")
Back to Journals » OncoTargets and Therapy » Volume 7
Study of the mechanism of sonodynamic therapy in a rat glioma model
Authors Song D, Yue W, Li Z, Li J, Zhao J, Zhang N
Received 2 August 2013
Accepted for publication 13 May 2014
Published 30 September 2014 Volume 2014:7 Pages 1801—1810
DOI https://doi.org/10.2147/OTT.S52426
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Dayong Song,1 Wu Yue,2 Zhiqiang Li,1 Jianhua Li,2 Jun Zhao,1 Ning Zhang1
1Department of Neurosurgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital South Campus, Shanghai Fengxian District Central Hospital, Shanghai, 2Department of Neurosurgery, The Fourth College Hospital of Harbin Medical University, Harbin, People's Republic of China
Purpose: The study reported here examined the effect of hematoporphyrin monomethyl ether (HMME)-mediated sonodynamic therapy (SDT) on C6 gliomas implanted in rat brains.
Methods: Two weeks after inoculation, glioma development was evaluated by measuring tumor volume using a 1.5 T magnetic resonance imager. Rats that had a well-developed C6 glioma (usually when the tumor diameter reached 3–5 mm) were used to test SDT, ultrasound-alone, and HMME-alone treatments. Rats both administered and not administered intravenous HMME 10 µg/mL were insonated by a 1 MHz ultrasound at a dose of 0.5 W/cm2.
Results: SDT treatment could effectively inhibit the expansion of intracranial gliomas in vivo. The treatment with ultrasound alone could inhibit glioma growth within 1 week; however, 1 week later, the tumor started growing again. In contrast, the effect of SDT could last at least 2 weeks. Injection of HMME alone had no effects on inhibiting glioma growth, suggesting the sonosensitizer HMME has no antitumor effect. Both SDT and ultrasound-alone treatment could extend the survival of rats implanted with a C6 glioma. Pathological and electron microscopic examinations suggested SDT and ultrasound-alone treatment could induce glioma necrosis by way of triggering glioma-cell apoptosis, which was confirmed by immunohistological examination with cytochrome-c and caspase-3 antibodies. Most importantly, we found that the sonosensitizer HMME could enhance the ultrasound-induced antitumor effect by selectively assisting ultrasound targeting of glioma angiogenesis inhibition.
Conclusion: This study with a rat C6 glioma experimental model showed that SDT can potentially be useful in the treatment of deep-seated malignant gliomas.
Keywords: antitumor mechanism, ultrasound, hematoporphyrin monomethyl ether
Introduction
A “glioma” is a type of tumor that starts in the brain or spine. The most common site of a glioma is the brain,1 and gliomas comprise ~30% of all brain and central-nervous-system tumors and 80% of all malignant brain tumors.2 Traditionally, surgery is the most common initial definitive treatment option for a glioma. Radiotherapy, chemotherapy, or the combination of these treatments is also widely used. However, malignant glial tumors, especially glioblastomas, may strongly invade neighboring tissues and may not be resected completely. Due to their nonselective killing of malignant and normal cells, both chemotherapy and radiotherapy can cause a range of side effects and may seriously lower the quality of the patient’s life. As tumor cells cannot be completely removed by these mentioned therapies, the recurrence rate of malignant gliomas is very high, and survival time of glioma patients is very short. Thus, to more efficiently kill tumor cells and extend patient survival, alternative or complementary glioma treatments are being sought. One tried treatment is noninvasive sonodynamic therapy (SDT).
The original idea of SDT was raised in 1989 by Umemura et al.3 A new approach to cancer treatment, “SDT” is based on the ultrasonic activation of a sonosensitizer.4,5 One of the main features of SDT is its ability to focus ultrasound (US) energy on malignant sites buried deep in tissues and locally activate the cytotoxicity of the sonosensitizer that resides in the tumor sites. By adjusting the acoustic intensity of US, its effect on the surrounding peripheral tissues can be greatly decreased,6 minimally damaging peripheral healthy tissues.7 Thus, it is believed that SDT can provide localized glioma treatment with a great many fewer side effects.8
The mechanisms involved in activating sensitizers by US irradiation are still not very clear yet. It has been demonstrated that the possible mechanisms of SDT may include the generation of sonosensitizer-derived radicals, which initiate the chain peroxidation of membrane lipids via peroxyl and/or alkoxyl radicals. The physical destabilization of the cell membrane by the sonosensitizer thereby either renders the cell more susceptible to shear forces or assists US in enhancing drug transport across the cell membrane (“sonoporation”).7,9,10 It has also been reported that SDT may enhance the apoptosis rate of cancer cells.11,12 A previous study by members of our group indicated that hematoporphyrin monomethyl ether (HMME)-mediated SDT can significantly inhibit the growth rate of C6 glioma cells in vitro and kill C6 glioma cells by inducing apoptosis and necrosis.13 Recently, a study showed that the intracellular uptake of HMME was time and dose dependent and that HMME can selectively accumulate in tumor cells, whereas normal cells absorb less HMME.14 This important finding suggests that HMME-mediated SDT can selectively kill tumor cells in vivo and have little effect on normal cells.
Many studies on the in vitro SDT of gliomas have been published in the past decade. However, there are almost no reports on the clinical application of SDT in gliomas. The in vivo study of SDT in animal models has been rarely reported.15,16 Clearly, to enhance SDT efficiency and understand the mechanism of SDT in the treatment of gliomas, it is necessary to extensively study the SDT of gliomas using in vivo animal models. Therefore, in the study reported here, we investigated the antitumor effect of SDT and its possible in vivo mechanism in a C6 glioma rat model.
