")
Back to Journals » International Journal of Nanomedicine » Volume 12
Styrene maleic acid-encapsulated RL71 micelles suppress tumor growth in a murine xenograft model of triple negative breast cancer
Authors Martey O, Nimick M, Taurin S, Sundararajan V , Greish K , Rosengren RJ
Received 11 August 2017
Accepted for publication 9 September 2017
Published 4 October 2017 Volume 2017:12 Pages 7225—7237
DOI https://doi.org/10.2147/IJN.S148908
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Orleans Martey,1 Mhairi Nimick,1 Sebastien Taurin,1 Vignesh Sundararajan,1 Khaled Greish,2 Rhonda J Rosengren1
1Department of Pharmacology and Toxicology, University of Otago, Dunedin, New Zealand; 2Department of Molecular Medicine, College of Medicine and Medical Sciences, Arabian Gulf University, Manama, Kingdom of Bahrain
Abstract: Patients with triple negative breast cancer have a poor prognosis due in part to the lack of targeted therapies. In the search for novel drugs, our laboratory has developed a second-generation curcumin derivative, 3,5-bis(3,4,5-trimethoxybenzylidene)-1-methylpiperidine-4-one (RL71), that exhibits potent in vitro cytotoxicity. To improve the clinical potential of this drug, we have encapsulated it in styrene maleic acid (SMA) micelles. SMA-RL71 showed improved biodistribution, and drug accumulation in the tumor increased 16-fold compared to control. SMA-RL71 (10 mg/kg, intravenously, two times a week for 2 weeks) also significantly suppressed tumor growth compared to control in a xenograft model of triple negative breast cancer. Free RL71 was unable to alter tumor growth. Tumors from SMA-RL71-treated mice showed a decrease in angiogenesis and an increase in apoptosis. The drug treatment also modulated various cell signaling proteins including the epidermal growth factor receptor, with the mechanisms for tumor suppression consistent with previous work with RL71 in vitro. The nanoformulation was also nontoxic as shown by normal levels of plasma markers for liver and kidney injury following weekly administration of SMA-RL71 (10 mg/kg) for 90 days. Thus, we report clinical potential following encapsulation of a novel curcumin derivative, RL71, in SMA micelles.
Keywords: curcumin derivatives, nanomedicine, EGFR, biodistribution
Introduction
Breast cancer can be classified into several different subtypes and all of these have different molecular signatures, prognoses, and thus a variable response to therapies.1–3 Triple negative breast cancers (TNBCs), which account for 10%–17% of all breast cancers, are largely classified by the absence of three key receptors, namely, the estrogen receptor (ER), progesterone receptor, and epidermal growth factor receptor (EGFR)-2.4,5 TNBCs are predominantly diagnosed in younger women as well as women who are either African-American,6,7 Hispanic,8 or obese.9,10 Furthermore, these cancers are more aggressive and poorly differentiated, and thus women with TNBC have a worse prognosis compared to other types of breast cancer.4,11 These patients usually respond well to standard chemotherapy. However, they also have a high rate of relapse as well as metastases that are unresponsive to the available drug treatments.12 Thus, there is a need to develop new drugs for this aggressive form of the disease.
In the search for new drug treatments for TNBC, our laboratory and others have produced synthetic analogs structurally based on the natural compound curcumin (diferuloylmethane). Curcumin, which is one of the aromatic compounds contained in turmeric (Curcuma longa Linn), has been extensively used in the treatment of a variety of diseases.13 However, a downfall of this natural product is its low bioavailability resulting in part from its rapid metabolism and elimination.14,15 Synthetic analogs of curcumin have been produced by many groups around the world, and these compounds consistently show superior potency to curcumin.16–22 Various groups have also attempted to improve the pharmacokinetic profile of curcumin by encapsulating it into micelles,23 liposomes,24 or by forming conjugates.25 However, clinical usefulness has still not been achieved. Therefore, further work should concentrate on encapsulation of curcumin analogs with improved potency. To this end, our laboratory has previously shown that a second-generation methylpiperidine-4-one curcumin analogue, 3,5-bis(3,4,5-trimethoxybenzylidene)-1-methylpiperidine-4-one (RL71), had a cytotoxic potency that was 20-fold greater than curcumin.17 Low concentrations (1 μM) also induced apoptosis and elicited G2/M phase cell cycle arrest in several TNBC cell lines.18 This potent cytotoxicity was mechanistically driven by a significant decrease in Akt, mechanistic target of rapamycin (mTOR), and nuclear factor-kappa B (NF-κB) protein levels.18 Therefore, of all the analogs we evaluated in vitro, RL71 was the leading candidate for further encapsulation experiments. To improve the tumor targeting effect of RL71, we used styrene maleic acid (SMA) micelles as the drug carrier system. SMA micelles are amphiphilic in nature and can be used to increase the solubility, stability, and pharmacokinetics of lipophilic drugs. The nanosize of the micelles also improves drug half-life as well as its passive accumulation in the tumor via the enhanced permeability and retention (EPR) effect.26,27 Furthermore, the safety of SMA has been shown in humans, as it has been approved for use in the clinic, conjugated to neocarzinostatin, as a treatment for hepatocellular carcinoma.28,29 Lastly, using SMA to form micelles has multiple advantages over other nanoformulations. These benefits include a simple and economical preparation, high recovery, and the ability to control not only the quantity of drug encapsulated but also its release rate.
We have previously prepared SMA-RL71 micelles with 15% loading and characterized their solubility, size (181.6 nm), and charge (−0.0432 mV).30 We have shown in vitro that the micellar formulation of RL71 elicited a favorable release rate, cytotoxicity profile, and disrupted tumor spheroid integrity to a greater degree than free RL71.30 Thus, SMA-RL71 micelles exhibited the characteristics suitable for passive tumor accumulation and targeting via the EPR effect. Thus, the in vivo efficacy of this novel formulation was examined to evaluate its potential as a novel drug formulation for the treatment of TNBC. In this study, we present the biodistribution, tumor targeting, tumor growth suppression, and potential mechanisms for this effect as well as subchronic toxicity of SMA-RL71.
