")
Back to Journals » International Journal of Nanomedicine » Volume 11
Styrene maleic acid-encapsulated paclitaxel micelles: antitumor activity and toxicity studies following oral administration in a murine orthotopic colon cancer model
Authors Parayath NN , Nehoff H, Norton SE, Highton AJ, Taurin S, Kemp RA, Greish K
Received 10 April 2016
Accepted for publication 16 June 2016
Published 17 August 2016 Volume 2016:11 Pages 3979—3991
DOI https://doi.org/10.2147/IJN.S110251
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Neha N Parayath,1 Hayley Nehoff,1 Samuel E Norton,2 Andrew J Highton,2 Sebastien Taurin,1,3 Roslyn A Kemp,2 Khaled Greish1,4
1Department of Pharmacology and Toxicology, 2Department of Microbiology and Immunology, University of Otago, Dunedin, New Zealand; 3Department of Obstetrics and Gynecology, University of Utah, Salt Lake City, UT, USA; 4Princess Al-Jawhara Centre for Molecular Medicine, Arabian Gulf University, Manama, Kingdom of Bahrain
Abstract: Oral administration of paclitaxel (PTX), a broad spectrum anticancer agent, is challenged by its low uptake due to its poor bioavailability, efflux through P-glycoprotein, and gastrointestinal toxicity. We synthesized PTX nanomicelles using poly(styrene-co-maleic acid) (SMA). Oral administration of SMA-PTX micelles doubled the maximum tolerated dose (60 mg/kg vs 30 mg/kg) compared to the commercially available PTX formulation (PTX [Ebewe]). In a murine orthotopic colon cancer model, oral administration of SMA-PTX micelles at doses 30 mg/kg and 60 mg/kg reduced tumor weight by 54% and 69%, respectively, as compared to the control group, while no significant reduction in tumor weight was observed with 30 mg/kg of PTX (Ebewe). In addition, toxicity of PTX was largely reduced by its encapsulation into SMA. Furthermore, examination of the tumors demonstrated a decrease in the number of blood vessels. Thus, oral delivery of SMA-PTX micelles may provide a safe and effective strategy for the treatment of colon cancer.
Keywords: oral delivery, anticancer nanomedicine, CT-26, enhanced permeability and retention (EPR) effect, HUVEC, antiangiogenic
Introduction
Oral administration of chemotherapeutics is a largely unmet clinical need that could improve the quality of life for cancer patients.1 So far, there is no single anticancer nanoformulation that is approved for oral use. In contrast to the intravenous administration, oral delivery can be tailored to maintain prolonged plasma drug concentrations, above the therapeutic level, in cancer cells. This prolonged exposure is especially beneficial for cell-cycle-specific chemotherapeutic agents such as paclitaxel (PTX).
PTX, a microtubule stabilizing agent, is currently approved by the US Food and Drug Administration for its broad spectrum efficacy in advanced and refractory non-small cell lung, ovarian, and breast cancers and Kaposi’s sarcoma.2 PTX as a single agent administered through intravenous infusion failed to show effective antitumor efficacy in a Phase II clinical trial for patients with colorectal adenocarcinoma.3 Many colorectal cancers are characterized by chromosomal instability and APC mutations. However, PTX (microtubule stabilizing agent) might be an effective anticancer agent in the subset of colorectal cancers lacking chromosomal instability and APC mutations.4 Furthermore, studies have shown that PTX inhibits cell migration and VEGF-induced neovascularization and has an antiproliferative effect toward activated endothelial cells.4
The antiangiogenic activity of PTX2 was demonstrated at low concentrations and thus it is considered a suitable candidate for metronomic chemotherapy.
Colon tumors exhibit increased permeability of the intestinal epithelium due to tight junction modifications.5,6 Thus, oral administration would facilitate as an effective strategy for the chemotherapeutic agent to reach the tumor site. However, oral administration of PTX has several limitations. The hydrophobicity of PTX affects the bioavailability of the drug.7,8 Additionally, PTX is a substrate of P-glycoprotein and cytochrome P450 monooxygenase 3A4,9 which promotes its efflux and metabolism, respectively, and limits its absorption into the systemic circulation. Furthermore, oral administration of PTX is associated with side effects such as nausea, vomiting, and diarrhea.8
The nanocarrier-based approach for oral drug delivery will increase solubility of highly hydrophobic drugs, limit toxicity to the gastrointestinal (GI) tract, reduce metabolism,10 and overall improve the bioavailability.11 Furthermore, the nanosized carriers show an enhanced permeability and retention (EPR) effect12,13 by taking advantage of the wide fenestrations of the tumor vasculature to leak out into the surrounding tumor tissue, and the impaired lymphatic drainage14,15 leads to retention in the tumor tissue.
We had previously shown that poly(styrene-co-maleic acid) (SMA) micelles can traverse the intestinal epithelium and accumulate at the tumor site following oral administration.16 In this study, we evaluated the anticancer activity of PTX encapsulated into SMA nanomicelles in a murine orthotopic colon cancer model. We show that oral administration of SMA-PTX micelles showed a significant reduction in tumor weight without any toxicity and thus could provide an effective strategy for treatment of colon tumors.
Materials and methods
Cumene-terminated poly(styrene-co-maleic anhydride) (Mn ~1,600) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDAC) were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). The Infinity Alanine Aminotransferase (ALT) Colorimetric Assay kit and Roswell Park Memorial Institute (RPMI) 1640 medium were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Diaminobenzidine (DAB) substrate kit and antibody to CD-105 was purchased from antibodies-online Inc. (Atlanta, GA, USA). Goat antirabbit secondary antibody was obtained from Calbiochem (San Diego, CA, USA). PTX was obtained from LC Laboratories (Woburn, MA, USA). Matrigel was obtained from BD Biosciences (San Jose, CA, USA). Creatinine assay kit was obtained from Cayman Chemical (Ann Arbor, MI, USA).
Synthesis of SMA micelles
SMA micelles were prepared as described previously.17 Briefly, SMA was hydrolyzed in 1 M NaOH at 70°C. Once soluble, the pH was adjusted to 7.0. SMA solution (8 mL of 10 mg/mL) was adjusted to pH 5, and the volume was made up to 50 mL using deionized water. Next, 20 mg of PTX solubilized in 1 mL of dimethyl sulfoxide (DMSO) and EDAC solubilized in distilled water (80 mg/mL) were added to the SMA solution under constant stirring and left for 20 minutes at pH 5. The solution was then adjusted to pH 11 with 0.1 N NaOH and stirred for 30 minutes to allow the formation of the SMA micelles and encapsulation of PTX. The pH was readjusted to 7.4, and the solution was ultrafiltered four times using a Labscale™ ultrafiltration system with a Pellicon® XL filter 10 kDa (EMD Millipore, Billerica, MA, USA). The concentrated micelle solution was lyophilized to obtain the SMA-PTX micelles powder.
Loading of the SMA micelles
SMA-PTX micelles were solubilized at 1 mg/mL in methanol, and the loading was measured by high-performance liquid chromatography (HPLC) using a BM-20Alite Prominence HPLC system (Shimadzu Corporation, Kyoto, Japan) on a reversed-phase Gemini 3 μm C18, 110A, 150×2 mm column (Phenomenex Gemini-NX) with a 2×4 mm C18 guard column. The mobile phase was acetonitrile and 0.03 M phosphate buffer (pH 3; 1:1 ratio). The flow rate was 1 mL/min with an injection volume of 10 μL, and the column temperature was set at 30°C. PTX peaks were detected at 226 nm with the retention time of 8.5 minutes.