Materials and methods
C6 glioma cell culture
The C6 glioma cell line was purchased from the Neurosurgery Institute of Harbin Medical University (Harbin, People’s Republic of China) and was cultured in RPMI (Roswell Park Memorial Institute) 1640 medium (HyClone™; GE Healthcare, Little Chalfont, UK) containing 10% fetal bovine serum (TBD, Tianjin, People’s Republic of China) in an incubator containing 5% CO2 and 95% O2 at 37°C. Cells were used after reaching the logarithmic phase.
C6 glioma animal model preparation
Adult male Wistar rats (body weight 250–350 g) were purchased from the Experimental Animal Center of the First Hospital of Harbin Medical University and fasted to food and water for 12 hours before surgery. Animals were anesthetized intraperitoneally with sodium pentobarbital (40 mg/kg). After being immobilized in a stereotaxic apparatus (Jiangwan type II, Shanghai Jiangwan Medical Instruments Factory, Shanghai, People’s Republic of China), an incision was made in the skin and a burr hole made in the skull. Gliomas were made by inoculating 5×106 glioma cells stereotaxically into the brain. Three days after surgery, animals were continuously injected with penicillin (40,000 U/day) and were housed routinely.
SDT of rat brain tumor
Fourteen days after injection of the C6 glioma cells, magnetic resonance imaging (MRI) (1.5T VISRT; Toshiba, Tokyo, Japan) was performed on the implanted rats to confirm the formation of gliomas. Rats were to be used only when the tumor diameter reached 3–5 mm. The implanted rats were anesthetized with 1% sodium pentobarbital (40 mg/kg). The implanted rats that were used for SDT were injected with HMME (10 ng/kg) (Beijing Yingfa Kangmei Technology Development Co Ltd, Beijing, People’s Republic of China) via the tail vein. Two hours later, the injected rats were exposed to US for 120 seconds. A schematic diagram of the experimental device for insonation is shown in Figure 1. US waves were generated by a US therapy device (Beijing Tianshi Technology Co Ltd, Beijing, People’s Republic of China). The frequency and sound strength of the US was 1.0 MHz and 0.5 W/cm2, respectively. For the next 7 days following US irradiation, the rats were housed routinely in the dark.
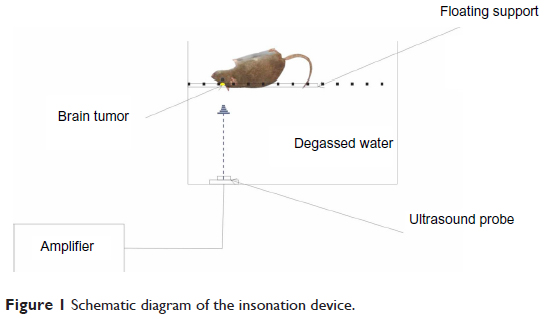 | Figure 1 Schematic diagram of the insonation device. |
Measurement of tumor volume
The rats to be examined were anesthetized intraperitoneally with 1% sodium pentobarbital (40 mg/kg). Before scanning with MRI (1.5 T), rats were injected with gadolinium-diethylenetriaminepentaacetic acid via the tail vein to enhance contrast. The tumor nidus was examined under conventional light microscopy. The tumor volume was evaluated by the maximum coronal plane and sagittal plane. Tumor length (L), width (W), and height (H) were measured for calculation of tumor volume (V) according to the formula V = (L × W × H) × π/6.
Detection of in situ apoptosis
After treatment with SDT, at the examined time points, the rat tumors were excised then fixed in 4% paraformaldehyde, paraffin embedded, and sectioned through the area of US irradiation. TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay was performed using an In Situ Cell Death Detection Kit, peroxidase (Roche Diagnostics, Basel, Switzerland). Sections were de-waxed in xylene, rehydrated in descending concentrations of ethanol at room temperature, then rinsed three times in double-distilled water. After rehydration, sections were treated with proteinase K (50 μL/section) for 30 minutes in a humidified chamber then rinsed three times with double-distilled water. After the sections were permeabilized, a mixture of terminal deoxynucleotidyl transferase, Biotin-11-dUTP, and labeling buffer solution was added to the sections (20 μL/section) and incubated at 37°C for 60 minutes in a humidified chamber. After washing three times with phosphate-buffered saline (PBS), blocking reagent was added (50 μL/section) for 30 minutes at room temperature. After the blocking reagent was removed, avidin-horseradish peroxidase was added (50 μL/section) at 37°C for 60 minutes in a humidified chamber before washing three times with PBS. Labeled apoptotic cells were identified by treating the sections with the peroxidased chromogenic substrate 3,3′-diaminobenzidine (DAB), as recommended in the kit. Counterstaining of nuclei was performed using hematoxylin and eosin stain (HE) by incubating the sections with the stain at room temperature for 10 minutes. After conventional mounting, the slides were observed under conventional light microscope.
Sepia staining in cell nuclei was judged a positive result. To avoid false-positive results, the necrosis cells, which show positive HE staining, were not counted. Under light microscope, magnification ×400, five view fields were randomly chosen from each slide. Two people double blindly counted positive cells from a total of 500 cells. The counts of positively stained cells from five observed fields were averaged. Areas of high inflammation and necrosis were excluded from the counted fields. The apoptosis percentage was calculated according to the formula: apoptosis percentage = positive cell number/total counted cells × 100%.