Materials and methods
Materials
MDA-MB-231 cells were purchased from American Type Culture Collection (Manassas, VA, USA). Cumene terminated poly (styrene-co-maleic anhydride) with an average Mn ~1,700, N-(3-dimethylaminopropyl)-N-ethyl-carbodiimide hydrochloride (EDAC), and β-tubulin were purchased from Sigma-Aldrich (St Louis, MO, USA). Matrigel, DAB substrate kit, monoclonal CD-105, protein kinase B (Akt), and streptavidin-horseradish peroxidase were purchased from BD Biosciences (San Diego, CA, USA). Cryomatrix, Vector hematoxylin quick stain (QS) (modified Mayer’s formula), avidin–biotin blocking kit were purchased from Vector Laboratories (Burlingame, CA, USA). Mini complete ethylenediaminetetraacetic acid (EDTA) free protein inhibitor cocktail, Tween 20, goat anti-rabbit IgG peroxidase conjugate, goat anti-mouse IgG peroxidase conjugate, ApopTag peroxidase in situ apoptosis detection kit, and polyvinylidene fluoride (PVDF) membrane were purchased from Merck (Billerica, MA, USA). Dulbecco’s Modified Eagle’s Medium nutrient mixture F-12 (DMEM/Ham’s F12), fetal bovine serum (FBS), L-glutamine, penicillin, and streptomycin were purchased from Life Technologies (Auckland, New Zealand). 4E-binding protein-1 (4EBP1) phosphorylated 4EBP1 (p4EBP1), mTOR, phosphorylated mTOR (pmTOR), cleaved caspase-3, phosphorylated Akt (pAkt), EGFR, phosphorylated EGFR at amino acid Tyr1148 (pEGFR), NF-κB were purchased from Cell Signaling Technology (Beverly, MA, USA). Alanine aminotransferase (ALT) colorimetric assay kit and cryomatrix were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Creatinine colorimetric assay kit was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). RL71 was a gift from Bill Hawkins from the Department of Chemistry, University of Otago, Dunedin, New Zealand.
Methods
Preparation of SMA-RL71 micelles
SMA-RL71 micelles were prepared as described previously.30 Hydrolyzed SMA was mixed with RL71 to achieve a targeted loading of 15%. Briefly, the hydrolyzed SMA solution was adjusted to pH 5; RL71 was dissolved in a minimum volume of dimethyl sulfoxide and added to the SMA solution with stirring. EDAC was solubilized in distilled water, added to the mixture, and allowed to stir for 20 min at pH 5. The solution was adjusted to pH 11 by the addition of 0.1 N NaOH and stirred for 30 min to allow micelle formation. The clear micelle suspension was ultrafiltered four times using a lab-scale ultrafiltration system mounted with a Pellicon XL filter 10 kDa (Merck Millipore, Billerica, MA, USA). The concentrated micelle solution was lyophilized to obtain the final SMA-RL71 powder. SMA was used as a vehicle control by dissolving the SMA in NaOH and adjusting the pH to 7.4.
Biodistribution of RL71 and SMA-RL71
All animal protocols were approved by the University of Otago Animal Ethics Committee. All drug administration, animal monitoring, necropsy, and establishment of the in vivo model of breast cancer were conducted in the protocols provided in the approved application (#41/11). Female severe combined immunodeficient (SCID) mice (7–8 weeks) were purchased from Animal Resources Centre (Murdoch, Perth, Australia). After acclimatizing for 1 week, the mice were inoculated subcutaneously into the rear flank with MDA-MB-231 cells (1×106/0.1 mL Matrigel 50%). Once the palpable tumors reached 100 mm3, the mice were randomly allocated into treatment groups (five animals per group). The mice were administered a single dose intravenously (iv) via the tail vein of 10 mg/kg of free RL71 or an equivalent dose of SMA-RL71 and were euthanized 6 h post-injection. Another group of mice were administered 10 mg/kg of free RL71 or an equivalent dose of SMA-RL71 twice a week for 2 weeks and were euthanized 3 days after the final dose. Full necropsies were then performed. Blood was immediately drawn from the inferior vena cava, centrifuged, and plasma collected and stored at −20°C until required for analysis. Tumors and major organs were collected, snap frozen, and stored at −80°C.
HPLC analysis
In order to extract RL71 from the various tissues, 50 mg of ground tissue was mixed with 0.5 mL of methanol, sonicated and incubated for 1 h on ice. The homogenate was then centrifuged at 14,000 rpm for 10 min and the supernatant reduced down to 100 μL using low-flow nitrogen to evaporate the methanol. Samples were centrifuged again and transferred to a high-performance liquid chromatography (HPLC) vial. The analysis was performed on a Shimadzu Prominence HPLC system using Chromeleon software and a Phenomenex Gemini® 5 μm C18 110 Å LC Column 150×4.6 mm. A flow rate of 1 mL/min was used with an 80 μL injection volume. Peaks were eluted with 10 mM sodium acetate pH 5.0 as mobile phase A and acetonitrile as mobile phase B. An isocratic flow of 35% A and 65% B was maintained over 10 min. The eluted peak for RL71 was monitored at 361 nm with a retention time of 4.4 min.
Xenograft model of TNBC
Female SCID mice (7–8 weeks) were inoculated subcutaneously into the rear flank with MDA-MB-231 cells (1×106/0.1 mL Matrigel 50%). Once the palpable tumors reached 100 mm3, the mice were randomly allocated into treatment groups (eight animals per group). Two different studies were conducted to determine the efficacy of RL71 as a free drug and to determine if this could be improved by the SMA-RL71 formulation. Study 1 – the mice were dosed intravenously twice per week for 2 weeks via the tail vein with either 10 mg/kg of free RL71, 10 mg/kg of SMA-RL71, or vehicle control and euthanized on day 24. Study 2 – the mice were dosed intravenously twice a week for 3 weeks via the tail vein with 10 mg/kg of SMA-RL71 or SMA control and were euthanized on day 24. Two independent measurements of tumor volume (L × W × H) were performed bi-weekly using electronic calipers. Following the treatment period, the mice were euthanized and full necropsies were performed. Blood was immediately drawn from the inferior vena cava, centrifuged, and plasma was collected and stored at −20°C until required for analysis. Plasma samples were analyzed for ALT activity using the commercially available Infinity ALT colorimetric assay kit and performed according to the manufacturer’s instructions. Plasma creatinine concentration was quantified by the Creatinine Colorimetric Assay kit and performed according to the manufacturer’s instructions. Major organs and tumors were collected, weighed, fixed in 4% paraformaldehyde, and frozen in cryomatrix compound until required for histological analysis.
Immunohistochemistry of tumor sections
Tissue sections were analyzed for microvessel density (MVD) as previously described.18 Briefly, tumors were embedded in cryomatrix, sectioned (12 μm), and fixed in acetone. Sections were then washed with PBS and treated with hydrogen peroxide (0.3% in methanol) for 20 min at room temperature. Angiogenesis was estimated by determining MVD using anti-rabbit CD105 monoclonal antibody. Sections were incubated (1 h) with blocking buffer (0.2% bovine serum albumin [BSA]/PBS, 1.5% goat serum with avidin) in a humidified chamber and were stained for endoglin with anti-CD105 (1:100 with biotin) overnight at 4°C in a humidified chamber. For negative controls, antibodies were replaced with PBS. When the staining protocol was complete, sections were counterstained with hematoxylin QS, dehydrated, and a mixture of distyrene, plasticizer and xylene mounting medium (Sigma-Aldrich, St Louis, MO, USA) was used to mount cover slips. Slides were scanned with Aperio Image ScanScope System (Leica, Chicago, IL, USA) and analyzed by an individual who was blinded to the treatment groups. The microvessel analysis algorithm was used to quantify the MVD at a dark- and light-staining threshold of 185 and 210, respectively.