Size, PDI, and zeta potential determination of SMA micelles
Lyophilized SMA-PTX micelles was solubilized in either NaHCO3 (0.1 M, pH 7.4) to determine the size and polydispersity index (PDI) or in distilled water or 10 mM KCl to estimate the charge. All measurements were done using the Malvern ZEN3600 Zetasizer Nano series (Malvern Instruments, Malvern, UK). The results were obtained from three independent experiments.
Release rate of PTX from SMA micelles at physiological pH and in simulated gastric fluid
Using a dialysis bag with a 12 kDa molecular weight cutoff, 1.5 mL of SMA-PTX micelles solution in distilled water (1 mg/mL) was dialyzed against 15 mL of distilled water (pH 7.4 or 6.8) or simulated gastric fluid (SGF; with pepsin, pH 1.6). At specified time points, 1 mL of the solution outside the dialysis bag was used to measure the PTX released by HPLC (as described earlier). The percentage of release was determined by the ratio of the amount of drug in the solution outside the bag at defined time points and that within the bag at t=0 minutes as follows:
|
|
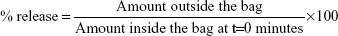
In vitro cytotoxicity of SMA micelles
CT-26.luc cells (gift from Professor Leonard W Seymour) were maintained in RPMI medium supplemented with 10% of fetal bovine serum, 2 mM of L-glutamine, 400 μg/mL of geneticin, 12.5 μg/mL of puromycin, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 2.2 g/L of NaHCO3. The cells were harvested using TrypLE Express (Thermo Fisher Scientific). To determine the cytotoxicity, CT-26.luc cells were seeded into 96-well plates (8,000 cells/well). After a 24-hour incubation, the cells were treated for 72 hours with PTX or SMA-PTX micelles at the equivalent concentrations indicated. SMA and DMSO were used as control. Following the incubation, the cells were fixed using 10% trichloroacetic acid solution. Cytotoxicity was determined using the sulforhodamine B assay.18 The concentration required to decrease the cell number by 50% (IC50) was determined by nonlinear regression using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA).
Endothelial tube formation assay
Matrigel (reduced growth factors) reduced (40 μL) was added to each well of a 96-well plate and incubated for 45 minutes at 37°C to allow solid gelation. Human umbilical vein endothelial cells (HUVECs) (2×104 cells/well) were seeded on the top of Matrigel matrix layer. To establish a coculture, HUVECs (1.2×104 cells/well) and CT-26.luc cells (1.6×104 cells/well) were seeded on Matrigel. PTX or SMA-PTX micelles (0.1 nM or 1 nM, respectively) was added into the wells and incubated for 20 hours at 37°C in 5% CO2 atmosphere. Representative images were taken from HUVECs alone and coculture with CT-26.luc cells following treatment with PTX or SMA-PTX micelles.
Animal housing and care
BALB/c mice were purchased from the Hercus Taieri Resource Unit (Dunedin, New Zealand). The mice were housed in pathogen-free conditions at 21°C–24°C on a scheduled 12-hour light/dark cycle, with access to sterile food (Reliance Rodent Diet, Dunedin, New Zealand) and water ad libitum. All the animal procedures were approved and followed as per the guidelines of the animal ethics committee of the University of Otago.
Determination of maximum tolerated dose
Female BALB/c mice (4–8 weeks) were dosed by oral gavage, with either SMA-PTX micelles (in 0.1% NaHCO3) or commercial PTX formulation (PTX [Ebewe]) or PTX solubilized in 10% DMSO (n=6 per group). A total of 25 mg/kg equivalent dose of PTX was orally administered, and body weight was measured for 4 days. The dose was escalated by 25 mg/kg, until either a maximum of 150 mg/kg or body weight loss of >10% was observed. Repeated maximum tolerated dose (MTD) was determined using healthy female BALB/c mice (4–8 weeks; n=4 per group). The mice were orally administered either SMA-PTX micelles (in 0.1% NaHCO3) or PTX (Ebewe) or PTX solubilized in DMSO. A dose of 30 mg/kg equivalent of PTX was administered every alternate day for 2 weeks. The dose was escalated by 30 mg/kg until the animals showed a body weight loss of >10%, which was deemed the repeated MTD. The animal body weight was measured daily.
Anticancer activity of SMA-PTX micelles following oral administration in CT-26.luc murine orthotropic colon cancer model
Surgery for intracecal injection was carried out according to Tseng et al.19 BALB/c mice were anesthetized to expose the abdominal cavity through an incision in the abdominal region. The cecum was lifted from beneath the small intestine using forceps. CT-26.luc cells (2×105 cells suspended in 50 μL of the RPMI medium) were injected subserosally in the cecal wall, and the abdominal incision was sutured. The mice were subcutaneously injected with carprofen (5 mg/kg) and amphoprim (30 mg/kg) twice daily for 3 days post-surgery. Eight days after the surgery, the 28 mice were randomly divided into control, SMA-PTX micelles (30 mg/kg and 60 mg/kg, po), and PTX (30 mg/kg, po) groups (seven mice per group). The doses were repeated every alternate day for 21 days post-surgery. The animal weight was assessed every day.
Toxicity analysis: body and organ weight, plasma ALT activity, and plasma creatinine levels
Organs such as liver, spleen, kidney, heart, and lungs were weighed at the end of the experiment (day 24) and weight was expressed as the ratio of organ weight to body weight of the mice and compared to the control group. The ALT activity in mice plasma samples was quantified using the ALT colorimetric assay kit (Thermo Fisher Scientific). Plasma samples and ALT reagent (37°C) were added to a 96-well plate in 1:10 ratio, respectively. Absorbance was measured at 340 nm using a microplate spectrophotometer at 37°C every 30 seconds for 1 minute. The ALT activity was expressed as international units per liter (IU/L) using the following equation:
|
|
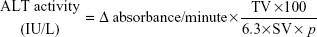
where Δ absorbance/minute is equal to (absorbance at t=0 minutes − absorbance at t=3 minutes)/3, TV is the total volume/well, 6.3 is the mM coefficient of NADH, SV is the sample volume, and p is the path length (0.3 cm).
Plasma creatinine levels were measured using a creatinine assay kit. Either plasma samples or standards were added to the 96-well plate. Creatinine reaction buffer and creatinine color reagent were added to these wells. The absorbance values were measured at 490 nm at t=1 minute and t=7 minutes. The creatinine concentration was determined by calculating optical density (OD) using the following equation:
|
|
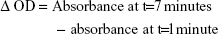
Δ OD of standard was subtracted from all readings to obtain adjusted Δ OD.
The standard adjusted Δ OD was then plotted to fit a linear regression. The change in absorbance was calculated and compared to a standard curve to determine the amount of creatinine present in the sample.
|
|

Immunohistochemistry of tumor sections
Tumors were embedded in the optimal cutting temperature compound, sectioned (10 μm), and air-dried overnight. The sections were then washed in phosphate-buffered saline, fixed in acetone, and incubated using hydrogen peroxide (3%) to decrease endogenous peroxidase activity. The tissue sections were then blocked in the bovine serum albumin solution supplemented with serum (1.5%) and avidin for 1 hour at room temperature (RT) and then incubated overnight with CD105 antibody with biotin at 4°C. The slides were then rinsed with phosphate-buffered saline and incubated with goat antirabbit secondary antibody for 30 minutes at RT in a humidified chamber, followed by incubation with streptavidin for 30 minutes at RT before development with 3,3′-diaminobenzidine tetrahydrochloride, followed by counterstaining with Hematoxylin QS. Once slides were dehydrated, DPX mounting medium and coverslips were applied. The images were obtained using Aperio ScanScope CS digital pathology system (Aperio, Vista, CA, USA). The tumor sections from each mouse were analyzed.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism™. Data were assessed using either one-way or two-way ANOVA with a Bonferroni post hoc test. The significance was set at P<0.05.
