")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Steric environment around acetylcholine head groups of bolaamphiphilic nanovesicles influences the release rate of encapsulated compounds
Authors Stern A, Guidotti M, Shaubi E, Popov M, Linder C, Heldman E, Grinberg S
Received 26 August 2013
Accepted for publication 4 October 2013
Published 20 January 2014 Volume 2014:9(1) Pages 561—574
DOI https://doi.org/10.2147/IJN.S53563
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Avital Stern,1 Matteo Guidotti,2 Eleonora Shaubi,1 Mary Popov,3 Charles Linder,4 Eliahu Heldman,3 Sarina Grinberg1
1Department of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel; 2Istituto di Scienze e Tecnologie Molecolari-CNR, Milano, Italy; 3Department of Clinical Biochemistry and Pharmacology, Ben-Gurion University of the Negev, Beer-Sheva, Israel; 4Zuckerberg Water Institute and Department of Biotechnology Engineering, Ben-Gurion University of the Negev, Beer-Sheva, Israel
Abstract: Two bolaamphiphilic compounds with identical acetylcholine (ACh) head groups, but with different lengths of an alkyl chain pendant adjacent to the head group, as well as differences between their hydrophobic skeleton, were investigated for their ability to self-assemble into vesicles that release their encapsulated content upon hydrolysis of their head groups by acetylcholinesterase (AChE). One of these bolaamphiphiles, synthesized from vernolic acid, has an alkyl chain pendant of five methylene groups, while the other, synthesized from oleic acid, has an alkyl chain pendant of eight methylene groups. Both bolaamphiphiles formed stable spherical vesicles with a diameter of about 130 nm. The ACh head groups of both bolaamphiphiles were hydrolyzed by AChE, but the hydrolysis rate was significantly faster for the bolaamphiphile with the shorter aliphatic chain pendant. Likewise, upon exposure to AChE, vesicles made from the bolaamphiphile with the shorter alkyl chain pendant released their encapsulated content faster than vesicles made from the bolaamphiphile with the longer alkyl chain pendant. Our results suggest that the steric environment around the ACh head group of bolaamphiphiles is a major factor affecting the hydrolysis rate of the head groups by AChE. Attaching an alkyl chain to the bolaamphiphile near the ACh head group allows self-assembled vesicles to form with a controlled release rate of the encapsulated materials, whereas shorter alkyl chains enable a faster head group hydrolysis, and consequently faster release, than longer alkyl chains. This principle may be implemented in the design of bolaamphiphiles for the formation of vesicles for drug delivery with desired controlled release rates.
Keywords: bolaamphiphiles, vesicles, drug delivery, controlled release, acetylcholine, acetylcholinesterase
Introduction
A variety of drug delivery systems (DDSs) are being developed with the goal of controlling the release rate of encapsulated drugs at a target site.1 A popular DDS, in clinical use, is based on phospholipids that form liposomes with bilayer membranes.2–3 Liposomes that are currently used in clinical applications passively deliver the drug to target organs, particularly tumors, due to the increased porosity of the blood vessels at the target site,4–7 and release their encapsulated drug in a noncontrolled manner. There are, however, continued efforts to develop improved liposomes that release their encapsulated drug as a result of a triggering event, such as increased temperature,8–10 changes in pH,11 alternating magnetic field,12 exposure to light,13 and exposure to specific enzymes.14–16 However, all these approaches involve DDSs in which the release rate is not controlled. There is a need for DDSs that will release drugs at their site of action at a desired rate, which would vary depending on the encapsulated drug and the medical indication treated by the drug. Optimal liposomes, or vesicles for drug delivery, should be stable so that the encapsulated material will not leak out at nonrelevant sites, but they have to be destabilized in a controlled manner at the target site. Bolaamphiphiles, compounds with two hydrophilic head groups connected to the two ends of a hydrophobic alkyl chain, are good building block candidates for such vesicles, since a) they could form encapsulating monolayer membranes17,18 that are characteristically more stable than bilayer membranes due to a high energy barrier for lipid exchange, and b) it is possible to provide such vesicles with a controlled release mechanism due to their tendency to switch from a vesicular structure to nanotubes or cylinders upon changes in their head groups.19,20 Indeed, vesicles made from a variety of bolaamphiphiles have been prepared for various applications, such as for molecular recognition,21 controllable release,22 targeted drug delivery, and imaging.23
Recently, we have described novel nanovesicles made of synthetic bolaamphiphiles with acetylcholine (ACh) head groups16,24 that are capable of releasing their encapsulated content upon exposure to acetylcholinesterases (AChEs).25–27 When AChE hydrolyzes the ACh surface groups of such vesicles, the vesicular structure is disrupted, resulting in a release of the encapsulated material. Since the hydrolysis of the head groups causes the disruption of the vesicular structure,25 we postulated that the rate of the head group hydrolysis would determine the release rate of the encapsulated drug. We further hypothesized that the rate of the head group hydrolysis may be varied by changing the molecular environment around the ACh head group, and thus affecting the interaction between the ACh head group and the hydrolyzing enzyme, AChE.28,29 One way of changing the molecular environment of the ACh head group is to change the alkyl chain pendant near the head groups of these bolaamphiphiles, and thus interfere sterically with the interaction between the substrate and the enzyme. To test this possibility, we synthesized two bolaamphiphiles with different lengths of alkyl chain pendants adjacent to the ACh head group and compared them with respect to the hydrolysis rate of the ACh head group by AChE. Then we examined the capability of these bolaamphiphiles to form vesicles that release their encapsulated material at different rates, according to the rate of the head group hydrolysis.
Materials and methods
Materials
Standard chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA). AChE (from electric eel, type V-S) was purchased from Sigma-Aldrich (catalog no C2888) and was dissolved in distilled water at a concentration of 1 mg solid/mL (1,047 units/mg solid), and adjusted to a concentration of 200 units per mL just before the experiment. GLH-20 was synthesized from vernonia oil (containing about 70% vernolic acid with naturally occurring epoxy groups),30 that was obtained from Ver Tech International (Rockville, MD, USA), according to the procedure described by us previously.16
Synthesis of GLH-32
Synthesis of 9,10-epoxy methyl stearate (1)
Methyl oleate (Sigma-Aldrich, purity 99%) was used as received. Ti-MCM-41 (1.8 wt% Ti) was prepared as described previously,31 and calcinated at 773 K under dry air before use. The epoxidation reaction was carried out in a glass batch reactor (stirring rate 500 rpm) at 363 K in dry ethyl acetate (Carlo Erba, Cornaredo, Italy). Anhydrous tert-butyl hydroperoxide (TBHP; Sigma-Aldrich, 5 M solution in decane) was used as oxidant for the fatty acid methyl esters (FAME). The molar ratio of the TBHP/FAME was 1.4. The reaction was followed by Gas Chromatography with a Flame Ionization Detector (GC-FID) analysis (HP5890; HP-5 column, 30 m × 0.25 mm). The desired 9,10-epoxy methyl stearate (compound 1 in Figure S1) was isolated from the final mixture, after filtration of the catalyst, by treatment with sodium thiosulfate and concentration under vacuum.32
Synthesis of 9,10-epoxy stearic acid (2)
A mixture of 2.4 g (7.7 mmol) of 9,10-epoxy methyl stearate (1), 0.8 g of KOH (85%), 1 mL of H2O, and 6 mL of ethanol (95%) was refluxed for 1 hour. The reaction mixture was cooled to 0°C, and 50 mL of water and 150 mL of diethyl ether were added and acidified with a solution of 16% HCl until pH=3.8. After separation of the phases, the organic phase was washed with water and brine, dried over anhydrous MgSO4, filtered, and the solvent removed under reduced pressure. The product was purified by flash chromatography on silica gel 60 (40–63 micron) using a mixture of hexane:ether:acetic acid (8:2:0.05) as the eluent. The yield of 9,10-epoxy stearic acid (compound 2 in Figure S1) was 1.5 g (5.0 mmol, 65.4%), purity 97% (determined by high-performance liquid chromatography [HPLC]). Fourier transform infrared spectroscopy (FT-IR) (neat, cm−1): 1700 (COOH), 845 and 820 (epoxy group) 1H NMR (nuclear magnetic resonance) (500 MHz δ ppm): 2.93–2.91 (2H, m, epoxy protons), 2.37–2.34 (2H, t, J=7.45 Hz CH2COOH), 13C NMR (500 MHz, CDCl3) δ ppm: 175.78 (COOH), 57.27 (epoxy carbons), 33.89 (CH2COOH), 24.14 (CH2CH2COOH). Anal Calcd (analysis calculated) for C19H36O3: epoxy group, 14.1%; Found: epoxy group 14.6%. Electrospray ionization (ESI) with mass spectrometry (MS) (negative mode): m/z=296.8 [M–1]–.
