, COX-2, and IL-1β through activating NF-κB signaling in lung and BALF.31 Enhancement of vascular endothelium cells permeability contributes to the secretion of inflammatory factors from injury to the endothelium and epithelial alveolar cells in lung, leading to inflammatory responses.3 Endothelial barrier strongly depends on the concentration of NO,32 which is required for vasodilation, platelet aggregation, and adhesion of inflammatory cells to the endothelium.33,34 DXM can attenuate the increased inflammatory factors such as IL-6, TNF-α, and COX-2 in LPS-stimulated ALI,17 potentially through affecting NO-dependent vascular endothelium permeability. In our research, we observed the increased production of inflammatory factors, formation of SOD and NO in BALF of LPS-treated WT and TLR4−/- mice. TLR4 knockout observably restrained the biotoxicity of LPS in mice model of ALI. In addition, SU5416 significantly suppressed LPS-triggered inflammatory responses in mice, characteristic with the decreases of inflammatory cells, production of inflammatory cytokines, SOD and NO in BALF. Combination of SU5416 and DXM appeared additive inhibitory effects on LPS-stimulated ALI. Furthermore, we found SU5416 might acted as an anti-ALI drug independent of TLR4.
Injury of alveolar epithelial cells and infiltration of inflammatory cells are the main hallmarks of LPS-induced ALI. LPS-TLR4 signaling activates NF-κB and Nlrp3 inflammasome, resulting in damage to alveolar epithelial cells and inflammation infiltration.35 A variety of studies have verified that targeting LPS/TLR4/NF-κB signaling pathway is crucial for treating ALI disease. It is reported that numerous Chinese traditional medicines can protect against LPS-induced ALI via inhibiting TLR4/NF-κB signaling, which includes Lianqinjiedu (LQJD),36 Mogroside IIIE,37 Artesunate,38 and so on. Activation of NF-κB promotes the production of inflammatory cytokines mainly through the resynthesis of IκB,39,40 leading to diffuse alveolar damage in the lung.41 Similar to these reports, our data illustrated SU5416 could inhibit LPS-evoked injury to alveolar epithelial cells and area of inflammation infiltration in lung tissues. VEGF/VEGFR, TLR4/NF-κB, and downstream signaling cascade were restrained in LPS-stimulated WT mice after treatment with SU5416, DXM, and co-treatment of SU5416 and DXM. And this inhibitory effect was more obvious in TLR4−/- mice. Possibly, SU5416 affected LPS-induced ALI partly through VEGF/VEGFR and TLR4/NF-κB signaling pathway. Other signals involving the progression of ALI independent of TLR4, might be disturbed by SU5416 administration.
Endothelial cell integrity and vascular permeability are crucial for the function of the endothelial barrier, which maintains tissue fluid homeostasis. Increases of vascular permeability is the main phenomenon of many inflammatory diseases. LPS decreases p53 expression in lung, while nutlin protects against LPS-evoked dysfunction of lung endothelial barrier via increases p53.42 ROS formatted by neutrophils or other inflammatory cells activates calcium signaling mechanisms to control endothelial barrier function and the progression of ALI.43 Therefore, inhibition of generation of oxygen radical could be beneficial during the pathologies of ALI and SOD, which might play an anti-inflammatory role. All these reports suggest that the status of the endothelial barrier is a key indicator of lung function. Here, similar to the positive control DXM, SU5416 inhibited vascular permeability via promoting endothelial cell integrity in LPS-stimulated primary PMVECs from WT and TLR4−/- mice. In accordance with the in vivo experiments, the inhibitory effect of SU5416 or DXM is more noticeable in primary PMVECs isolated from TLR deletion mice. Thus, SU5416-mediated protective effects on LPS-evoked ALI was potentially independent of TLR4. In addition, it should be noted that targeting drugs, including antioxidants and in particular SOD, to specific cells is being pursued in animals using ICAM-1, ACE, APP-1, GP80, CD31 and PV1-directed carriers.44–47 However, the present study is lacking in investigating the lung-specific treatment strategy. Thus, further studies will be needed to explore the effects of cell-specific intervention in the lung.