Results
Synthesis and characterization of SMA-PTX micelles
The SMA micelles synthesized had a recovery of 97%. The loading was 18.2% and is expressed as weight percentage of PTX in the final micelles compared to the total weight of recovered SMA micelles. The mean diameter of the SMA-PTX micelle was 195±12.3 nm as determined by dynamic light scattering. The PDI of SMA-PTX micelles was 0.34±0.02 and zeta potential was −0.06 mV in deionized water and 0.02 mV in 10 mM KCl (Table 1).
 | Table 1 Recovery, loading, size, PDI, and zeta potential of SMA-PTX micelles |
SMA-PTX micellar stability
The release rate of PTX from the SMA micelle was evaluated at physiological pH 7.4, intestinal pH 6.8, and in SGF (pH 1.6). Approximately 10% of PTX was released during the first 4 hours, at physiological pH, intestinal pH, and in SGF (Figure 1). After 12 hours, the release at physiological pH, intestinal pH, and in SGF increased to 24%, 19%, and 27%, respectively. From 12 hours to 24 hours, the release at physiological pH and intestinal pH was constant, increasing to 32% and 25%, respectively, at 36 hours. However, the PTX released in SGF was 50% and 73% after 24 hours and 36 hours, respectively. The time required by the SMA micelles to transit through the stomach (pH 1.6) to the small intestine (pH 6.8) was ~2 hours. The release rate of PTX from SMA-PTX micelles in SGF (pH 1.6) for the initial 2 hours was less than 10% (Figure 1); thus, based on the stability in gastric pH, SMA-PTX micelles were suitable for oral delivery.
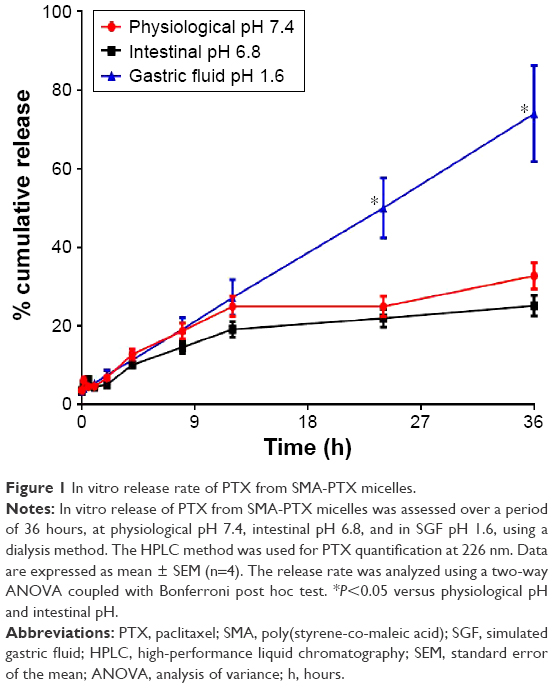 | Figure 1 In vitro release rate of PTX from SMA-PTX micelles. |
Cytotoxicity of SMA-PTX micelles
The cytotoxicity of SMA-PTX micelles was compared to commercially available PTX (Ebewe) and free PTX solubilized in 0.2% DMSO using CT-26.luc mice colon cancer cells (Figure 2A). The IC50 values for SMA-PTX micelles, free PTX, and PTX (Ebewe) were 104.8 nM, 84.7 nM, and 101.5 nM respectively (Figure 2B). Thus, the commercially available PTX and SMA-PTX micelles showed approximately similar cytotoxicity toward CT-26.luc cells.
 | Figure 2 Cytotoxicity of SMA-PTX micelles against CT-26.luc cells. |
Effect of SMA-PTX micelles on endothelial tube formation
In vitro models of angiogenesis, using HUVECs and coculture with CT-26.luc cells, were employed to ascertain if the SMA-PTX micelles showed potential efficacy. As seen in Figure 3A, PTX and SMA-PTX micelles (0.1 nM and 1 nM) completely inhibited endothelial tube formation by HUVECs. Since advanced colorectal tumors are often associated with tumor cells exhibiting vascular mimicry,20 the effect of SMA-PTX micelles was evaluated on coculture of HUVECs and CT-26.luc cells. PTX (1 nM) moderately inhibited, while 1 nM of SMA-PTX micelles completely abolished vascular mimicry (Figure 3B). SMA alone had no effect on endothelial tube formation.
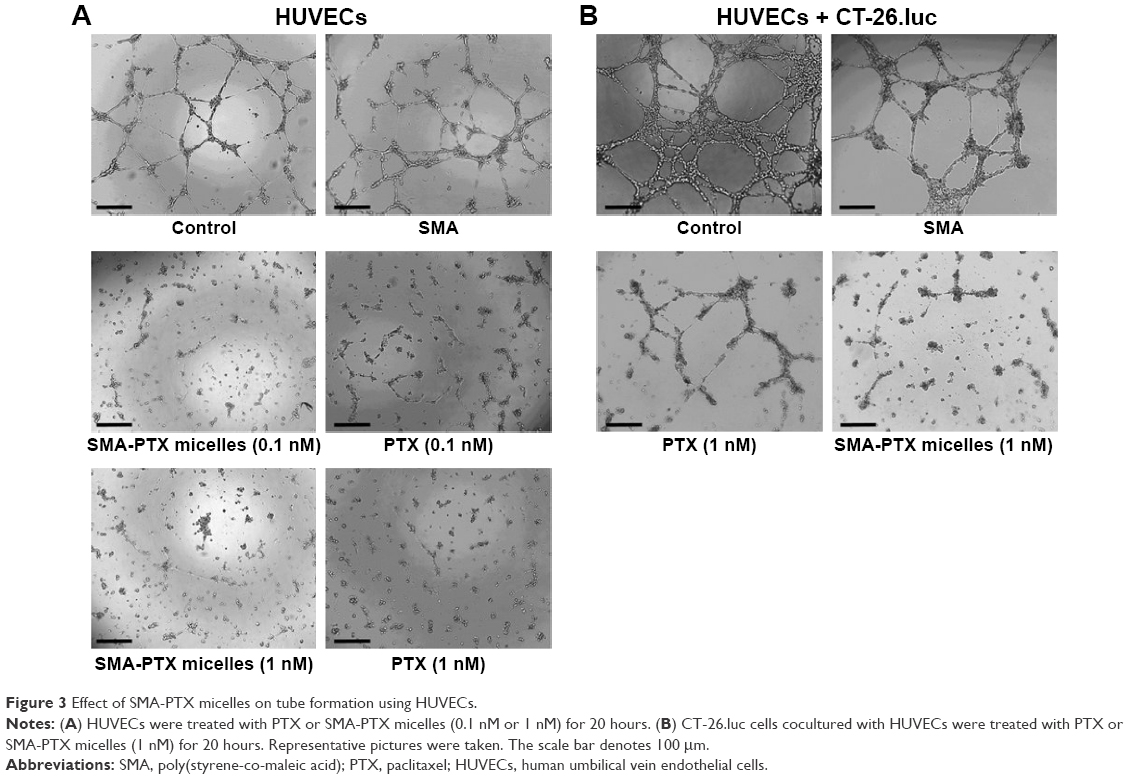 | Figure 3 Effect of SMA-PTX micelles on tube formation using HUVECs. |
MTD of SMA-PTX micelles following oral administration
Furthermore, to evaluate the safe doses of SMA-PTX micelles for oral administration, an MTD study was performed. In the single-dose study, oral administration of SMA-PTX micelles 120 mg/kg and free PTX 120 mg/kg solubilized in 10% DMSO resulted in <10% body weight loss, as observed over a period of 4 days (Figure 4A). However, in the group of six mice administered with PTX (Ebewe), two mice died following an oral dose of 80 mg/kg of PTX (Ebewe) due to the neurotoxicity associated with the PTX formulation. Thus, single MTD in the female BALB/c mice for PTX (Ebewe) was 60 mg/kg and 120 mg/kg for SMA-PTX micelles (Table 2). Free PTX administered with 10% DMSO did not show any weight loss at 120 mg/kg similar to SMA-PTX micelles. However, at this dose, free PTX was not completely soluble in 10% DMSO. In the repeated dose study, 30 mg/kg dose showed <10% body weight loss in both SMA-PTX micelles and PTX (Ebewe) groups as observed over a period of 2 weeks. However, with the dose of 60 mg/kg, the mice in the PTX (Ebewe) group showed >20% body weight loss (Figure 4B). Thus, the repeated MTD in the BALB/c mice was 60 mg/kg for SMA-PTX micelles and 30 mg/kg for PTX (Ebewe; Table 2).