Synthesis of decane diepoxy distearate (3)
A mixture of 1.5 g (5 mmol) of 9,10-epoxy stearic acid (compound 2 in Figure S1), 0.43 g (2.5 mmol) of 1,10 decandiol, and 0.043 g of immobilized Candida antarctica lipase B (Novozym 435) in 100 mL of toluene was refluxed with azeotropic distillation under reduced pressure (130–170 mmHg) for about 7 hours. After cooling the reaction mixture, the lipase was filtered off and most of the solvent was removed under reduced pressure. Then 200 mL of methanol was added and the obtained suspension was left overnight in the refrigerator. The precipitate was filtered and washed with cold methanol to give 0.94 g (1.4 mmol, yield 25%) of product 3, with a 99.3% purity (determined by HPLC).
Thin-layer chromatography (TLC) – eluent system – petroleum ether (60°C–80°C): diethyl ether–7: 3.
IR (neat, cm−1): 1727 (RCOOR), 1,178 cm−1(CH2O(CO)), 845, 821 (epoxy group), Anal Calcd for C46H86O6 Epoxy group, 11.4%, Found: epoxy group, 12.9%.

Synthesis of the dichloroacetate of diepoxy distearate (4)
A mixture of 0.92 g of diepoxy distearate (compound 3 in Figure S1) (1.25 mmol) and 1.37 g (14.4 mmol) of chloroacetic acid in 25 mL of toluene was heated at 85°C for 48 hours. After cooling, toluene (150 mL) was added and the reaction mixture was washed first with a solution of 6% NaHCO3 and then water. The organic phase was separated and dried over anhydrous MgSO4, and the solvent was removed under reduced pressure to give 1.0 g (1.1 mmol, 86.8% yield) purity 92% (determined by HPLC) of product 4.
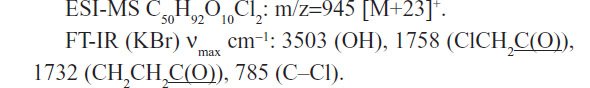

Quaternization stage for the bolaamphiphile GLH-32
A mixture of 1 g (1 mmol) of dichloroacetate diepoxystearate (compound 4 in Figure S1) and 1.8 g (13.7 mmol) of N,N-dimethylaminoethyl acetate was stirred at 65°C–75°C for about 6 hours.
After cooling the reaction mixture, diethyl ether was added and the mixture was left to stand overnight in the freezer. After decantation of the solvent, the product was dissolved in chloroform, diethyl ether was added, and the solvent was decanted again. The procedure was repeated three more times to give 1.1 g of bolaamphiphile GLH-32 in a 92.3% yield.

Characterization of synthesized compounds
Epoxy groups were determined by potentiometric titration.33 FT-IR analysis was carried out on a Nicolet Impact 410 spectrometer (Nicolet Instrument Corporation/Thermo Scientific, Waltham, MA, USA). 1H and 13C NMR (500 MHz) spectra were recorded on Brucker WP-500 SY spectrometers (Bruker Biosciences Corporation, Billerica, MA, USA) in CDCl3 with tetramethylsilane (TMS) as the internal standard or d6 DMSO (deuterated dimethyl sulfoxide) solutions. HPLC analysis was carried out on a C18RP column (Macherey-Nagel ES 250/4 Nucleosil 100-5C18AB; Macherey-Nagel, Düren, Germany) with an evaporative light scattering detector (evaporation temperature 46°C; mobile phase methanol:water [9:1, v/v]; flow rate 0.5 mL/minute). MS analysis was carried out on a Waters Micromass Q-TOF Premier Mass spectrometer (Waters, Milford, MA, USA).
Vesicle formation
Vesicles were prepared from the bolaamphiphiles by film hydration followed by sonication or consecutive extrusions through microporous membranes with pores of 200 nm and 100 nm, respectively, using common methods for the preparation of liposomes.34,35 Briefly, the vesicle formulation contained 10 mg/mL bolaamphiphile, 1.6 mg/mL cholesterol, and 2.1 mg/mL cholesteryl hemisuccinate (a molar ratio of 2:1:1, respectively). These components were dissolved in chloroform and the organic solvent was evaporated under vacuum to form a thin film. Hydration of the thin film was done by phosphate buffered saline (PBS). For encapsulation of carboxyfluorescein (CF) the hydrating solution contained 1 mg/mL of the fluorescent dye.
Vesicle characterization
Fluorimetric measurements were performed with a Cary Eclipse spectrofluorimeter, and spectrophotometric measurements were performed with a Cary 50 scan ultraviolet (UV)/visible spectrophotometer (both Varian, Palo Alto, CA, USA).
Dynamic light scattering (DLS) was measured by a High Performance Particle Sizer-Non Invasive Back-Scatter (HPPS-NIBS), light scattering apparatus (ALV-Laser, Langen, Germany) with the laser powered at 3 mW helium–neon (HeNe) laser line (632.8 nm).
For transmission electron microscopy (TEM), a drop of vesicle suspension was placed on a Formvar-coated copper grid, air-dried, negatively stained with a drop of 0.5% uranyl acetate solution in water, and dried again at room temperature. Visualization of the samples was performed by JEOL JEM-1230 TEM (JEOL, Tokyo, Japan) at 80 kV and TEM micrographs were taken using TemCam-F214 (Tietz Video and Image Processing Systems, Gauting, Germany).
Determination of vesicle stability
Vesicles with encapsulated CF were incubated for various times in PBS and the amount of the encapsulated CF was measured by determining fluorescence of the vesicle suspension, as a function of incubation time, before and after the addition of Triton X100.24 When AChE-induced release of encapsulated CF was measured, the fluorescence of the vesicle suspension was measured for several minutes until a stable fluorescence reading was obtained. Then, the enzyme AChE was added to the vesicle suspension and the fluorescence measurements continued for an additional 7–8 minutes, monitoring the rise in the fluorescence as an indication for CF release (the encapsulated CF is quenched and is dequenched upon its release, emitting a fluorescent signal). After that, Triton X100 was added to disrupt the remaining intact vesicles, in order to determine the total CF encapsulation. The percentage of CF released was determined at different time points after the addition of AChE, by calculating the amount of CF released at a given time compared to the total encapsulated CF.
Measurements of ACh head group hydrolysis
The rate of the ACh head group hydrolysis was determined by a modification of the pH-stat method previously used by us.25 Briefly, the pH of the bolaamphiphile, suspended in water, was measured for several minutes until a stable reading was achieved. Then 0.1 unit of AChE was added with a continuous pH measurement for 11 minutes. The change in the pH was transformed to moles of H+ released as a function of incubation time.
