")
Back to Journals » International Journal of Nanomedicine » Volume 14
Starch nanoparticles for delivery of the histone deacetylase inhibitor CG-1521 in breast cancer treatment
Authors Alp E , Damkaci F , Guven E, Tenniswood M
Received 23 October 2018
Accepted for publication 24 January 2019
Published 20 February 2019 Volume 2019:14 Pages 1335—1346
DOI https://doi.org/10.2147/IJN.S191837
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Esma Alp,1–4 Fehmi Damkaci,2 Eylem Guven,1 Martin Tenniswood3,4
1Department of Nanotechnology and Nanomedicine, Hacettepe University, Beytepe, Ankara 06800, Turkey; 2Department of Chemistry, State University of New York at Oswego, Oswego, NY 13126, USA; 3Cancer Research Center, Rensselaer, NY 12144, USA; 4Department of Biomedical Sciences, State University of New York, University at Albany, Rensselaer, NY 12144, USA
Background: The efficacy of epigenetic drugs, such as histone deacetylase inhibitors, is often diminished by poor aqueous solubility resulting in limited bioavailability and a low therapeutic index. To overcome the suboptimal therapeutic index, we have developed a biocompatible starch nanoparticle formulation of CG-1521, a histone deacetylase inhibitor in preclinical development for hard-to-treat breast cancers, which improves its bioavailability and half-life.
Methods: The physicochemical parameters (size, zeta potential, morphology, loading, and release kinetics) of these nanoparticles (CG-NPs) have been optimized and their cytotoxic and apoptotic capacities measured in MCF-7 breast cancer cell line. The mechanism of action of the encapsulated drug was compared with the free drug at molecular level.
Results: We show that encapsulation of CG-1521 substantially reduces the release rate of drug and provides a significantly enhanced cytotoxic ability of nanoparticles compared with equivalent dose of free CG-1521. CG-NPs induced cell cycle arrest and significant apoptosis in MCF-7 cells in vitro. The biological action of encapsulated drug has the similar impact with free drug on gene expression.
Conclusion: The findings suggest that encapsulation of CG-1521 into starch nanoparticles can improve drug delivery of histone deacetylase inhibitors for breast cancer therapy without interfering with the mechanism of action of the drug.
Keywords: cell cycle, apoptosis, DNA fragmentation, gene expression, epigenetics, MCF-7
Introduction
Breast cancer is the second leading cause of mortality among all cancers in women with an estimated 40,610 deaths in the USA in 2017.1 The majority of deaths are associated with metastatic dissemination of the disease to distant sites. Conventional treatments for localized breast cancer include surgery and radiotherapy followed by targeted therapies for estrogen receptor-positive and human epidermal receptor growth factor 2 (ERBB2/HER2)-positive tumors. In contrast, triple negative breast cancer and metastatic breast cancers are usually treated systemically with chemotherapy. Standard chemotherapies and many newly developed systemic therapies designed to treat aggressive breast cancer are hampered by rapid metabolism or dose-limiting off-target toxicity. Nanotechnology-based drug delivery may offer more effective formulations for the delivery of therapeutic agents that can overcome biological barriers and selectively target tumor tissues, improving efficacy and reducing toxicity.2
Several different kinds of nanocarriers are currently being developed as potential delivery vehicles, including liposomes,3 hydrogels,4 solid lipid nanoparticles (NPs),5 and synthetic polymeric NPs.6,7 Polymers derived from natural sources are also being explored as drug delivery systems due to their biochemical similarity with extracellular matrix components, ease of isolation and chemical modifications, and biocompatibility after enzymatic and hydrolytic degradation in the body.8 Polysaccharide-based nanocarriers are particularly useful due to their structural flexibility for modifications, varied composition, and properties. These NPs can be formulated to improve the solubility of hydrophobic drugs, leading to prolonged half-life and increased drug concentration in the targeted tumor as a result of enhanced permeability and retention (EPR).9,10 Formulating cytotoxic agents into polymeric NPs provides a significant reduction in side effects while increasing the therapeutic index of antitumor drugs.11 For example, NPs composed of poly(lactic-co-glycolic) acid (PLGA) loaded with cisplatin have been shown to be taken up efficiently by LNCaP prostate cancer cells, resulting in a greater cytotoxic effect compared with free cisplatin,12 and dextran sulfate-chitosan encapsulation of the tyrosine kinase inhibitor, lapatinib, is more effective against BT474 HER2-positive breast cancer cells than the free drug.13
Polysaccharides are relatively understudied as drug delivery vehicles, even though they possess many advantages such as preventing nonspecific protein adsorption, providing neutral coating with low surface energy,14 and allowing modification with various ligands due to high proportion of reactive groups in their backbone.15 Starch has been used successfully for drug delivery since it is biocompatible and biodegradable.16 Starch NPs, modified by oxidation, cross-linking, or hydroxypropylation,17 have been used to encapsulate ciprofloxacin,18 curcumin,19 and sodium diclofenac20 among others. However, few studies have focused on fabrication of NPs using starch without any chemical modification.
Acetylation and deacetylation of histones play a key role in the regulation of chromatin structure and transcription and are controlled by histone acetyltransferase and histone deacetylase (HDAC) enzymes. The alterations (mutation, translocation, amplification) in the activities of these enzymes have been reported in various cancers.21,22 Histone deacetylase inhibitors (HDACi) represent a class of promising therapeutics currently in clinical trials as monotherapies or combined therapies against multiple cancers.23 CG-1521 (7-phenyl-2, 4, 6-hepta-trienoyl hydroxamic acid) is an HDAC inhibitor in preclinical development. Several studies have shown that CG-1521 inhibits cell proliferation and stabilizes the acetylation of tumor suppressor, p53, particularly in breast and prostate cancer cell lines, leading to cell cycle arrest and apoptosis.24–26 However, the therapeutic potential of CG-1521 and most other HDACi is limited by insolubility and rapid metabolism in vivo.27,28 Thus, there is a need to develop novel strategies to overcome these limitations and improve the therapeutic effects of HDACi for both solid tumors and for hematological cancers.
The aim of this study was to determine whether encapsulation in starch NPs improves the efficacy of CG-1521 for the treatment of breast cancer. This would represent a significant therapeutic advance for triple negative and inflammatory breast cancers, given the current paucity of chemotherapeutic treatment options.
The data presented in this manuscript demonstrate that encapsulating CG-1521 into starch NPs increases the efficacy of the drug and increases the half-life of the drug in vitro without altering the biological targets of CG-1521.
Experimental procedures
Methods
α-MEM cell culture media, trypsin-ETDA, Glutamax, and glutaraldehyde were purchased from Life Technologies Corporation (Thermo Fisher Scientific, Waltham, MA, USA). FBS was obtained from Atlanta Biologicals (Norcross, GA, USA). Triton X-100, BSA, dimethyl sulfoxide (DMSO), polyvinyl alcohol (PVA), rhodamine 6C, propodium iodide, and starch (unmodified corn starch containing ~73% amylopectin and 27% amylose, product number: S4126, CAS number: 9005-25-8) were purchased from Sigma-Aldrich (St Louis, MO, USA). Formaldehyde was purchased from Polysciences, Inc. (Warrington, PA, USA). Bromodeoxy-uridine triphosphate (Br-dUTP) was purchased from Phoenix Flow Systems (San Diego, CA, USA). Terminal transferase, cobalt chloride, and RNase were purchased from Roche Applied Science (Indianapolis, IN, USA). Hexadeutero dimethylsulfoxide (DMSO-d6) was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA). Crystal violet was purchased from Acros Organics (Morris Plains, NJ, USA). Reagents for Muse kit analysis were purchased from EMD Millipore (Billerica, MA, USA). CG-1521 was a gift from Errant Gene Therapeutics, LLC (Chicago, IL, USA).
