")
Back to Journals » Cancer Management and Research » Volume 11
Statins may have double-edged effects in patients with lung adenocarcinoma after lung resection
Authors Nishikawa S , Menju T , Takahashi K, Miyata R , Chen-Yoshikawa TF, Sonobe M, Yoshizawa A, Sabe H, Sato T , Date H
Received 8 January 2019
Accepted for publication 21 March 2019
Published 18 April 2019 Volume 2019:11 Pages 3419—3432
DOI https://doi.org/10.2147/CMAR.S200819
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Shigeto Nishikawa,1 Toshi Menju,1 Koji Takahashi,1 Ryo Miyata,1 Toyofumi Fengshi Chen-Yoshikawa,1 Makoto Sonobe,1 Akihiko Yoshizawa,2 Hisataka Sabe,3 Tosiya Sato,4 Hiroshi Date1
1Department of Thoracic Surgery, Graduate School of Medicine, Kyoto University, Kyoto, Japan; 2Department of Diagnostic Pathology, Kyoto University Hospital, Kyoto, Japan; 3Department of Molecular Biology, Faculty of Medicine, Hokkaido University, Sapporo, Japan; 4Department of Biostatistics, Kyoto University School of Public Health, Kyoto, Japan
Purpose: The epithelial to mesenchymal transition (EMT) is pivotal for driving metastasis and recurrence in lung cancer. Some in vitro reports have shown that statins suppress EMT by inactivating mutant p53 functions. Several clinical trials of conventional treatments with statins have been performed, but the effect of these drugs on prognosis is still uncertain. The purpose of this study is to examine the impact of statins on EMT and the prognosis of patients with lung adenocarcinoma.
Materials and methods: Morphological changes were evaluated and EMT markers (E-cadherin, vimentin) were analyzed by Western blotting in p53-overexpressing H1650 and mutant p53-harboring H1975 lung adenocarcinoma cells, with and without simvastatin administration. The invasive ability of these cells was analyzed in a Matrigel chemoinvasion assay. A total of 250 lung adenocarcinoma specimens were also collected from patients who underwent surgery in our institute. EMT markers in these tumor specimens were evaluated by immunostaining and p53 mutation status was determined by direct sequencing. Associations among EMT status, p53 mutation status, and statin use were evaluated, and prognosis was analyzed using a marginal structural model.
Results: Mutant p53 induced EMT and increased the invasive ability of H1650 cells. Simvastatin restored the epithelial phenotype and decreased the invasive ability of both H1650 and H1975 cells. Statin administration was associated with inactivation of EMT only in patients with mutant p53, which was consistent with the in vitro results. Moreover, in patients with mutant p53, statin users had significantly better survival than non-statin users. In contrast, statins significantly worsened the prognosis of patients with wild type p53 (HR 2.10, 95% CI 1.14–3.85).
Conclusion: Statins suppress EMT and change the prognosis of patients with lung adenocarcinoma in a p53 mutation-dependent manner.
Keywords: p53, epithelial to mesenchymal transition, statin, survival analysis, non-small cell lung cancer
Introduction
Lung cancer is a leading cause of cancer death worldwide.1,2 Recent advances in cancer therapy, including postoperative adjuvant chemotherapy and use of immune checkpoint inhibitors (ICIs), have led to dramatic clinical responses.3 However, the postoperative 5-year survival rates in lung cancer are still unsatisfactory due to metastasis and recurrence, even in operable stages.4,5 To address this problem, extensive research has been performed on the mechanisms of metastasis and recurrence.
The epithelial–mesenchymal transition (EMT) is pivotal for driving metastasis and recurrence in lung cancer, and has been widely studied in recent years.6–8 Various factors, including mutant p53, can induce EMT,9–11 and suppression of EMT activation has become an important target in cancer therapy. Some reports have shown that statins have an anticancer ability and suppress functions of mutant p53 in vitro.12–15 Several clinical trials of conventional treatments with statins have been performed,16–20 but there is little literature on the effects of statins on early stage lung adenocarcinoma. Additionally, the impact of statins on prognosis is unclear because these reports did not investigate the p53 mutation status.
We hypothesized that the effects of statins may depend on the p53 mutation status, and we analyzed cancer cell lines and patient survival with a specific focus on this status. The purpose of this study is to examine the impact of statins on EMT and the prognosis of patients with lung adenocarcinoma harboring p53 mutations.
Materials and methods
Cell culture
Human non-small cell lung cancer cell lines, NCI-H1650 and NCI-H1975, were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). H1650 has wild-type p53 with EGFR mutation (del E746-A750), whereas H1975 has mutant p53 (R273H) with EGFR mutations (L858R, T790M). Cells were maintained in the ATCC-recommended medium (RPMI 1640; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS (HyClone, Thermo Fisher Scientific K.K., Kanagawa, Japan) and penicillin/streptomycin in standard culture conditions (5% CO2, 100% humidity, 37°C). Mycoplasma negativity was confirmed for these cell lines before use.
p53 manipulation
For the generation of cells stably expressing recombinant p53, lentivirus plasmids were generated as follows. pBabe-hygro vector-based retrovirus plasmids encoding wild type or mutant p53 (R175H, R273H) were kindly provided by Professor Sabe (Hokkaido University).13 A cDNA encoding a wild type or mutant p53 (R175H, R273H) was generated by PCR-based cloning independently. The oligonucleotide primers were as follows: forward: 5ʹ-ACT GGA TCC ATG GAG GAG CCG CAG-3ʹ; reverse: 5ʹ-CGC GAA TTC TCA GTC TGA GTC AGG CCC TTC-3ʹ. After double restriction digestion with EcoRI and BamHI (TaKaRa, Japan), each cDNA fragment was ligated into an equally cut recipient plasmid, pENTR2B (Thermo Fisher Scientific K.K.) using DNA Ligation Kit Mighty Mix (TaKaRa). The three independent cDNA fragments were cloned into a CSII-CMV-RfA-IRES2-venus plasmid (RIKEN BioResource Center, Tsukuba, Japan) through a LR Clonase Reaction using Gateway® LR Clonase® II enzyme mix (Thermo Fisher Scientific K.K.). Lentivirus plasmids containing wild type or mutant p53 (R175H, R273H) and control plasmid were transfected into 293T cells independently, together with the envelope plasmid pCMV-VSV-G-RSV-Rev and the packaging plasmid pCAG-HIVgp (both from RIKEN BioResource Center) using Lipofectamine 2000 (Invitrogen). The cultured supernatants were harvested 48 hrs after transfection and filtered through 0.45-μm filters (Millipore, Billerica, MA, USA), and the resultant lentivirus preparations were then applied to target cells in the presence of 8 μg/mL polybrene (Santa Cruz Biotechnology, Dallas, TX, USA). Stable transformants of venus-infected cancer cells were selected using a FACS Aria III cell sorter (BD Biosciences, Franklin Lakes, NJ, USA).
Chemicals
Erlotinib (Wako Pure Chemical Industries, Osaka, Japan), osimertinib (Selleck Chemicals, Houston, TX, USA), and simvastatin (S6196, Sigma-Aldrich) were used in the study. Simvastatin was activated by alkaline hydrolysis to the acidic form prior to usage, as previously described.12
Western blot analysis
Whole cell lysates were resolved by 4–20% SDS-PAGE and transferred onto Immobilon-P PVDF membrane (Millipore). Blots were incubated overnight (4°C) with anti-E-cadherin (TaKaRa), anti-vimentin (Leica Biosystems, Wetzlar, Germany), anti-p53 (Santa Cruz Biotechnology), anti-pERK, anti-ERK (Cell Signaling Technology, Danvers, MA, USA), anti-ZEB1 (Abcam, Cambridge, UK), and anti-β-actin (Sigma-Aldrich). Membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA), and then visualized using an EzWestLumi Plus detection kit (Atto, Tokyo, Japan), with detection of luminescence using the LuminoGraph II imaging system (Atto).