Spectroscopic determination of acetylthiocholine hydrolysis
Hydrolysis of acetylthiocholine (ATC) by AChE yields thiocholine and acetic acid. The thiocholine reacts with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) to give a yellow product that was determined quantitatively by absorbance measurements at 412 nm.36 For this determination, 0.64 mM ATC, 0.3 mM DTNB, 0.8 μL AChE (0.1 u/μL), and 5 μL ethanol were added to 1 mL of low ionic strength PBS and placed in a spectrophotometer. The change in absorbance at 412 nm as a function of time was measured continuously, on line, for 300 seconds. To test the effect of bolaamphiphiles on the rate of ATC hydrolysis, 5 μL of the tested derivative dissolved in ethanol was added to the reaction mixture.
Lineweaver–Burk analysis
Lineweaver–Burk plots were calculated using the data obtained from measurements of the hydrolysis rates of ATC by AChE (as described above) at several concentrations of ATC in the presence or in the absence of three different concentrations of the tested bolaamphiphile.
Results and discussion
Synthesis of bolaamphiphiles
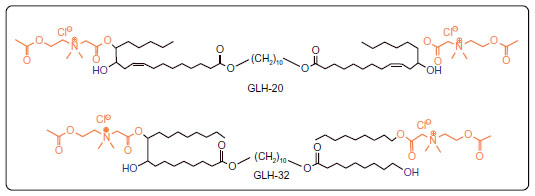
The starting materials for the synthesis of the bolaamphiphiles GLH-20 and GLH-32 (Figure 1) were vernolic acid (cis-12,13 epoxy-cis-9-octadecenoic acid)37 and oleic acid,32 respectively. The ACh head groups of GLH-20 were previously shown by us to be hydrolyzed by AChE.16 In order to compare the hydrolysis rate of GLH-20’s head group to a similar ACh head group of a bolaamphiphile with a longer alkyl chain pendant adjacent to the ACh head group, we had to synthesize a novel bolaamphiphile with the required features. We used oleic acid to synthesize the new bolaamphiphile, which was named GLH-32 (Figure 1).
 | Figure 1 Chemical structures of GLH-20 and GLH-32. |
For the synthesis of GLH-32, the methyl ester of oleic acid was first epoxidized, using an approach previously described by Guidotti et al,31,32 based on the acid-free epoxidation with tert-butyl hydroperoxide on a heterogeneous catalyst, and yielding 9,10-epoxy methyl stearate. The synthetic strategy comprised two main steps: a) synthesis of the bolaamphiphile’s skeleton by elongation of the corresponding fatty acid through its carboxylic group in an esterification reaction; and b) head group addition through the functional groups on the fatty acid aliphatic chain.
Bolaskeleton formation
To synthesize skeleton (3) (Figure S1A) of the bolaamphiphile, GLH-32, we used the epoxy methyl stearate (1) synthesized by epoxidation of methyl oleate in the presence of grafted titanium-containing silica materials as the catalyst.31 The epoxy methyl stearate (1) was hydrolyzed to obtain the mono epoxy stearic acid (2). MS in negative mode showed m/z=296.8 [M–1]+. A peak at 1,700 cm−1 appeared in the infrared (IR) spectra, indicating the presence of the carboxylic group. NMR spectroscopy showed that the epoxy group remained unreacted (2.93–2.91 ppm), and the α-methylene group of the carboxylic acid CH2COOH appeared at 2.37–2.34 ppm. The epoxy stearic acid (2) was reacted, using a chemo enzymatic reaction, with stoichiometric amounts of an aliphatic diol, 1,10-decanediol, in toluene, in the presence of immobilized Candida antarctica lipase as the catalyst.38 The product, decane diepoxy distearate (3), is the skeleton of the bolaamphiphilic compound.
The FT-IR spectrum of the diester (3) showed the disappearance of the absorption band at 1,700 cm−1, which is related to the carboxylic acid group, and the appearance of the absorption band at 1,727 cm−1, characteristic of the new ester group. The new alkoxy methylene group CH2OC(O) appeared at 4.04 ppm in the 1H-NMR spectrum and at 64.4 ppm in the 13C-NMR. The epoxy group remained unchanged.
Attachment of the head group
After synthesizing the decane diepoxy distearate as the bolaamphiphile’s skeleton, the head groups were attached in a two-stage reaction (Figure S1B): 1) Opening the epoxy ring with chloroacetic acid to give the dichloroacetate derivative (4); and 2) quaternization stage of N,N-dimethylaminoethyl acetate with compound 4 (Figure S1) to give the bolaamphiphile GLH-32 with two ACh head groups bound on opposite ends of the hydrophobic chain through the nitrogen of the choline moiety.
The diepoxy distearate (3) was reacted with an excess of chloroacetic acid in dry toluene at 85°C for 48 hours. The reaction progress was followed by TLC and HPLC. To remove the excess chloroacetic acid, the reaction mixture was washed with a concentrated solution of NaHCO3, and the product purified by flash chromatography. The FT-IR spectrum of the dichloroacetate derivative (4) showed a new absorption band of the chloroacetate group at 1,758 cm−1 and carboxylic ester absorption band at 1732 cm−1 of the starting diester. In the 1H NMR spectrum the following new signals appeared: a peak at 4.09 ppm of the methylene protons of the chloroacetate group (CH2Cl); a peak at 4.87–4.91 ppm of the proton of the new ester group (CHOC(O)); and a peak at 3.60 ppm of the proton near the hydroxyl group CHOH group (Figure 2). The corresponding chemical shifts in the 13C NMR spectrum appeared at 40.96 ppm (CH2Cl) for the chloroacetate group, at 72.32 ppm for the carbon near the hydroxyl group (CHOH), at 78.81 and 78.89 ppm for the carbon adjacent to the new ester group (CHOCOCH2Cl) and at 167.20 ppm for the carbonyl carbon of this new ester group. The formation of structural isomers in compound 4 (Figure S1) was identified by the two peaks at 78.81, 78.89 for CHOC(O) in the 13C NMR spectrum, confirming previously reported data regarding the presence of structural isomers for the chloroacetate of methyl vernolate.37 The presence of structural isomers can also be followed from the terminal methyl group at 0.87 ppm and the α-methylene carbonyl group CH2C(O)O at 2.27 ppm; both appear as two triplets.
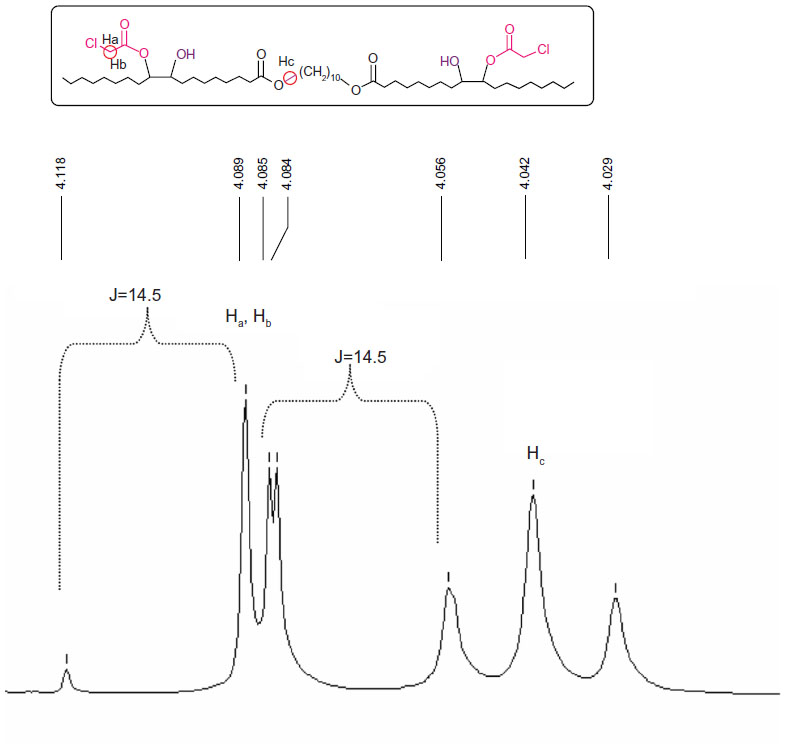 | Figure 2 Chemical shifts of the chloromethylene (CH2Cl) and alkoxymethylene (C(O)OCH2) groups of compound 4. |
Figure 2 shows that due to the proximity of the chiral carbon, the two protons of the chloromethylene group are diastereotopic hydrogens, and they split each other. Two doublets were obtained, one for the Ha proton and the second one for the Hb proton, with one of the signals overlapping with the triplet of the alkoxy methylene group of the original ester.