Cell culture
The MCF-7 breast cancer cell line was obtained from American Type Culture Collection (Rockville, MD, USA). Cells were grown and maintained in alpha-MEM medium supplemented with 10% FBS and 1% Glutamax under conditions of 5% CO2 and 85% humidity at 37°C. The cells were passaged every 4–5 days when they reach ~80% confluence.
Preparation of CG-1521-loaded starch NPs
CG-1521-loaded starch NPs (CG-NPs) were prepared using the emulsion-solvent diffusion technique.29 One hundred milligrams starch and 25 mg CG-1521 were dissolved in 10 mL of DMSO as the organic phase and 1 g of PVA was dissolved in 50 mL distilled water as the aqueous phase. The starch and CG-1521 solution were added to the aqueous PVA solution using a homogenizer (IKA T-18 Ultra-Turrax, Wilmington, NC, USA) under nitrogen at room temperature. The resulting biphasic system was stirred for 5 minutes to form droplets, which were converted to NPs when DMSO was diffused by addition of 30 mL distilled water, followed by stirring on a magnetic stirrer for 2 hours. NPs were separated by centrifugation at 12,000 rpm for 30 minutes in an Eppendorf (5810R) centrifuge, washed with distilled water, lyophilized (Labconco, Kansas City, MO, USA), and stored at 4°C. Void NPs (VD-NPs) were prepared using the same method as CG-NPs except that CG-1521 was not added to organic phase.
Characterization of NPs
The average particle size, size distribution, polydispersity index (PDI), and zeta potential of VD-NPs and CG-NPs were characterized using light scattering particle size analyzer (Zeta Sizer 3000 HSA, Malvern Instruments, Malvern, UK). NPs were dispersed in distilled water before size and potential measurements.
The morphological analyses of the prepared NPs were performed using scanning electron microscopy (SEM; JOEL 6100LV, Peabody, MA, USA) and atomic force microscopy (AFM; NanoMagnetics EzAFM, Oxford, UK). For SEM analysis, NPs were dispersed in distilled water, dropped on a glass slide, air dried at room temperature, placed onto an aluminum stub, and sputter-coated with gold. For AFM imaging, a drop of NP suspension was placed on a silicon wafer and air-dried. Sample was scanned in tapping mode with standard Si cantilever at 310 kHz resonance frequency.
The drug entrapment efficiency of CG-1521 in NPs was determined by NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Grand Island, NY, USA). Starting with a known starting concentration of CG-1521, NPs were separated from dispersion by centrifugation at 12,000 rpm. After centrifugation, the supernatant was analyzed for the amount of free CG-1521 by NanoDrop spectrophotometer at 335 nm using standard calibration curve of CG-1521. The percentage of encapsulated CG1-521 was estimated by subtracting the drug present in the supernatant from the starting concentration.
FTIR spectra of NPs were obtained by FTIR spectrometry (Nicolet™ iS™ 5, Thermo Fisher Scientific, Madison, WI, USA). The lyophilized samples were placed on a diamond cell and scanned through the range of 600–4,000 cm−1 to investigate chemical characterization of drug-loaded NPs. 1H-NMR spectra were obtained using Bruker 300 MHz UltraShield (Billerica, MA, USA) instrument. The lyophilized samples were dissolved in 1 mL DMSO-d6 for NMR analysis.
In vitro CG-1521 release
In vitro CG-1521 release from NPs was measured in PBS buffer at pH 6.0 and pH 7.4. NPs were dispersed in PBS buffers at different pH levels to the final concentration 0.3 mg CG-1521/mL per 1 mL buffer. The samples of NP suspension or free CG-1521 solution in methanol were sealed in dialysis membranes (Spectra Por, 12,000–14,000 Da MWCO, Spectrum Laboratories, Rancho Dominguez, CA, USA) placed separately in 50 mL of PBS and agitated constantly at 300 rpm. At specified time intervals, 0.5 mL samples were withdrawn from PBS medium and replaced with 0.5 mL release medium. The CG-1521 concentration of each sample was diluted with methanol and measured at 335 nm using NanoDrop spectrophotometer. The release of free CG-1521 solution from dialysis membrane was used as control group.
The cumulative release (%) of CG-1521 was determined using the following formulae:
|
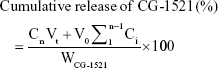
where Vt: total volume of PBS (50 mL); V0: replaced volume of buffer (0.5 mL); WCG-1521: total amount of CG-1521 loaded in NPs; Ci and Cn: concentrations of CG-1521 at different times (mg/mL).
In vitro cytotoxicity assay
Crystal violet assays were used to measure the effect of free CG-1521 and NP-encapsulated CG-1521 on cell number. MCF-7 cells 1×103 were seeded in 150 μL of alpha-MEM supplemented with 5% FBS and 1% Glutamax in each 96-well format plates. After 48 hours of incubation, cells were treated with free CG-1521 at different concentrations (0–7.5 μM) and equivalent CG-1521-loaded NPs for 24, 48, and 72 hours. Controls included VD-NPs that were added at the same particle density as CG-NPs and DMSO, the vehicle for the delivery of free CG-1521. After treatment, the medium was gently aspirated, and 200 μL of 2% glutaraldehyde in PBS was added to each well for fixation, followed by incubation for 20 minutes at room temperature. The glutaraldehyde solution was replaced with 150 μL of 0.1% Crystal violet and the plate incubated for 30 minutes at room temperature and destained. After air drying, 200 μL of 0.2% Triton X-100 was added to each well to solubilize the dye and the absorbance was measured at 590 nm (Victor3 V 1420 Multilabel Counter, PerkinElmer Inc., Waltham, MA, USA).
Apoptosis and DNA fragmentation assays
The Annexin V dead cell assay was performed for the quantitative analysis of live, dead, early, and late apoptotic cell populations. 7-amino-actinomycin D was used as a marker of dead cells. Briefly, MCF-7 treated with free CG-1521, CG-NPs, and vehicle controls (DMSO and VD-NPs) for 48 hours was prepared as described above. One hundred microliters of cell suspension were diluted with Muse Annexin reagent (1:8), incubated for 20 minutes in the dark at room temperature according to the manufacturer’s protocol, and analyzed using the Muse cell analyzer.
Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) assay was used to detect DNA fragmentation after treatment with free CG-1521, CG-NPs, and vehicle controls. Briefly, MCF-7 cells were treated with free 1.5 μM CG-1521 and equivalent amount of drug-loaded CG-NPs for 72 hours and vehicle controls (DMSO and VD-NPs). After treatment, cells were trypsinized for 5 minutes at 37°C, washed with PBS, suspended in 3 mL of 1% FBS/PBS, fixed with 4% formaldehyde for 15 minutes at 4°C, and permeabilized with ice-cold 90% ethanol overnight at −20°C. Cells 1×106 were transferred to microfuge tube, washed with PBS/0.2% BSA, and suspended in 50 μL DNA labeling solution (20% TdT reaction buffer, 16% Br-dUTP, 10% cobalt chloride, 2.5% TdT enzyme, and 51.5% distilled water). Cells were incubated in the DNA labeling solution for 1 hour at 37°C, washed with PBS/0.2% BSA and rinse buffer (PBS/0.1% Triton X-100/1% BSA), and centrifuged at 1,200 rpm at 4°C for 3 minutes. The cell pellet was suspended in 100 μL FITC-conjugated anti-BrdU monoclonal antibody and incubated in the dark for 30 minutes. Cells were analyzed using BD LSR II Flow Cytometer (BD Biosciences, San Jose, CA, USA) within 1 hour.
Cell cycle analysis
To determine the effect of free CG-1521 and CG-NP on cell cycle, cells were treated with 3 μM CG-1521 (DMSO control) and CG-NPs (VD-NP control) for 72 hours. After the treatment period, medium was collected and cell monolayers were washed with PBS. The cells were trypsinized to collect cells and debris by combining the medium and washes, centrifuged at 1,200 rpm for 3 minutes at 4°C, and resuspended in 3 mL 1% FBS/PBS. The cells were fixed overnight in ice-cold 90% ethanol at −20°C. Fixed cells were centrifuged at 1,200 rpm for 3 minutes at 4°C and suspended in 600 μL PBS. Cells 3×105 were transferred to a microfuge tube, washed with PBS/0.2% BSA, and suspended in 200 μL Muse cell cycle reagent. After 30 minutes of incubation in the dark at room temperature, the cells were analyzed using the Muse cell analyzer.
Real-time quantitative reverse transcriptase PCR
MCF-7 cells were plated at a density of 3×105 cells/dish for 48 hours of incubation. After 48 hours of treatment, cells were harvested by trypsinization. Total RNA was extracted from free CG-1521 and CG-NP-treated (equivalent to 3 μM CG-1521) MCF-7 cells, using the Qiagen RNeasy kit (Qiagen, Valencia, CA, USA). The concentration of total RNA was determined using a NanoDrop spectrophometer (Thermo Fisher Scientific); 1.5 μg of total RNA was used to synthesize cDNA using MultiScribe™ Reverse Transcriptase (Applied Biosystem, TX, USA) and reverse transcriptase reagents. The reaction of cDNA synthesis was incubated in Mastercycler (Eppendorf) for 10 minutes at 25°C, 60 minutes at 37°C, and 5 minutes at 95°C. Gene transcript levels were determined using a QuantStudio™ 12K Flex Real-Time PCR System (Fisher Scientific) using SYBR Green PCR Master mix (Applied Biosystems), consisting of 40 cycles (15 seconds, 95°C; 1 minute, 60°C). The changes in the mRNA level of each transcript were normalized to endogenous control GAPDH for each sample. The data were analyzed using 2−ΔΔCt method. The primer sequences (Integrated DNA Technologies, Coralville, IA, USA) used are shown in Table 1.
 | Table 1 Primer sequences for RT-PCR |
Statistical analyses were performed using GraphPad Prism Software using one-way ANOVA (Tukey’s test) and significance was defined as P-value of <0.05. The data are expressed as mean ± SD of three independent biological replicates.
Results
Size and zeta potential distributions of NPs were determined using a Zeta Sizer 3000 HSA as described in the Methods section. As shown in Figure 1, the average particle size (hydrodynamic diameter) of VD-NPs in aqueous solution is 180 nm with a PDI of 0.14 (Figure 1A). The low PDI value (<0.4) indicates that the NPs are in narrow size range, which plays important role in tissue accumulation and renal clearance.30 The particle size distribution of CG-1521-loaded NPs (CG-NP) (200 nm) with a PDI of 0.12 demonstrates that the CG-NPs have a similar size as the VD-NPs (Figure 1B). The zeta potential (net surface charge) is another physical characteristic that plays a role in the stability of the NPs in the circulation and accumulation of NPs at the site of interest.31 The average zeta potential of VD-NPs and CG-NPs were −16.1 mV (Figure 1C) and −10.2 mV (Figure 1D), respectively, suggesting that for both NPs, the hydroxyl groups of starch are predominantly localized on the outer surface of NPs.
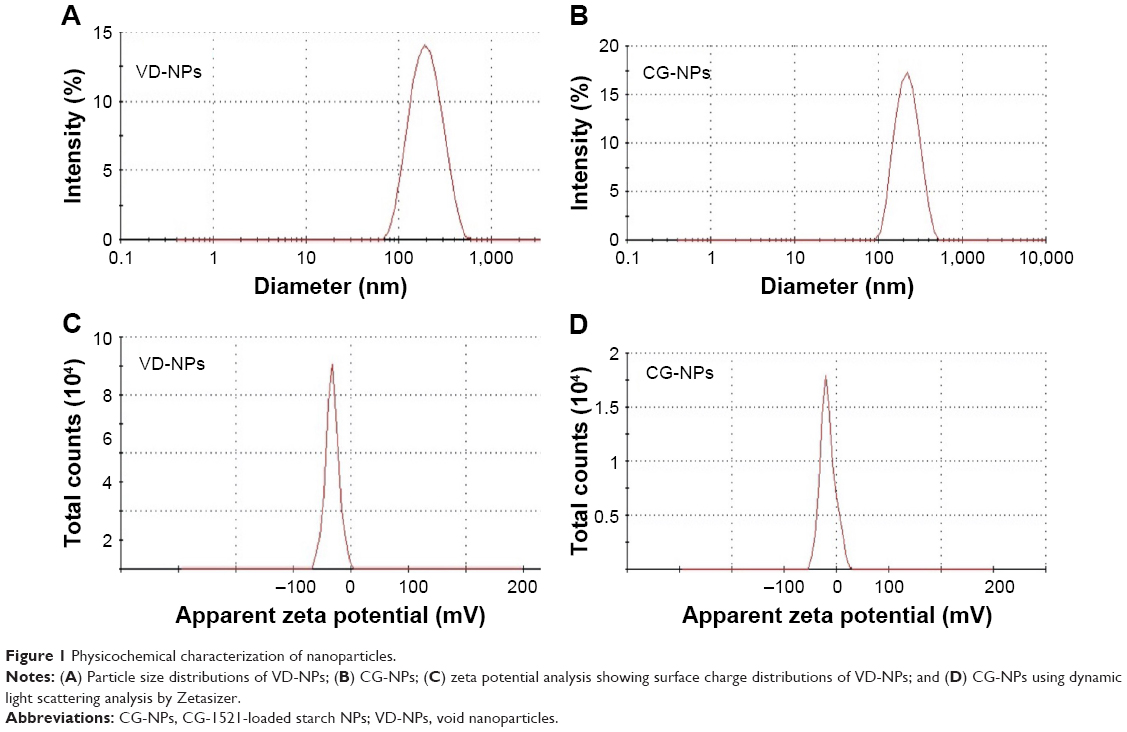 | Figure 1 Physicochemical characterization of nanoparticles. |
The morphological characteristics of VD-NPs were visualized by SEM (Figure 2A) and AFM (Figure 2B). Both AFM and SEM demonstrate that the starch NPs have spherical topographies and homogeneous distributions. The particle sizes of VD-NPs determined by SEM and AFM are consistent with those measured by Zetasizer. Chemical characterization of VD-NPs and CG-NPs was evaluated by FTIR (Figure 2C). In the spectrum of CG-NPs, a peak at 3,011 cm−1 is attributed to stretching vibration of the −C=C from aromatic group of CG-1521, the peaks at 1,580 and 1,600 cm−1 are assigned to the stretching vibration of C=C, the peak at 2,923 cm−1 corresponding to the vibration of C−H, and another peak at 1,632 cm−1 is attributed to carbonyl stretching of the −C=O. The results confirm the encapsulation of CG-1521 without any chemical alteration.