Matrigel chemoinvasion assay
The Matrigel chemoinvasion assay was performed using Matrigel chambers (Biocoat; BD). In brief, 2.5×104 cells were seeded on the upper wells of 24-well chambers with 0.1% FBS containing medium, with the lower wells filled with 10% FBS containing medium. After incubation for 14 hrs, cells were fixed in methanol, and the number of cells that transmigrated through the chamber filter to the lower surface of the filters was counted by staining with Diff-Quick. Data were collected from three independent experiments.
Drug sensitivity assay
Cell viability was determined using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Cell viability was assessed 72 hrs after drug treatment, using three wells for each drug concentration. Experiments were performed in triplicate. The half-maximum inhibitory concentration (IC50) was calculated using Prism7 (GraphPad, La Jolla, CA, USA) with a three-parameter sigmoidal curve fit. The p-values for the two-curve comparisons were calculated using an extra sum of squares F test.
Combination index
The combination effect of two drugs was evaluated based on the combination index.21–23 The combination index was calculated using Compusyn software (ComboSyn, Inc. Paramus, NJ, USA). The index <1 indicated a synergistic effect, the index =1 indicated an additive effect, and the index >1 indicated an antagonistic effect.
Specimens from lung cancer patients
A total of 282 lung adenocarcinoma specimens were collected from patients who underwent surgery at our hospital from January 2001 to December 2007. Among these specimens, 32 were excluded from analysis because the specimen on the tissue microarray was inappropriate for evaluation or cDNA was not available. The median follow-up time was 59.3 months (range, 1–129). Overall survival (OS) times and outcomes data were available for all patients. The Kyoto University Graduate School and Faculty of Medicine Ethics Committee approved the study (approval number: G0028-7, R1706) and written informed consent in accordance with the Declaration of Helsinki for tumor tissue use was obtained preoperatively from all patients. All tumors were reviewed by a pathologist (A.Y.) who confirmed predominant tumor subtypes, node status, and local lymph-vascular involvements. The tumors were restaged based on the TNM classification of the International Union Against Cancer, 7th edition.
Tissue microarrays
Tissue microarrays were assembled from paraffin-embedded tumor blocks by pathologists in the Department of Diagnostic Pathology at Kyoto University Hospital, using the approach described by Kononen et al24. The most representative areas of the tumors were selected based on the morphology on hematoxylin and eosin-stained slides. Tissue cores measuring 2 mm in diameter were arrayed in a paraffin block. Non-neoplastic lung tissue cores from selected patients were arrayed in the same block as controls.
Immunohistochemical (IHC) analysis
IHC staining for E-cadherin and vimentin was performed with mouse an anti-human E-cadherin monoclonal antibody (36B5, dilution 1:300, Leica Biosystems, Newcastle upon Tyne, UK) and a mouse anti-human vimentin monoclonal antibody (SRL33, dilution 1:300, Leica Biosystems) using the standard avidin-biotin-peroxidase complex method. Visualization was performed with 3,3′-diaminobenzidine tetrahydrochloride (Dojindo Laboratories), with hematoxylin counterstaining. After immunostaining, each specimen was categorized as negative or positive for E-cadherin and vimentin expression. EMT status was classified into three categories:6 full EMT (E-cadherin negative, vimentin positive), partial EMT (both E-cadherin and vimentin positive or both negative), and null EMT (E-cadherin positive, vimentin negative).
PCR and DNA sequencing
Using a RNeasy Plus mini kit (QIAGEN, Valencia, CA, USA), total RNA was extracted from tumor samples that had been frozen and stocked within 30 mins after resection. Total RNA was reverse transcribed to cDNA using a Ready-To-Go You-Prime First-Strand Beads (GE Healthcare Life Sciences, Pittsburgh, PA, USA). For PCR amplification, each cDNA was diluted to 10 ng/μL. PCR conditions were as follows: p53 exon 4 forward: 5ʹ-CCC AAG CAA TGG ATG ATT TG-3ʹ; p53 exon 10 reverse: 5ʹ-AGC CTG GGC ATC CTT GAG-3ʹ. The PCR assay was carried out in a 15μL volume that contained 15 ng of cDNA and 1 unit of Taq PCR Master Mix Kit (QIAGEN). Each PCR reaction was started at 95°C for 5 mins, and then cDNA was amplified for 40 cycles at 95°C for 30 s, 54.7°C for 30 s, and 72°C for 90 s, with a final extension time of 7 mins at 72°C. Each amplicon was purified using a QIAquick Gel Extraction Kit (QIAGEN) after agarose gel electrophoresis. Purified PCR products were sequenced in forward and reverse directions using a 3130xl Genetic Analyzer (Thermo Fisher Scientific K.K.). We detected p53 mutations in exons 5 through 8, similar to previous reports.25,26
Statistical analysis
Baseline characteristics were compared between statin users and non-statin users by Student’s t-test for continuous variables and chi-square test for categorical variables. Propensity scores were used to adjust for potential biases that may have influenced the prognosis. These scores were estimated using a logistic model that included age, gender, smoking history, EGFR mutation status, p53 mutation status, and pathological stage, which was recategorized as IA, IB, II, III, IV due to small frequencies. Patients were analyzed using a marginal structural model with standardized mortality ratio weights (SMRWs) to estimate the effects of statins.27 SMRWs were calculated as 1 for statin users and PS/(1-PS) for non-statin users, and comparable statin and non-statin users were created. Time-to-event curves for OS were estimated using the Kaplan–Meier method, differences in time-to-event curves were evaluated by log-rank test, and HRs were estimated using a Cox regression model. A p-value <0.05 was considered significant. Statistical analyses were conducted using JMP Pro 13 and 14, and SAS 9.4 (SAS Institute, Cary, NC, USA).
Results
Mutant p53 induces EMT and simvastatin suppresses induction of EMT
To investigate the impact of mutant p53 on EMT in lung cancer cells, we established a H1650 cell line that stably overexpressed mutant p53. Two months after mutant p53 (R175H, R273H) transfection, H1650 cells showed morphological changes (Figure 1A) of spindles and increased intercellular spaces. After simvastatin treatment, these morphological changes were diminished. To examine whether simvastatin can suppress EMT in a cell line with a mesenchymal phenotype, we used H1975 cells, which have endogenous mutant p53 (R273H). After simvastatin treatment, H1975 cells changed morphology from a mesenchymal to epithelial type, as for the H1650 cells overexpressing mutant p53, in a dose-dependent manner (Figure 1B). The simvastatin-treated cells showed reversed EMT changes 2 months after withdrawal of simvastatin (data not shown).