In another set of slides, apoptotic cells were visualized using the ApopTag in situ detection kit. The staining procedure followed the manufacturer’s instructions. Briefly, sections were fixed in ethanol–acetic acid solution (2:1) and treated with 3% hydrogen peroxide. Each section was incubated with equilibrating buffer and terminal deoxynucleotidyl transferase (TdT) enzyme was pipetted into the sections. The reaction was stopped using a stop/wash buffer. Negative control slides substituted TdT enzyme with PBS and anti-digoxigenin peroxidase was allowed to bind to labeled nucleotides. Finally, 3,3′-diaminobenzidine substrate was used for color development and slides were counterstained with hematoxylin QS. DPX mounting medium was used to mount cover slips. Slides were scanned with Aperio Image ScanScope System (Leica, Chicago, IL, USA) and analyzed by an individual blinded to the treatment groups. The nuclear image analysis algorithm was used to quantify apoptotic stained cells as the percentage of positively stained nuclei.
Tumor lysate preparation and immunoblot analysis
Protein extracts from tumors were prepared as previously described.18 Briefly, tumors were snap frozen, grounded into a fine powder, homogenized, and sonicated in 250 μL of lysis buffer (20 mM Tris–HCl, pH 8.0, 137 mM NaCl, 10% glycerol, 1% NP-40, 2 mM EDTA, 0.5% sodium deoxycholate, supplemented with protease inhibitors [10 mM NaF, 10 mM Na3VO4, 0.1 mM PMSF, 1 mM Na4P2O7] and mini complete EDTA free protease inhibitor cocktail) and centrifuged at 20,000× g for 8 min. After boiling in Laemmli buffer, lysates were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (40 μg protein/well) and then transferred to a PVDF membrane. Protein levels were analyzed with the indicated primary antibodies (ie, cleaved caspase 3, EGFR, pEGFR, Akt, pAkt, mTOR, pmTOR, 4EBP1, p4EBP1, and NF-κB), followed by horseradish peroxidase-conjugated secondary antibodies. Membranes were developed with SuperSignal substrate, which was visualized on radiographic film and images were scanned using a GS-710 densitometer (Bio-Rad). Lysates from individual mice were examined in triplicate. The density of each band was normalized to a β-tubulin loading control.
Subchronic toxicity of SMA-RL71
Female BALB/c mice (5–6 weeks old) were purchased from Hercus Taieri Resource Unit (Dunedin, NZ) and were allowed to acclimatize for 3 days prior to experimentation. Mice (five per treatment group) were injected once per week with SMA-RL71 (10 mg/kg, iv) for 90 days. Mice weight was recorded twice weekly for the duration of the study as an indicator of the general health. Twenty-four hours after the last dose, the mice were euthanized by CO2 inhalation and blood was collected for determination of alanine amino transferase and creatinine as we have described previously.31 Major organs were removed and the weight was expressed as a percentage of the animal’s body weight and this was compared to the control group.
Statistical analysis
Tumor growth experiments were analyzed using a two-way repeated-measures analysis of variance (ANOVA) coupled with a Bonferroni post-hoc test, where p<0.05 was required for statistical significance. Analyses that were independent of time were analyzed using a one-way ANOVA coupled with a Bonferroni post-hoc test, where p<0.05 was required for statistical significance. All data with unequal variances were log transformed and reanalyzed with the appropriate ANOVA.
Results
Biodistribution of SMA-RL71
We recently developed a polymeric micelle assembled from amphiphilic SMA copolymers in order to encapsulate the synthetic curcumin derivative, RL71.30 SMA-RL71 was characterized by a 15% loading as determined by UV spectrophotometry and expressed as a percentage of the weight of RL71 over SMA. SMA-RL71 was highly stable and had a diameter of 181.6 nm in buffer and 275.1 nm in serum as measured by dynamic light scattering and a near neutral charge of −0.0432 mV.30 We first examined the tissue distribution pattern of SMA-RL71 and compared it to free RL71. For these studies, we used a xenograft model of TNBC. Tumor-bearing mice were intravenously injected with an equivalent dose of 10 mg/kg of either free RL71 or SMA-RL71. After 6 h, the mice were euthanized and major organs were collected. The results showed that there was a significant 1.7-fold increase in the amount of RL71 detected in the tumor 6 h after treatment with SMA-RL71 compared to free drug (Figure 1A). The drug was also detected in the liver, kidney, and spleen of treated mice, with more RL71 reaching the liver and spleen following administration of SMA-RL71 compared to free drug. This was expected because nanoparticles in the 100–200 nm range will also accumulate in organs with larger fenestrations.32,33 Since the results showed that SMA-RL71 increased drug accumulation in the tumor after a single dose, another group of mice received SMA-RL71 (10 mg/kg, iv) twice a week for 2 weeks. In these mice, more drug accumulated in the tumor compared to a single dose (5.3±0.75 vs 59±10.3 μg/g, for single dose and repeat dose, respectively). Additionally, more RL71 accumulated in the tumor following SMA-RL71 compared to free drug. On day 17, drug concentration in the tumor was 16-fold higher in the SMA-RL71 treatment group compared to mice treated with RL71 (Figure 1B). SMA-RL71 treatment also resulted in more drug accumulating in the spleen, kidney, and liver compared to mice administered free RL71.
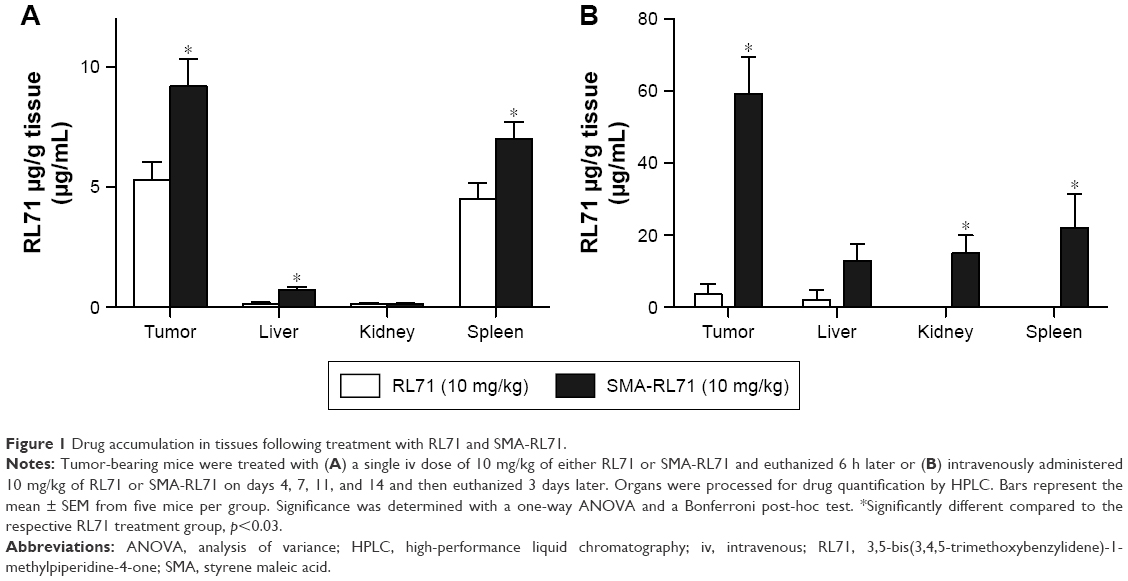 | Figure 1 Drug accumulation in tissues following treatment with RL71 and SMA-RL71. |
Efficacy of RL71 in a xenograft model of TNBC
Since we had shown that SMA-RL71 (15% loading) accumulated in the xenograft tumors following repeated administration, we investigated whether this formulation and drug administration regimen could elicit superior in vivo efficacy. For this study, female mice bearing MDA-MB-231 xenografts were administered RL71 (10 mg/kg, iv), SMA-RL71 (10 mg/kg, iv), and vehicle control (100 μL, iv), twice a week for 2 weeks. The results showed that RL71 as a free drug given iv was unable to suppress tumor growth. However, by day 17, SMA-RL71 significantly decreased tumor growth compared to both the free drug and vehicle control, and this effect continued for 10 days following the last iv dose of SMA-RL71 (Figure 2A). To determine if more frequent drug administration would produce a superior effect, another group of tumor-bearing mice were administered SMA-RL71 (10 mg/kg, iv) or SMA control (100 μL, iv) twice a week for 3 weeks. SMA-RL71 elicited a similar level of tumor suppression by day 17. However, the additional drug administration did not improve drug efficacy as tumors from mice treated with SMA-RL71 continued to grow after day 17 (Figure 2B).