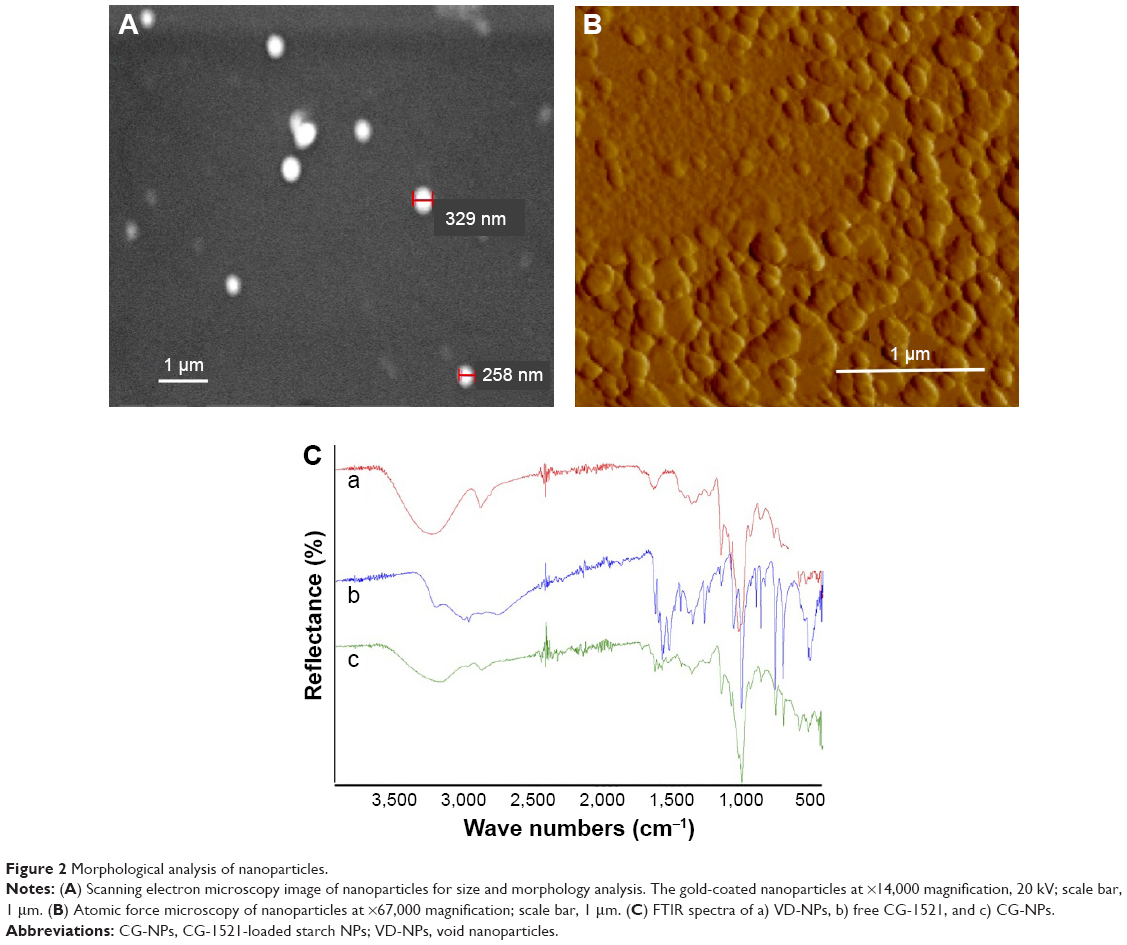 | Figure 2 Morphological analysis of nanoparticles. |
The cumulative release of CG-NPs and free CG is compared in Figure 3. Approximately 95% of free CG-1521 was released within 4 hours, whereas the release curve of CG-NPs was characterized by an initial rapid release during the first 10 hours, followed by slower and continuous increase over 120 hours. The effect of pH on release pattern of encapsulated CG-1521 in PBS (pH 6.0 and pH 7.4) was also determined. Over the first 10 hours, the release of CG-1521 from the CG-NPs at pH 6.0 and pH 7.4 is not significantly different. The release of the CG-1521 from the NPs incubated at pH 7.4 (curve A) does not increase further after 24 hours. In contrast, the release of CG-1521 continues to increase when the NPs are incubated at pH 6.0 from ~48% (at 24 hours) to 64% (at 120 hours). This represents an additional release of ~18% of the encapsulated CG-1521, demonstrating that nearly 30% of the encapsulated CG-1521 is released in a sustained manner.
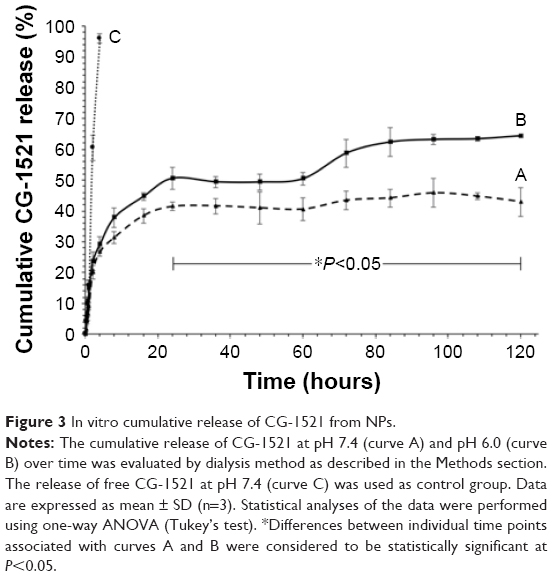 | Figure 3 In vitro cumulative release of CG-1521 from NPs. |
Confocal microscopy of rhodamine 6G-labeled VD-NPs demonstrates the localization of NPs in the cytoplasm and perinuclear region of MCF-7 cells indicating that they are taken up efficiently into the cells by 12 hours (data not shown).
The dose-dependent effects of free CG-1521 and CG-NPs on MCF-7 human breast cancer cells were evaluated using the crystal violet assay. The results demonstrate that at equivalent drug concentrations CG-NPs are more effective at inducing cell death than the free drug in a dose-dependent manner (Figure 4).