Immunochemical staining
The expression levels of cytochrome-c (Cyto-C), caspase-3, vascular endothelial growth factor (VEGF), and CD34 were analyzed by immunohistochemical staining. After SDT, at the examined time points, the rat tumors were excised then fixed in 4% paraformaldehyde, paraffin embedded, and sectioned through the area of US irradiation. Commercially available antibodies to Cyto-C, VEGF, Caspase-3, and CD34 (Wuhan Boster Biological Technology, Wuhan, People’s Republic of China) were used. Immunohistochemical staining was carried out using the avidin-biotin method and a commercially available kit (AbC™ Anti-Mouse Bead Kit; Thermo Fisher Scientific, Waltham, MA, USA). Briefly, the slides were baked at 60°C for 2 hours before being de-waxed in xylene and rehydrated in descending concentrations of ethanol. De-paraffinized sections were treated with methanol containing 3% hydrogen peroxide for 10 minutes at room temperature to quench the endogenous peroxidase. After washing with PBS, blocking serum (5%–10% goat serum) was applied for 10 minutes at room temperature. The sections were incubated with an anti-Cyto-C antibody (1:100), anti-caspase-3 antibody (1:100), anti-VEGF antibody (1:100), and anti-CD34 antibody (1:100) overnight at 4°C. Negative control sections were incubated with PBS instead of the primary antibody. After washing in PBS, a biotin-marked secondary antibody was applied for 10 minutes followed by a peroxidase-marked streptavidin for an additional 10 minutes. The reaction was visualized using 3, 3′-diaminobenzidine tetrahydrochloride. The nuclei were counterstained with HE. Positive and negative immunohistochemistry controls were routinely used. The reproducibility of the staining was confirmed by re-immunostaining via the same method in multiple, randomly selected specimens.
Determination of positive expression of Cyto-C, Caspase-3, VEGF, and CD34 protein
Under light microscope, magnification ×400, five view fields were randomly chosen from each slide. Two people double blindly counted positive cells from a total of 500 cells. The counts of positively stained cells from five observed fields were averaged then the express percentage was calculated using the formula: expression percentage = positive cell number/(positive + negative) staining cells × 100%.
Calculation of microvessel density (MVD)
Since CD34 proteins are mainly expressed in the endothelial cells of blood vessels, in cell matrix and cancer nests, tube-like, line-like or dot-like microvessels can usually be found. One or a group of CD34-labeled endothelial cells can be taken as one microvessel. Thus, the MVD can be calculated by measuring CD34 expression. Under regular light microscope with a magnification of ×40, a blood-vessel-abundant area was chosen, then the stained cells were observed under the microscope with a magnification of ×400. Five visual fields were randomly selected, the microvessels stained with CD34 were counted, then the average number of the five counts was taken as the MVD.
Evaluation of survival time
Fourteen days after injection of the C6 glioma cells, MRI was performed on the implanted rats to confirm the formation of gliomas. Four groups of implanted rats after different treatments (an additional eight mice in each group) were raised until anesthetized. The death time was calculated as the duration from the date of tumor implanting to the date of natural death or the date of reaching point of death. For example, if the implanted rats survived 60 days, survival time was calculated as 60 days.
Statistical analysis
Data are presented as the mean value ± standard deviation (SD). Statistical analysis was performed with SAS9.1.3 software (SAS Institute Inc., Cary, NC, USA). MRI results were analyzed with mixed linear models for repeated measures and compared one by one. Analysis of the survival data was performed by completely randomly designed variance analysis and use of the Student–Newman–Keuls method to make paired comparisons. Factorial analysis was applied to analyzed protein expression levels of Cyto-C, Caspase-3, VEGF, and CD34. Pearson correlation analysis was also performed with protein expression level data. Values <0.05 were considered statistically significant.
Results
Effect of SDT on rat gliomas
Figure 2A shows the tumor volume change after different treatments. The tumor volumes were measured with MRI and calculated in the way described in the “Materials and methods” section.
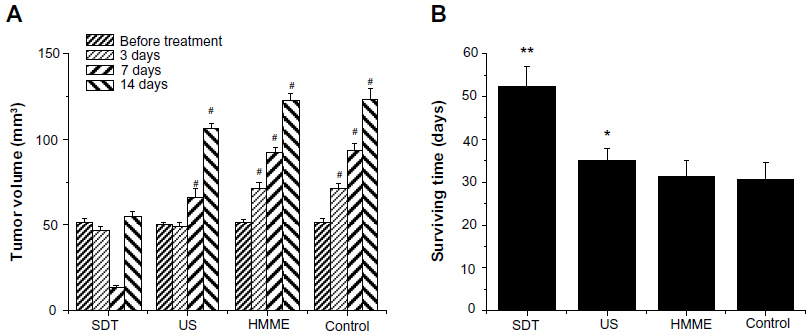 | Figure 2 Effect of sonodynamic therapy (SDT) treatment on tumor volume and animal survival time. The tumor volume was measured with conventional light microscopy before treatment and after treatment on Days 3, 7, and 14 (A). Animal survival time was calculated as the number of days until the animal died of natural causes or were anesthetized (B). If a glioma had developed for more than 60 days, survival time was counted as 60 days. |
Before the treatment, there was no significant difference in tumor size. Three days after treatment, the tumor size of control group and HMME-treated group was greatly increased and the tumors kept continuously growing, suggesting that they could not be effectively controlled by the injection of HMME alone. In the US-treated group, even though the tumor size was well controlled in the first week, the tumors eventually grew significantly again. When implanted rats were injected HMME then exposed to US, tumors could be well controlled. While, two weeks after treatment, the tumors were found to be still growing, they were much smaller than those in the rats treated with other treatments. This strongly implies that SDT may effectively control tumor growth and increase survival time (Figure 2B).