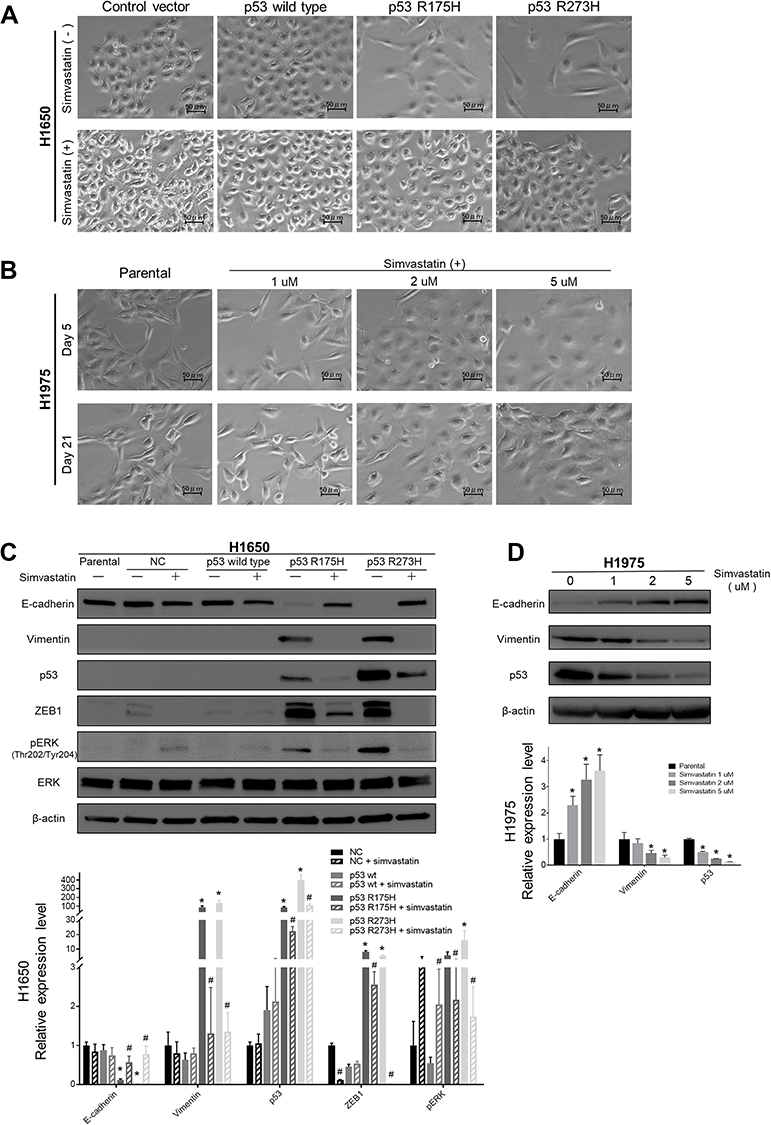 | Figure 1 Microscopic and Western blot analysis after p53 transfection and simvastatin treatment of lung cancer cells. (A) In microscopic analysis, wild type p53-overexpressing H1650 cells did not show morphological changes. Two months after mutant p53 (R175H, R273H) transfection, H1650 cells showed morphological changes to spindles with increased intercellular space. At 35 days after addition of 1 μM simvastatin to these cells, these morphological changes were diminished. No distinct morphological changes occurred in wild type p53-overexpressing H1650 cells after simvastatin treatment. (B) Simvastatin was administered to H1975 cells at the indicated concentrations (vehicle, 1 , 2, and 5 μM), after which the cells changed morphological shape form the mesenchymal to epithelial type in a dose-dependent manner. (C) Western blot analysis of whole cell lysate from H1650 cells (parental, control vector-, wild-type p53-, p53 R175H-, and p53 R273H-transfected) with or without simvastatin treatment. Membranes were blotted with ZEB1, E-cadherin, vimentin, p53, pERK (Thr202/Thr204), and total ERK antibodies. β-actin was used to confirm equal protein loading. (n=3, * p<0.05, p53-wild type/R175H/R273H vs control group. # p<0.05, p53-wild type/R175H/R273H with simvastatin vs without simvastatin) (D) Western blot analysis of whole cell lysate from H1975 cells with or without simvastatin added at the indicated concentrations. Membranes were blotted with E-cadherin, vimentin, and p53 antibodies. β-actin was used to confirm equal protein loading. (n=3, * p<0.05, simvastatin 1/2/5 μM vs H1975 parental). |
Western blotting of whole cell lysates was performed to confirm the EMT changes observed microscopically in the two cell lines. Mutant p53-overexpressing H1650 cells showed significant E-cadherin suppression and vimentin overexpression accompanied by ZEB1 overexpression, which indicates induction of EMT, and simvastatin eliminated these EMT-related changes (Figure 1C). Similarly, H1975 cells had significantly restored E-cadherin and decreased vimentin expression after simvastatin treatment in a dose-dependent manner (Figure 1D). Moreover, these EMT changes observed by Western blotting were correlated with the p53 expression level. Again, we confirmed using Western blotting that simvastatin-treated cells regained EMT changes after withdrawal of simvastatin (data not shown). Interestingly, pERK and ZEB1 were significantly downregulated in simvastatin-treated mutant p53-overexpressing H1650 cells, whereas pERK was significantly upregulated and ZEB1 was not downregulated in simvastatin-treated wild type p53-overexpressing H1650 cells (Figure 1C).
Mutant p53 increases invasive ability and simvastatin suppresses this invasive ability
The effects of mutant p53 and simvastatin on the invasion ability of lung cancer cells were examined in a Matrigel chemoinvasion assay. This assay showed that mutant p53 significantly increased the invasion ability of H1650 cells, and this was diminished by simvastatin treatment (Figure 2A–E). In contrast, simvastatin increased the invasion ability of wild type p53-overexpressing H1650 cells (Figure 2C–E). Consistent with the results in H1650 cells with mutant p53, the invasion ability of H1975 cells was significantly suppressed by simvastatin treatment (Figure 2F).
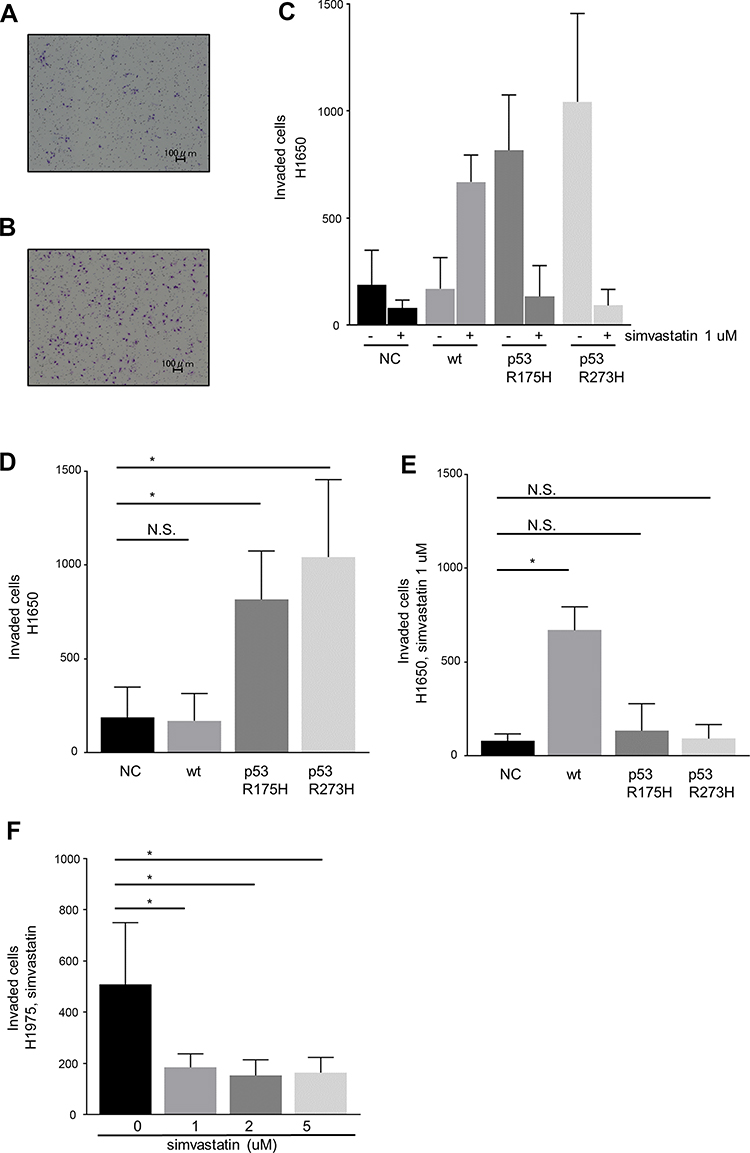 | Figure 2 A Matrigel chemoinvasion assay was performed using Matrigel chambers. Images of invading cells were captured and counted using a dynamic cell counter BZ-H1C. Representative photographs of (A) wild type p53- and (B) p53 R273H-transfected H1650 cells. (C) Mutant p53 significantly promoted invasive ability. (D, E) Quantification of invasion of H1650 cells without (D) and with (E) simvastatin treatment in control vector-, wild type p53-, p53 R175H-, and p53 R273H-transfected H1650 cells. Simvastatin significantly suppressed invasion ability in mutant p53-transfected H1650 cells, but increased invasion by wild type p53-transfected H1650 cells. (F) Simvastatin significantly suppressed invasion of H1975 cells. Data are from three independent experiments. Each bar indicates the number of invading cells. The error bar indicates the standard deviation. Data sets were analyzed by one-way ANOVA followed by a Dunnett multiple comparisons test. * p<0.05 vs control. Abbreviations: NC, negative control; wt, wild type; N.S., not significant. |