In summary, our findings demonstrated that SU5416 significantly inhibited LPS-induced lung injury and may serve as an efficient small molecule inhibitor in the treatment of patients with ALI.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yingkun N, Zhenyu W, Jing L, Xiuyun L, Huimin Y. Stevioside protects LPS-induced acute lung injury in mice. Inflammation. 2013;36(1):242–250. doi:10.1007/s10753-012-9579-6
2. Dengler V, Downey GP, Tuder RM, Eltzschig HK, Schmidt EP. Neutrophil intercellular communication in acute lung injury. Emerging roles of microparticles and gap junctions. Am J Respir Cell Mol Biol. 2013;49(1):1–5. doi:10.1165/rcmb.2012-0374OC
3. Kosutova P, Mikolka P, Balentova S, et al. Intravenous DXM attenuated inflammation and influenced apoptosis of lung cells in an experimental model of acute lung injury. Physiol Res. 2016;65(Supplementum 5):S663–S672.
4. Aujla SJ, Chan YR, Zheng M, et al. IL-22 mediates mucosal host defense against gram-negative bacterial pneumonia. Nat Med. 2008;14(3):275–281. doi:10.1038/nm1710
5. Kim KY, Lee HS, Seol GH. Eucalyptol suppresses matrix metalloproteinase-9 expression through an extracellular signal-regulated kinase-dependent nuclear factor-kappa B pathway to exert anti-inflammatory effects in an acute lung inflammation model. J Pharm Pharmacol. 2015;67(8):1066–1074. doi:10.1111/jphp.12371
6. Rice P, Martin E, He J-R, et al. Febrile-range hyperthermia augments neutrophil accumulation and enhances lung injury in experimental gram-negative bacterial pneumonia. J Immunol. 2005;174(6):3676–3685.
7. Chen G, Sun X, Dong C. RhoA regulates lipopolysaccharide‑induced lung cell injury via the Wnt/β‑catenin pathway. Mol Med Rep. 2017;16(6):8501–8506. doi:10.3892/mmr.2017.7662
8. Ding Q, Liu G, Zeng Y, et al. Glycogen synthase kinase‑3β inhibitor reduces LPS‑induced acute lung injury in mice. Mol Med Rep. 2017;16(5):6715–6721. doi:10.3892/mmr.2017.7469
9. Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi:10.1126/science.282.5396.2085
10. Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13(11):460–469. doi:10.1016/j.molmed.2007.09.002
11. Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. 2014;5:316. doi:10.3389/fimmu.2014.00316
12. Broad A, Kirby JA, Jones DE. Applied immunology and transplantation research group: toll-like receptor interactions: tolerance of MyD88-dependent cytokines but enhancement of MyD88-independent interferon-beta production. Immunology. 2007;120(1):103–111. doi:10.1111/j.1365-2567.2006.02485.x
13. Hoth JJ, Wells JD, Brownlee NA, et al. Toll-like receptor 4-dependent responses to lung injury in a murine model of pulmonary contusion. Shock. 2009;31(4):376–381. doi:10.1097/SHK.0b013e3181862279
14. Villar J, Cabrera N, Casula M, et al. Mechanical ventilation modulates Toll-like receptor signaling pathway in a sepsis-induced lung injury model. Intensive Care Med. 2010;36(6):1049–1057. doi:10.1007/s00134-010-1799-3
15. Liu W, Shan L-P, Dong X-S, Liu Z. Toll-like receptor 4 implicated in acute lung injury induced by paraquat poisoning in mice. Int J Clin Exp Med. 2014;7(10):3392–3397.