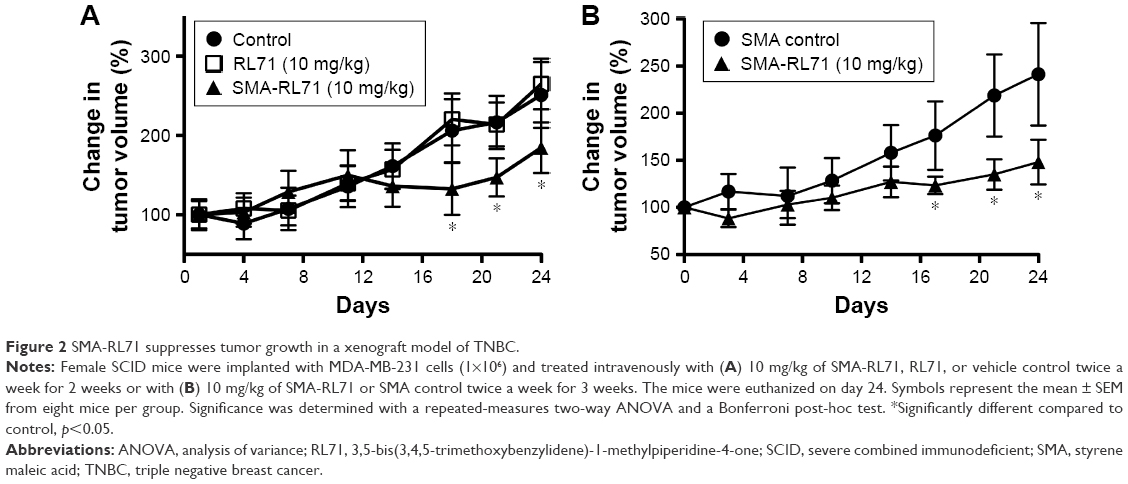 | Figure 2 SMA-RL71 suppresses tumor growth in a xenograft model of TNBC. |
Mechanism(s) of tumor suppression
Tumors from SMA control and SMA-RL71-treated mice were processed for immunohistochemistry and immunoblotting to determine mechanisms responsible for tumor suppression elicited by SMA-RL71. Analysis of tumors from mice treated with SMA-RL71 twice a week for 3 weeks showed that the number of cells undergoing apoptosis was increased 24% (Figure 3A and B) and cleaved caspase-3 in the tumor increased 51% (Figure 3C and D) compared to SMA control. Furthermore, analysis of MVD via the quantification of CD105 staining showed that SMA-RL71 treatment reduced microvessels in tumors from mice treated with SMA-RL71 by 26% compared to control (Figure 3E and F). Previous studies in various TNBC cell lines revealed that RL71 increased the number of cells undergoing apoptosis and decreased markers of angiogenesis as well as decreased phosphorylated protein levels of the EGFR, Akt, NF-κB, mTOR, and 4EBP1.18 Thus, the role of these proteins as mechanistic drivers of the tumor suppressive actions of SMA-RL71 was examined in tumors from treated mice. The results showed that, compared to vehicle-treated mice, pEGFR/EGFR was decreased 72% (Figure 4A and B), pAkt/Akt was decreased 57% (Figure 4A and C), pmTOR/mTOR was decreased 34% (Figure 4A and D), and p4EBP1/4EBP1 was decreased 65% (Figure 4A and E), while NF-κB was unchanged (Figure 4A and F).