Mutant p53 decreases EGFR-TKI sensitivity and simvastatin restores this sensitivity
The influence of mutant p53 and simvastatin on EGFR-TKI sensitivity was examined using a drug sensitivity assay. Mutant p53 significantly decreased the sensitivity of H1650 cells to osimertinib (Figure 3A), and this acquired drug resistance was reversed by simvastatin treatment (Figure 3B). To reveal whether simvastatin and EGFR-TKIs have a synergistic effect, we performed drug sensitivity assay with simvastatin without EGFR-TKIs. There was no significant effect on cell viability at a concentration of 1 μM simvastatin monotherapy (Figure 4A, B). Calculated combination indices ranged from 0.38 to 0.75, which showed synergism in osimertinib with simvastatin in H1650 cells.21 The sensitivity of H1650 cells with wild-type p53 was slightly decreased by simvastatin and combination index was calculated as 2.17 (Figure 3B). Consistent with the results for H1650 cells with mutant p53, the sensitivity of H1975 cells to osimertinib was significantly recovered by simvastatin administration (Figure 3C). Calculated combination indices ranged from 0.15 to 0.27, which showed strong synergism in osimertinib with simvastatin in H1975 cells.21 However, simvastatin did not recover the sensitivity of H1975 cells to erlotinib and synergism was not observed (Figure 3D).
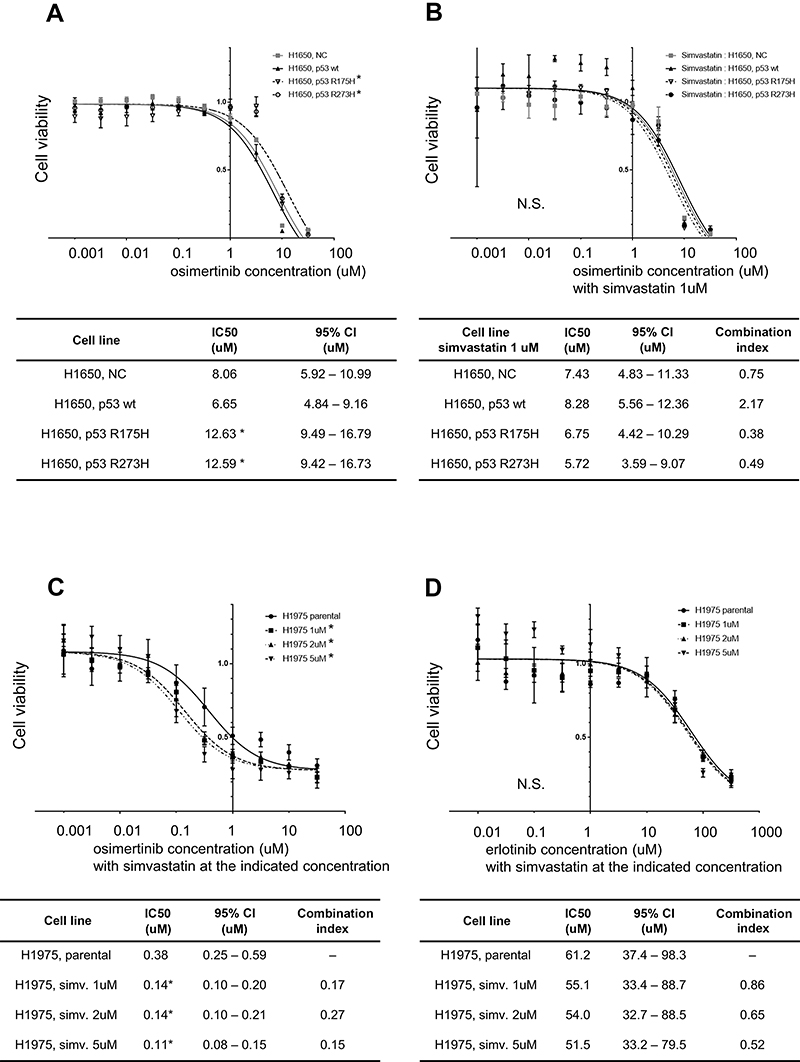 | Figure 3 Drug sensitivity assay in H1650 and H1975 cells. Cell viabilities in response to osimertinib in control vector-, wild-type p53-, p53 R175H-, and p53 R273H-transfected H1650 cells without (A) and with (B) simvastatin treatment. Mutant p53 significantly decreased sensitivity to osimertinib and this mutant p53-induced acquired drug resistance was eliminated by simvastatin. (C) Simvastatin-treated H1975 cell viabilities in response to osimertinib. The loss of sensitivity of H1975 cells to osimertinib was significantly reversed by simvastatin. (D) Simvastatin-treated H1975 cell viabilities in response to erlotinib. Simvastatin did not reverse the loss of sensitivity of these cells to erlotinib. Experiments were performed in triplicate. IC50 values were calculated using Prism software. n=3, * p<0.05 vs control (extra sum of squares F test). Combination indices were calculated using Compusyn software. Combination index <1 indicated a synergistic effect, and index >1 indicated am antagonist effect. Abbreviations: NC, negative control; wt, wild type. |
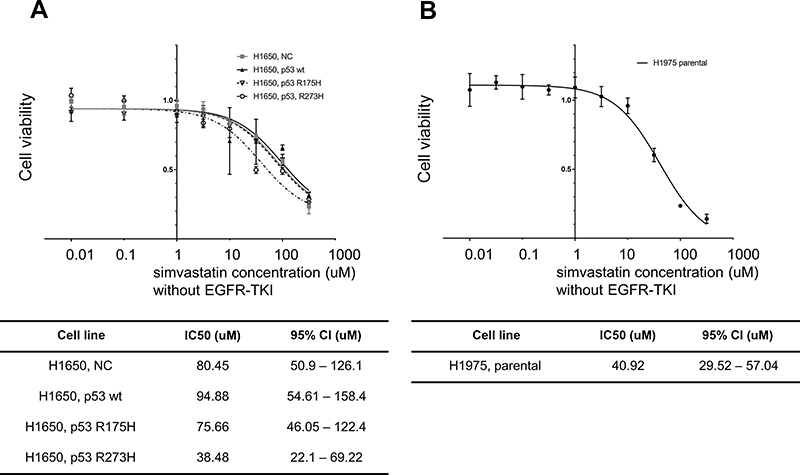 | Figure 4 Drug sensitivity assay in H1650 and H1975 cells without EGFR-TKIs. Cell viabilities in response to simvastatin monotherapy in control vector-, wild-type p53-, p53 R175H-, and p53 R273H-transfected H1650 cells (A) and parental H1975 cells (B). No difference in cell viability was observed at the concentration of 1 μM of simvastatin. Experiments were performed in triplicate. IC50 values were calculated using Prism software. n=3, * p<0.05 vs control (extra sum of squares F test). Abbreviations: NC, negative control; wt, wild type. |
Statin administration is associated with EMT status in a p53-mutation dependent manner
The clinicopathological characteristics of the 250 patients are summarized in Table 1. Of these patients, 51 (20.4%) had taken statins (simvastatin, atorvastatin, pravastatin) for hyperlipidemia or coronary artery disease. The doses of statin and their intensity are listed in Table 2. Statin intensity was categorized based on the guideline on the statin treatment.28 Most of these patients took lower potency statins with their minimum dose. No patients died of cardiovascular events. p53 mutation was positive in 70 patients (28.0%), including 13 with p53 R175H and 9 with p53 R273H. The p53 mutation status was associated with gender (male) (risk ratio (RR) =1.68, 95% CI 1.37–2.07, p<0.01), smoking history (current and former) (RR=1.45, 95% CI 1.19–1.77, p<0.01), wild-type EGFR (RR=1.51, 95% CI 1.18–1.92, p<0.01), and pathological stage (p<0.01, Fisher exact test).
