")
Back to Journals » OncoTargets and Therapy » Volume 13
SSPH I, a Novel Anti-Cancer Saponin, Inhibits Autophagy and Induces Apoptosis via ROS Accumulation and ERK1/2 Signaling Pathway in Hepatocellular Carcinoma Cells
Authors Zhou J, Huang X, Qiu H, Gan R, Zhou H, Zhu H, Zhang X, Lu G , Liang G
Received 10 March 2020
Accepted for publication 1 June 2020
Published 23 June 2020 Volume 2020:13 Pages 5979—5991
DOI https://doi.org/10.2147/OTT.S253234
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Jin-ling Zhou,1,* Xiu-ying Huang,2,* Han-chen Qiu,3 Ri-zhi Gan,1 Huan Zhou,1 Hong-qing Zhu,1 Xuan-xuan Zhang,1 Guo-dong Lu,4 Gang Liang1
1School of Pharmacy, Guangxi Medical University, Nanning, People’s Republic of China; 2Liuzhou Employment Service Centre for the Disabled, Liuzhou, People’s Republic of China; 3Department of Pharmacy, The People’s Hospital of Hezhou, Hezhou, People’s Republic of China; 4School of Public Health, Guangxi Medical University, Nanning, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Gang Liang
Department of Pharmacology, School of Pharmacy, Guangxi Medical University, Nanning 530021, Guangxi Province, People’s Republic of China
Tel + 86 771 5302271
Fax + 86 771 5358272
Email [email protected]
Guo-dong Lu
Department of Toxicology, School of Public Health, Guangxi Medical University, Nanning 530021, Guangxi Province, People’s Republic of China
Tel + 86 771 535 8114
Fax + 86 771 5350823
Email [email protected]
Introduction: Saponin of Schizocapsa plantaginea Hance I (SSPH I), a novel bioactive phytochemical isolated from the rhizomes of Schizocapsa plantaginea, has been demonstrated to exhibit anti-cancer activity against various tumors in preclinical studies. However, the molecular mechanisms involved in the suppression of hepatocellular carcinoma (HCC) are poorly understood. The present study aimed at analyzing the effects of SSPH I on autophagy and apoptosis in vitro.
Methods: MTT and colony forming assays were used to detect cell viability and cell proliferation. Hoechst 33,258 staining and flow cytometry were used to determine apoptosis and ROS production. The apoptosis and autophagy-related protein expression levels were evaluated via Western blot assay. Characteristics of autophagy and apoptosis were observed by transmission electron microscopy. Lysosomal activity was stained with Lyso-Tracker Red and Magic Red Cathepsin B.
Results: The results showed that SSPH I exhibited potent anti-cancer activity and proliferation in HepG2 and BEL-7402 cells and inhibited HepG2 cells through inhibiting autophagy and promoting apoptosis. The mechanistic study indicated that the inhibition of autophagy of SSPH I was mediated by blocking autophagosome–lysosome fusion. Additionally, we found that SSPH I could mediate the activation of MAPK/ERK1/2 signaling pathway, and the use of NAC (ROS inhibitor) and U0126 (MEK1/2 inhibitor) converted the effect of SSPH I on apoptosis and autophagy in HepG2 cells.
Conclusion: These data suggest that SSPH I induces tumor cells apoptosis and reduces autophagy in vitro by inducing ROS and activating MAPK/ERK1/2 signaling pathway, indicating that SSPH I might be a novel agent for the treatment of HCC.
Keywords: saponins of Schizocapsa plantaginea Hance I, SSPH I, autophagy, hepatocellular carcinoma cells, apoptosis, ROS, MAPK
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors around the world, which is the main cause of cancer-related death. The incidence rate of liver cancer is increasing worldwide. More than 800 thousand new cases are added each year, causing 78,000 deaths per year.1 Surgical treatment is only applicable to 15–25% of patients. Although new treatment schemes are adopted, including systemic chemotherapy, radiotherapy, immunotherapy and targeted therapy, the 5-year survival rate of liver cancer is still lower than 15%.2 Therefore, in order to improve the prognosis of patients with liver cancer, more efforts are needed to find new and effective anti-tumor drugs and reveal their mechanisms of action.