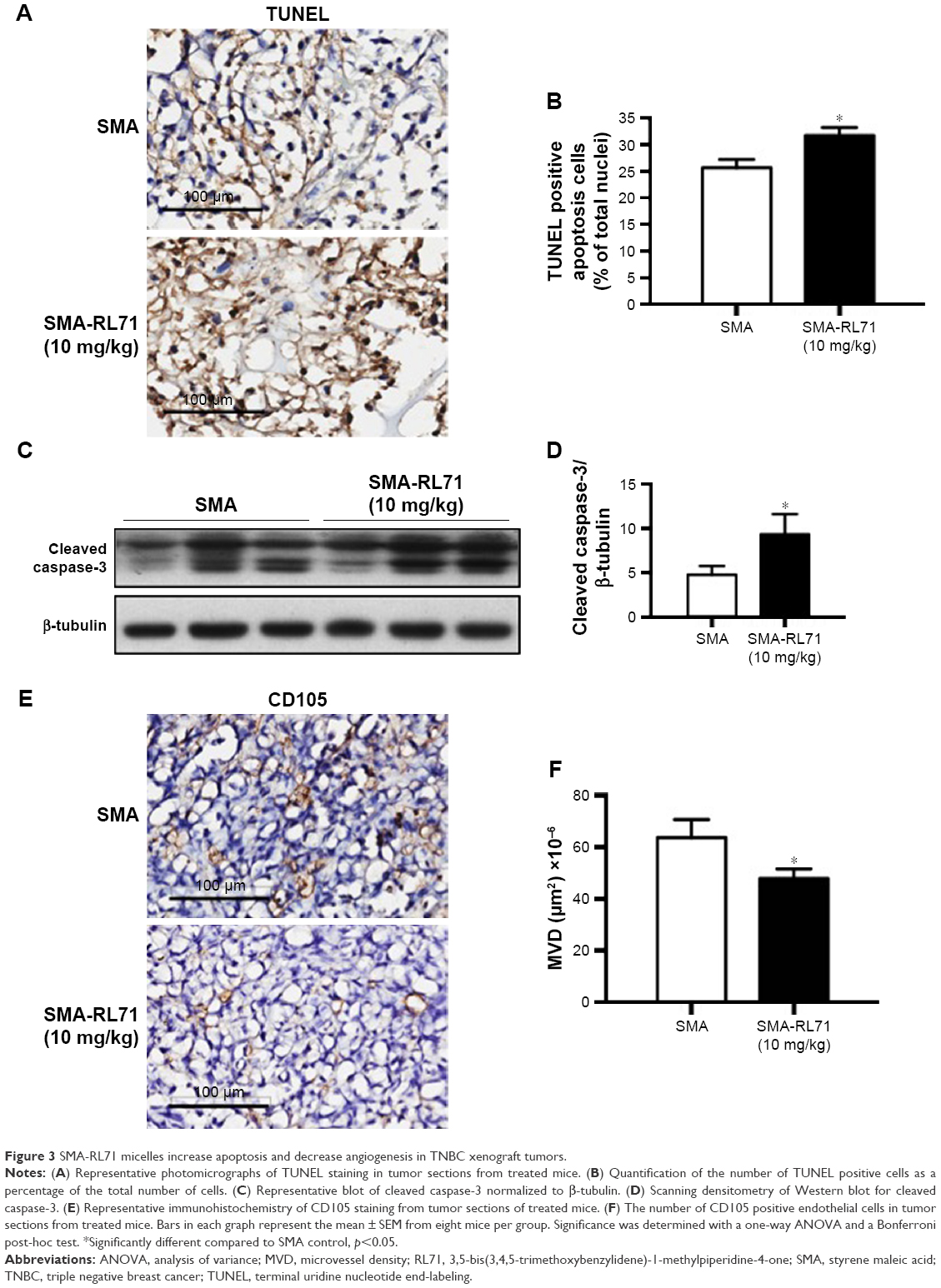 | Figure 3 SMA-RL71 micelles increase apoptosis and decrease angiogenesis in TNBC xenograft tumors. |
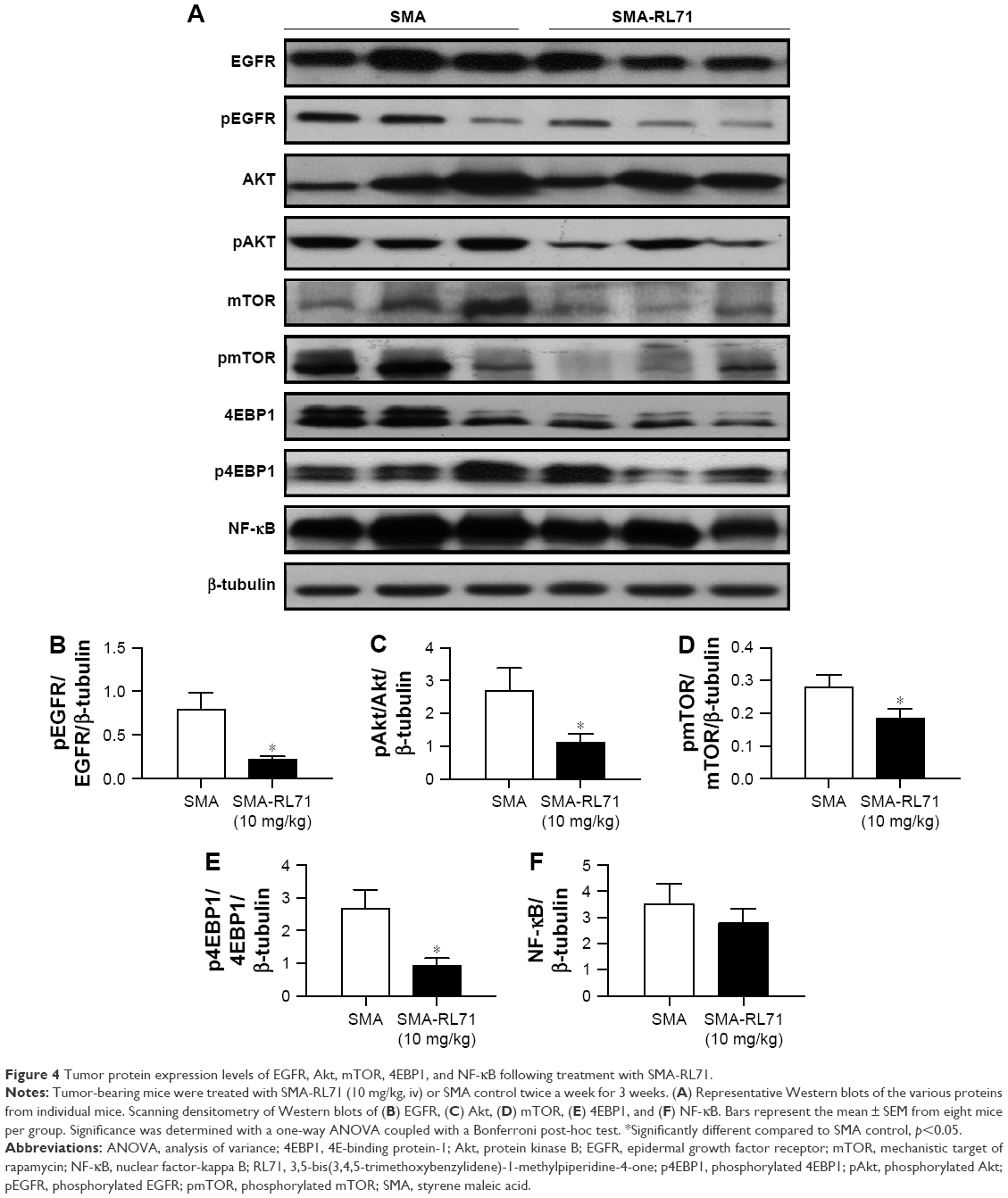 | Figure 4 Tumor protein expression levels of EGFR, Akt, mTOR, 4EBP1, and NF-κB following treatment with SMA-RL71. |
Subchronic toxicity of SMA-RL71
Since SMA-71 showed superior tumor targeting and elicited efficacy in a xenograft model, it was important to determine any potential drug induced toxicity. During all of the tumor suppression studies, all mice gained a similar amount of weight compared to control. Furthermore, drug treatment had no significant effect on liver or kidney toxicity as measured by ALT activity and creatinine in the plasma (Table S1). Furthermore, organ weight was not different between the treatment groups for all organs except the spleen, which was significantly elevated above control in SMA-RL71-treated mice (Table S1). To evaluate this further, a subchronic toxicity analysis of SMA-RL71 was performed using female Swiss Webster mice. The mice were treated with SMA-RL71 (10 mg/kg, once per week, 90 days, iv) and compared to a control group. During the treatment period, the mice gained a normal amount of body weight and plasma markers for ALT activity and creatinine were not different to control (Table 1). The weight of the spleen was significantly increased 1.4-fold compared to control, while histologically, the spleen appeared normal. The weight of other major organs (liver, kidney, and heart) was not different to control following subchronic administration of SMA-RL71 (Table 1).