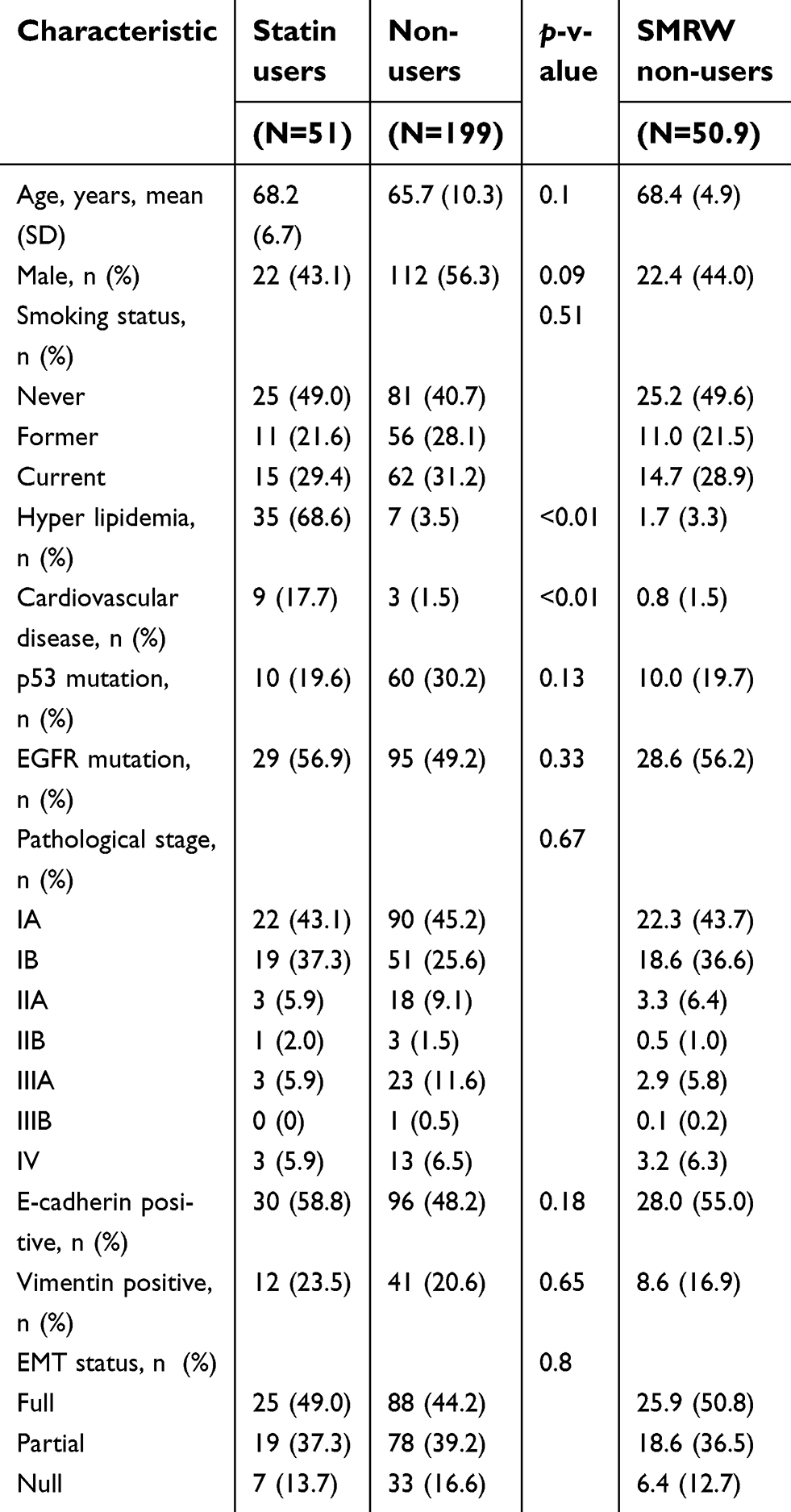 | Table 1 Characteristics of the patients |
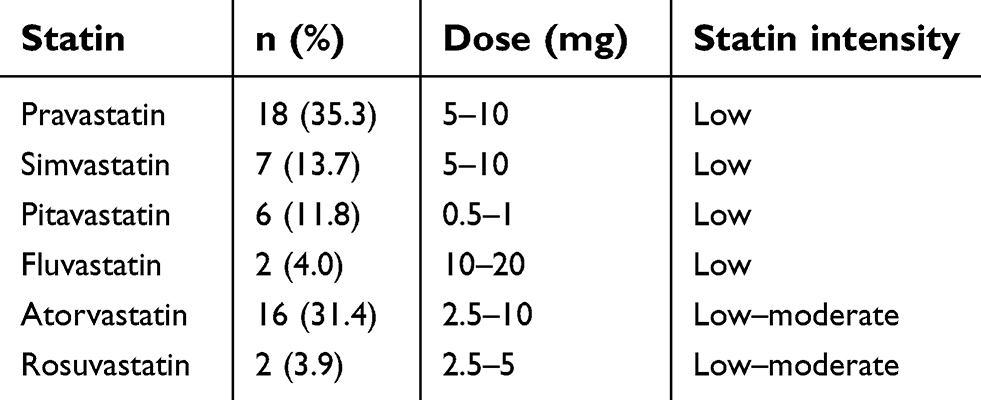 | Table 2 Statin dose and intensity |
Expression of E-cadherin and vimentin were analyzed using IHC of the tissue microarray. E-cadherin was positive in 126 cases (50.4%) and vimentin was positive in 53 cases (21.2%). A full, partial, and null EMT status was found in 40 (16.0%), 97 (38.8%), and 113 (45.2%) cases, respectively. There was no significant association of EMT status with p53 mutation, but the EMT status tended to be higher in patients with mutant p53. In all patients, there was also no significant association of statin use with EMT status. In subgroup analyses, statin use was not significantly associated with inactivation of EMT in patients with mutant p53 (p=0.06, Fisher exact test), but none of these patients had full EMT activation. Statin use did not have an effect on EMT status in patients with wild-type p53 (Figure 5A). Consistent with these results, statin use had no significant association with vimentin positivity in patients with mutant p53 (p=0.07, Fisher exact test), but none of these patients were vimentin-positive, in contrast to patients with wild-type p53 (Figure 5B).