Fundamental pathological observations
To understand how the different treatments work, we further examined the fundamental pathological changes. HE-stained sections of untreated gliomas showed heteromorphism characteristic of glioma cells, infiltration of tumor cells into normal tissues, blurred boundaries, and the formation of new blood vessels within the tumors (Figure 3H). No clear necrosis was found in the gliomas from the control rats or the rats injected with HMME alone at each examined time point. Six hours after SDT, irregular lamellar necrosis was observed; however, only sporadic punctiform necrosis cells were found inside gliomas after 6 hours’ treatment with US alone (data not shown). Twenty-four hours after SDT, the amount of irregular lamellar necrosis had clearly increased. In the center of the tumor necrosis, no cell structures were found; instead, large amounts of debris and shrunken glioma cells were discovered at the boundary of the necrosis and normal tumor tissue (Figure 3E), suggesting tumor necrosis reached its peak at 24 hours after treatment with SDT. Interestingly, a small area of irregular lamellar necrosis was also observed in the rats treated with US alone, 24 hours later. At the same time, sporadic punctiform necrosis could still be observed in the transitional area between the necrosis and normal tumor tissue (Figure 3F). At 3 days after treatment, in gliomas treated with SDT, the area of lamellar necrosis was much smaller than that at 24 hours, and little sporadic punctiform necrosis was observed in gliomas treated with US alone. At 7 days after treatment, there was no necrosis found in gliomas treated with SDT or US alone. No necrosis was observed in the control or HMME groups at each of the observed time points.
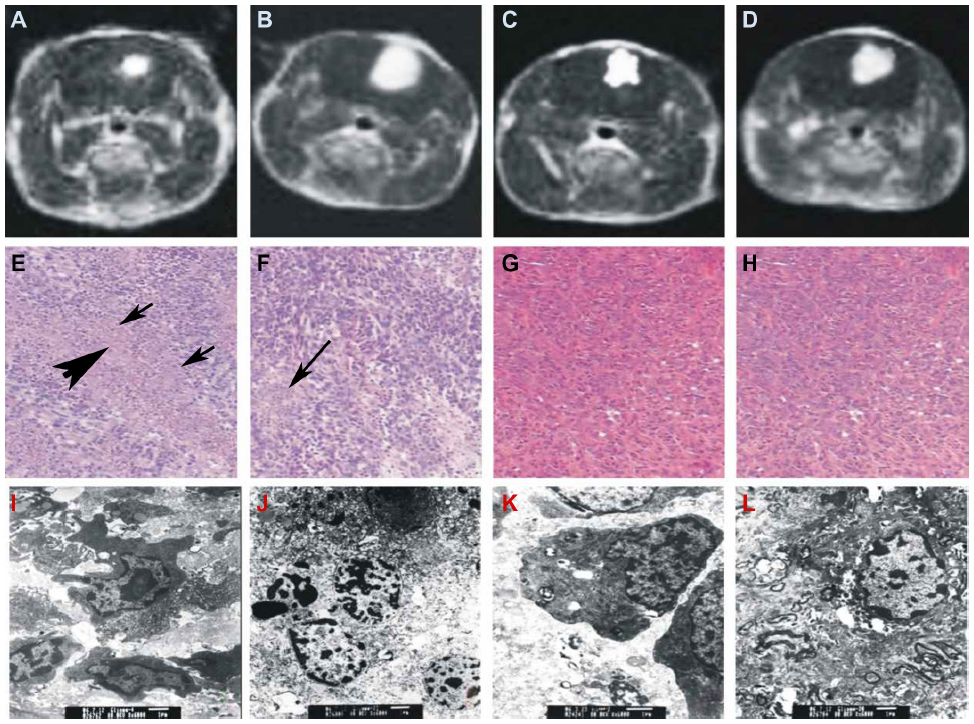 | Figure 3 Evaluation of treatments by magnetic resonance imaging (MRI) (A–D), hematoxylin and eosin (HE) staining (E–H), and transmission electron microscopy (TEM) (I–L). Treated by sonodynamic therapy (SDT) (A, E, I), ultrasound (B, F, J), hematoporphyrin monomethyl ether (HMME) (C, G, K), and untreated control (D, H, L). |
Electron microscopy images showed that the cells of the control rats and rats treated with HMME alone displayed normal structure. With US treatment alone, slightly punctiform necrotic cells with some morphological features of apoptosis were found. However, with SDT, a large number of necrosis cells with morphological apoptotic features were discovered (Figure 3I–L), suggesting SDT is much more effective in killing cancer cells than the other treatments.
Examination of SDT-induced apoptosis
“Apoptosis” is gene-coded programmed cellular death. Cell apoptosis is believed to be closely related to tumor development. In multicellular organisms, the total number of cells is a balance between the cell-generating effects of mitosis and cell death that is induced through apoptosis. A disruption of this delicate balance can lead to the development of cancer. Thus, any approaches to induce or initiate apoptosis may be an effective way to treat cancer, like gliomas.17 A study by members of our group indicated that apoptosis is an important mechanism of SDT in killing in vitro C6 gliomas.13 To explore whether this is also the mechanism for SDT in treating in vivo gliomas, we explored how SDT affects apoptosis and the relevant protein expression.
At 24 hours after treatment with SDT, in situ apoptosis assay showed a marked increase in the cell apoptosis rate. The apoptotic rate of glioma cells with SDT was much higher than that with the other treatments (Figure 4), which strongly suggests that SDT can induce apoptosis. TUNEL results showed that there were few apoptotic cells in the HMME treatment group and in the control rats. Further, there was no statistical difference in apoptotic rate between each time point in these two groups. At 24 hours after treatment, the apoptotic rate in rats treated with US alone was higher than that of rats that received HMME treatment alone and control rats. However, there was no statistical difference in apoptotic rate between the mentioned groups at 3 or 7 days after treatment.