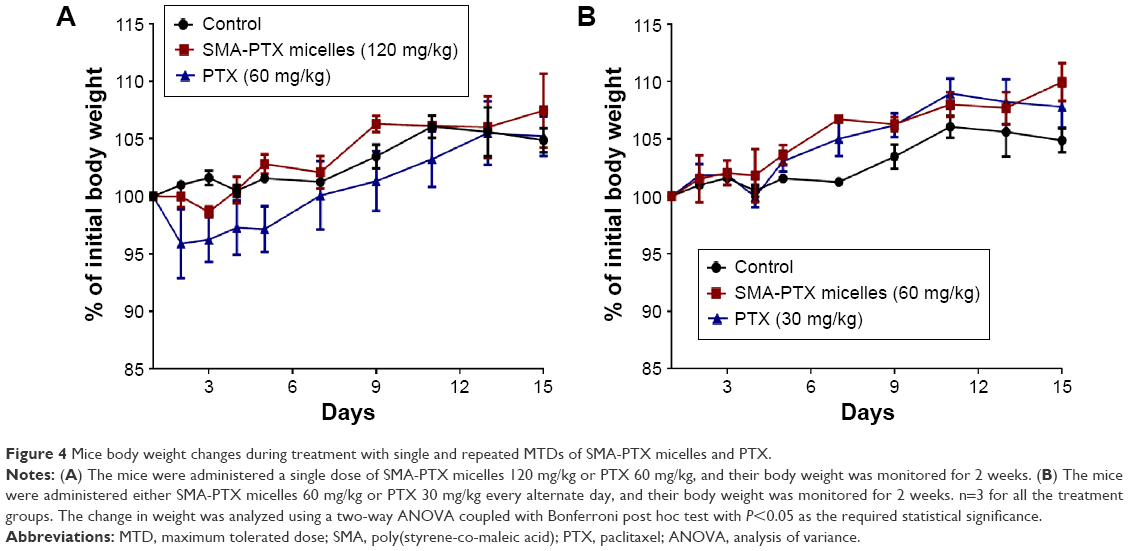 | Figure 4 Mice body weight changes during treatment with single and repeated MTDs of SMA-PTX micelles and PTX. |
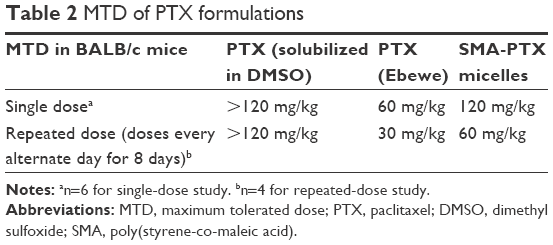 | Table 2 MTD of PTX formulations |
Furthermore, the body weights of mice treated with SMA-PTX and PTX as single and repeated doses were not significantly different from the weight at day 1, suggesting that the treatments did not have any effect on the general health of mice (Figure 4). A single dose of 60 mg/kg PTX resulted in a significant increase in the liver weight (7.44%±0.53% of the body weight) and spleen weight (0.86%±0.08% of the body weight) compared to the control liver weight (6.11%±0.33% of the body weight) and control spleen weight (0.61%±0.08% of the body weight; Figure S1). No significant differences in the plasma ALT activity or creatinine levels were observed between any of the treatment groups (Table S1). Overall, the MTDs of SMA-PTX micelles and PTX (Ebewe) did not elicit any significant systemic toxicity.
Antitumor efficacy and antiangiogenic potential of SMA-PTX micelles following oral administration in an orthotopic colon cancer model
To determine the antitumor efficacy of orally administered SMA-PTX micelles, an orthotopic colon cancer mouse model was utilized. Tumor size reduction was clearly observed following oral administration of SMA-PTX micelles (60 mg/kg and 30 mg/kg) compared to PTX (Ebewe; 30 mg/kg; Figure 5A and B). Furthermore, oral administration of SMA-PTX micelles 30 mg/kg resulted in an average tumor weight of 187.8±52.8 mg and SMA-PTX micelles 60 mg/kg resulted in an average tumor weight of 126.4±27.8 mg, showing 54.8% and 69.6% reduction, respectively in the tumor weight as compared to the tumors of the control group (416.4±53.3 mg; Figure 5B). Additionally, SMA-PTX micelles 60 mg/kg resulted in 63.3% tumor weight reduction as compared to the PTX (Ebewe) group (344.5±76.7 mg; Figure 5B), without any toxic effects (Figure 5C and D). To determine if the tumor reduction shown by SMA-PTX micelles was accompanied with a decrease in microvessel density, tumors were stained for the blood vessel marker CD105. As seen in Figure 6A, administration of SMA-PTX micelles (30 mg/kg and 60 mg/kg, po) was associated with significantly decreased CD105 staining, by 56% compared to control tumors. Furthermore, administration of SMA-PTX micelles (60 mg/kg, po) resulted in significantly decreased CD105 staining (50%) compared to mice treated with PTX (30 mg/kg, po; Figure 6B).