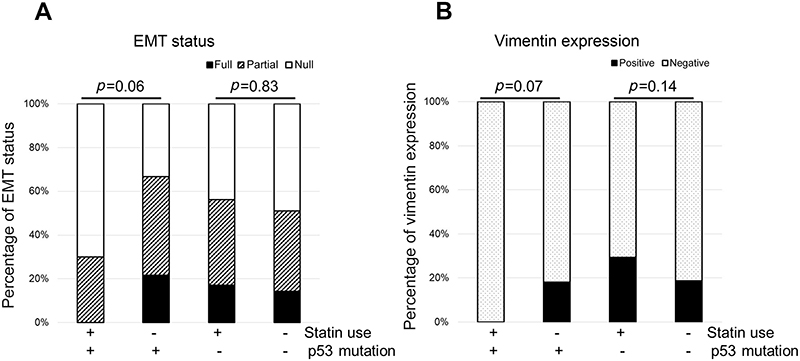 | Figure 5 Association between EMT status and p53 mutation with or without statin use. EMT status was categorized into three groups: full (E-cadherin negative, vimentin positive), partial (both E-cadherin and vimentin positive or both negative), and null (E-cadherin positive, vimentin negative). (A) No statin users had full EMT activation. (B) No statin users with p53 mutation were vimentin-positive. Abbreviation: EMT, epithelial to mesenchymal transition. |
Statins change the prognosis of patients in a p53 mutation-dependent manner
The effects of mutant p53 and statin use on OS were analyzed. In unadjusted analysis in all cases, median OS did not differ for statin users and non-users (103.9 vs.124.0 months, p=0.70) (Figure 6A, F), and statin use did not affect the prognosis (HR: 1.12, 95% CI: 0.64–1.95) (Figure 6F). However, in subgroup analyses, statin use was associated with better survival in patients with mutant p53 (median OS and HR were not estimated) (Figure 6B, F), and with poor survival in those with wild-type p53 (HR: 1.83, 95% CI: 1.01–3.34) (Figure 6C, F). To control for potential bias, we used a marginal structural model with SMRWs for further evaluation.27 A total of 199 non-statin users were condensed to 50.9 patients who were comparable to the 51 statin users using this method. Baseline characteristics were well balanced after adjustment (Table 1). In this analysis, statin use improved OS for patients with mutant p53 (HR not estimated, p=0.00), but reduced OS for those with wild-type p53 (HR: 2.10, 95% CI: 1.14–3.85) (Figure 6D–F).
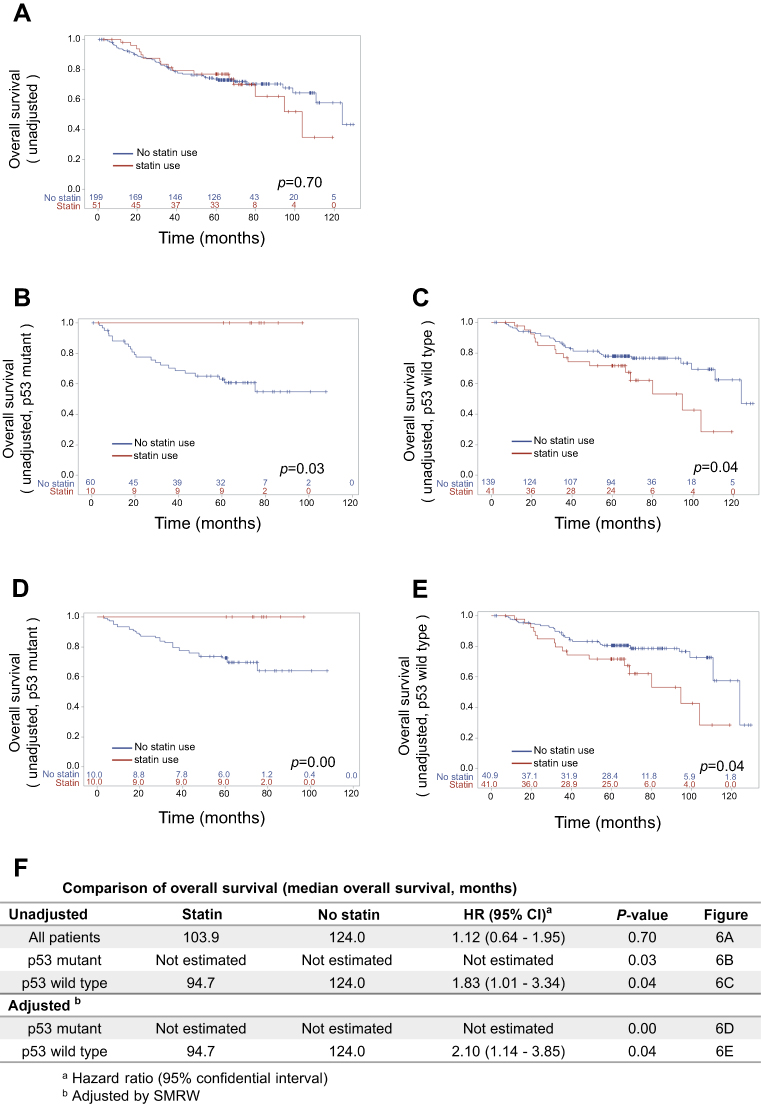 | Figure 6 Analysis of associations among p53 mutation status, statin use, and prognosis. Kaplan–Meier overall survival (OS) curves for (A) all patients and for patients with (B, D) mutant p53 and (C,E) wild-type p53. Curves are shown (B, C) before and (D, E) after standardization by the SMRW method. (F) Comparison of OS for patients treated with and without statins. Median OS and HR with 95% CI are listed. Abbreviation: SMRW, standardized mortality ratio weight. |
Discussion
Drugs that affect metabolic pathways, including statins, have been suggested to have additional benefits in cancer treatment.29,30 However, the results in clinical trials are controversial,16–20 and this may partly be because these studies have not taken p53 mutation status into account. Various compounds have been tested for suppression of EMT,31 but drugs targeting EMT are not available at present. This is the first report to investigate the clinical effects of statins on EMT and prognosis with a focus on p53 mutation status in lung adenocarcinoma after lung resection.
In previous reports,32–34 p53 mutations have been divided into two representative categories: conformational mutations and DNA-contact mutations. Among these mutations, we selected R175H (conformational mutation) and R273H (DNA-contact mutation) respectively, because these two mutations are the most common in each category35 and have different features.36
Simvastatin restored an epithelial phenotype in mutant p53-overexpressing H1650 cells and parental H1975 cells, which indicates that simvastatin affects both endogenous and exogenous mutant p53. Additionally, based on the mutant p53-overexpressing H1650 cells regaining a mesenchymal phenotype after withdrawal of simvastatin, the mutant p53-induced EMT appears to be reversible. Restoration of an epithelial phenotype in H1975 cells was dependent on the p53 expression level, which implies that simvastatin had anti-EMT effects through degradation of mutant p53, rather than wild-type p53. This finding is similar to that in a previous report.37
Simvastatin did not enhance sensitivity of H1975 cells to erlotinib, which suggests that simvastatin affected mutant p53 instead of having a direct effect on the gatekeeper in EGFR. Since statins act differently from EGFR-TKIs, synergistic effects of a statin and an EGFR-TKI on EGFR mutants concomitant with p53 mutations may be promising clinically. In the present study, a simvastatin concentration of 1 μM was sufficient for suppressing invasion ability and restoring sensitivity to osimertinib in both cell lines. This result suggests that these inhibitory effects of statins can be obtained in clinical practice because concentrations from 100 nM to 1 μM of simvastatin are clinically achievable.12,38,39 The difference in sensitivity in H1650 and H1975 cells was significant, even at a statin concentration of 1 μM, but this difference may not be large enough to have an additional effect in clinical practice. We used high concentrations of simvastatin of 2 μM and 5 μM to investigate the dose-response relationship in H1975 cells, even though these high concentrations may be toxic in clinical practice.