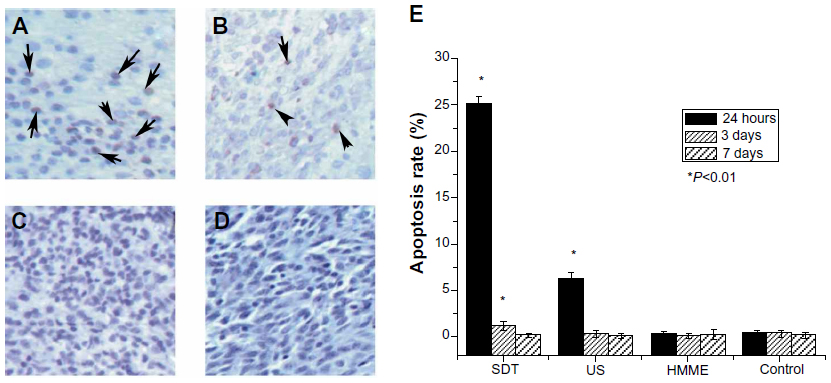 | Figure 4 Effect of sonodynamic therapy (SDT) on apoptosis detected by TUNEL assay. The images were taken at 24 hours after treatment with SDT (A), ultrasound (US) (B), and hematoporphyrin monomethyl ether (HMME) (C), or after no treatment (D). Summary of in situ apoptosis assay at different times (24 hours, 3 days, and 7 days) after the indicated treatments (E). |
Cyto-C is a component of the electron transport chain in mitochondria. It is believed that Cyto-C is released by the mitochondria in response to pro-apoptotic stimuli. Upon the release of Cyto-C into the cytoplasm, the protein binds apoptotic protease activating factor-1.18 This release of Cyto-C in turn activates “caspase 9”, a cysteine protease. Caspase 9 can then go on to activate caspase 3 and caspase 7, which are responsible for destroying the cell from within. Thus, to explore how SDT induces apoptosis, we examined the expression of Cyto-C and caspase-3 inside the gliomas.
Twenty-four hours after treatment (Figure 5), the expression rate of both Cyto-C protein and caspase-3 protein was much higher in rats treated with SDT than in those treated with any of the other treatments. However, at this time point, the protein expression rate in rats treated with US alone was also higher than that of rats treated with HMME alone and control rats. Few cells expressed Cyto-C and Caspase-3 in the HMME-alone and control groups at different time points, and no difference in the expression rate of Cyto-C and caspase-3 protein was found between the two groups. At 3 and 7 days after treatment, the protein expression rate was obviously decreased with all treatments. No statistical difference in protein expression rate was found between each of the treatments. This suggests that both SDT and US alone can trigger apoptosis, although SDT can prolong this initiating effect much longer. HMME treatment alone had no effect on initiating apoptosis.
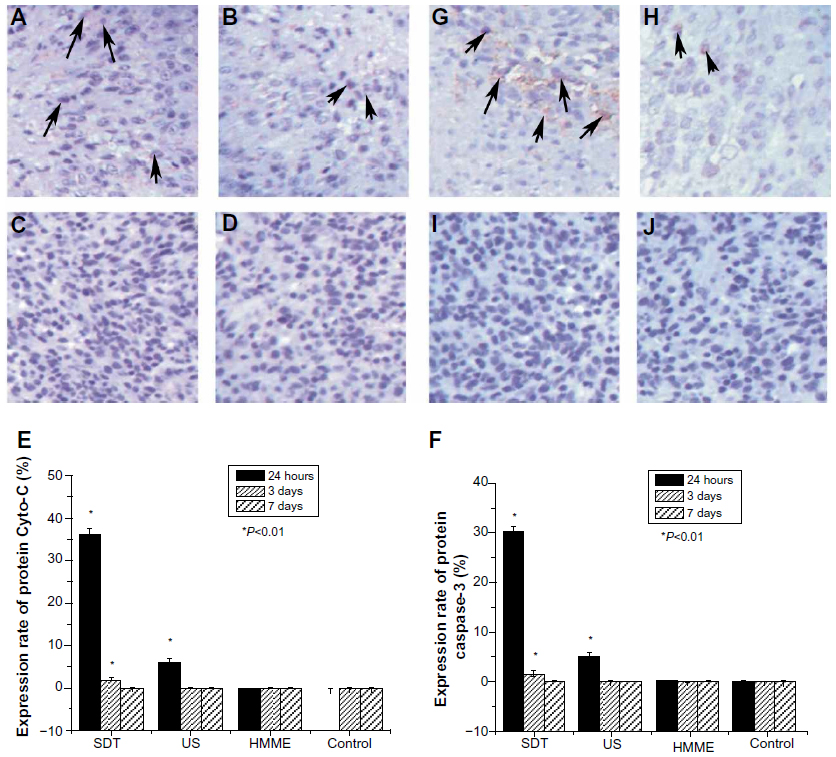 | Figure 5 Effect of sonodynamic therapy (SDT) treatment on the expression of protein cytochrome-c (Cyto-C) and protein caspase-3. Immunohistochemical staining with Cyto-C antibody at 24 hours after treatment with SDT (A), ultrasound (US) (B), and hematoporphyrin monomethyl ether (HMME) (C), or after no treatment (D). Summary of the effect of SDT on protein Cyto-C (E). Immunohistochemical staining with caspase-3 antibody at 24 hours after treatment with SDT (G), US (H), and HMME (I), or no treatment (J). Summary of the effect of SDT on protein caspase-3 (F). |
Determination of CD34 and VEGF expression
The inhibition of angiogenesis has been shown to be clinically successful in patients with cancer. Thus, the detection of response to antiangiogenic treatment is of high clinical relevance. To evaluate the clinical potential of SDT, in this study, the anti-angiogenic effects of different treatments were investigated. The “CD34” protein is a member of a family of single-pass transmembrane sialomucin proteins that show expression on early hematopoietic and vascular-associated tissue.19 Cells expressing CD34 are normally found in the endothelial cells of blood vessels and other tissues. In cell matrix and cancer nests, tube-like, line-like, or dot-like microvessels can usually be found. If one or a group of CD34-labeled endothelial cells is considered to be one microvessel, then MVD can be determined by measuring CD34 expression. “VEGF”, a signal protein produced by cells, stimulates vasculogenesis and angiogenesis.