Autophagy and apoptosis are two main mechanisms of programmed cell death. Autophagy, also known as type II cell death, is a dynamic degradation process. Autophagy provides survival advantages for cells in nutritional deficiency or other stress states, and maintains intracellular homeostasis.3,4 Autophagy plays a crucial role in many physiological and pathological processes such as apoptosis, cell death and cell survival.5,6 Apoptosis (also known as type I cell death) is characterized by nuclear division, chromatin concentration, cell contraction and apoptotic body formation.7 In general, apoptosis can be triggered by endogenous mitochondria or exogenous death receptor mediated pathways.8,9 Once there are some abnormal conditions, such as ROS overload, apoptosis will occur.10 Due to the regulatory mechanisms are interrelated in many aspects, autophagy and apoptosis are not independent processes, there exists overlap between them.11 Mitochondria provide energy and material for the survival of cancer cells, but also produce a small amount of ROS, which will be eliminated by autophagy. Damaged mitochondria will produce a large amount of ROS, and excessive ROS will attack nucleic acids and mitochondria, resulting in a vicious cycle, eventually leading to apoptosis. Recent study has shown that Ubenimex exerts anti-cancer efficacy and induces apoptosis and autophagy via the ROS/ERK1/2 signaling pathway.12 Thus, ROS and MAPK/ERK1/2 play important roles in the interaction between autophagy and apoptosis of tumor cells.13,14
Traditional Chinese medicine (TCM) has the characteristics of multi-target, multi-step and multi-level synergism. They are both historically important therapeutic agents and important source of new drugs.15,16 The most famous examples of TCM are artemisinin and its derivatives, they are the gifts from TCM not only for malaria control but also for schistosomiasis control.17 In addition, it has been widely reported that many kinds of TCM have the effects of anti-proliferation, anti-inflammatory, anti-oxidant, pro-apoptosis and antiangiogenic activities in vitro and vivo.18–20 Researches show that Saponins inhibit the development of various tumors. For example, aescin, a mixture of triterpenoid saponins, has anti-cancer effects in a variety of tumor models in vitro and in vivo, including colorectal cancer, liver cell cancer and bladder cancer,21–24 etc. Astragaloside II, a monomer extracted from Astragalus, suppresses autophagy by interfering with Beclin-1 and LC3 via MAPK-mTOR pathway, through which sensitized human cancer resistant cells to 5-FU-induced cell death.9 Our previous studies have found that SSPH is a mixture of steroidal saponins (including SSPH I, SSPH II and stigmasterol glycosides), which possess antitumor effect on hepatoma cells and less toxicity on normal human hepatocytes.25 However, the main bioactive steroidal saponin SSPH I, which was isolated and purified from SSPH, its anti-hepatoma effect and mechanism of autophagy and apoptosis are not clear.
In the current study, we were the first to find that SSPH I inhibits autophagy and induces apoptosis by inhibiting autophagy lysosomal fusion. Besides, we attempted to elucidate the potential mechanisms of SSPH I on autophagy and apoptosis using HepG2 cell line in vitro. Therefore, the NAC (ROS inhibitor) and U0126 (MEK1/2 inhibitor) were used to determine the potential mechanisms of action. Our results indicated that the effects of SSPH I on autophagy and apoptosis might be associated with inducing ROS accumulation and activating the MAPK/ERK1/2 signaling pathway.
Materials and Methods
Preparation and Storage of SSPH I
SSPH I isolated from the rhizomes of Schizocapsa plantaginea, was prepared, purified and identified by following the procedures of our previous study.26 Briefly, the tubers of Schizocapsa plantaginea were pieced and extracted using 80% ethanol. The ethanol extract was further treated with petroleum ether, ethyl acetate and n-butanol successively. Saponins were separated from the butanol soluble portion by adsorption process with macroporous resin D101, followed by ethanal elution. The 60% ethanal soluble portion was separated by silica gel column chromatography following elution processes with gradient chloroform/methanol/water (8/1/0.05, 6/1/0.05, 4/1/0.05 in v/v). The method of chloroform-methanol and Sephadex LH-20 column was adopted in turn to obtain SSPH I. The chemical structure of SSPH I was identified by 1H NMR and 13C NMR spectra analyses. SSPH I was dissolved in dimethyl sulfoxide (DMSO) at a final concentration of 10 mM and stored in −20 °C until use.