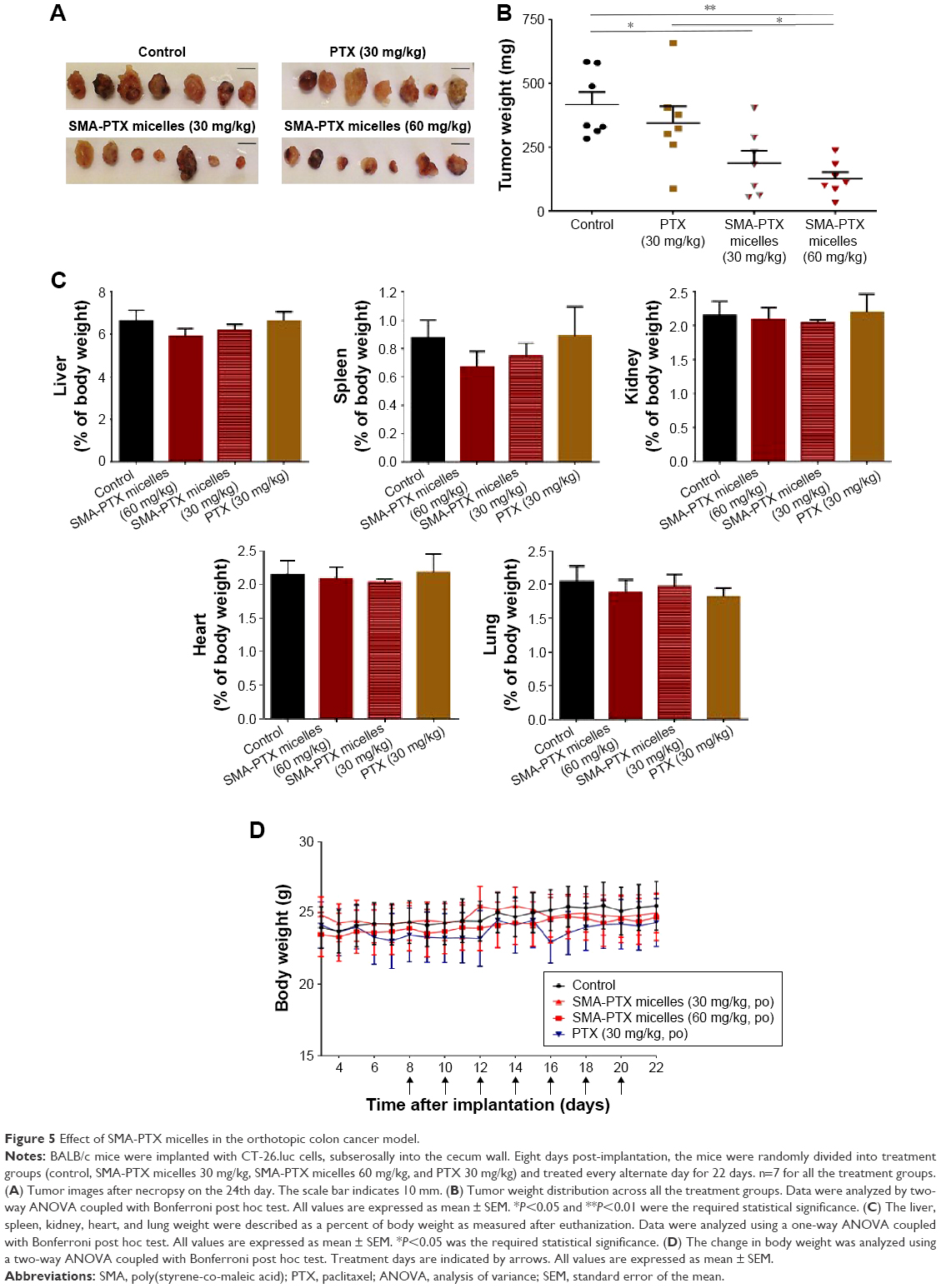 | Figure 5 Effect of SMA-PTX micelles in the orthotopic colon cancer model. |
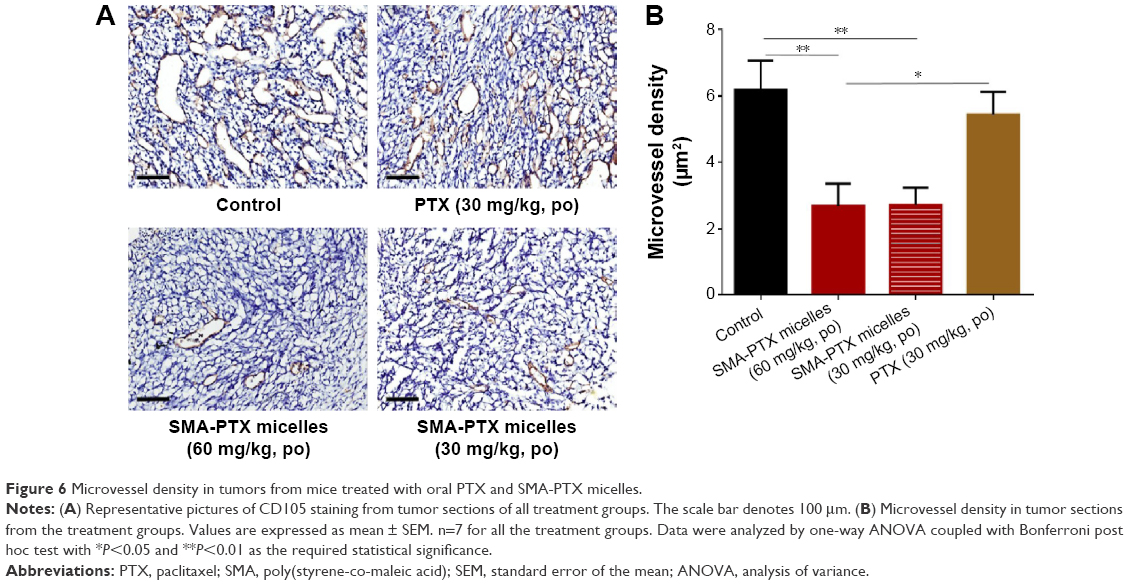 | Figure 6 Microvessel density in tumors from mice treated with oral PTX and SMA-PTX micelles. |
Toxicity analysis of SMA-PTX micelles in the orthotopic colon cancer model
To determine the general health of the animal, body weight was measured thrice weekly during the treatment period (Figure 5D). The body weight of all four groups was not significantly different from the weight at day 1, suggesting that the treatments did not have any effect on the general health of mice. The liver, kidney, spleen, heart, and lungs from all the mice were collected and evaluated for toxicity. No difference was observed in the weight of the organs between the treatment groups (Figure 5C). To further investigate any treatment-induced toxicity to the liver and kidney, the plasma ALT activity and creatinine levels were measured at the end of the treatment period. Collected blood showed no evidence of hemolysis and the plasma was clearly separated.
No significant differences in the plasma ALT or creatinine levels were observed between any of the treatment groups. Additionally, plasma ALT activity and creatinine levels were within the normal range for laboratory mice.21 Overall, the dosing regimen for oral administration of SMA-PTX micelles at 30 mg/kg and 60 mg/kg and PTX at 30 mg/kg elicited no significant systemic toxicity (Table 3).