The MVD and VEGF protein expression level were similar in rats treated with HMME alone and control rats at each of the examined time points (Figure 6). At 24 hours after treatment, MVD and VEGF protein expression showed a decreased trend in the US treatment alone group compared with in control rats and those treated with HMME alone; however, there was no statistical difference between these groups at 3 and 7 days after treatment. At 24 hours after SDT, the MVD and VEGF protein expression was much lower than at the same time point with any other treatment. The lamellar necrosis or sporadic punctiform necrosis cells were found in slices. By 3 days after treatment, the protein expression level of VEGF with SDT increased a little, but was still lower than with other treatments. At the same time, the size of the area of lamellar necrosis was observed to be clearly smaller than that at 24 hours after treatment, and the number of necrosis cells also decreased substantially. At 7 days after treatment, no statistical difference in the expression level of VEGF or MVD was found between each treatment group.
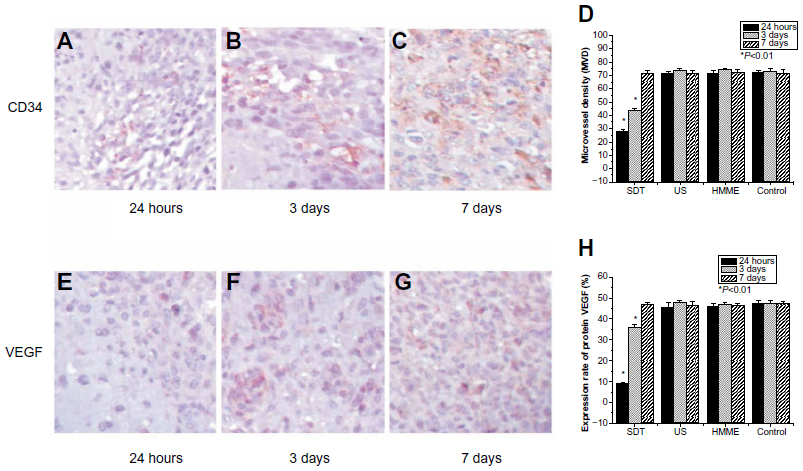 | Figure 6 Effect of sonodynamic therapy (SDT) treatment on glioma angiogenesis. Immunohistochemical staining with CD34 antibody at 24 hours (A), 3 days (B), and 7 days (C) after treatment with SDT. Summary of the effect of SDT, US and HMME on microvessel density (MVD) evaluated by counting CD34-positive microvessels (D). Immunohistochemical staining with vascular endothelial growth factor (VEGF) antibody at 24 hours (E), 3 days (F), and 7 days (G) after treatment with SDT. Summary of the effect of SDT, US and HMME on VEGF levels (H). |
Discussion
Malignant gliomas are one of the most aggressive forms of brain cancer. They are rather refractory to current therapeutic approaches including surgery, radiotherapy, chemotherapy, and immunotherapy.17 Recently, many new modalities have been clinically applied to the treatment of gliomas. SDT, one of the new approaches for cancer treatment, involves having a synergistic effect on cell damage through the combination of a sonosensitizer and US.20 SDT has been studied for more than 10 years. However, except in cell cultures, there are very few recent studies of US-mediated antitumor effects in combination with a sonosensitizer that have been performed on animal models.15,16,21 A previous study by our group showed that HMME-mediated SDT could effectively kill C6 glioma cells in vitro. In the study reported here, we used a C6 glioma rat in vivo model to investigate the antitumor effect of SDT and its possible mechanism.
“HMME” is a second-generation porphyrin-related photosensitizer. Experimental studies and clinical trials have demonstrated that HMME, which can be selectively taken up by tumor tissues has a stronger photodynamic effect, lower toxicity, and short-term skin photosensitizations. After injection, the HMME is unevenly distributed among different tissues, perhaps because of possible uneven absorbance and excretion rates. Although the mechanisms by which HMME selectively accumulates in tumors are complex and not fully understood, it is presumably because of the high vascular permeability of the agents, as well as their affinity for proliferating endothelia and the lack of lymphatic drainage in tumors. The fact that HMME accumulates well in glioma tissues presents the opportunity to use SDT on these tumors.13
In our study, the MRI results demonstrated that all the test animals had tumors of similar size prior to administration of the different study treatments, suggesting all the implanted individual rats were comparable (Figure 2). Three days after initial treatment, the volume of the tumors had clearly got smaller with the SDT and US-alone treatment, indicating that SDT and US alone can inhibit tumor growth in the early stages. However, no clear difference was observed with MRI between SDT and US-alone treatment (data not shown). This might be due to the apoptosis cells and necrotic tissue not having been replaced by normal tissue. Yet, this does not indicate SDT and US had similar inhibitive effect on tumor growth. At the interim stage following treatment (7 days), due to fast absorbance and tumors replaced by normal tissue, the size of the tumors had greatly decreased with SDT. However, US-alone treatment was unable to stop tumor growth after a while, indicating the inhibitive effect of US alone is weaker than that of SDT. At 14 days after cessation of treatment, all tumors in the different treatment groups had begun growing again. Some of them even grew bigger than what they were before treatment, suggesting that, even with SDT, multiple treatments may be needed to effectively control the tumor. With SDT, the antitumor effect reached its peak after 24 hours, implying that a 24-hour interval between each treatment might potentially be a good choice. The animal survival time data following each treatment also support the results evaluated by tumor volume change. The average survival time in the SDT group was 52.25 days, which was much longer than with the other treatments (Figure 2B), indicating that SDT not only inhibits tumor growth but may also have little impact on normal tissues. These C6 glioma rat model data demonstrate that SDT could be a potentially useful treatment for deep-seated malignant gliomas.