The last stage of the synthesis is the quaternization reaction of N,N-dimethylamino ethyl acetate with the dicholoro acetate 4 (Figure S1B) that yields the final bolaamphiphile, GLH-32 with two ACh head groups. The reaction was carried out with a large excess of the amine at 45°C for 6 hours, followed up by repeated washings with ether to remove the excess of the tertiary amine. The desired bolaamphiphile was obtained as a yellow viscous product.
The 1H-NMR of the bolaamphiphilic compound (Figure 3) can distinguish the new peaks of the ACh head group. The methyl (24) of the acetate CH3C(O)O appeared as a singlet at 2.12 ppm. The methylene group (22) N+CH2CH2O near the quaternary nitrogen appeared at 4.25 ppm and the methylene group (23) N+CH2CH2OC(O) near the oxygen appeared at 4.60 ppm. The two methyl groups (21) of the quaternary nitrogen appeared as two singlets at 3.61 and 3.62 ppm, while the two different protons of the methylene group (20) between the quaternary nitrogen and the carbonyl OC(O)CH2N+ both appeared as a multiplet at 4.79 and 5.46 ppm.
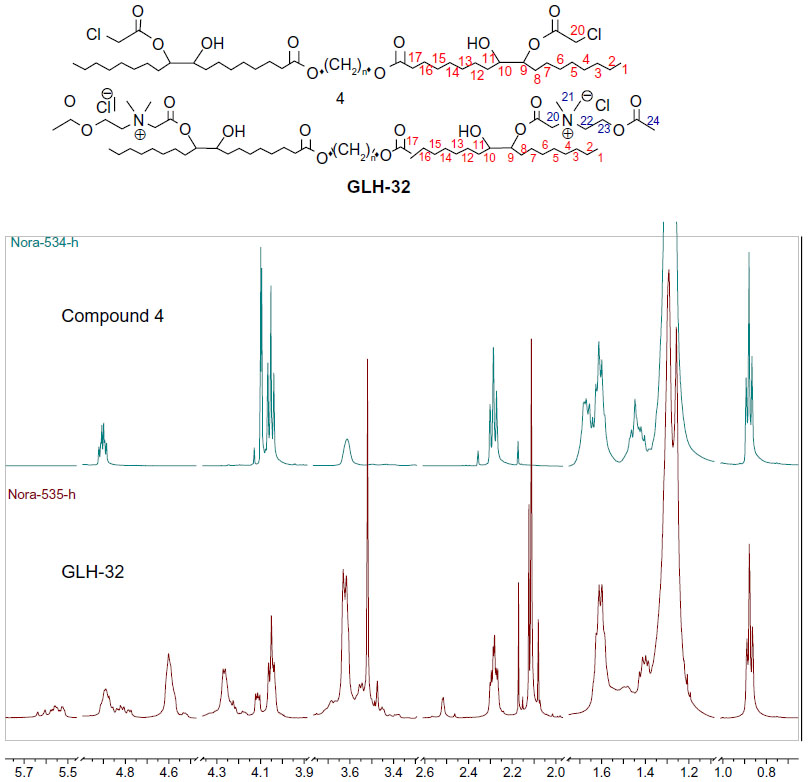 | Figure 3 Comparison of the nuclear magnetic resonance (NMR) spectrum in CDCl3 of the dichloroacetate intermediate compound 4 and the bolaamphiphile GLH-32. |
Enzymatic hydrolysis of the ACh head groups by AChE
In a previous study we demonstrated that choline esters, covalently attached to the two ends of a bolaamphiphilic skeleton via the nitrogen atom of the choline moiety, are hydrolyzed when exposed to the enzyme AChE.25,39 ACh head groups attached to bolaamphiphiles via the nitrogen atom of the choline moiety were hydrolyzed regardless if the alkyl chain of the skeleton contained 30 atoms,39 or 38 atoms,25 suggesting that long alkyl chains that contain 30 atoms or more enable the ACh head group (which is covalently attached to them), to reach the bottom of the catalytic gorge of AChE so that they can be hydrolyzed. Due to steric considerations, it is expected that the interaction between the ACh head group and the active site of AChE would be influenced by chemical moieties, which are situated near the head group. In fact, it is well known that because of the nature of the acyl pocket in the enzyme, the bulky acyl group of the substrate (eg, acetyl, propionyl, butyryl, etc) affects its hydrolysis rate.40 Thus, larger or longer alkyl groups cannot fit into the acyl pocket subsite and be accommodated correctly for nucleophilic attack by the active site’s serine.41,42 In addition, the choline group of the substrate has to be stabilized by the choline binding site at the bottom of the catalytic gorge of AChE and the presence of a substituent near the ACh head group could interfere with this stabilization by preventing efficient binding of the substrate.43 We hypothesized that an alkyl chain, with one end free, which will be situated near the head group, may be a substituent that will affect the interaction of the head group with the enzyme, either similarly to the action of bulky acyl moieties, or by interfering with the stabilization of the choline moiety of the head group at the choline binding site of AChE. We further hypothesized that the length of the alkyl chain adjacent to the ACh head group may influence the degree of the interference with the interaction between the ACh head group and the active site of AChE; therefore, it should also affect the rate of the head group hydrolysis. To test our hypothesis, we used the head groups of GLH-20 and GLH-32 as substrates and measured their hydrolysis, using concentrations of the bolaamphiphiles below their critical aggregation concentration (CAC). Below the CAC, the bolaamphiphiles are at their monomeric form, and therefore aggregated structures do not interfere with the access of the entire bolaamphiphile to the enzyme. The reaction was carried out in water so that changes in the pH could be easily detected and ions did not interfere with the hydrolysis. As can be seen from Figure 4, the head groups of both bolaamphiphiles (GLH-20 and GLH-32) were hydrolyzed by AChE, but the rate of the hydrolysis of GLH-20’s head group was faster than that of GLH-32’s head group, indicating that a longer alkyl chain near the ACh moiety retards the rate of the head group’s hydrolysis more than a shorter alkyl chain. In comparison, the hydrolysis of free ATC, an analog of ACh that like ACh does not contain an adjacent alkyl chain, is much faster than the hydrolysis rate of the ACh head groups of both bolaamphiphiles (Figure 4). ATC was used in this experiment, rather than ACh, since its hydrolysis could be determined by two independent methods: the pH stat method;29 and the Elman reagent.29,36 Therefore, our measurements with the pH stat method could have been confirmed by the spectroscopic method, and also compared to the results of the experiment in which we investigated possible competition between ATC and the ACh head groups of both bolaamphiphiles (see Figure 5). Since the hydrolysis rates of ACh and ATC by AChE and their Michaelis–Menten constant (Km) values are similar,29 our comparison of ATC to ACh head group is legitimate and valid. Our results showed that the order of the hydrolysis rates is as follows: ATC>ACh head group of GLH-20>ACh head group of GLH-32. This order of the hydrolysis rates is consistent with our hypothesis that an alkyl chain adjacent to the ACh head group interferes with the interaction of the head group with AChE, and thus affects the hydrolysis rate. The longer the alkyl chain, the slower the hydrolysis of the adjacent ACh head group by AChE.