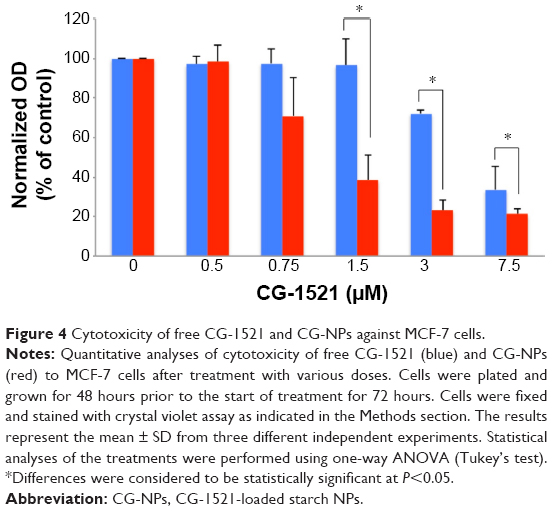 | Figure 4 Cytotoxicity of free CG-1521 and CG-NPs against MCF-7 cells. |
Cell cycle analysis was performed to determine cell populations in different phases of the cell cycle after treatment with free CG-1521 and CG-NPs (Figure 5). In cells treated with CG-NPs, there was a significant increase in proportion of cells in G1 compared with free CG-1521.
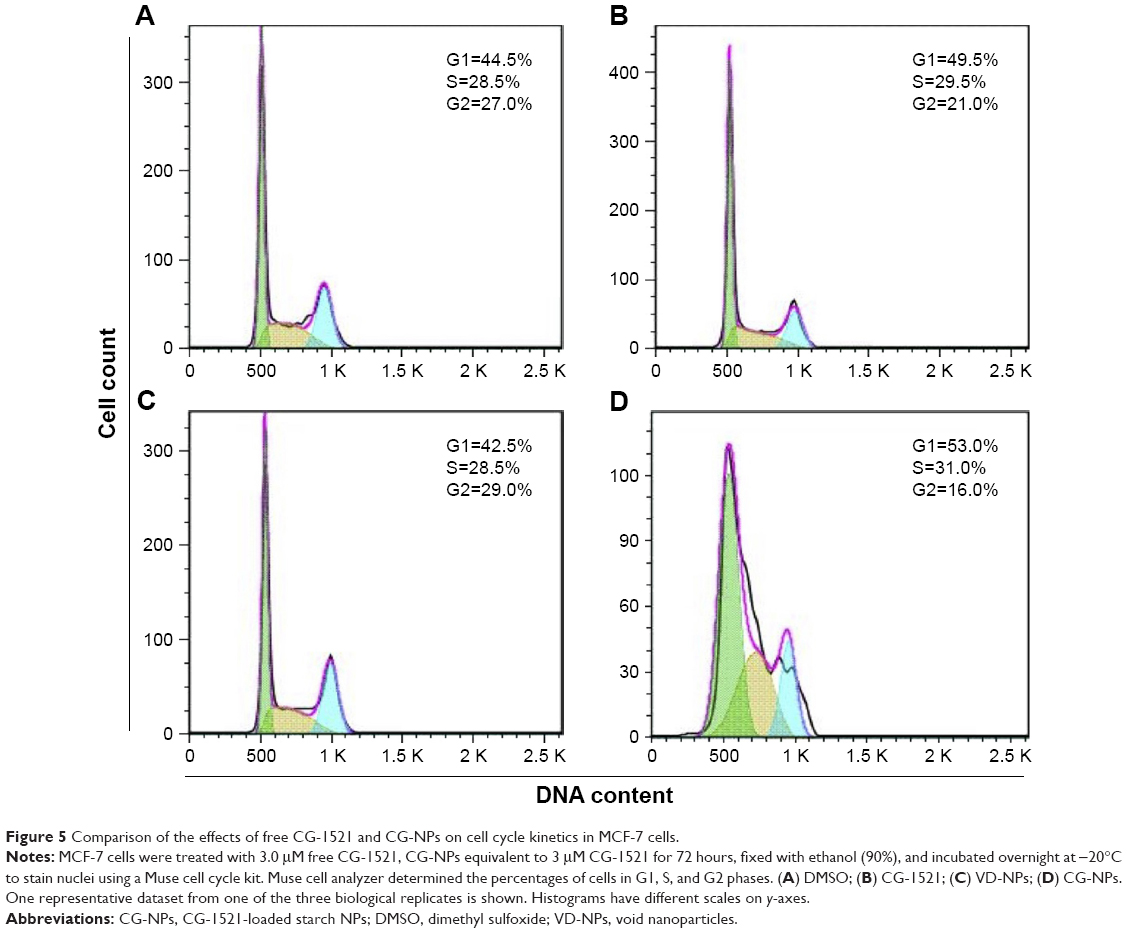 | Figure 5 Comparison of the effects of free CG-1521 and CG-NPs on cell cycle kinetics in MCF-7 cells. |
Treatment of MCF-7 cells with CG-NPs significantly increases the proportion of the cells undergoing apoptotic cell death compared with CG-1521 alone. As shown in Figure 6A, the percentage of dead cell population treated with CG-NPs (48.5%) is significantly higher than those treated with free CG-1521 (17.9%). Figure 6B shows the scatterplots of DNA strand breaks in apoptotic cells treated with free CG-1521 and CG-NPs, as well as control groups. There is an increase in PI-positive population of cells treated with CG-NPs (Q2 – 9.29%) in comparison with free CG-1521 (Q2 – 3.40%) demonstrating that CG-NPs are more effective than free CG-1521 at equivalent concentrations (1.5 μM).