Previous studies have shown that SDT can kill C6 glioma cells in vitro through induction of apoptosis and necrosis.22 The HE staining and electron microscopy results of the present study suggest that apoptosis and necrosis may also be a way to kill glioma cells in vivo and control tumor growth (Figure 3).
The immunohistological and in situ apoptosis assay data show that tumor necrosis and the rate of apoptosis reached their peak at 24 hours after treatment with SDT. Afterwards, the area of tumor necrosis and rate of apoptosis gradually decreased. At about 7 days following treatment, tumor necrosis and apoptosis were found to have completely disappeared. This may indicate that apoptosis change complies well with tumor necrosis, and that apoptosis might be one of the ways that the glioma cells were killed by SDT.
Treatment with US alone can also induce tumor necrosis and apoptosis. The peak of the treatment effect is at around 24 hours after treatment. But the area of necrosis and apoptosis rate were clearly lower with US alone than with SDT, suggesting that US alone can still produce a weak antitumor effect by inducing apoptosis. In contrast, treatment with HMME alone did not show any antitumor effect.
Currently, the mechanism through which SDT induces apoptosis is far from clear. One possible reason for this is that US and SDT physically destroy tumor cell mitochondria and, further, release Cyto-C. Upon the release of Cyto-C into the cytoplasm, the protein binds apoptotic protease activating factor-1.23 In turn, this Cyto-C release activates caspase 9, a cysteine protease. Caspase 9 can then go on to activate caspase 3 and caspase 7, which are responsible for destroying the cell from within. Immunohistochemistry staining results showed that the levels of Cyto-C and caspase 3 greatly increased within 24 hours after treatment with SDT. US alone can also produce similar but weaker effect on Cyto-C and caspase 3 expression. This strengthens the suggestion that SDT or US-alone treatment can kill glioma cells by triggering the process of apoptosis in tumor cells.
In malignant gliomas, the blood vessels are extremely abundant. The activation and growth of endothelial cells are the prerequisite of angiogenesis in tumors. It is well known that angiogenesis is a fundamental process in tumor growth and metastasis. “VEGF”, a main regulator of angiogenesis in tumors, is a signal protein produced by cells that stimulates vasculogenesis and angiogenesis. Its expression level is positively correlated with the density of newly formed microvessels in gliomas.18 VEGF production can be induced in cells that are not receiving enough oxygen.24 When a cell is deficient in oxygen, it produces hypoxia-inducible factor (HIF), a transcription factor. Among its other functions (including modulation of erythropoiesis), HIF stimulates the release of VEGF. Circulating VEGF then binds to VEGF receptors on endothelial cells, triggering a tyrosine kinase pathway leading to angiogenesis. HIF1 alpha and HIF1 beta are constantly being produced, but HIF1 alpha is highly O2 labile, so, in aerobic conditions, it is degraded. When the cell becomes hypoxic, HIF1 alpha persists and the HIF1 alpha/beta complex stimulates VEGF release. In recent years, much effort has been put into anti-VEGF therapies for the treatment of certain cancers, such as gliomas. Thus, regulating VEGF expression may be a good approach to treating gliomas.
In our study, in the early phase of SDT (first 24 hours), the expression level of VEGF and MVD were significantly decreased. Three days after SDT, both the VEGF expression level and MVD were increased slightly, but these were still much lower than those in the control group. By 1 week after SDT, VEGF expression had returned to the normal level. This is probably because the implanted rats were only treated with SDT once. One SDT treatment would have been unable to completely destroy the tumor blood vessels or, further, induce necrosis of the whole tumor and all blood vessels. This might be improved if multiple SDT treatments were applied.
One of the interesting questions we sought to answer in this study was whether there was any difference between SDT and US-alone treatment. Is the role of HMME only to enhance the effects of US? In terms of animal survival time, apoptosis rate, and Cyto-C and caspase-3 expression levels, it appears that the sonosensitizer HMME enhanced the antitumor effect of US. However, with SDT, the gliomas grew much slower than with any of the other treatments. Even over a period such as 2 weeks, tumor volume remained almost constant with SDT (Figure 2), suggesting HMME may do more than just enhancing US effect. Some potential functions of sonosensitizers may still have not been discovered yet. In addition, to assist US to kill cancer cells, sonosensitizer HMME may also take a role on assisting US to inhibit glioma tissue angiogenesis in order to control glioma growth, since neither US alone or HMME has any effect on angiogenesis (Figure 6).
Conclusion
In our study, we found that SDT could effectively inhibit the expansion of intracranial gliomas in vivo, and that the corresponding mechanism was closely related to mechanical injury and the induction of apoptosis, which was also proved in vitro. US alone slightly inhibited the expansion of intracranial gliomas; however, it exhibited a markedly weaker inhibition effect compared with SDT. HMME alone had no inhibitive effect on intracranial gliomas.