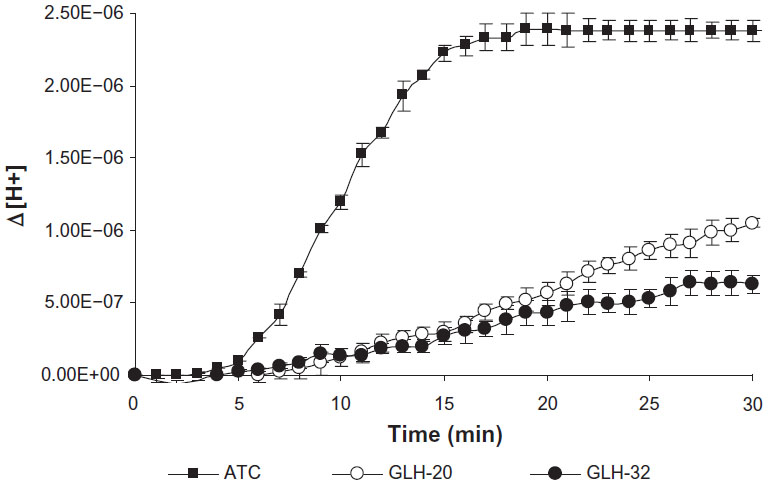 | Figure 4 Hydrolysis of the ACh head groups of GLH-20 and GLH-32 by AChE. Hydrolysis was determined by measuring the pH change after the addition of AChE (which was added 4 minutes after the beginning of the pH measurements) to the incubation medium and was converted to a change in the proton concentration. |
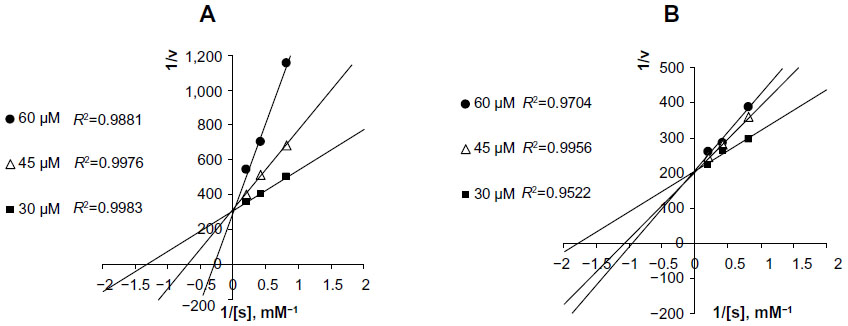 | Figure 5 Lineweaver–Burk plots of ATC hydrolysis by AChE in the presence of several concentrations of GLH-20 (A) and GLH-32 (B). |
The difference in the hydrolysis rates between GLH-20 and GLH-32 can be attributed to several differences that exist between these two bolaamphiphiles. As can be seen from Figure 1, GLH-20 is different from GLH-32 in the following three molecular features: a) the alkyl chain pendant (with a free end), which is bound to the bolaamphiphile near the head group is of five carbon members in GLH-20 and of eight carbon members in GLH-32; b) the alkyl chain skeleton that connects the two ACh head groups contains 38 atoms (36 carbon atoms and two oxygen atoms) in GLH-20 and 32 atoms (30 carbon atoms and two oxygen atoms) in GLH-32; and c) the alkyl chain of the skeleton of GLH-20 contains two double bonds and the alkyl chain of the skeleton of GLH-32 is fully saturated. In principle, every one of these differences, separately or in combination, may account for the different hydrolysis rates of the head groups of the two bolaamphiphiles. With respect to the length of the alkyl chain that connects the two ACh head groups (in fact, the alkyl chain of the bolaamphiphilic skeleton), it seems that both alkyl chains of GLH-20 and GLH-32 are long enough to enable their ACh head group to reach the bottom of the gorge, as shorter alkyl chains such as those of decamethonium and BW284C51 (inhibitors of ACh that bind to the active site of the enzyme) enable their quaternary moiety to reach the bottom of the gorge of the catalytic site.42 Also, our previous studies have shown that the ACh head groups of bolaamphiphiles with hydrophobic skeleton shorter than that of GLH-32 are also well hydrolyzed by AChE. These results argue that the difference in the length of the alkyl chain of the skeleton is not a major factor responsible for the difference in the hydrolysis rates. Also, the presence of the double bonds in the alkyl chain of the bolaamphiphilic skeleton of GLH-20 does not seem to significantly affect the hydrolysis rate, as bolaamphiphiles with different moieties within the alkyl chain of their skeleton are well hydrolyzed by AChE.25,39 Moreover, the surface head group of GLH-20 is also hydrolyzed by AChE when the bolaamphiphile is aggregated into vesicles,29 and the alkyl chain of the bolaamphiphilic skeleton has no access to the enzyme as it is buried deep in the monolayer membrane (see below results obtained with intact vesicles described in Figure 9). These results exclude a significant contribution of the alkyl chain of the bolaamphiphilic skeleton to the difference in the hydrolysis rate of the ACh head group of GLH-20 and GLH-32 and leave the alkyl chain pendant, which is adjacent to the head group, as the most likely candidate that affects the hydrolysis rate.
The ability of AChE to hydrolyze the ACh head groups of both bolaamphiphiles (GLH-20 and GLH-32) suggests that the ACh head groups of both bolaamphiphiles are substrates for the enzyme, and thus both head groups bind to the active site of AChE. We assumed that as a substrate, the ACh head group of the bolaamphiphiles reacts with AChE in much the same way as does the substrate ACh, binding at both the anionic and the esteratic sites on the enzyme. Therefore, the two substrates, the free ACh and the ACh head group, are expected to compete for the same enzyme form, suggesting a mode of competitive inhibition. If the alkyl chain pendant, which is adjacent to the ACh head group, has an effect on the interaction between the head group and the enzyme, then bolaamphiphiles that contain alkyl chain pendant of different lengths (as GLH-20 and GLH-32) should differently affect the Km of the substrate with which they compete.
To compare between the inhibitory potentials of GLH-20 and GLH-32, the rate of ATC hydrolysis was measured in the presence of three concentrations of each of these bolaamphiphiles, and the kinetics were analyzed using a Lineweaver–Burk plot. The results (Figure 5) show that both GLH-20 and GLH-32 inhibit the hydrolysis of ATC in a competitive manner, as increasing their concentrations affected the Km, but not the maximum rate (Vmax) of ATC hydrolysis. However, GLH-20 (Figure 5A) affects the Km significantly more than the same concentrations of GLH-32 (Figure 5B), indicating that GLH-20’s head group has higher affinity to the enzyme than GLH-32. This finding suggests that the head group of GLH-20 is a better substrate for AChE than that of GLH-32; therefore, the hydrolysis rate of its head group is faster than the hydrolysis rate of GLH-32’s head groups.