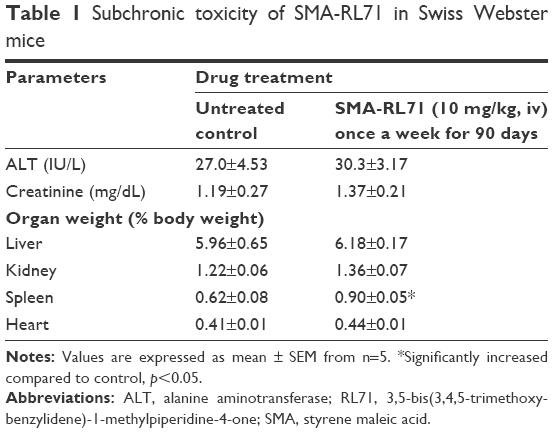 | Table 1 Subchronic toxicity of SMA-RL71 in Swiss Webster mice |
Discussion
The most common form of cancer diagnosed in women is breast cancer, and there are extremely limited drug treatment options for the 10%–17% of patients with TNBC.4,5 In the search for new drug candidates for TNBC, our lab has synthesized a variety of curcumin derivatives and of these RL71 was the lead drug candidate due to its potent actions in vitro.18 To improve the in vivo potential of this drug, we encapsulated it in SMA micelles. We chose to use SMA because polymeric micelles are the second most commonly examined drug delivery system34 and can deliver single as well as multiple chemotherapeutic agents in a targeted manner toward the tumor.34–36 Amphiphilic nanocarrier systems, such as SMA micelles, are able to encapsulate various hydrophobic drugs into the core of the micelles whereby the hydrophilic carboxyl groups of maleic acid form the outer shell and the hydrophobic styrene rings form the inner core. This results in a nanoformulation with high aqueous solubility. It also led to a high degree of RL71 loading (15%).30 This drug loading is an improvement compared to liposomes encapsulating the synthetic curcumin analog EF24, which achieved only 5% drug loading.37 The higher drug loading of SMA-RL71 coupled with its slow release rate, namely, 30% after 84 h,30 was expected to result in a more favorable biodistribution profile compared to the free drug. This was seen not only in the biodistribution results but also in the tumor suppression results, which occurred on day 17.
Our previous work demonstrated that SMA-RL71 micelles had the physiochemical properties required for passive tumor accumulation via the EPR effect. Specifically, their size and charge predict a prolonged circulatory half-life due to their ability to escape renal clearance38 and recognition by the reticuloendothelial system.39 Encapsulation also protected the active drug, RL71, from degradation and improved its cytotoxic properties. This was confirmed in TNBC cells and tumor spheroids in vitro, where SMA-RL71 had a superior ability to disrupt the integrity of TNBC tumor spheroids as shown by disruption of the spheroid as well as a 59% decrease in alkaline phosphatase in the spheroids.30 Thus, we hypothesized that the micelles would accumulate at the tumor site and increase the in vivo efficacy of RL71, a synthetic curcumin analog with high cytotoxic potency toward TNBC cell lines.
Encapsulating the curcumin analog RL71 into SMA micelles resulted in a drug formulation that enhanced all of the properties of RL71 in vivo. Specifically, SMA-RL71 increased drug accumulation in the tumor 16-fold compared to control, and significantly suppressed tumor growth after four intravenous doses. This was a vast improvement on the free drug, which was unable to alter tumor growth in a TNBC model. However, another group has reported suppression of tumor growth by free RL71. Specifically, RL71 (4 mg/kg/day) when administered intraperitoneally for 14 days significantly suppressed tumor growth in a xenograft model of colon cancer.40 However, the colon tumors still exhibited an aggressive growth rate of 400% over 15 days following treatment with RL71.40 Additionally, intravenous drug administration is a more clinically relevant route of drug administration compared to intraperitoneal injection. It is also worth noting that the mice treated with RL71 (4 mg/kg) had a nonsignificant loss of body weight after 15 days.40 Importantly, RL71 did not alter liver or spleen weight following intraperitoneal administration for 2 weeks.40 Thus, there is a consistent body of work showing that RL71 is nontoxic toward mice. However, our study shows that encapsulation of RL71 into SMA micelles is required for in vivo efficacy in a TNBC model.
Previous studies have shown a more favorable pharmacokinetic profile and increased efficacy with nanoformulations of curcumin.41–44 However, the lowest dose of curcumin that showed efficacy was 20 mg/kg.43,44 Since synthetic curcumin analogs, when used as free drugs, have already shown increased potency compared to curcumin, these analogs are good candidates for encapsulation. However, a liposomal formulation of the curcumin analog EF-24 failed to decrease tumor growth in a xenograft model of pancreatic cancer when administered at 10 mg/kg intravenously every other day for 3 weeks.37 Therefore, our results with SMA-RL71 are more promising than those reported with either nanoformulations of curcumin41–44 or other curcumin derivatives.37 Importantly, curcumin has a broad range of targets, and studies with synthetic analogs show that they act in a similar manner to curcumin. Specific actions include: arresting cells in the G2/M phase of the cell cycle, inhibition of the EGFR, Akt, mTOR as well as NF-κB.16–22 Additionally, the synthetic analogs are potent inducers of apoptosis and also inhibit angiogenesis in a variety of in vitro assays.16–22 This is important, as we have previously shown that the concomitant suppression of the EGFR, Akt, and mTOR are key mechanistic drivers that result in significant tumor suppression in in vivo models of TNBC.31 Since not all drugs can be encapsulated into SMA micelles, it is crucial that our SMA drug formulation contains a drug that can elicit these effects upon delivery to the tumor. Thus, RL71 is a promising drug for eliciting efficacy toward TNBCs. Our study showed this to be true because the micellar formulation, SMA-RL71, retained its mechanistic actions in vivo as it increased apoptosis; inhibited angiogenesis; and inhibited EGFR, Akt, mTOR, and its downstream effector 4EBP1 in the tumors of treated mice compared to tumors from control mice. Furthermore, we have shown that SMA-RL71 is not only an improvement on free RL71, but it is also more efficacious than other nanoformulations of curcumin41–44 and other curcumin analogs.37
The EPR effect is designed to exploit the larger gaps between the endothelial cells that comprise the tumor vasculature. These gaps can vary from only a few nanometers to as large as 400 nm.45,46 Thus, nanomedicines within this range can passively accumulate in tumor tissues but tumors are not the only organs with larger fenestrations. Noteworthy are the spleen and liver as they both exhibit similar characteristics. In humans, liver sinusoids can have fenestration of ~100 nm and the spleen has a sinusoid lumina of ~5 μm in order to provide extravasation of red blood cells.32,33 Therefore, these two organs receive a larger amount of nanosized drugs compared to other major organs. This was effectively shown in a meta-analysis of 73 studies investigating the EPR effect of various nanoconstructs (conjugates, micelles, and liposomes). The meta-analysis revealed that the liver and the spleen were the two primary organs along with tumors to accumulate the various drugs.47 In the spleen, this was observed to a greater extent following treatment with liposomes and conjugates compared to micelles.47 Specifically, with regard to drug content reaching the spleen, liposomes and conjugate formulations resulted in 20-fold more drug accumulation compared to free drug, while micelles had a maximum increase of 10-fold above control.47 Thus, it is not surprising that in subchronic drug administration of SMA-RL71, the spleen was the only organ with an increase in weight compared to untreated control.