Reagents
Magic Red Cathepsin B Assay was purchased from Immunochemistry Technologies (Bloomington, MN, USA). Lyso-Tracker Red and chloroquine (CQ) were from Sigma-Aldrich (St. Louis, MO, USA). U0126 and NAC (N-acetyl-cysteine) were obtained from Med Chem Express (Monmouth Junction, NJ, USA). GAPDH antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). All remaining cell culture reagents were purchased from Life Technologies (Rockville, MD, USA). PVDF membranes were from Millipore (Burlington, MA, USA).
Cell Culture
The cell lines were obtained from the Shanghai Cell Bank of the Chinese Academy of Sciences and propagated in our laboratory. The cells were maintained in 75 cm2 flasks in RPMI-1640 supplemented with 10% (v/v) FBS, 100 units/mL penicillin, and 100 µg/mL streptomycin. The cultures were incubated at 37°C in an atmosphere containing 5% CO2.
Treatment of Drugs and Reagents
Cells were seeded and incubated for 24 h. SSPH I, chloroquine and/or U0126 were added to treat the cells alone or combined for another 24 h. For groups of NAC pretreated, NAC was added for 1 h in prior and removed before any treatment that mentioned above.
Cell Viability Assay
Methyl thiazolyl tetrazolium (MTT) and colony formation assays were employed to examine the effects of SSPH I on the proliferation of HepG2 and BEL-7402 cells. Briefly, cells were seeded in 96-well plates at a density of 7000 cells per well and incubated for 24 h. Subsequently, cells were treated for 24 h with varying concentrations of SSPH I diluted in RPMI-1640 (Gibco, Gaithersburg, MD, USA) medium containing 10% FBS. The supernatant was then removed, 100 µL of RPMI-1640 medium containing 1 mg/mL of MTT was added each well, and the cells were incubated for an additional 4 h. Subsequently, 100 µL of DMSO was added, and the samples were shaken for 10 min in the dark to solubilize formazan. To determine cell proliferation, the absorbance at 570 nm was recorded using a microplate reader (Molecular Devices, Hongkong, China).
Cell Proliferation Assay
As to colony formation assay, cells were seeded in six-well plates (500 cells/well) and incubated for 24 h. Subsequently, cells were treated for 24 h with varying concentrations of SSPH I diluted in RPMI-1640 and refreshed the new culture medium every 2–3 days. After incubation for 14 days, the colonies were fixed with 4% paraformaldehyde, and stained with 0.2% (w/v) crystal violet. Colonies were quantified with the Image J software.
Hoechst 33,258 Staining
Hoechst 33,258 staining was used to evaluate cell apoptosis. HepG2 cells (105/dish) were seeded in glass bottom dishes for 24 h and treated with SSPH I and U0126 for 24 h. Cells were then washed twice with PBS and then stained with Hoechst 33,258 (10 μg/mL in PBS) at 37 °C for 15 min in dark, and observed under a fluorescent inverted microscope (BX61W1, OLYMPUS, Japan).
Measurement of Intracellular ROS
Prior to measurement of Intracellular ROS level, the cells were cultured in the absence or presence of 10 mM NAC for 1 h. Subsequently, SSPH I with series of concentrations (2.9~5.8 μM, diluted in RPMI 1640) was added to the cell culture for 24 h. The fluorescent staining of cells was performed with DCFH-DA using the ROS Assay kit (Sigma, USA) for ROS measurement, according to the manufacturer’s instruction. The fluorescence intensity of each lysate was measured using a flow cytometric machine (FC500, Beckman Coulter, USA).
Annexin V/PI Staining
Apoptosis was measured by flow cytometry after staining with Alexa Flour 488 Annexin V and Propidium Iodide (PI) using Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific). The staining was performed according to the manufacturer’s instructions. For each sample, at least 1×104 cells were analyzed by flow cytometry (BD, USA). Flow Jo (Tree Star Inc., Ashland, OR, USA) software was used for quantitative analysis.