The results described in Figure 5 are consistent with results published in a previous study,25 where similar competitive inhibition was described for bolaamphiphiles with ACh head groups. In fact, unlike the mixed type of inhibition shown for ACh inhibitors, such as decamethonium and BW284C51,42 competitive inhibition for compounds that are hydrolyzed by AChE in a similar mode of action to that of ACh has been described for several substrates of AChE that also act as inhibitors of the enzyme. For example, in neostigmine (a reversible inhibitor of AChE), hydrolysis occurs in a similar manner to that of ACh, splitting off the quaternary ammonium phenol and leaving the carbamoylated enzyme, which is hydrolyzed to regenerate the free enzyme. The hydrolysis of the carbamoylated enzyme is similar in mechanism to that of the acylated enzyme (a form of the enzyme produced when the enzyme reacts with ACh), but is considerably slower than the acylated enzyme. This mode of action agrees well with competitive inhibition, as has been described for neostigmine,44 as well as for the bolaamphiphiles, as described in Figure 5.
Vesicle formation and characterization
Amphiphiles in general, and specifically bolaamphiphiles, can aggregate into micelles, multilayered sheets, spherical vesicles, rings, or a variety of nanostructures with cylindrical geometry, such as rods, tubules, ribbons, and helices.18 The morphology of the self-assembled structure is a function of the molecular parameters of the specific bolaamphiphile. As can be seen in Figure 6, both bolaamphiphiles, GLH-20 and GLH-32, formed spherical vesicles, with an average diameter of about 130 nm.
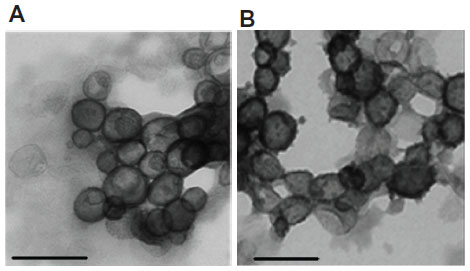 | Figure 6 TEM micrographs of vesicles formed from GLH-20 (A) and GLH-32 (B). |
Size distribution determined by DLS showed an average diameter of 125±3.7 nm for GLH-20 vesicles and 140±8.2 nm for GLH-32 vesicles. The vesicle size did not change during a 2-week incubation at room temperature (Figure 7), indicating that no aggregation nor fusion occurred during storage.
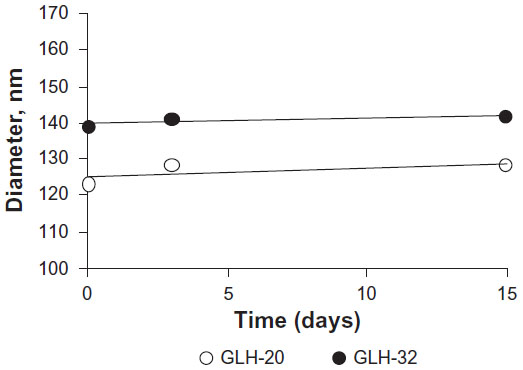 | Figure 7 Vesicle size as a function of time in storage as determined by DLS. |
Both GLH-20 and GLH-32 vesicles were positively charged due to their cationic ACh head groups. Accordingly, the zeta potentials of GLH-20 and GLH-32 vesicles were 55.7±2.25 mV and 41.9±1.13 mV, respectively. The slightly lower zeta potential of the GLH-32 vesicle is consistent with its larger outer surface area compared to GLH 20 vesicles.
Vesicle stability
Vesicle stability was evaluated by measuring the amount of encapsulated CF as a function of various incubation times in PBS at room temperature. Vesicles from formulations that contained GLH-20 and GLH-32 remained stable for at least 16 days (the last time point of our measurements), without changes in the amount of their encapsulated CF (Figure 8).
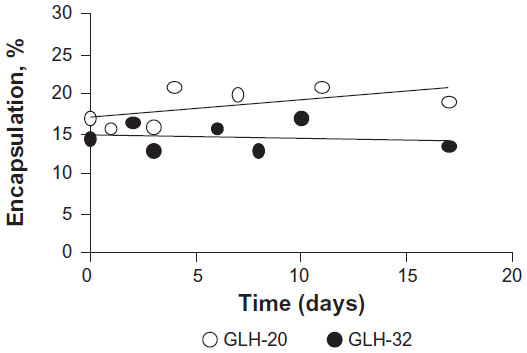 | Figure 8 Vesicle stability determined by monitoring the changes in percent encapsulation as a function of time. |
Release of encapsulated CF from vesicles by AChE
In a previous study, we have shown that the hydrolysis of the surface head groups of bolaamphiphilic vesicles leads to destabilization of the vesicular structure and release of the encapsulated CF to the medium.25 The fluorescence of the encapsulated CF, which is present in relatively high concentrations in the vesicle’s core, is quenched, but when it is released, it emits a fluorescence signal due to dequenching that occurs following its dilution in the bathing medium.Using the same technique, we measured the rate of CF release from GLH-20 and GLH-32 vesicles following their exposure to AChE, and determined if the release rate correlates to the rate of the hydrolysis of the ACh head group. Figure 9 shows that upon exposure of CF-encapsulated vesicles to the enzyme AChE, an increase in the fluorescent signal is observed for both GLH-20 and GLH-32 vesicles, suggesting that the vesicles released their encapsulated CF into the medium.
For both GLH-20 and GLH-32 vesicles, the release rates were biphasic, with an initial rapid release immediately after the addition of the enzyme, followed by a slower and constant rate after about 20–50 seconds. To ensure that the biphasic release rate is not due to insufficient enzyme, which may have been depleted during the incubation period, we added ten times more enzyme and obtained very similar results, indicating that the amount of the enzyme that was used in the experiment described in Figure 9 was saturating. One possible explanation for the biphasic feature of the release rate might be related to the mechanism of the release, which most probably involves a transition of the vesicular structure to ribbons or cylinders,18,19 and may occur in an initial rapid rate, which then slows down. We found that the biphasic curve was not seen when the hydrolysis of the ACh head groups was measured with the monomeric form of the bolaamphiphiles, at a concentration below the CAC (Figure 4). This suggests that the biphasic feature of the release is related to the changes in the vesicular structure and not to the hydrolysis of the bolaamphiphiles’ head groups, and supports the notion that the biphasic release rate is related to the mechanism of release and not to the hydrolysis process itself,
The results shown in Figure 9 also indicate that the release rate from GLH-20 vesicles was more rapid for both phases than the release rate from GLH-32 vesicles. Since the release was measured from vesicles in which the concentrations of the bolaamphiphiles were above the CAC, the question is whether changes in the CAC due to effects of ions or proteins may influence the rate of ACh-induced hydrolysis. In a previous study, we measured the CAC of six different bolaamphiphiles,20 including that of GLH-20. From the results of this study, it can be concluded that GLH-32 has a similar CAC to that of GLH-20, and since the conditions under which the release from the vesicles were identical for both bolaamphiphiles, it is unlikely that the difference in the hydrolysis rate could be attributed to differences in the CAC. Moreover, at the concentration of the bolaamphiphiles that was used to form the vesicles (10 mg/mL), which is much higher than the CAC, no effect of ion strength and proteins was seen on the aggregation properties of the bolaamphiphiles.20,25,26 Since AChE-induced release from the vesicles was carried out under conditions that do not affect the aggregation properties of both bolaamphiphiles, the difference in the release rate that was observed between GLH-20 vesicles and GLH-32 vesicles cannot be attributed to the effects of ions and/or proteins (ie, the protein of the hydrolyzing enzyme) on the CAC of the bolaamphiphiles.