While spleen function can be compensated for by other lymphatic organs, liver injury could be a possibility if significant concentrations of a cytotoxic nanomedicine accumulated in the liver, as has been reported with polymeric cisplatinum micelles.48 Importantly, this was not an issue in this study as the subchronic administration of SMA-RL71 also showed that the nanoformulation was nontoxic to the mice as plasma indicators of liver and kidney function were in the normal range and were not different to untreated control. This was not surprising, since RL71 as a free drug was not toxic to the liver37 or any other organ. Additionally, we have previously shown that30 in serum the size of SMA-RL71 increases to 275.1 nm and thus less of the drug is expected to accumulate in the liver. Thus, the SMA formulation is a safe and effective way to elevate the concentration of RL71 in the tumor and improve drug efficacy. However, improvements to drug efficacy are still required due to the fact that the xenograft tumors continued to grow following drug administration of two times a week for 3 weeks. One possibility would be to co-encapsulate RL71 with another chemotherapeutic agent. This approached has shown promise in models of breast and prostate cancer. For example, highly potent tumor suppression was reported when doxorubicin (10 mg) and curcumin (100 mg) were combined in transferrin-targeted nanoparticles,49 as well as following the co-encapsulation of docetaxel (5 mg/kg) and curcumin (10 mg/kg) in lipid–polymer hybrid nanoparticles.50 Alternatively, the micelles could undergo further modifications to their exterior including the addition of folate51 or other active targeting moieties. Therefore, there is potential to improve on the efficacy elicited by SMA-RL71. However, RL71 is a good drug candidate for patients with TNBC due to its ability to inhibit three pathways critical for tumor growth, namely, EGFR, Akt, and mTOR. Thus, further studies with SMA-RL71 may produce a nanomicellar formulation that provides a safe and efficacious single or combination treatment for patients with TNBC.
Conclusion
The SMA micellar encapsulation provided an effective mechanism for delivering a novel hydrophobic drug (ie, RL71) to TNBC tumors without producing liver or kidney toxicity. In a murine xenograft model, intravenous administration of SMA-RL71 micelles elicited significantly higher drug accumulation in the tumor and greater antitumor efficacy compared to free RL71. Furthermore, SMA-RL71 micelles induced apoptosis, reduced angiogenesis, and inhibited EGFR function along with various other downstream proteins involved in cell proliferation within the tumor. The nanomicelles also had an excellent safety profile following 90-day subchronic toxicity analysis. Thus, this novel drug formulation has the potential for further development for use in the treatment of TNBC.
Acknowledgments
This work was supported by grants from The New Zealand Breast Cancer Foundation and the Maurice and Phyllis Paykel Trust. The authors would like to thank B.C. Hawkins from the Department of Chemistry for synthesizing RL71, R.E. Alumbaugh from the Department of Pharmacology and Toxicology for technical assistance, and A.G. Cridge from the Department of Biochemistry for editorial assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. | ||
Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100(14):8418–8423. | ||
Sotiriou C, Neo SY, Mcshane LM, et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A. 2003;100(18):10393–10398. | ||
Rakha EA, El-Sayed ME, Green AR, Lee AH, Robertson JF, Ellis IO. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109(1):25–32. | ||
Carey LA, Dees EC, Sawyer L, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13(8):2329–2334. | ||
Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52(1):108–118. | ||
Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: therapeutic options. Lancet Oncol. 2007;8(3):235–244. | ||
Lara-Medina F, Perez-Sanchez V, Saavedra-Perez D, et al. Triple-negative breast cancer in Hispanic patients: high prevalence, poor prognosis, and association with menopausal status, body mass index, and parity. Cancer. 2011;117(16):3658–3669. | ||
Vona-Davis L, Rose DP, Hazard H, et al. Triple-negative breast cancer and obesity in a rural Appalachian population. Cancer Epidemiol Biomarkers Prev. 2008;17(12):3319–3324. | ||
Kwan ML, Kushi LH, Weltzien E, et al. Epidemiology of breast cancer subtypes in two prospective cohort studies of breast cancer survivors. Breast Cancer Res. 2009;11(3):R31. | ||
Nielsen TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10(16):5367–5374. | ||
Metzger-Filho O, Tutt A, De Azambuja E, et al. Dissecting the heterogeneity of triple-negative breast cancer. J Clin Oncol. 2012;30(15):1879–1887. | ||
Chainani-Wu N. Safety and anti-inflammatory activity of curcumin: a component of tumeric (Curcuma longa). J Altern Complement Med. 2003;9(1):161–168. | ||
Cheng AL, Hsu CH, Lin JK, et al. Phase I clinical trial of curcumin, a chemo-preventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001;21(4B):2895–2900. | ||
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4(6):807–818. | ||
Yadav B, Taurin S, Rosengren RJ, et al. Synthesis and cytotoxic potential of heterocyclic cyclohexanone analogues of curcumin. Bioorg Med Chem. 2010;18(18):6701–6707. | ||
Yadav B, Taurin S, Larsen L, Rosengren RJ. RL66 a second generation curcumin analog has potent in vivo and in vitro anticancer activity in ER negative breast cancer models. Int J Oncol. 2012;41(5):1723–1732. | ||
Yadav B, Taurin S, Larsen L, Rosengren RJ. RL71, a second generation curcumin analog, induces apoptosis and down regulates AKT in ER-negative breast cancer cells. Int J Oncol. 2012;41(3):1119–1127. | ||
Hutzen B, Friedman L, Sobo M, et al. Curcumin analogue GO-Y030 inhibits STAT3 activity and cell growth in breast and pancreatic carcinomas. Int J Oncol. 2009;35(4):867–872. | ||