Western Blot Analysis
Cells were lysed with RIPA Lysis Buffer (Beyotime, Shanghai, China) containing PMSF (1:100 v/v, Solarbio, Beijing, China) and phosphatase inhibitors (1:100 v/v, Solarbio, Beijing, China). Protein concentration in the supernatant was measured using the BCATM Protein Assay Kit (Beyotime, Shanghai, China). Approximately 50 µg of total protein was subjected to SDS-PAGE, and transferred onto PVDF membranes. The membranes were washed, blocked with a 5% skim milk for 1 h. then incubated with rabbit monoclonal antibodies (1:1000 dilution; Cell Signaling, Danvers, USA) and GAPDH antibody (1:1000 dilution; Santa Cruz Biotechnology, CA, USA), overnight at 4 °C on a shaker. The membranes were then incubated with the fluorescent labeled secondary antibodies (1:10,000 dilution; Cell Signaling, Danvers, USA), at room temperature for 1h. The intensity of the bands was determined using an Odyssey infrared fluorescence scanning imaging system (LI-COR, Lincoln, NE, USA).
Transmission Electron Microscopy (TEM)
HepG2 cells were plated into 6-well plates at a density of 2×105 cells/well. Cells were treated with vehicle or SSPH I for 24 h. Subsequently, the cells were harvested, fixed overnight in 2.5% glutaraldehyde, and incubated with osmium tetroxide for two hours at 4°C. Specimens were embedded in epoxy resin. The embedded cells were sliced into 100 nm and stained with uranyl acetate and lead citrate. Ultrastructure of cells was imaged by a Hitachi HT7700 transmission electron microscope (Hitachi, Tokyo, Japan).
Lysosomes Detection with Lyso-Tracker Red Staining
After the incubation with the tested drug or reagent, cells were stained with lysosome-specific red fluorescent dye Lyso-Tracker Red (50 nM) for 45 min at 37°C and photographed using a fluorescence microscope. The intensity of red fluorescence represents the number of lysosomes.
Cathepsin B Detection with Magic Red Cathepsin B Staining
Magic Red® Cathepsin assay kits enable researchers to quantitate and monitor intracellular cathepsin-B, -K, or -L activity over time in vitro. The Magic Red (MR) reagent is a non-cytotoxic substrate that fluoresces red upon cleavage by active cathepsin enzymes. Because MR is cell-permeant, it easily penetrates the cell membrane and the membranes of the internal cellular organelles – no lysis or permeabilization steps are required. If cathepsin enzymes are active, they will cleave off the two dipeptide cathepsin targeting sequences and allow the cresyl violet fluorophore to become fluorescent upon excitation. After incubating with the tested drugs or reagents, cells were stained with 1×Magic Red Cathepsin B dye for 30 min at 37°C, according to the manufacturer’s instruction. The fluorescence of cathepsin B in the cells was observed under a fluorescence microscope and imaged. The intensity of the red fluorescence represents the activity of intracellular cathepsin B.
Statistical Analysis
SPSS software version 16.0 (IBM, Armonk, NY, USA) was used for statistical analysis. The data were expressed as mean ± standard deviation (SD). The least significant difference (LSD) method of one-way ANOVA was used to compare the independent groups. The difference was statistically significant (P < 0.05). The figure shows the significance level of P-value lower than 0.05, 0.01 and 0.001.
Results
SSPH I Inhibited Viability and Proliferation of HepG2 and BEL-7402 Cells
SSPH I [structural formula as (25S)-spirost-5-en-3 beta-yl-o-alpha-rhamnopyranosyl-(1→2) -o- [o-beta-d-glucopyranosyl-(1→4) -alpha-l-rhamnopyranosyl-(1→3)]-beta-d-glucopyranoside], is a spirostano-type steroidal saponin with four sugar moieties (Figure 1A). The molecular formula is C51H82O21, and molecular weight is 1030. According to the results of our identification, SSPH I prepared in our study was with a purity of over 96% (Supplementary Figure S1), suggesting the successful isolation of SSPH I for the use of the following tests. We investigated the effect of SSPH I on the growth of HepG2 and BEL-7402 cells by evaluating cell viability using MTT assay for 24 h. As shown in Figure 1B, SSPH I inhibited the viability of HepG2 and BEL-7402 cells in a dose-dependent manner. At 24 h, the IC50 value of SSPH I was 1.14 ± 0.09 μM for HepG2 and 2.89±0.13 μM for BEL-7402 cells. Anti-proliferation of SSPH I on HepG2 and BEL-7402 cells, was further confirmed by colony formation assay (Figure 1C), where SSPH I with concentration ranging from 1.75 to 3.5 μM significantly prohibited the colony number of HepG2 cells or BEL-7402 cells, comparing with the vehicle control group.