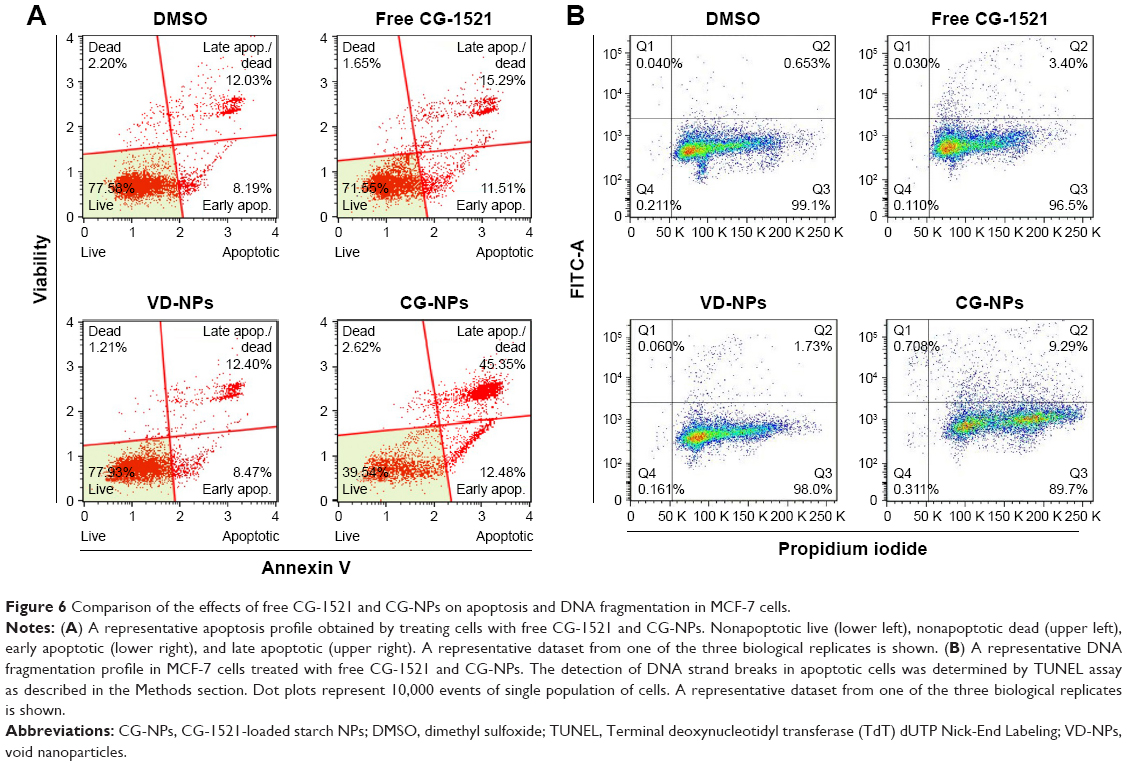 | Figure 6 Comparison of the effects of free CG-1521 and CG-NPs on apoptosis and DNA fragmentation in MCF-7 cells. |
To compare the underlying biological mechanism of free and CG-NPs, the transcript levels of several genes associated with ontologies associated with cell death, cell cycle arrest, and spindle formation were measured 48 hours after treatment. These sequences were selected based on previous studies from the laboratory describing the effects of free CG-1521 in MCF-7 cells.32 Figure 7A compares the effects of free and encapsulated CG-1521 on the expression of four transcripts implicated in cell death in MCF-7 cells. Transcript levels of Bnip3 are significantly downregulated after 48 hours of treatment by both free CG-1521 and CG-NPs. In contrast, Bnip3L levels are not significantly altered by either formulation. Gdf15 and survivin are significantly upregulated and downregulated, respectively, after treatment with CG-NPs but not by free CG-1521.
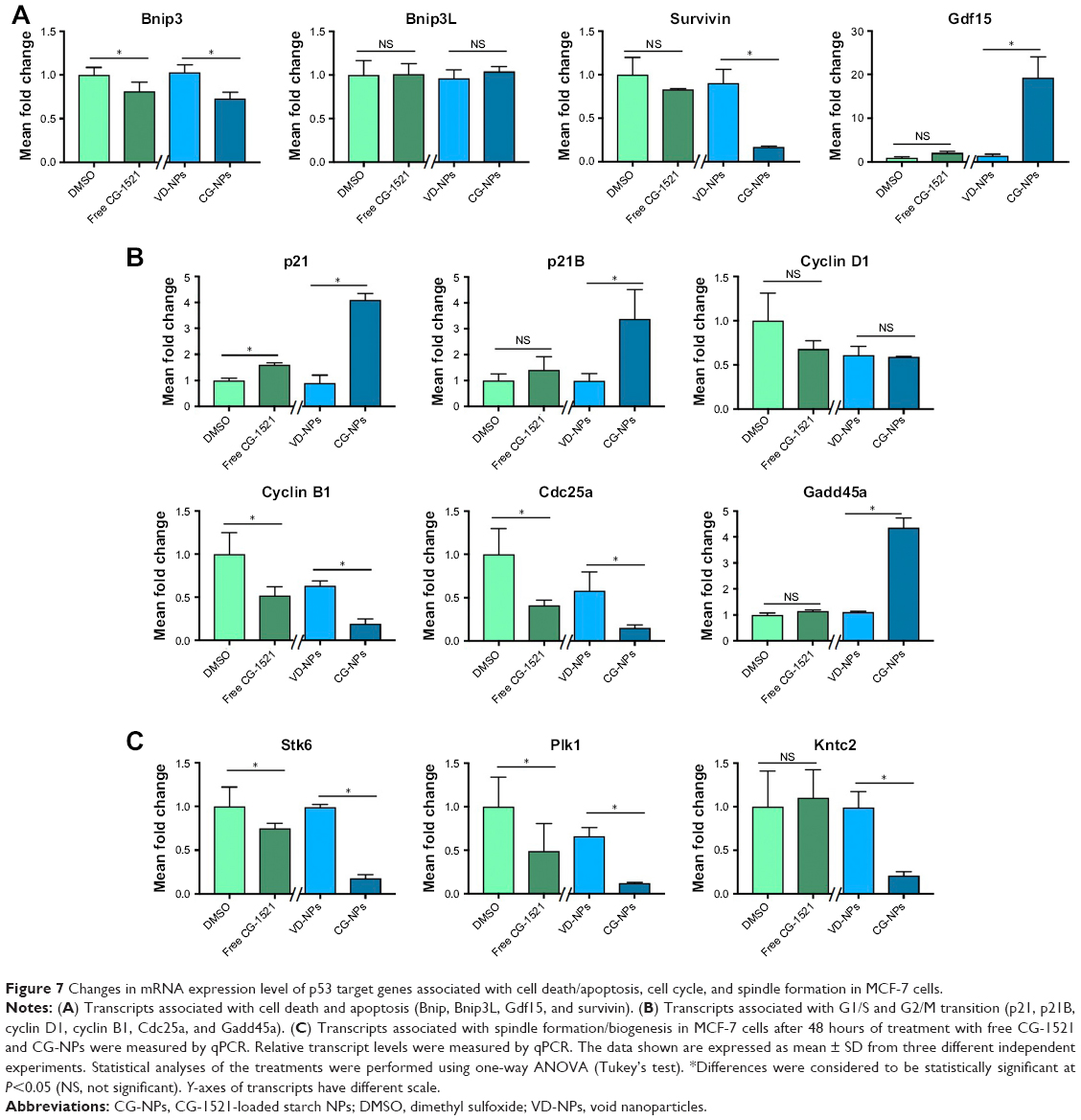 | Figure 7 Changes in mRNA expression level of p53 target genes associated with cell death/apoptosis, cell cycle, and spindle formation in MCF-7 cells. |
Figure 7B shows that p21, its variant p21B, and Cdc25a, major targets of p53 associated with cell cycle arrest, are upregulated significantly after 48 hours of treatment by both free CG-1521 and CG-NPs compared with vehicle controls, while the expression level of cyclin D1 (which is associated with G1/S transition) is not significantly altered. The transcript level of cyclin B1, a G2/M transition-associated gene, is downregulated significantly by both treatments. The mRNA level of Gadd45a, which is also associated with G2/M transition, is increased by CG-NP treatment significantly, while free CG-1521 has a less profound effect on Gadd45a transcript levels.
Changes in the transcript level of three genes (Stk6, Plk1, Kntc2) involved in spindle and centrosome formation were examined after treatment with free CG-1521 and CG-NPs (Figure 7C). The transcript levels of both Stk6 and Plk1 are downregulated significantly with similar fold change after treatment with free CG-1521 and CG-NPs. The transcript level of Kntc2 gene is also downregulated significantly by CG-NPs; however, the free drug does not affect the Kntc2 mRNA levels significantly.