Interestingly, simvastatin increased the invasion ability of wild type p53-overexpressing H1650 cells. This result might explain worsening of the prognosis by statins in patients with lung cancer harboring wild-type p53. Kim et al mentioned about functional link between p53 and PTEN.40 Park et al reported that PTEN loss can cause resistance to simvastatin treatment because this loss strongly induces pAkt in triple-negative breast cancer.41 Wang et al also showed that simvastatin suppresses pAkt by enhancing PTEN expression.42 On the other hand, H1650 cells lose PTEN expression so that Akt is highly activated.43 Also, Robin et al reported that statins may accelerate Wnt/β-catenin signaling,44 and some reports have shown that statins upregulate phosphorylation of ERK.45–47 In the present study, Western blot analysis revealed that pERK was significantly upregulated and ZEB1 was not downregulated in H1650 cells overexpressing wild-type p53. Akt was also highly activated in H1650 cells (data not shown). Previous reports and these results might explain why simvastatin increases invasion ability; however, this explanation is still limited because the expression levels of pERK and ZEB1 were not proportionate to the invasion ability in comparison of H1650 cells overexpressing wild type and mutant p53. Thus, the mechanism through which simvastatin increases invasion ability is still unclear. As a next step, additional investigations of PTEN and other candidate factors should be performed in H1650 cells and in tissue from patients with wild type p53.
IHC showed that EMT tended to be highly activated only in patients with mutant p53, which was consistent with the results of Western blotting. The reason why there was also no significant association of statin use with EMT status may be related with the statin doses. Most of these patients took statins with their minimum doses necessary to control hyperlipidemia or coronary artery disease, which may be much lower than that used to produce anticancer effect. We found the dose-response relationship in H1975 cells with simvastatin, which can also support this idea. Statin use seemed to be related to vimentin suppression more than E-cadherin expression, which may be due to the different interactions of mutant p53 with each protein. Recent reports have suggested that mutant p53 downregulates E-cadherin through interactions with Slug and ZEB1.9,10 Little is known about the interaction between mutant p53 and vimentin, but this interaction may be an interesting therapeutic target.
The poor prognosis of lung cancer with mutant p53 is well known.25,48,49 Statin use did not affect this prognosis in an unadjusted all-case analysis, but survival of statin users with mutant p53 was significantly better than that of non-statin users with wild-type p53. This result suggests that statins may have other effects, in addition to degradation of mutant p53, and is consistent with a previous report.50 Kodach et al51 suggested that statins might directly augment chemosensitivity in p53 mutant cases; that is, statins might have other positive effects on adjuvant chemotherapy and treatment after recurrence. Moreover, statins have recently been shown to affect the immune system.52 Use of an ICI may improve the prognosis of patients with lung cancer, but no patients took an ICI in this study. It will be of interest to investigate ICI use in a future study.
There are some limitations in this study. First, it was not possible to adjust for comorbidities of hyperlipidemia and cardiovascular disease because these conditions are highly correlated with statin use. Serum cholesterol levels on admission were also not available, since hyperlipidemia is not a risk factor for lung resection, and this prevented investigation of the direct impact of serum cholesterol on prognosis. However, there were no cardiovascular disease-related deaths in the study, and cardiovascular disease did not affect the prognosis. Still, there is a possibility that the effects of other comorbidities similar to cardiovascular disease were not excluded. Second, the study was performed retrospectively at a single institute, and statistical analyses were limited by the number of statin users (51, including 10 with mutant p53 and 41 with wild-type p53). Given the low number of statin users with mutant p53, one event could influence results for OS. Lin et al reported that lower potency statins, such as simvastatin, fluvastatin, and pravastatin conferred slightly better survival,16 but we could not examine differences between lower and higher potency statins due to the shortage of cases. As Turrell et al have demonstrated genotype-specific statin sensitivity, there might be an influence from other mutation type.36 In consistent with this report, the change of protein expression level was observed greater in R273H than R175H. Genotype-specific analysis may help us to elucidate it, but we were not able to perform this analysis due to a small number in our patients’ group. However, we found that statins may benefit patients with p53 mutation regardless of their genotype in this study. Third, the minimum duration of statin use was unknown. Microscopic and Western blot analyses showed that mutant p53 induced EMT in H1650 cells and simvastatin restored the epithelial phenotype, with >21 days required for this reversal. The appropriate duration for statin administration is still unclear in clinical practice.
The mechanism of how statins exert anticancer effects is an interesting area for further study because statins may have double-edged effects on the prognosis of patients according to p53 mutation status. Therefore, we strongly recommend examining the p53 mutation status before using statins as an additional anticancer treatment. Further accumulation of data is needed to permit identification of patients who will and will not benefit from statins in this context, and a future prospective study with a specific focus on p53 mutation status should be performed.
Conclusion
Our results show that statins suppress EMT and change the prognosis of patients with lung adenocarcinoma after lung resection in a p53 mutation-dependent manner.
Acknowledgments
We would like to thank our department members (Terumasa Sowa, Shinya Neri, Takao Nakanishi, Hiroyuki Cho, Hiroyuki Ishikawa, Misa Noguchi, Yojiro Yutaka, Daisuke Nakajima, Akihiro Ohsumi, Masatsugu Hamaji, and Toshihiko Sato) for useful discussions and collaboration on the early stages of this work. DNA sequencing analysis was performed at the Medical Research Support Center, Graduate School of Medicine, Kyoto University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387
2.
3. Brahmer JR, Kim ES, Zhang J, Smith MM, Rangwala RA, O′Brien MER. KEYNOTE-024: phase III trial of pembrolizumab (MK-3475) vs platinum-based chemotherapy as first-line therapy for patients with metastatic non-small cell lung cancer (NSCLC) that expresses programmed cell death ligand 1 (PD-L1). J Clin Oncol. 2015;33(15_suppl):TPS8103–TPS8103.
4. Sawabata N, Miyaoka E, Asamura H, et al. Japanese lung cancer registry study of 11,663 surgical cases in 2004: demographic and prognosis changes over decade. J Thorac Oncol. 2011;6(7):1229–1235.
5. Gwozdz P, Pasieka-Lis M, Kolodziej K, et al. Prognosis of patients with stages I and II non-small cell lung cancer with nodal micrometastases. Ann Thorac Surg. 2018;105(5):1551–1557.
6. Sowa T, Menju T, Sonobe M, et al. Association between epithelial-mesenchymal transition and cancer stemness and their effect on the prognosis of lung adenocarcinoma. Cancer Med. 2015;4(12):1853–1862.
7. Sabe H. Cancer early dissemination: cancerous epithelial-mesenchymal transdifferentiation and transforming growth factor beta signalling. J Biochem. 2011;149(6):633–639.
8. Menju T, Sowa T, Sonobe M, et al. Clinicopathologic significance of epithelio-mesenchymal transition in human lung adenocarcinomas: an integrative analysis, inclusive of genetic alterations, on 256 surgically resected cases. Cancer Treat Res Commun. 2017;12:62–68.
9. Wang SP, Wang WL, Chang YL, et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of slug. Nat Cell Biol. 2009;11(6):694–704.