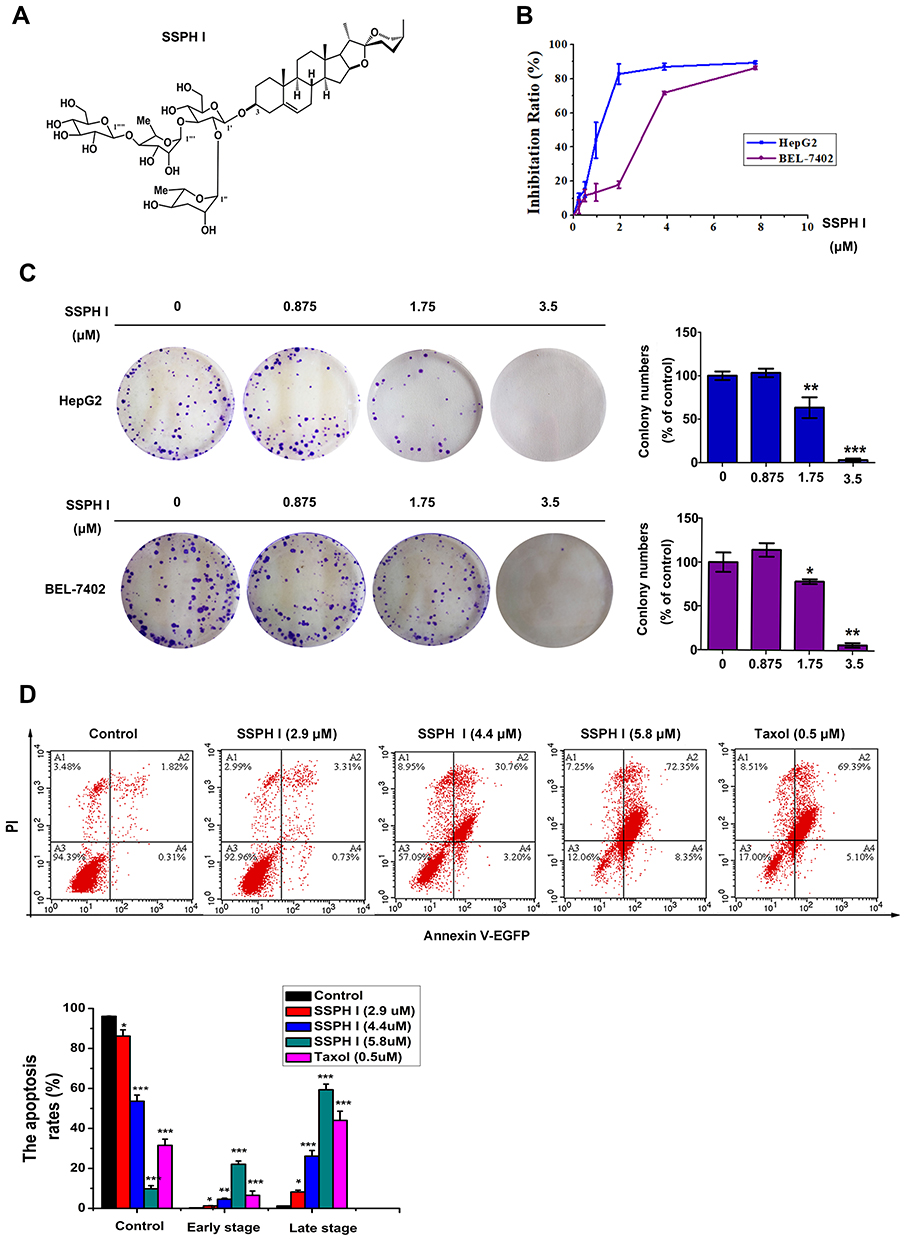 |
Figure 1 The effects of SSPH I on the proliferation and apoptosis of human hepatocellular carcinoma cell lines HepG2 and BEL-7402. (A) The molecular structure of SSPH I. (B) Inhibition ratio of SSPH I on HepG2 and BEL-7402 cells for 24 h by MTT assay. (C) Anti-proliferation of SSPH I on HepG2 and BEL-7402 cells for 24 h by colony formation assay. (D) The effects of SSPH I on apoptosis of HepG2 cells. Taxol was used as a positive control. Experiments were performed for three independent times. *P < 0.05, **P < 0.01, ***P < 0.001 vs untreated cells. Abbreviations: PI, propidium iodide; EGFP, enhanced green fluorescent protein. |
SSPH I Increased Apoptosis in HepG2 Cells
To underlie the molecular mechanism of the anti-cancer cytotoxicity of SSPH I, we thereupon investigated whether this agent induces apoptosis. As shown in Figure 1D, treatment with SSPH I triggered HepG2 cell apoptosis in a concentration-dependent manner. By contrast, apoptosis rate of HepG2 cell treated with SSPH I (5.8 μM) is quite closed to that with Taxol treatment (0.5μM). This result suggests that cell apoptosis induced by SSPH I is involved in the cytotoxic effect against HCC cells.