Discussion
HDACi have shown promising antitumor properties in breast cancer.32,33 However, as free drugs these compounds have limited clinical utility because they are rapidly metabolized in the peripheral circulation. Encapsulating antitumor agents in polymeric NPs offers several advantages, including improving drug solubility, protection from systemic metabolism, increased drug exposure time, and reduction of systemic toxicity. The small size of NPs allows them to accumulate at the tumor site by taking advantage of the EPR when intravenously injected.34 The pathophysiological properties of tumors, including leaky vasculature and poor lymphatic drainage, provide the opportunity to improve distribution and target NP-encapsulated therapeutics to solid tumors.35,36 However, despite these obvious advantages, there have been limited efforts to improve the delivery of HDACi using nanoformulation. Some studies have described the synthesis and applications for the delivery of HDACi by nanoformulations including PLGA-based NPs for the delivery of vorinostat to prostate and colorectal cancers37 and a polyethylene oxide-norbornene macromonomer platform for the delivery of different HDACi prodrugs for mesothelioma models.38 In another study, lipid–polymer conjugated based NPs were utilized for the delivery of two HDACi, vorinostat and quisinostat, as radiosensitizers to investigate their therapeutic potentials in xenograft models of colorectal and prostate carcinomas.39 The studies outlined here represent the first study using polymeric starch NPs prepared using standard emulsification methods for the delivery of therapeutic drugs to breast cancer cells. The data demonstrate that encapsulation of CG-1521 enhances the therapeutic efficacy toward MCF-7 breast cancer cells and suggest that this encapsulation technology may be useful for other hormone-dependent cancers.
Characterization of the CG-NPs demonstrates that they are spherical NPs (<400 nm), which based on their zeta potential have a slightly overall negative charge. These NPs have excellent drug encapsulation capacity and a relatively slow release at physiological pH. However, the drug release is enhanced at the more acidic pH commonly associated with the tumor microenvironment and early endosomal pathway. This provides a prolonged half-life in the circulation and enhanced uptake and release in tumors compared with normal tissues. The delivery of CG-1521 encapsulated in starch NPs to MCF-7 cells is significantly better than that achieved by liposomal delivery. This is reflective of the enhanced uptake of starch NPs, which are not as hydrophobic as the liposomal carriers and therefore interact more effectively with the cellular uptake machinery and the early endocytic pathway.40 It should be noted that the delivery of other chemotherapeutic drugs, including simvastatin using lipid nanoencapsulation,41 and cholamine surface-modified gelatin NPs for siRNA delivery have also shown encouraging efficacy.42 Furthermore, the release of CG-1521 from the NPs at more acidic pH suggests that CG-NPs are most likely to release the drug in tumor microenvironment and early endosomes.
In MCF-7 cells, both free CG-1521 and CG-NPs induce cell cycle arrest in G2/M phase and an increase in the population of cells in the G1/S phase of the cell cycle. The G1/S arrest is most likely due to the stabilization of the acetylated isoforms of p53 that are induced by CG-1521.25 The increase of cells in G2/M has been described by our laboratory and others and associated with the stabilization of acetylated isoforms of the proteins associated with mitotic spindle assembly and mitotic catastrophe.33 CG-NPs induce cell death in a dose-dependent manner more effectively than CG-1521 alone as measured by both Annexin V positivity and DNA fragmentation. This is probably due to the limited uptake of free CG-1521 compared with the uptake and the release of the encapsulated drug. To establish that encapsulation of CG-1521 into NPs does not alter the biological pathways responsible for the onset of cell cycle arrest and apoptosis, we compared the effects of free and encapsulated CG-1521 on the expression of a cohort of genes previously shown to be responsive to CG-1521 in MCF-7 cells.32 This cohort contains genes that are modulated in response to the stabilization of acetylated p53 and a number of genes that are modulated by CG-1521 in a p53-independent manner in inflammatory breast cancer cells.26 The modulation of these sequences by CG-1521 in SUM 159PT breast cancer cells has previously been shown to induce mitotic catastrophe, suggesting that the encapsulation of CG-1521 enhances G2/M-associated cell death.26
With the exception of three transcripts, the expression of the majority of the transcripts is concordantly in response to free CG-1521 and CG-NP. Free CG-1521 does not modulate the expression of three genes (Gdf15, Gadd45a, and Kntc2), but encapsulated delivery significantly induces Gdf15 and Gadd45a while repressing the expression of Kntc2. This study represents a single time point, as a result it is not possible to determine whether the differences in the expression of these three transcripts represent differences in the induction of the biological pathways involved or differences in time course of regulation of these transcripts. In either case, the anticipated consequence of these changes in response to CG-NP would be a prolonged enhancement of G2/M-associated cell death.
Conclusion
Taken together, these data confirm that NP-assisted delivery of CG-1521 does not alter the mechanism of action of the HDACi and induce cell cycle arrest and apoptosis by the same mechanism previously reported for free CG-1521.26,32 Encapsulation of CG-1521 in polymeric starch NPs enhances the therapeutic efficacy by allowing a steady sustained release of the drug and improving its bioavailability. This could be clinically significant for the treatment of breast cancer and other solid tumors. Further modification of the starch monomers with targeting moieties such as folic acid for breast cancer or prostate-specific membrane antigen inhibitors for prostate cancer may further increase the specificity of the NPs and enhance the therapeutic response to CG-NPs. These encouraging data can be applied for targeting the delivery of HDACi or other drugs to improve their cancer cell selectivity with appropriate targeting moieties and could result in further enhanced therapeutic efficacy.
Acknowledgments
The research team would like to acknowledge Errant Gene Therapeutics for supplying CG-1521. The abstract of this paper was presented at the Applied Pharmaceutical Nanotechnology as a poster presentation with preliminary data at Broad Institute, Cambridge, MA, USA (October 2017). The poster abstract was published in APN 2017 Conference Agenda. (https://www.bostonsociety.org/APN/images/APN/pdfs/2017/APN2017Program.pdf).
Author contributions
EA, EG, and MT designed the project. EA performed all synthesis and characterization experiments with advice from FD. MT provided guidance for all cell studies, biostatistical analyses, and interpretation of the data. EA and MT wrote the manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflict of interest in this work.