The results shown in Figure 9 strongly suggest that the alkyl chain adjacent to the ACh head group significantly affects the hydrolysis rate of the head group; hence, affecting the rate of release of encapsulated compounds from the vesicles. When vesicles from both bolaamphiphiles are exposed to AChE, only the head groups and the alkyl chain (which is attached to the bolaamphiphile near the head group), have access to the enzyme, whereas other components of the bolaamphiphiles are unexposed to AChE as they are part of the hydrophobic region of the monolayer membrane. Between the two moieties that are exposed to the enzyme, only the alkyl chain pendant is different in GLH-20 and GLH-32 vesicles, suggesting that the alkyl chain adjacent to the head group is primarily responsible for the different hydrolysis rates, and consequently for the different release rates.
To quantify the differences in the release rates, the percent release from each vesicle preparation was calculated for several time points during the second release phase (the data used for the calculation were taken from Figure 9). This analysis (Figure 10) shows that the slope of the curve, which represents the release rate at the second phase, was significantly greater for GLH-20 vesicles, compared to GLH-32 vesicles.
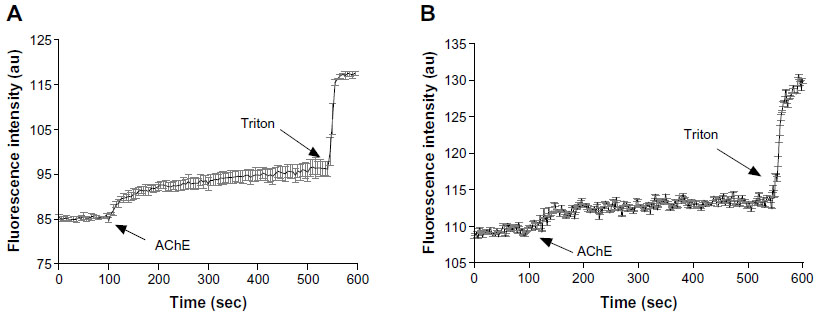 | Figure 9 The effect of AChE on the release of CF from vesicles made from GLH-20 (A) and GLH-32 (B). |
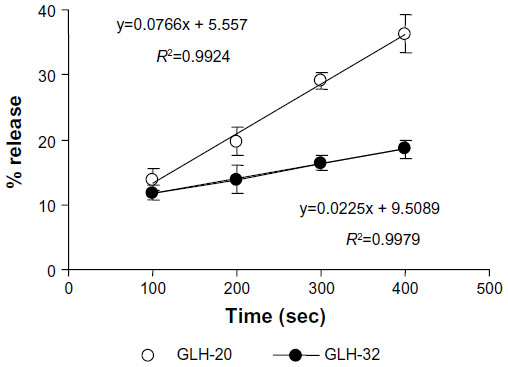 | Figure 10 Percent release of encapsulated CF at different times after exposing bolaamphiphilic vesicles to AChE. |
The steeper slope represents a faster release rate; indeed, at 400 seconds after the addition of AChE to the vesicles, about 35% of the CF was released from GLH-20 vesicles, whereas less than 20% of the encapsulated CF was released from GLH-32 vesicles at the same time point (Figure 10). It should be noted, however, that differences in the hydrolysis rates of the head groups of GLH-20 and GLH-32 were seen only 15–20 minutes after exposure of the bolaamphiphiles to AChE, whereas differences in the rates of release were already observed 3 minutes after the addition of AChE to the vesicles. This difference could be due to the time resolution of the two methods (ie, determination of head group hydrolysis rate by the pH-stat method and determination of release rate by monitoring the increase in fluorescence); one determines the hydrolysis rate, while the other determines the release rate. Apparently, vesicles start to disrupt after the removal of only a few ACh head groups from the surface of the vesicles, whereas in order to see a significant hydrolysis by a pH change, many more head groups have to be hydrolyzed. Yet vesicles that were made from bolaamphiphiles with head groups that are hydrolyzed faster by AChE also released their encapsulated material faster, suggesting that there is a correlation between the rate of the head groups hydrolysis and the rate of release. This correlation implies that the release rate can be varied in a controlled manner by using bolaamphiphiles with head groups that are designed to be hydrolyzed by AChE at different rates. This principle can be implemented in the design of vesicles that will release their content at organs or tissues that highly express the hydrolyzing enzyme. This design may be based on bolaamphiphiles with head groups such that their interaction with the hydrolyzing enzyme is modified by adjacent alkyl chains of various lengths, as described here.
Conclusion
In the present study we have shown that the release of encapsulated material from bolaamphiphilic vesicles can be varied by attaching alkyl chains of different lengths to the bolaamphiphile in the vicinity of the ACh head group. The results of this study show that when the alkyl chain in the vicinity of the ACh moiety is longer, the hydrolysis rate is slower, possibly because of steric hindrance that reduces the substrate’s affinity to the enzyme active site. Vesicles made from bolaamphiphiles, such as those described in the present study, show release rates that correlate with the rate of hydrolysis of the head groups by AChE. This study demonstrates the potential value of bolaamphiphilic vesicles as candidates for designing drug delivery systems with controlled release of the encapsulated drug. Bolaamphiphiles are particularly suitable as building blocks for constructing DDS with controlled release mechanisms, due to the high stability of their self-assembled vesicles and the tendency of the vesicles to disrupt upon head groups’ hydrolysis. The concept presented in the present study may be implemented in the design of bolaamphiphilic nanovesicles with various release rates for controlled drug delivery.
Acknowledgments
This study was supported in part by the Israel Science Foundation, 478 Grant No 973/11.
Disclosure
The authors report no conflicts of interest in this work.
References
Pavlukhina S, Sukhishvili S. Polymer assemblies for controlled delivery of bioactive molecules from surfaces. Adv Drug Deliv Rev. 2011;63(9):822–836. | |
Sharma G, Anabousi S, Elrhardt C, Ravi Kumar MN. Liposomes as targeted drug delivery systems in the treatment of breast cancer. J Drug Target. 2006;14(5):301–310. | |
Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1(3):297–315. | |
Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–3756. | |
Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A. 1988;85(18):6949–6953. | |
Gabizon AA. Pegylayed liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest. 2001;19:424–436. | |
Harrington KJ, Syrigos KN, Vile RG. Liposomally targeted cytotoxic drugs for the treatment of cancer. J Pharm Pharmacol. 2002;54(12):1573–1600. | |
Bikram M, West JL. Thermo-responsive systems for controlled drug delivery. Expert Opin Drug Deliv. 2008;5(10):1077–1091. | |
Kono K. Thermosensitive polymer-modified liposomes. Adv Drug Deliv Rev. 2001;53(3):307–319. | |
Grüll H, Langereis S. Hyperthermia-triggered drug delivery from temperature-sensitive liposomes using MRI-guided high intensity focused ultrasound. J Control Release. 2012;161(2):317–327. | |