16. Joh E-H, Gu W, Kim D-H. Echinocystic acid ameliorates lung inflammation in mice and alveolar macrophages by inhibiting the binding of LPS to TLR4 in NF-κB and MAPK pathways. Biochemical Pharmacology. 2012;84(3):331–340. doi:10.1016/j.bcp.2012.04.020
17. Al-Harbi NO, Imam F, Al-Harbi MM, et al. DXM attenuates LPS-induced acute lung injury through inhibition of NF-κB, COX-2, and pro-inflammatory mediators. Immunol Invest. 2016;45(4):349–369. doi:10.3109/08820139.2016.1157814
18. Papaioannou AI, Kostikas K, Kollia P, Gourgoulianis KI. Clinical implications for vascular endothelial growth factor in the lung: friend or foe? Respir Res. 2006;7:128. doi:10.1186/1465-9921-7-128
19. Mura M, Santos Dos CC, Stewart D, Liu M. Vascular endothelial growth factor and related molecules in acute lung injury. J Appl Physiol. 2004;97(5):1605–1617. doi:10.1152/japplphysiol.00202.2004
20. Kaner RJ, Crystal RG. Compartmentalization of vascular endothelial growth factor to the epithelial surface of the human lung. Mol Med. 2001;7(4):240–246. doi:10.1007/BF03401843
21. Fujita N, Imai J-I, Suzuki T, et al. Vascular endothelial growth factor-A is a survival factor for nucleus pulposus cells in the intervertebral disc. Biochem Biophys Res Commun. 2008;372(2):367–372. doi:10.1016/j.bbrc.2008.05.044
22. Zhang Y, Lu Y, Ma L, et al. Activation of vascular endothelial growth factor receptor-3 in macrophages restrains TLR4-NF-κB signaling and protects against endotoxin shock. Immunity. 2014;40(4):501–514. doi:10.1016/j.immuni.2014.01.013
23. Takyar S, Zhang Y, Haslip M, et al. An endothelial TLR4-VEGFR2 pathway mediates lung protection against oxidant-induced injury. FASEB J. 2016;30(3):1317–1327. doi:10.1096/fj.15-275024
24. Litz J, Sakuntala Warshamana-Greene G, Sulanke G, Lipson KE, Krystal GW. The multi-targeted kinase inhibitor SU5416 inhibits small cell lung cancer growth and angiogenesis, in part by blocking kit-mediated VEGF expression. Lung Cancer. 2004;46(3):283–291. doi:10.1016/j.lungcan.2004.05.005
25. Yu X, Lin Q, Qin X, et al. ACE2 antagonizes VEGFa to reduce vascular permeability during acute lung injury. Cell Physiol Biochem. 2016;38(3):1055–1062. doi:10.1159/000445551
26. Takao Y, Mikawa K, Nishina K, Maekawa N, Obara H. Lidocaine attenuates hyperoxic lung injury in rabbits. Acta Anaesthesiol Scand. 1996;40(3):318–325.
27. Vlaar APJ, Juffermans NP. Transfusion-related: a clinical review. Lancet. 2013;382(9896):984–994. doi:10.1016/S0140-6736(12)62197-7
28. Jernigan NL, Naik JS, Weise-Cross L, et al. Contribution of reactive oxygen species to the pathogenesis of pulmonary arterial hypertension. PLoS One. 2017;12(6):e0180455. doi:10.1371/journal.pone.0180455
29. Kim EH, Kim M-S, Jeong YK, et al. Mechanisms for SU5416 as a radiosensitizer of endothelial cells. Int J Oncol. 2015;47(4):1440–1450. doi:10.3892/ijo.2015.3127
30. Ou XM, Li WC, Liu DS, et al. VEGFR-2 antagonist SU5416 attenuates bleomycin-induced pulmonary fibrosis in mice. Int Immunopharmacol. 2009;9(1):70–79. doi:10.1016/j.intimp.2008.10.002
31. Chen XY, Dou YX, Luo DD, et al. β-Patchoulene from patchouli oil protects against LPS-induced acute lung injury via suppressing NF-κB and activating Nrf2 pathways. Int Immunopharmacol. 2017;50:270–278. doi:10.1016/j.intimp.2017.07.001
32. Liu XJ, Zhang ZD, Ma XC. High glucose enhances LPS-stimulated human PMVEC hyperpermeability via the NO pathway. Exp Ther Med. 2013;6(2):361–367. doi:10.3892/etm.2013.1145