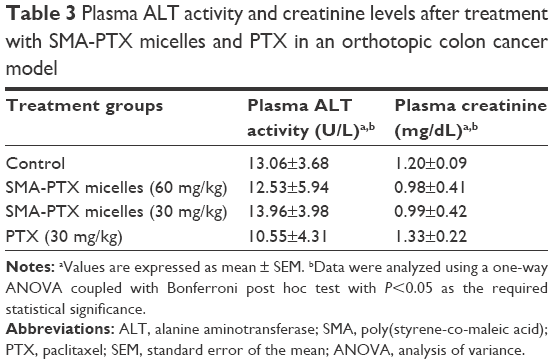 | Table 3 Plasma ALT activity and creatinine levels after treatment with SMA-PTX micelles and PTX in an orthotopic colon cancer model |
Discussion
Colon tumors exhibit increased permeability of the intestinal epithelium due to tight junction modifications.5,6 Thus, oral administration would be an efficient strategy for treatment of colorectal cancer, since the chemotherapeutic agent could effectively reach the tumor site. However, oral administration of PTX is associated with a number of limitations. PTX is poorly absorbed by the intestinal epithelium22 and shows oral bioavailability of <1% in the murine model23 and <6% in human beings.8
Polymeric micelles are the second most commonly studied drug delivery system in cancer therapeutics24 and have been evaluated to deliver single or multiple chemotherapeutic agents, targeted toward the tumor tissue.25–27 However, their use as an oral drug delivery system is limited. The use of amphiphilic nanocarrier systems, such as SMA micelles allows the encapsulation of these hydrophobic drugs into the core of the micelles with the hydrophilic carboxyl groups of maleic acid forming the outer shell and hydrophobic styrene rings forming the inner core, enabling the formulation to have a high aqueous solubility. Furthermore, SMA micelles are not a substrate for P-glycoprotein, and it has been shown that intracellular accumulation of SMA micelles is essentially dependent on the endocytosis process and is not subjected to membrane transporters.28 Thus, encapsulation of PTX into SMA micelles would avoid the efflux from the intestinal epithelium cell. The amphiphilic nature of SMA micelles was effective in bringing about the high loading of PTX (18%) and thus this formulation is advantageous compared to other polymeric micellar formulations such as pegylated poly(trimethylene carbonate) or cholesteryl 2-(5-methyl-2-oxo-1,3-dioxane-5-carboxyloyloxy)ethyl carbamate, which require hydrophobic modifications to achieve higher loading of the drugs.29 The currently available commercial PTX formulations in Cremophor EL and dehydrated ethanol (1:1) induce hypersensitivity reactions attributed to the Cremophor. Additionally, the amount of Cremophor necessary to solubilize the therapeutic doses of PTX is significantly higher than that of any other marketed drug.30 SMA-PTX micelles formulation showed good aqueous solubility and no toxicity or hypersensitivity associated with the copolymer and therefore could be advantageous over the commercially available PTX formulations.
The nanosize of the SMA-PTX micelles (195 nm) would favor the tumor accumulation due to the EPR effect and evasion of renal clearance (size >7 nm of the glomerular slits).31 The SMA-PTX micelles showed PDI of 0.34, thereby suggesting that SMA micelles had low aggregation.32 The SMA micelles displayed near-neutral surface charge. Carboxylated poly(styrene) nanoparticles show a significantly decreased affinity to intestinal epithelia, compared to positively charged and uncharged poly(styrene) nanoparticles.33 However, negatively charged poly(anhydride) copolymers of fumaric and sebacic acids were detected in paracellular spaces, enterocytes, and Peyer’s patches and showed a twofold increase in the absorption rates of dicoumarol.34 Thus, the near-neutral charge of SMA micelles is beneficial for increased affinity toward the intestinal epithelium as well as accumulation in Peyer’s patches.
The SMA micelles are stable at physiological pH and intestinal pH. However, at gastric pH, there is a steady rise in the release rate of the drug from the micelles. The relatively higher release rate under acidic conditions could be attributed to the protonation of the maleate component of the SMA copolymer, which plays a critical role in micellar stability. At alkaline pH, the micelle becomes slowly ionized, which increases its hydrodynamic volume, possibly due to the repulsive force between negative COO− groups. At acidic pH, the unionized maleic acid leads to a smaller hydrodynamic volume of the micelle.35 This effect could result in the release of PTX that is not in hydrophobic interaction with the styrene core, resulting in burst release. The acidic pH of the stomach can impede the stability of polymeric drug delivery systems.36 However, SMA micelles showed a release of <12% of PTX in SGF for the initial 2 hours, which is the time required for gastric transit. PEG2000-DSPE TPGS1000 and dequalinium micelles encapsulating PTX showed 60% and 50% release in gastric fluid and at physiological pH, respectively, at 24 hours.37 Thus, SMA-PTX micelles display higher stability with 50% and 20% release in gastric fluid and at physiological pH, respectively at this time point. Furthermore, PAH-PAA-PTX-layersomes encapsulating PTX showed 70% release at physiological pH at 24 hours.38 These lipid-based formulations display poor stability to aggregation of the constituent phospholipids10,36,39 and require surface modifications for maintaining stability under gastric conditions.40 This indicates an advantage of SMA micelles over other lipid-based nanoformulations.
The encapsulation of PTX into SMA micelles did not alter the cytotoxic effect of PTX. SMA-PTX micelles showed comparable IC50 values as compared to the free PTX and PTX (Ebewe) in CT-26.luc colon cancer cells. Furthermore, the SMA copolymer did not affect the cell viability at the mentioned concentrations, which is in agreement with the previous study41 and supports the inert nature of SMA copolymer previously described.42 PTX exhibits antiangiogenic properties that were shown by the reduction of proliferation, mobility, and cord formation of endothelial cells. Additionally, PTX was shown to inhibit VEGF-induced neovascularization and in vivo angiogenesis.43,44 SMA-PTX micelles showed similar antiangiogenic potential as PTX by inhibiting the ability of HUVECs to form tube-like networks.
Colorectal cancers usually show no phenotypic symptoms at early stages45 and thus are often detected at advanced stages due to lack of screening and relevant biomarkers. Liu et al identified the presence of vascular mimicry in 19.2% of colorectal cancer patients, which was associated with induction of epithelial mesenchymal transition by zinc finger E-box binding homeobox 1.46 Baeten et al showed a correlation between vascular mimicry and cancer stage and that the incidence of vascular mimicry is associated with the shortened survival time within the intermediate cancer stages.20 Thus, vascular mimicry is an important target in current antiangiogenic therapies for colorectal cancers. In our study, in a coculture model of HUVECs and CT-26.luc cells, the ability of PTX to inhibit formation of tube-like network was reduced, which could be attributed to the interaction with the cancer cells. However, the interaction of CT-26.luc cells with HUVECs was significantly reduced by the treatment with SMA-PTX micelles, which inhibits the tube-like formation exhibiting the antiangiogenic effect of SMA-PTX micelles by targeting vascular mimicry. Furthermore, in a murine orthotopic colon cancer model, a significant reduction of microvessel density was observed in SMA-PTX micelles-treated tumor sections as compared to PTX (Ebewe)-treated tumor sections exhibiting improved antiangiogenic effect of SMA-PTX micelles.
The single and repeated MTDs of SMA-PTX micelles (120 mg/kg and 60 mg/kg, respectively) following oral delivery showed a twofold increase in the therapeutic window as compared to that of the commercially available PTX (Ebewe; 60 mg/kg and 30 mg/kg, respectively). In contrast, the free PTX group showed no toxic effect at 120 mg/kg since PTX is not soluble at such a high dose2 and thus is less likely to be absorbed by the intestinal epithelium. In a Phase II clinical trial for patients with colorectal adenocarcinoma, PTX as a single agent administered through intravenous infusion failed to show effective antitumor efficacy.3 However, since PTX is a microtubule-stabilizing agent, it might be an effective anticancer agent in the subset of colorectal cancers lacking chromosomal instability and APC mutations. In this study, the CT-26.luc colon carcinoma model was chosen since it does not show APC mutations (although it can exhibit chromosomal instability)47 and because it shows sensitivity toward PTX.48 Oral administration of SMA-PTX micelles at 30 mg/kg and 60 mg/kg led to 54% and 69% reduction in the tumor weight, respectively, as compared to the control group, while no significant reduction in tumor weight was observed with administration of 30 mg/kg of PTX (Ebewe). There are two possible routes by which SMA-PTX micelles would reach the tumors following oral administration. First, SMA-PTX micelles could be absorbed through the small intestinal epithelium, and reaching the systemic circulation, would accumulate at the tumor site of the cecum due to the EPR effect. The second mechanism is associated with the histology of the intestinal epithelium of colon tumors. Since the colon tumors can exhibit increased permeability of the intestinal epithelium due to tight junction modifications,5,6 SMA-PTX micelles can pass paracellularly due to the modified tight junctions and reach the tumor site beneath the epithelium. Thus, together these pathways could contribute to the enhanced uptake of SMA-PTX micelles by the colon tumors, resulting in a greater therapeutic effect.