SDT effectively induced glioma-cell apoptosis. The two possible mechanisms might be: 1) promoting mitochondria to release Cyto-C and activate Caspase-3, then to initiate apoptosis; 2) the destroying of microvessels, inhibition of angiogenesis, and, further, the induction of ischemia and anoxia of glioma cells, resulting in increased expression of HIF1 in glioma cells and apoptosis of glioma cells via the B-cell lymphoma 2 pathway. Most importantly, in addition to killing glioma cells, SDT also displayed a vessel-targeting effect that inhibited intracranial gliomas.
Disclosure
Our study was supported by National Natural Science Foundation of China (81072079), Research of Health Department of Heilongjiang Province (2013102) and Science, and Technology Commission of Fengxian District of Shanghai. The authors declare no conflicts of interest in this work.
References
Mamelak AN, Jacoby DB. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601). Expert Opin Drug Deliv. 2007;4(2):175–186. | |
Goodenberger ML, Jenkins RB. Genetics of adult glioma. Cancer Genet. 2012;205(12):613–621. | |
Umemura S, Yumita N, Nishigaki R, Umemura K. Sonochemical activation of hematoporphyrin: a potential modality for cancer treatment. IEEE Ultrasonic Symp. 1989;2:955–960. | |
Yumita N, Nishigaki R, Umemura K, Umemura S. Hematoporphyrin as a sensitizer of cell-damaging effect of ultrasound. Jpn J Cancer Res. 1989;80(3):219–222. | |
Kessel D, Lo J, Jeffers R, Fowlkes JB, Cain C. Modes of photodynamic vs sonodynamic cytotoxicity. J Photochem Photobiol B. 1995;28(3):219–221. | |
Sasaki K, Yumita N, Nishigaki R, Umemura S. Antitumor effect sonodynamically induced by focused ultrasound in combination with Ga-porphyrin complex. Jpn J Cancer Res. 1998;89(4):452–456. | |
Rosenthal I, Sostaric JZ, Riesz P. Sonodynamic therapy – a review of the synergistic effects of drugs and ultrasound. Ultrason Sonochem. 2004;11(6):349–363. | |
DeVita VT Jr, Rosenberg SA. Two hundred years of cancer research. N Engl J Med. 2012;366(23):2207–2214. | |
Yumita N, Nishigaki R, Umemura K, Umemura S. Synergistic effect of ultrasound and hematoporphyrin on sarcoma 180. Jpn J Cancer Res. 1990;81(3):304–308. | |
Miyoshi N, Igarashi T, Riesz P. Evidence against singlet oxygen formation by sonolysis of aqueous oxygen-saturated solutions of Hematoporphyrin and rose bengal. The mechanism of sonodynamic therapy. Ultrason Sonochem. 2000;7(3):121–124. | |
Dai S, Hu S, Wu C. Apoptotic effect of sonodynamic therapy mediated by hematoporphyrin monomethyl ether on C6 glioma cells in vitro. Acta Neurochir (Wien). 2009;151(12):1655–1661. | |
Tang W, Liu Q, Wang X, Wang P, Zhang J, Cao B. Potential mechanism in sonodynamic therapy and focused ultrasound induced apoptosis in sarcoma 180 cells in vitro. Ultrasonics. 2009;49(8):786–793. | |
Li JH, Song DY, Xu YG, Huang Z, Yue W. In vitro study of haematoporphyrin monomethyl ether-mediated sonodynamic effects on C6 glioma cells. Neurol Sci. 2008;29(4):229–235. | |
Zeng H, Sun M, Zhou C, et al. Hematoporphyrin monomethyl ether-mediated photodynamic therapy selectively kills sarcomas by inducing apoptosis. PLoS One. 2013;8(10):e77727. | |
Nonaka M, Yamamoto M, Yoshino S, Umemura SI, Sasaki K, Fukushima T. Sonodynamic therapy consisting of focused ultrasound and a photosensitizer causes a selective antitumor effect in a rat intracranial glioma model. Anticancer Res. 2009;29(3):943–950. | |
Jeong EJ, Seo SJ, Ahn YJ, Choi KH, Kim KH, Kim JK. Sonodynamically induced antitumor effects of 5-aminolevulinic acid and fractionated ultrasound irradiation in an orthotopic rat glioma model. Ultrasound Med Biol. 2012;38(12):2143–2150. | |
Rieger L, Weller M, Bornemann A, Schabet M, Dichgans J, Meyermann R. BCL-2 family protein expression in human malignant glioma: a clinical-pathological correlative study. J Neurol Sci. 1998; 155(1):68–75. | |
Ye SF, Zhong XY. [VEGF receptors and glioma angiogenesis]. Journal of Jinan University Natural Science and Medicine Edition. 2004;21(2):122–125. Chinese. | |
Nielsen JS, McNagny KM. Novel functions of the CD34 family. J Cell Sci. 2008;121(Pt 22):3683–3692. | |
Umemura S, Yumita N, Nishigaki R. Enhancement of ultrasonically induced cell damage by a gallium-porphyrin complex, ATX-70. Cancer Res. 1993;84(5):582–588. | |
Ohmura T, Fukushima T, Shibaguchi H, et al. Sonodynamic therapy with 5-aminolevulinic acid and focused ultrasound for deep-seated intracranial glioma in rat. Anticancer Res. 2011;31(7):2527–2533. | |
Song DY, Yue W, Zhao T, et al. Effect of sonodynamic chemistry therapy on C6 glioma cells in vitro. Chinese J Neurosurg. 2007; 23(2):107-110. | |
Kumarswamy R, Chandna S. Putative partners in Bax mediated cytochrome-c release: ANT, CypD, VDAC or none of them? Mitochondrion. 2009;9(1):1–8. | |
Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19(10):2003–2012. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.