Felber AE, Dufresne MH, Leroux JC. pH-sensitive vesicles, polymeric micelles, and nanospheres prepared with polycarboxylates. Adv Drug Deliv Rev. 2012;64(11):979–992. | |
Nappini S, Bonini M, Baldelli Bombelli F, et al. Controlled drug release under a low frequency magnetic field: effect of the citrate coating on magnetoliposomes stability.. 2011;7:1025–1037. | |
Yavlovich A, Smith B, Gupta K, Blumenthal R, Puri A. Light-sensitive lipid-based nanoparticles for drug delivery: design principles and future considerations for biological applications. Mol Membr Biol. 2010;27(7):364–381. | |
Menger FM, Johnston DE Jr. Specific enzyme-induced decapsulation. J Am Chem Soc. 1991;113(14):5467–5468. | |
Andresen TL, Thompson DH, Kaasgaard T. Enzyme-triggered nanomedicine: drug release strategies in cancer therapy. Mol Membr Biol. 2010;27(7):353–363. | |
Grinberg S, Kolot V, Linder C, et al. Synthesis of novel cationic bolaamphiphiles from vernonia oil and their aggregated structures. Chem Phys Lipids. 2008;153(2):85–97. | |
Roussel M, Lognoné V, Plusquellec D, Benvegnu T. Monolayer lipid membrane-forming dissymmetrical bolaamphiphiles derived from alginate oligosaccharides. Chem Commun (Camb). 2006;(34):3622–3624. | |
Fuhrhop J-H, Wang T. Bolaamphiphiles. Chem Rev. 2004;104(6): 2901–2937. | |
Masuda M, Shimizu T. Lipid nanotubes and microtubes: experimental evidence for unsymmetrical monolayer membrane formation from unsymmetrical bolaamphiphiles. Langmuir. 2004;20(14):5969–5977. | |
Hutter T, Linder C, Heldman E, Grinberg S. Interfacial and self-assembly properties of bolaamphiphilic compounds derived from a multifunctional oil. J Colloid Interface Sci. 2012;365(1):53–62. | |
Chen ZX, Su XX, Deng SP. Molecular recognition of melamine by vesicles spontaneously formed from orotic acid derived bolaamphiphiles. J Phys Chem B. 2011;115(8):1798–1806. | |
Sun YL, Wang SS, Han X, Chen ZX. Realization of the reversible vesicle-micelle transition of vitamin-derived bolaamphiphiles by heat change monitoring. J Phys Chem B. 2012;116(40):12372–12380. | |
Philosof-Mazor L, Dakwar GR, Popov M, et al. Bolaamphiphilic vesicles encapsulating iron oxide nanoparticles: new vehicles for magnetically targeted drug delivery. Int J Pharm. 2013;450(1–2):241–249. | |
Popov M, Linder C, Deckelbaum RJ, et al. Cationic vesicles from novel bolaamphiphilic compounds. J Liposome Res. 2010;20(2):147–159. | |
Popov M, Grinberg S, Linder C, et al. Site-directed decapsulation of bolaamphiphilic vesicles with enzymatic cleavable surface groups. J Control Release. 2012;160(2):306–314. | |
Dakwar GR, Abu Hammad I, Popov M, et al. Delivery of proteins to the brain by bolaamphiphilic nano-sized vesicles. J Control Release. 2012;160(2):315–321. | |
Popov M, Abu Hammad I, Bachar T, et al. Delivery of analgesic peptides to the brain by nano-sized bolaamphiphilic vesicles made of monolayer membranes. Eur J Pharm Biopharm. 2013. pii: S0939-6411(13)00223-3. | |
Reiner E, Simeon V. Competition between substrates for acetylcholinesterase and cholinesterase. Biochim Biophys Acta. 1977;480(1):137–142. | |
Komersová A, Komers K, Zdrazilová P. Kinetics of hydrolysis of acetylthiocholine and acetylcholine by cholinesterases. Chem Biol Interact. 2005;157–158:387–388. | |
Gunstone FD. Fatty acids. Part II. The nature of the oxygenated acid present in Vernonia anthelmintica (Willd.) seed oil. J Chem Soc. 1954:1611–1616. | |
Guidotti M, Ravasio N, Psaro R, Gianotti E, Marchese L, Coluccia S. Heterogeneous catalytic epoxidation of fatty acid methyl esters on titanium-grafted silicas. Green Chem. 2003;5:421–424. | |
Guidotti M, Psaro R, Ravasio N, Sgobba M, Gianotti E, Grinberg S. Titanium-silica catalysts for the production of fully epoxidized fatty acid methyl esters. Catal Lett. 2008;122:53–56. | |
Jay RR. Direct titration of epoxy compounds and aziridines. Anal Chem. 1964;36:667–668. | |
New RRC, editor. Liposomes: A Practical Approach, Oxford: Oxford University Press; 1990. | |
Torchilin V, Weissig V, editors. Liposomes. 2nd ed. Oxford: Oxford University Press; 2003. | |
Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. | |
Carlson KD, Schneider WJ, Chang SP, Princen LH. Vernonia galamensis seed oil: a new source for epoxy coatings. In: Pryde EH, Princen LD, Mukherjee F, Mukherjee KD, editors. New Sources of Fats and Oils. Champaign, IL: American Oil Chemists’ Society; 1981:297–318. | |
Halldorsson A, Magnusson CD, Haraldsson GG. Chemoenzymatic synthesis of structured triacylglycerols by highly regioselective acylation. Tetrahedron. 2003;59(46):9101–9109. | |
Grinberg S, Kipnis N, Linder C, Kolot V, Heldman E. Asymmetric bolaamphiphiles from vernonia oil designed for drug delivery. Eur J Lipid Sci Technol. 2010;112(1):137–151. | |
Jung DI, Shin YJ, Lee ES, Moon TS, Yoon CN, Lee BH. Acetylcholinesterase(AChE)-catalyzed hydrolysis of long-chain thiocholine esters: shift to a new chemical mechanism. Bull Korean Chem Soc. 2003;24(1):65–69. | |
Vellom DC, Radic Z, Li Y, Pickering NA, Camp S, Taylor P. Amino acid residues controlling acetylcholinesterase and butyrylcholinesterase specificity. Biochemistry. 1993;32(1):12–17. | |
Radic Z, Pickering NA, Vellom DC, Camp S, Taylor P. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry. 1993;32(45):12074–12084. | |
Ordentlich A, Barak D, Kronman C, et al. Dissection of the human acetylcholinesterase active center determinants of substrate specificity. Identification of residues constituting the anionic site, the hydrophobic site, and the acyl pocket. J Biol Chem. 1993;268(23):17083–17095. | |
Armenta S, Blanco M. Ion mobility spectrometry: a valuable tool for kinetic studies in enzymology. Anal Chim Acta. 2011;685(1):1–8. |
Supplementary material
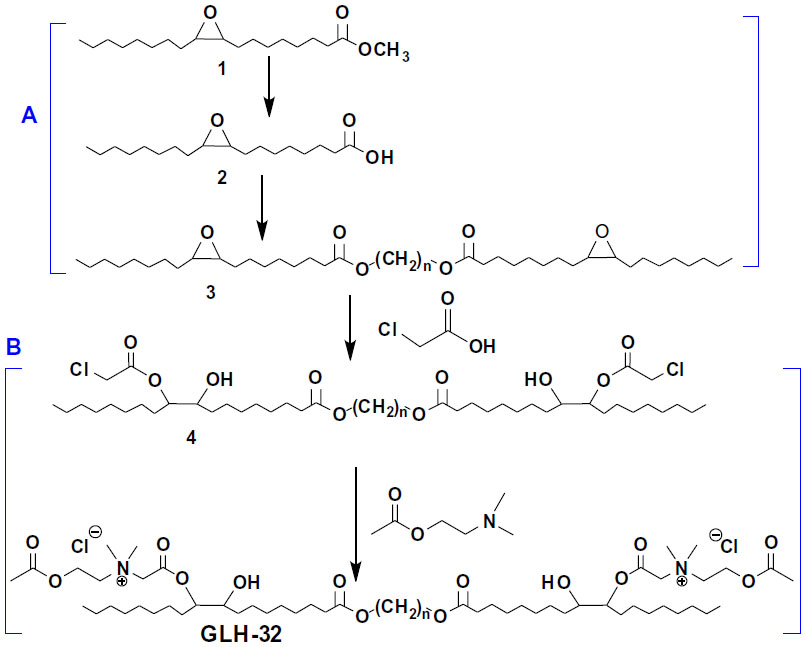 | Figure S1 Synthetic stages of the bolaamphiphile GLH-32: synthesis of the bolaamphiphilic skeleton (A) and the addition of the head groups (B). |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.