Abraxane (PTX nanoformulation) is under Phase II clinical trial for the treatment of metastatic colorectal cancer through intravenous delivery.49 However, Abraxane, which is an albumin-bound PTX nanoformulation, shows instability in the GI tract50 and thus is unfavorable for oral administration. Other oral PTX formulations, such as BMS-275183, in a Phase I clinical trial showed grade 1 and grade 2 liver-associated toxicity51 in four out of 20 patients with nonhematologic malignancy, when administered with an oral dose of 6 mg/m2/d of BMS-275183.52 Compared to these data, SMA-PTX micelles did not elevate plasma ALT levels or creatinine levels, suggesting that these SMA-PTX micelles are a safer system for oral PTX delivery.
Conclusion
SMA micellar formulations may provide an effective strategy for oral delivery of PTX with no liver or kidney toxicities. In a murine orthotopic colon cancer model, oral administration of SMA-PTX micelles showed significantly higher antitumor efficacy as compared to commercial PTX formulation. Furthermore, SMA-PTX micelles formulations show antiangiogenic effect associated with inhibiting vascular mimicry. Thus, oral administration of SMA-PTX micelles formulations could be an improved approach for colorectal cancer treatment.
Acknowledgment
This work has been supported by 2014 University of Otago Research Grant (110273.01.R.LM) to KG.
Disclosure
The authors report no conflicts of interest in this work.
References
Yamanaka YJ, Leong KW. Engineering strategies to enhance nanoparticle-mediated oral delivery. J Biomater Sci Polym Ed. 2008;19(12):1549–1570. | ||
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1–2):179–192. | ||
Einzig AI, Neuberg D, Wiernik PH, et al. Phase II trial of paclitaxel in patients with advanced colon cancer previously untreated with cytotoxic chemotherapy: an eastern cooperative oncology group trial (PA286). Am J Ther. 1996;3(11):750–754. | ||
Swanton C, Tomlinson I, Downward J. Chromosomal instability, colorectal cancer and taxane resistance. Cell Cycle. 2006;5(8):818–823. | ||
Mullin JM, Laughlin KV, Ginanni N, Marano CW, Clarke HM, Peralta Soler A. Increased tight junction permeability can result from protein kinase C activation/translocation and act as a tumor promotional event in epithelial cancers. Ann N Y Acad Sci. 2000;915(1):231–236. | ||
Soler AP, Miller RD, Laughlin KV, Carp NZ, Klurfeld DM, Mullin JM. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis. 1999;20(8):1425–1432. | ||
Wood AJ, Rowinsky EK, Donehower RC. Paclitaxel (taxol). N Engl J Med. 1995;332(15):1004–1014. | ||
Malingré MM, Terwogt JMM, Beijnen JH, et al. Phase I and pharmacokinetic study of oral paclitaxel. J Clin Oncol. 2000;18(12):2468–2475. | ||
DeMario MD, Ratain MJ. Oral chemotherapy: rationale and future directions. J Clin Oncol. 1998;16(7):2557–2567. | ||
Thanki K, Gangwal RP, Sangamwar AT, Jain S. Oral delivery of anticancer drugs: challenges and opportunities. J Control Release. 2013;170(1):15–40. | ||
Peltier S, Oger J-M, Lagarce F, Couet W, Benoît J-P. Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules. Pharm Res. 2006;23(6):1243–1250. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Zhao J, Liu J, Wei T, et al. Quercetin-loaded nanomicelles to circumvent human castration-resistant prostate cancer in vitro and in vivo. Nanoscale. 2016;8(9):5126–5138. | ||
Iwai K, Maeda H, Konno T. Use of oily contrast medium for selective drug targeting to tumor: enhanced therapeutic effect and X-ray image. Cancer Res. 1984;44(5):2115–2121. | ||
Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 2000;60(16):4324–4327. | ||
Parayath NN, Nehoff H, Müller P, Taurin S, Greish K. Styrene maleic acid micelles as a nanocarrier system for oral anticancer drug delivery–dual uptake through enterocytes and M-cells. Int J Nanomed. 2015;10:4653. | ||
Greish K, Sawa T, Fang J, Akaike T, Maeda H. SMA–doxorubicin, a new polymeric micellar drug for effective targeting to solid tumours. J Control Release. 2004;97(2):219–230. | ||
Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. | ||
Tseng W, Leong X, Engleman E. Orthotopic mouse model of colorectal cancer. J Vis Exp. 2007;10:484. | ||
Baeten CI, Hillen F, Pauwels P, de Bruine AP, Baeten CG. Prognostic role of vasculogenic mimicry in colorectal cancer. Dis Colon Rectum. 2009;52(12):2028–2035. | ||
AHC. Academic Health Centre, University of Minnesota; 2011. Available from: http://www.ahc.umn.edu/rar.refvalues.html. Accessed October 21, 2015. | ||
Moynihan H, Crean A. Physicochemical Basis of Pharmaceuticals. New York: Oxford University Press; 2009. | ||
Eiseman J, Eddington N, Leslie J, et al. Plasma pharmacokinetics and tissue distribution of paclitaxel in CD2F1 mice. Cancer Chemother Pharmacol. 1994;34(6):465–471. | ||
Taurin S, Nehoff H, Greish K. Anticancer nanomedicine and tumor vascular permeability; where is the missing link? J Control Release. 2012;164(3):265–275. | ||