SSPH I Induced Autophagosome Formation but Inhibited Autophagy Flux
To study the effects of SSPH I on autophagy, we tested autophagy-related proteins of HepG2 cells by Western blotting. The results showed that the protein expression of LC3-II increased with the increasing of SSPH I concentration, while the expression of LC3-I decreased accordingly, that suggested the LC3 protein was shifted from cytoplasmic form (LC3-I) to autophagosomal form (LC3-II) (Figure 2A and B). This effect of SSPH I was dose-dependent (Figure 2A) and time-dependent (Figure 2B). However, p62, whose expression is inversely proportional to the autophagy activity, showed a tendency of increasing (Figure 2A and B), indicating that the cells did not generate autophagy flux.
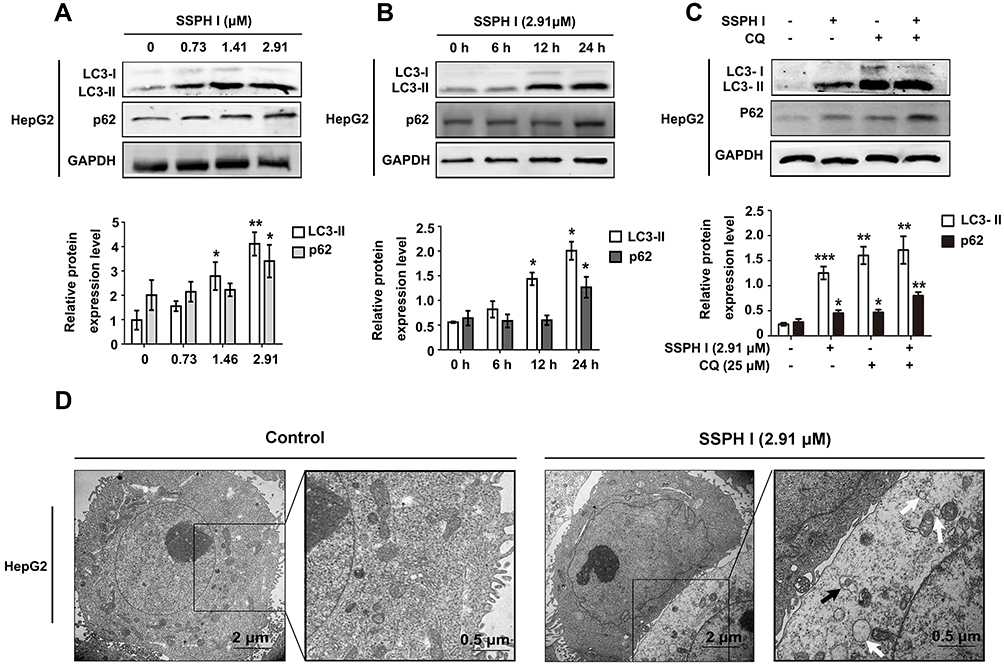 |
Figure 2 SSPH I induced dysregulation of autophagy in HepG2 cells. (A) Effects of different concentrations of SSPH I on LC3-II and p62 levels in HepG2 cells. (B) Effects of SSPH I on LC3-II and p62 levels in HepG2 cells at various time points. (C) Effect of SSPH I (2.91 μM) on the protein level of LC3-II and p62 in HepG2 cells in the presence or absence of CQ (25 μM). (D) TEM images illustrating the morphology of untreated HepG2 cells (left panels) and cells treated with 2.91 μM SSPH I (right panels). White arrows: vacuole-like structures; black arrows: autophagosomes. Experiments were performed for three independent times. *P < 0.05, **P < 0.01, ***P < 0.001 vs untreated cells. Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CQ, chloroquine; TEM, transmission electron microscopy. |
In order to further confirm the existence of specific solid autophagy flow, we detected the autophagic response of the cells to SSPH I in the presence or absence of a lysosomal acidification inhibitor CQ. Similar to SSPH I, CQ induced significant upregulation of LC3-II and p62 in HepG2 cells, and CQ and SSPH I had synergistic effect on LC3-II and p62 (Figure 2C). The result showed that the autophagy flux did not increase after SSPH I treatment.