References
National Cancer Institute [webpage on the Internet]. Cancer Stat Facts: Female Breast Cancer. Available from: https://seer.cancer.gov/statfacts/html/breast.html. Accessed December 12, 2017. | ||
Jain KK. Nanotechnology-based drug delivery for cancer. Technol Cancer Res Treat. 2005;4(4):407–416. | ||
Urbinati G, Marsaud V, Plassat V, Fattal E, Lesieur S, Renoir JM. Liposomes loaded with histone deacetylase inhibitors for breast cancer therapy. Int J Pharm. 2010;397(1–2):184–193. | ||
Fan T, Li M, Wu X, Li M, Wu Y. Preparation of thermoresponsive and pH-sensitivity polymer magnetic hydrogel nanospheres as anticancer drug carriers. Colloids Surf B Biointerfaces. 2011;88(2):593–600. | ||
Wang F, Li L, Liu B, Chen Z, Li C. Hyaluronic acid decorated Pluronic p85 solid lipid nanoparticles as a potential carrier to overcome multidrug resistance in cervical and breast cancer. Biomed Pharmacother. 2017;86:595–604. | ||
Lin G, Zhang H, Huang L. Smart polymeric nanoparticles for cancer gene delivery. Mol Pharm. 2015;12(2):314–321. | ||
Patel T, Zhou J, Piepmeier JM, Saltzman WM. Polymeric nanoparticles for drug delivery to the central nervous system. Adv Drug Deliv Rev. 2012;64(7):701–705. | ||
Beneke CE, Viljoen AM, Hamman JH. Polymeric plant-derived excipients in drug delivery. Molecules. 2009;14(7):2602–2620. | ||
Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47(1):113–131. | ||
Meng L, Huang W, Wang D, Huang X, Zhu X, Yan D. Chitosan-based nanocarriers with pH and light dual response for anticancer drug delivery. Biomacromolecules. 2013;14(8):2601–2610. | ||
Farokhzad OC, Langer R. Nanomedicine: developing smarter therapeutic and diagnostic modalities. Adv Drug Deliv Rev. 2006;58(14):1456–1459. | ||
Gryparis EC, Hatziapostolou M, Papadimitriou E, Avgoustakis K. Anticancer activity of cisplatin-loaded PLGA-mPEG nanoparticles on LNCaP prostate cancer cells. Eur J Pharm Biopharm. 2007;67(1):1–8. | ||
Mobasseri R, Karimi M, Tian L, Naderi-Manesh H, Ramakrishna S. Hydrophobic lapatinib encapsulated dextran-chitosan nanoparticles using a toxic solvent free method: fabrication, release property and in vitro anti-cancer activity. Mat Sci Eng C. 2017;74:413–421. | ||
Österberg E, Bergström K, Holmberg K, et al. Protein-rejecting ability of surface-bound dextran in end-on and side-on configurations: comparison to PEG. J Biomed Mater Res. 1995;29(6):741–747. | ||
Sarikaeo K. Starch: introduction and structure-property relationship. In: Visakh PM, Long Y, editors. Starch-Based Blends, Composites and Nanocomposites. London: The Royal Society of Chemistry; 2016:17–48. | ||
Marques AP, Reis RL, Hunt JA. The biocompatibility of novel starch-based polymers and composites: in vitro studies. Biomaterials. 2002;23(6):1471–1478. | ||
Wu X, Chang Y, Fu Y, Ren L, Tong J, Zhou J. Effects of non-solvent and starch solution on formation of starch nanoparticles by nanoprecipitation. Starch Stärke. 2016;68(3–4):258–263. | ||
Mahmoudi Najafi SH, Baghaie M, Ashori A. Preparation and characterization of acetylated starch nanoparticles as drug carrier: ciprofloxacin as a model. Int J Biol Macromol. 2016;87:48–54. | ||
Pang SC, Chin SF, Nadirah A, Tay SH, Yazid SNAM. Fabrication of polysaccharide-based nanoparticles as drug delivery nanocarriers. ECS Transactions. 2015;66(37):15–32. | ||
El-Naggar ME, El-Rafie MH, El-Sheikh MA, El-Feky GS, Hebeish A. Synthesis, characterization, release kinetics and toxicity profile of drug-loaded starch nanoparticles. Int J Biol Macromol. 2015;81:718–729. | ||
Pandolfi PP. Transcription therapy for cancer. Oncogene. 2001;20(24):3116–3127. | ||
Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci. 1998;95(6):3003–3007. | ||
Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27(32):5459–5468. | ||
de Ruijter AJM, Kemp S, Kramer G, et al. The novel histone deacetylase inhibitor BL1521 inhibits proliferation and induces apoptosis in neuroblastoma cells. Biochem Pharmacol. 2004;68(7):1279–1288. | ||
Roy S, Packman K, Jeffrey R, Tenniswood M. Histone deacetylase inhibitors differentially stabilize acetylated p53 and induce cell cycle arrest or apoptosis in prostate cancer cells. Cell Death Differ. 2005;12(5):482–491. | ||
Chatterjee N, Wang WL, Conklin T, Chittur S, Tenniswood M. Histone deacetylase inhibitors modulate miRNA and mRNA expression, block metaphase, and induce apoptosis in inflammatory breast cancer cells. Cancer Biol Ther. 2013;14(7):658–671. | ||
Elaut G, Török G, Vinken M, et al. Major phase I biotransformation pathways of trichostatin A in rat hepatocytes and in rat and human liver microsomes. Drug Metab Dispos. 2002;30(12):1320–1328. | ||
Vanhaecke T, Papeleu P, Elaut G, Rogiers V. Trichostatin A-like hydroxamate histone deacetylase inhibitors as therapeutic agents: toxicological point of view. Curr Med Chem. 2004;11(12):1629–1643. | ||
Niwa T, Takeuchi H, Hino T, Kunou N, Kawashima Y. In vitro drug release behavior of D,L-lactide/glycolide copolymer (PLGA) nanospheres with nafarelin acetate prepared by a novel spontaneous emulsification solvent diffusion method. J Pharm Sci. 1994;83(5):727–732. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. | ||
Knutson AK, Welsh J, Taylor T, Roy S, Wang WL, Tenniswood M. Comparative effects of histone deacetylase inhibitors on p53 target gene expression, cell cycle and apoptosis in MCF-7 breast cancer cells. Oncol Rep. 2012;27(3):849–853. | ||
Chatterjee N, Tenniswood M. The potential of histone deacetylase inhibitors in breast cancer therapy. Breast Cancer Manag. 2015;4(2):85–97. | ||
Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51(4):691–743. | ||
Maeda H, Fang J, Inutsuka T, Kitamoto Y. Vascular permeability enhancement in solid tumor: various factors, mechanisms involved and its implications. Int Immunopharmacol. 2003;3(3):319–328. | ||
Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42(6):463–478. | ||
Tran TH, Choi JY, Ramasamy T, et al. Hyaluronic acid-coated solid lipid nanoparticles for targeted delivery of vorinostat to CD44 overexpressing cancer cells. Carbohydr Polym. 2014;114:407–415. | ||
El Bahhaj F, Denis I, Pichavant L, et al. Histone deacetylase inhibitors delivery using nanoparticles with intrinsic passive tumor targeting properties for tumor therapy. Theranostics. 2016;6(6):795–807. | ||
Wang EC, Min Y, Palm RC, et al. Nanoparticle formulations of histone deacetylase inhibitors for effective chemoradiotherapy in solid tumors. Biomaterials. 2015;51:208–215. | ||
Sun Y, Zou W, Bian S, et al. Bioreducible PAA-g-PEG graft micelles with high doxorubicin loading for targeted antitumor effect against mouse breast carcinoma. Biomaterials. 2013;34(28):6818–6828. | ||
Safwat S, Hathout RM, Ishak RA, Mortada ND. Augmented simvastatin cytotoxicity using optimized lipid nanocapsules: a potential for breast cancer treatment. J Liposome Res. 2017;27(1):1–10. | ||
Abozeid SM, Hathout RM, Abou-Aisha K. Silencing of the metastasis-linked gene, AEG-1, using siRNA-loaded cholamine surface-modified gelatin nanoparticles in the breast carcinoma cell line MCF-7. Vol. 145. Colloids Surf B Biointerfaces. 2016:607–616. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.