Ramasamy T, Ruttala HB, Choi JY, et al. Engineering of a lipid-polymer nanoarchitectural platform for highly effective combination therapy of doxorubicin and irinotecan. Chem Commun. 2015;51(26):5758–5761. | ||
Ramasamy T, Kim J, Choi HG, Yong CS, Kim JO. Novel dual drug-loaded block ionomer complex micelles for enhancing the efficacy of chemotherapy treatments. J Biomed Nanotechnol. 2014;10(7):1304–1312. | ||
Ramasamy T, Kim JH, Choi JY, et al. pH sensitive polyelectrolyte complex micelles for highly effective combination chemotherapy. J Mater Chem B. 2014;2(37):6324–6333. | ||
Pritchard T, Rosengren RJ, Greish K, Taurin S. Raloxifene nanomicelles reduce the growth of castrate-resistant prostate cancer. J Drug Target. 2016;24(5):441–449. | ||
Lee AL, Venkataraman S, Sirat SB, Gao S, Hedrick JL, Yang YY. The use of cholesterol-containing biodegradable block copolymers to exploit hydrophobic interactions for the delivery of anticancer drugs. Biomaterials. 2012;33(6):1921–1928. | ||
Rowinsky EK, Onetto N, Canetta R, Arbuck S. Taxol: the first of the taxanes, an important new class of antitumor agents. Seminars in Oncology. 1992;19(6):646–662. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Dickinson E, Ettelaie R, Kostakis T, Murray BS. Factors controlling the formation and stability of air bubbles stabilized by partially hydrophobic silica nanoparticles. Langmuir. 2004;20(20):8517–8525. | ||
Jani P, Halbert GW, Langeidge J, Florence AT. Nanoparticle uptake by the rat gastrointestinal mucosa: quantitation and particle size dependency. J Pharm Pharmacol. 1990;42(12):821–826. | ||
Mathiowitz E, Jacob JS, Jong YS, et al. Biologically erodable microspheres as potential oral drug delivery systems. Nature. 1997;386(6623):410–414. | ||
Iyer AK, Greish K, Fang J, Murakami R, Maeda H. High-loading nanosized micelles of copoly(styrene–maleic acid)–zinc protoporphyrin for targeted delivery of a potent heme oxygenase inhibitor. Biomaterials. 2007;28(10):1871–1881. | ||
Hörter D, Dressman J. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 2001;46(1):75–87. | ||
Yao H-J, Ju R-J, Wang X-X, et al. The antitumor efficacy of functional paclitaxel nanomicelles in treating resistant breast cancers by oral delivery. Biomaterials. 2011;32(12):3285–3302. | ||
Jain S, Patil SR, Swarnakar NK, Agrawal AK. Oral delivery of doxorubicin using novel polyelectrolyte-stabilized liposomes (layersomes). Mol Pharm. 2012;9(9):2626–2635. | ||
Derycke AS, de Witte PA. Liposomes for photodynamic therapy. Adv Drug Deliv Rev. 2004;56(1):17–30. | ||
Iwanaga K, Ono S, Narioka K, et al. Oral delivery of insulin by using surface coating liposomes: improvement of stability of insulin in GI tract. Int J Pharm. 1997;157(1):73–80. | ||
Taurin S, Nehoff H, van Aswegen T, Rosengren RJ, Greish K. A novel role for raloxifene nanomicelles in management of castrate resistant prostate cancer. Biomed Res Int. 2014;2014(2014):323594. | ||
Müller B. Polymeric corrosion inhibitors for aluminium pigment. React Funct Polym. 1999;39(2):165–177. | ||
Belotti D, Rieppi M, Nicoletti MI, et al. Paclitaxel (Taxol (R)) inhibits motility of paclitaxel-resistant human ovarian carcinoma cells. Clin Cancer Res. 1996;2(10):1725–1730. | ||
Myoung H, Hong S-D, Kim Y-Y, Hong S-P, Kim M-J. Evaluation of the anti-tumor and anti-angiogenic effect of paclitaxel and thalidomide on the xenotransplanted oral squamous cell carcinoma. Cancer Lett. 2001;163(2):191–200. | ||
Wang S, Xiang J, Li Z, et al. A plasma microRNA panel for early detection of colorectal cancer. Int J Cancer. 2015;136(1):152–161. | ||
Liu Z, Sun B, Qi L, Li H, Gao J, Leng X. Zinc finger E-box binding homeobox 1 promotes vasculogenic mimicry in colorectal cancer through induction of epithelial-to-mesenchymal transition. Cancer Sci. 2012;103(4):813–820. | ||
Castle JC, Loewer M, Boegel S, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15(1):190. | ||
Yang X-Y, Li Y-X, Li M, Zhang L, Feng L-X, Zhang N. Hyaluronic acid-coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013;334(2):338–345. | ||
ClinicalTrials.gov [homepage on the Internet]. Available from: ClinicalTrials.gov. Accessed October 26, 2015. | ||
Yang F-H, Zhang Q, Liang Q-Y, et al. Bioavailability enhancement of paclitaxel via a novel oral drug delivery system: paclitaxel-loaded glycyrrhizic acid micelles. Molecules. 2015;20(3):4337–4356. | ||
NIH [webpage on the Internet]. livertox.nih; 2015. Available from: http://livertox.nih.gov/Severity.html. Accessed October 26, 2015. | ||
Heath EI, LoRusso P, Ramalingam SS, et al. A phase 1 study of BMS-275183, a novel oral analogue of paclitaxel given on a daily schedule to patients with advanced malignancies. Invest New Drugs. 2011;29(6):1426–1431. |
Supplementary materials
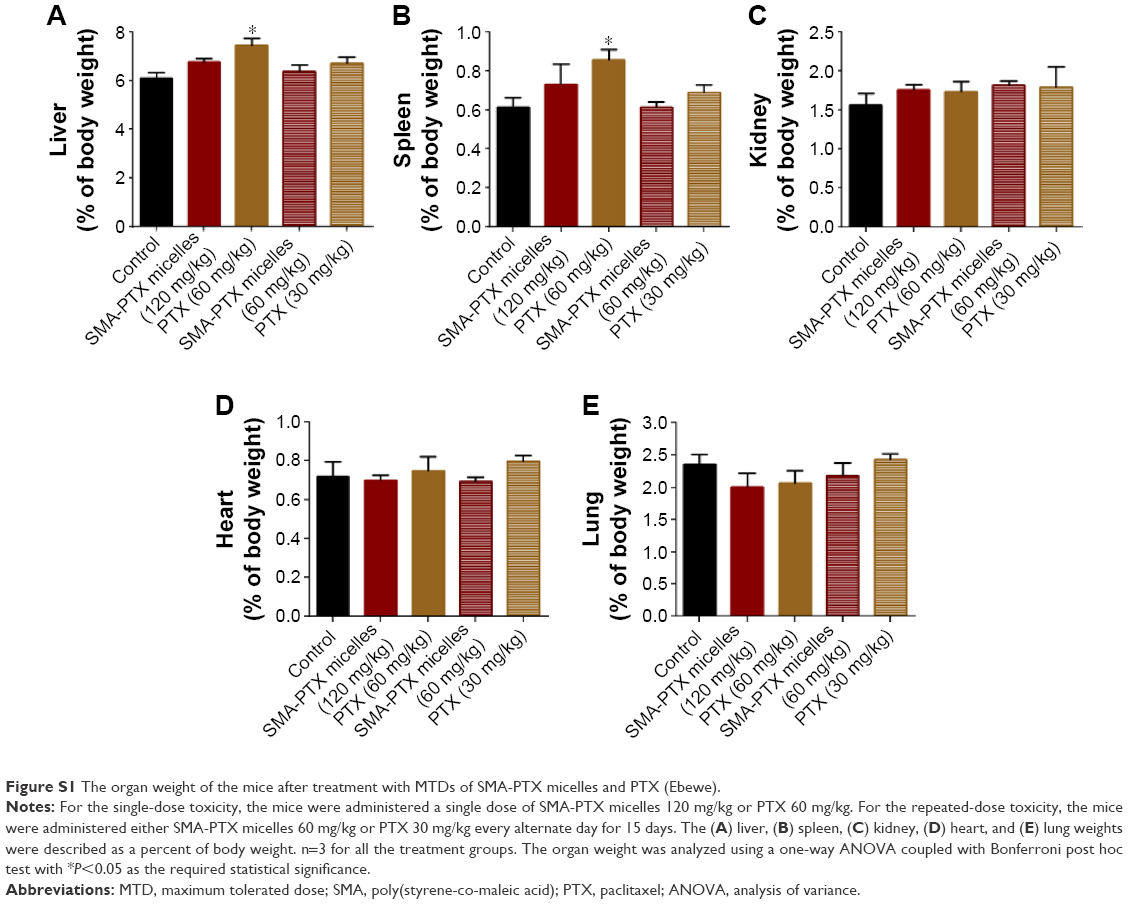 | Figure S1 The organ weight of the mice after treatment with MTDs of SMA-PTX micelles and PTX (Ebewe). |
 | Table S1 Plasma ALT activity and creatinine levels after treatment with single and repeated MTDs of SMA-PTX micelles and PTX |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.