33. Akaike T, Maeda H. Nitric oxide and virus infection. Immunology. 2000;101(3):300–308.
34. van der Vliet A, Eiserich JP, Shigenaga MK, Cross CE. Reactive nitrogen species and tyrosine nitration in the respiratory tract: epiphenomena or a pathobiologic mechanism of disease? Am J Respir Crit Care Med. 1999;160(1):1–9. doi:10.1164/ajrccm.160.1.9807044
35. He X, Qian Y, Li Z, et al. TLR4-upregulated IL-1β and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci Rep. 2016;6:31663. doi:10.1038/srep31663
36. Deng G, He H, Chen Z, et al. Lianqinjiedu decoction attenuates LPS-induced inflammation and acute lung injury in rats via TLR4/NF-κB pathway. Biomed Pharmacother. 2017;96:148–152. doi:10.1016/j.biopha.2017.09.094
37. Tao L, Cao F, Xu G, Xie H, Zhang M, Zhang C. Mogroside IIIE attenuates LPS-induced acute lung injury in mice partly through regulation of the TLR4/MAPK/NF-κB axis via AMPK activation. Phytother Res. 2017;31(7):1097–1106. doi:10.1002/ptr.v31.7
38. Zhao D, Zhang J, Xu G, Wang Q. Artesunate protects LPS-induced acute lung injury by inhibiting TLR4 expression and inducing Nrf2 activation. Inflammation. 2017;40(3):798–805. doi:10.1007/s10753-017-0524-6
39. Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38105–38108. doi:10.1074/jbc.R300028200
40. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25(51):6680–6684. doi:10.1038/sj.onc.1209954
41. Li D, Pan X, Zhao J, et al. Bone marrow mesenchymal stem cells suppress acute lung injury induced by lipopolysaccharide through inhibiting the TLR2, 4/NF-κB pathway in rats with multiple trauma. Shock. 2016;45(6):641–646. doi:10.1097/SHK.0000000000000548
42. Barabutis N, Dimitropoulou C, Birmpas C, Joshi A, Thangjam G, Catravas JD. p53 protects against LPS-induced lung endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2015;308(8):L776–87. doi:10.1152/ajplung.00334.2014
43. Di A, Mehta D, Malik AB. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium. 2016;60(3):163–171. doi:10.1016/j.ceca.2016.02.002
44. Brenner JS, Greineder C, Shuvaev V, Muzykantov V. Endothelial nanomedicine for the treatment of pulmonary disease. Expert Opin Drug Deliv. 2015;12(2):239–261. doi:10.1517/17425247.2015.961418
45. Brenner JS, Bhamidipati K, Glassman PM, et al. Mechanisms that determine nanocarrier targeting to healthy versus inflamed lung regions. Nanomedicine. 2017;13(4):1495–1506. doi:10.1016/j.nano.2016.12.019
46. Muro S, Dziubla T, Qiu W, et al. Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. J Pharmacol Exp Ther. 2006;317(3):1161–1169. doi:10.1124/jpet.105.098970
47. Danielyan K, Ding BS, Gottstein C, Cines DB, Muzykantov VR. Delivery of anti-platelet-endothelial cell adhesion molecule single-chain variable fragment-urokinase fusion protein to the cerebral vasculature lyses arterial clots and attenuates postischemic brain edema. J Pharmacol Exp Ther. 2007;321(3):947–952. doi:10.1124/jpet.107.120535
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.