10. Dong P, Karaayvaz M, Jia N, et al. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013;32(27):3286–3295. doi:10.1038/onc.2012.334
11. Alexandrova EM, Yallowitz AR, Li D, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015;523(7560):352–356. doi:10.1038/nature14430
12. Freed-Pastor WA, Mizuno H, Zhao X, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148(1–2):244–258. doi:10.1016/j.cell.2011.12.017
13. Hashimoto A, Oikawa T, Hashimoto S, et al. P53- and mevalonate pathway-driven malignancies require Arf6 for metastasis and drug resistance. J Cell Biol. 2016;213(1):81–95. doi:10.1083/jcb.201510002
14. Alizadeh J, Zeki AA, Mirzaei N, et al. Mevalonate cascade inhibition by simvastatin induces the intrinsic apoptosis pathway via depletion of isoprenoids in tumor cells. Sci Rep. 2017;7:44841. doi:10.1038/srep44841
15. Shi Y, Felley-Bosco E, Marti TM, Stahel RA, Aboussekhra A. Differential effects of lovastatin on cisplatin responses in normal human mesothelial cells versus cancer cells: implication for therapy. PLoS One. 2012;7(9):e45354. doi:10.1371/journal.pone.0045354
16. Lin JJ, Ezer N, Sigel K, Mhango G, Wisnivesky JP. The effect of statins on survival in patients with stage IV lung cancer. Lung Cancer. 2016;99:137–142. doi:10.1016/j.lungcan.2016.07.006
17. Cardwell CR, Mc Menamin U, Hughes CM, Murray LJ. Statin use and survival from lung cancer: a population-based cohort study. Cancer Epidemiol Biomarkers Prev. 2015;24(5):833–841. doi:10.1158/1055-9965.EPI-15-0052
18. Hung MS, Chen IC, Lee CP, et al. Statin improves survival in patients with EGFR-TKI lung cancer: A nationwide population-based study. PLoS One. 2017;12(2):e0171137. doi:10.1371/journal.pone.0171137
19. Lee Y, Lee KH, Lee GK, et al. Randomized phase II study of afatinib plus simvastatin versus afatinib alone in previously treated patients with advanced non-adenocarcinomatous non-small cell lung cancer. Cancer Res Treat. 2017. doi:10.4143/crt.2016.546
20. Seckl MJ, Ottensmeier CH, Cullen M, et al. Multicenter, phase III, randomized, double-blind, placebo-controlled trial of pravastatin added to first-line standard chemotherapy in small-cell lung cancer (LUNGSTAR). J Clin Oncol. 2017;35(14):1506–1514. doi:10.1200/JCO.2016.69.7391
21. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. doi:10.1124/pr.58.3.10
22. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. doi:10.1158/0008-5472.CAN-09-1947
23. Nakanishi T, Menju T, Nishikawa S, et al. The synergistic role of ATP-dependent drug efflux pump and focal adhesion signaling pathways in vinorelbine resistance in lung cancer. Cancer Med. 2018;7(2):408–419. doi:10.1002/cam4.1282
24. Kononen J, Bubendorf L, Kallionimeni A, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4:844.
25. Poeta ML, Manola J, Goldwasser MA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357(25):2552–2561. doi:10.1056/NEJMoa073770
26. Molina-Vila MA, Bertran-Alamillo J, Gasco A, et al. Nondisruptive p53 mutations are associated with shorter survival in patients with advanced non-small cell lung cancer. Clin Cancer Res. 2014;20(17):4647–4659. doi:10.1158/1078-0432.CCR-13-2391
27. Sato T, Matsuyama Y. Marginal structural models as a tool for standardization. Epidemiology. 2003;14(6):680–686. doi:10.1097/01.EDE.0000081989.82616.7d
28. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S1–S45. doi:10.1161/01.cir.0000437738.63853.7a
29. Courtin A, Smyth T, Hearn K, et al. Emergence of resistance to tyrosine kinase inhibitors in non-small-cell lung cancer can be delayed by an upfront combination with the HSP90 inhibitor onalespib. Br J Cancer. 2016;115(9):1069–1077. doi:10.1038/bjc.2016.294
30. Yang Z, Su Z, DeWitt JP, et al. Fluvastatin prevents lung adenocarcinoma bone metastasis by triggering autophagy. EBioMedicine. 2017;19:49–59.
31. Reka AK, Kuick R, Kurapati H, Standiford TJ, Omenn GS, Keshamouni VG. Identifying inhibitors of epithelial-mesenchymal transition by connectivity map-based systems approach. J Thorac Oncol. 2011;6(11):1784–1792.
32. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15(1):2–8.
33. Solomon H, Buganim Y, Kogan-Sakin I, et al. Various p53 mutant proteins differently regulate the Ras circuit to induce a cancer-related gene signature. J Cell Sci. 2012;125(Pt 13):3144–3152.
34. Salari A, Thomay K, Lentes J, et al. Effect of TP53 contact and conformational mutations on cell survival and erythropoiesis of human hematopoietic stem cells in a long term culture model. Oncotarget. 2018;9(52):29869–29876.
35. van Oijen MGCT, Slootweg PJ. Gain-of-function mutations in the tumor suppressor gene p53. Clin Cancer Res. 2000;6(6):2138.
36. Turrell FK, Kerr EM, Gao M, et al. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev. 2017;31(13):1339–1353.
37. Parrales A, Ranjan A, Iyer SV, et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol. 2016;18(11):1233–1243.
38. Soma MR, Pagliarini P, Butti G, Paoletti R, Paoletti P, Fumagalli R. Simvastatin, an inhibitor of cholesterol biosynthesis, shows a synergistic effect with N,N′-Bis(2-chloroethyl)-N-nitrosourea and β-interferon on human glioma cells. Cancer Res. 1992;52(16):4348.
39. Wong WWL, Dimitroulakos J, Minden MD, Penn LZ. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002;16:508.
40. Kim S. New and emerging factors in tumorigenesis: an overview. Cancer Manag Res. 2015;7:225–239.
41. Park YH, Jung HH, Ahn JS, Im YH. Statin induces inhibition of triple negative breast cancer (TNBC) cells via PI3K pathway. Biochem Biophys Res Commun. 2013;439(2):275–279.
42. Wang T, Seah S, Loh X, et al. Simvastatin-induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget. 2016;7(3):2532–2544.
43. Sos ML, Koker M, Weir BA, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69(8):3256–3261.
44. Robin NC, Agoston Z, Biechele TL, James RG, Berndt JD, Moon RT. Simvastatin promotes adult hippocampal neurogenesis by enhancing Wnt/beta-catenin signaling. Stem Cell Rep. 2014;2(1):9–17.
45. Chen J, Zhang ZG, Li Y, et al. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol. 2003;53(6):743–751.
46. Chen P-Y, Sun J-S, Tsuang Y-H, Chen M-H, Weng P-W, Lin F-H. Simvastatin promotes osteoblast viability and differentiation via Ras/Smad/Erk/BMP-2 signaling pathway. Nutr Res. 2010;30(3):191–199.
47. Lee DK, Park EJ, Kim EK, et al. Atorvastatin and simvastatin, but not pravastatin, up-regulate LPS-induced MMP-9 expression in macrophages by regulating phosphorylation of ERK and CREB. Cell Physiol Biochem. 2012;30(3):499–511.
48. Aisner DL, Sholl LM, Berry LD, et al. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations-the Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res. 2018;24(5):1038–1047.
49. Deben C, Deschoolmeester V, Lardon F, Rolfo C, Pauwels P. TP53 and MDM2 genetic alterations in non-small cell lung cancer: evaluating their prognostic and predictive value. Crit Rev Oncol Hematol. 2016;99:63–73.
50. Bockorny B, Dasanu CA. HMG-CoA reductase inhibitors as adjuvant treatment for hematologic malignancies: what is the current evidence? Ann Hematol. 2015;94(1):1–12.
51. Kodach LL, Jacobs RJ, Voorneveld PW, et al. Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness‘ via the bone morphogenetic protein pathway. Gut. 2011;60(11):1544–1553.
52. Bedi O, Dhawan V, Sharma PL, Kumar P. Pleiotropic effects of statins: new therapeutic targets in drug design. Naunyn Schmiedebergs Arch Pharmacol. 2016;389(7):695–712.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.