Lin L, Hutzen B, Ball S, et al. New curcumin analogues exhibit enhanced growth-suppressive activity and inhibit AKT and signal transducer and activator of transcription 3 phos-phorylation in breast and prostate cancer cells. Cancer Sci. 2009;100(9):1719–1727. | ||
Kasinski AL, Du Y, Thomas SL, et al. Inhibition of IkappaB kinase-nuclear factor-kappaB signaling pathway by 3,5-bis(2-urobenzylidene)piperidin-4-one (EF24), a novel monoketone analog of curcumin. Mol Pharmacol. 2008;74(3):654–661. | ||
Somers-Edgar TJ, Taurin S, Larsen L, Chandramouli A, Nelson MA, Rosengren RJ. Mechanisms for the activity of heterocyclic cyclohexanone curcumin derivatives in estrogen receptor negative human breast cancer cell lines. Invest New Drugs. 2011;29(1):87–97. | ||
Song Z, Feng R, Sun M, et al. Curcumin-loaded PLGA-PEG-PLGA triblock copolymeric micelles: preparation, pharmacokinetics and distribution in vivo. J Colloid Interface Sci. 2011;354(1):116–123. | ||
Rahman S, Cao S, Steadman KJ, Wei M, Parekh HS. Native and β-cyclodextrin-enclosed curcumin: entrapment within liposomes and their in vitro cytotoxicity in lung and colon cancer. Drug Deliv. 2012;19(7):346–353. | ||
Yang R, Zhang S, Kong D, Gao X, Zhao Y, Wang Z. Biodegradable polymer-curcumin conjugate micelles enhance the loading and delivery of low-potency curcumin. Pharm Res. 2012;29(12):3512–3525. | ||
Maeda H, Matsumura Y. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit Rev Ther Drug Carrier Syst. 1989;6(3):193–210. | ||
Greish K, Fang J, Inutsuka T, Nagamitsu A, Maeda H. Macromolecular therapeutics: advantages and prospects with special emphasis on solid tumour targeting. Clin Pharmacokinet. 2003;42(13):1089–1105. | ||
Maeda H, Takeshita J, Kanamaru R. A lipophilic derivative of neocarzinostatin. A polymer conjugation of an antitumor protein antibiotic. Int J Pept Protein Res. 1979;14(2):81–87. | ||
Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. | ||
Taurin S, Nehoff H, Diong J, Larsen L, Rosengren RJ, Greish K. Curcumin-derivative nanomicelles for the treatment of triple negative breast cancer. J Drug Target. 2013;21(7):675–683. | ||
Scandlyn MJ, Stuart EC, Somers-Edgar TJ, Menzies AR, Rosengren RJ. A new role for tamoxifen in estrogen receptor negative breast cancer when it is combined with epigallocatechin gallate. Br J Cancer. 2008;99(7):1056–1063. | ||
Horn T, Henriksen JH, Christoffersen P. The sinusoidal lining cells in “normal” human liver. A scanning electron microscopic investigation. Liver. 1986;6(2):98–110. | ||
Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2:14. | ||
Ramasamy T, Ruttala HB, Gupta B, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Control Release. 2017;258:226–253. | ||
Wang L, Zhang DZ, Wang YX. Bioflavonoid fisetin loaded alpha-tocopherol-poly(lactic acid)-based polymeric micelles for enhanced anticancer efficacy in breast cancers. Pharm Res. 2017;34(2):453–461. | ||
Ruttala HB, Ramasamy T, Poudal BK, et al. Molecularly targeted co-delivery of a histone deacetylase inhibitor and paclitaxel by lipid-protein hybrid nanoparticles for synergistic combinational chemotherapy. Oncotarget. 2017;8(9):14925–14940. | ||
Bisht S, Schlesinger M, Rupp A, et al. A liposomal formulation of the synthethetic curcumin analog EF24 (Lipo-EF24) inhibits pancreatic cancer progression: towards future combination therapies. J Nanobiotechnol. 2016;14(1):57. | ||
Choi HS, Liu W, Liu F, et al. Design considerations for tumour-targeted nanoparticles. Nat Nanotechnol. 2010;5(1):42–47. | ||
Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. | ||
Yang B, Zhang M, Gao J, et al. Small molecule RL71 targets SERCA2 at a novel site in the treatment of human colorectal cancer. Oncotarget. 2015;6(35):37613–37625. | ||
Allam AN, Komeil IA, Fouda MA, Abdallah OY. Preparation, characterization and in vivo evaluation of curcumin self-nano phospholipid dispersion as an approach to enhance oral bioavailability. Inter J Pharm. 2015;489(1–2):117–123. | ||
Wang J, Ma W, Tu P. The mechanism of self-assembled mixed micelles in improving curcumin oral absorption: in vitro and in vivo. Colloids Surface B Biointerfaces. 2015;133:108–119. | ||
Szymusiak M, Hu X, Leon Plata PA, Ciupinski P, Wang ZJ, Liu Y. Bioavailability of curcumin and cucumin glucuronide in the central nervous system of mice after oral delivery of nano-curumin. Int J Pharm. 2016;511(1):415–423. | ||
Ranjan AP, Mukerjee A, Gdowski MA, et al. Curcumin-ER prolonged subcutaneous delivery for the treatment of non-small cell lung cancer. J Biomed Nanotechnol. 2016;12(4):679–688. | ||
Yuan A, Yang PC, Yu CJ, et al. Tumor angiogenesis correlates with histologic type and metastasis in non-small-cell lung cancer. Am J Respir Crit Care Med. 1995;152(6 Pt 1):2157–2162. | ||
Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–4612. | ||
Taurin S, Nehoff H, Greish K. Anticancer nanomedicine and tumor vascular permeability; Where is the missing link? J Control Release. 2012;164(3):265–275. | ||
Uchino H, Matsumura Y, Negishi T, et al. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer. 2005;93(6):678–687. | ||
Cui T, Zhang S, Sun H. Co-delivery of doxorubicin and pH-sensitive curcumin prodrug by transferrin-targeted nanoparticles for breast cancer treatment. Oncol Report. 2017;37(2):1253–1260. | ||
Yan J, Wang Y, Zhnag X, Liu S, Tian C, Wang H. Targeted nanomedicine for prostate cancer therapy: docetaxel and curcumin co-encapsulated lipid-polymer hybrid nanoparticles for the enhanced anti-tumor activity in vitro and in vivo. Drug Deliv. 2016;23(5):1757–1762. | ||
Phan QT, Le MH, Le TT, Tran TH, Xuan PN, Ha PT. Characteristics and cytotoxicity of folate-modified curcumin-loaded PLA-PEG micellar nano systems with various PLA:PEG ratios. Inter J Pharm. 2016;507:32–40. |
Supplementary material
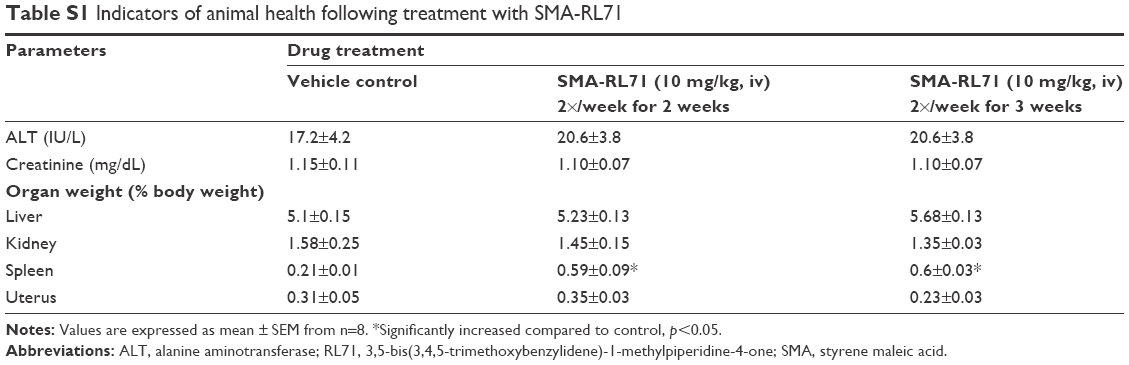 | Table S1 Indicators of animal health following treatment with SMA-RL71 |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.