In addition to testing the changes in protein expression, the morphology of cells and organelles was observed by TEM (Figure 2D). The obtained images showed the nuclear division, chromatin concentration and cell contraction induced by SSPH I, which are the characteristics of apoptosis. In addition, the accumulation of cytoplasmic vacuole and autophagy was also obvious. However, there is no autolysosome. Therefore, the increase of autophagosomes induced by SSPH I is probably due to the inhibition of autolysosome formation.
SSPH I Promoted the Lysosomal Degradation
For the purpose of determining the possible cause of downstream blockage of autophagosome formation by SSPH I, we examined the key proteins of the lysosomal membrane and the lysosomal cathepsins by Western blotting. As shown in Figure 3, we found that SSPH I inhibited the expression of lysosomal membrane proteins LAMP1 and LAMP2A in a dose-dependent manner (Figure 3A), as well as the lysosomal cathepsins proteins cathepsin B and cathepsin D (Figure 3B). The change pattern of protein expression in the cells indicated that the blockage of autolysosome formation might be related to the SSPH I mediated lysosomal degradation.
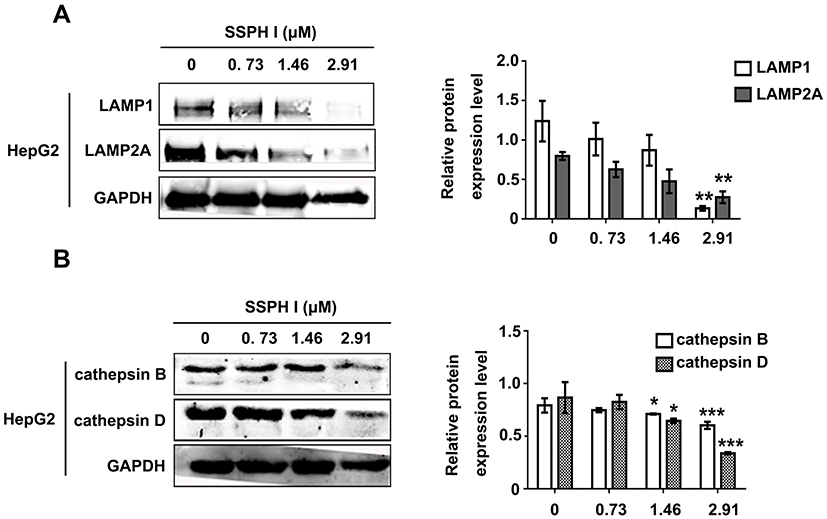 |
Figure 3 Effect of SSPH I on lysosomal membrane proteins and protein levels of lysosomal cathepsins in HepG2 cells. (A) Effect of SSPH I on the level of LAMP1 and LAMP2A. (B) Effect of SSPH I on the level of cathepsins B and D in HepG2 cells. *P < 0.05, **P < 0.01, ***P < 0.001 vs untreated cells. Abbreviation: GAPDH: glyceraldehyde-3-phosphate dehydrogenase. |
SSPH I Induced Intracellular ROS Accumulation
To investigate the mechanism of SSPH I in promoting apoptosis and suppressing autophagy, we examined the upstream of autophagy and apoptosis. As shown in Figure 4A, SSHP I treatment induced ROS generation in a concentration-dependent manner. When the increased ROS was blocked by the NAC pretreatment, the high expression of LC3-II and p62 proteins induced by SSPH I decreased (Figure 4B), and the high apoptosis rates and ROS generation induced by SSPH I were both decreased simultaneously (Figure 4C and D). Based on the above results, we believed that SSPH I could induce the accumulation of ROS, and thereby mediate the inhibition of autophagy and promotion of apoptosis.
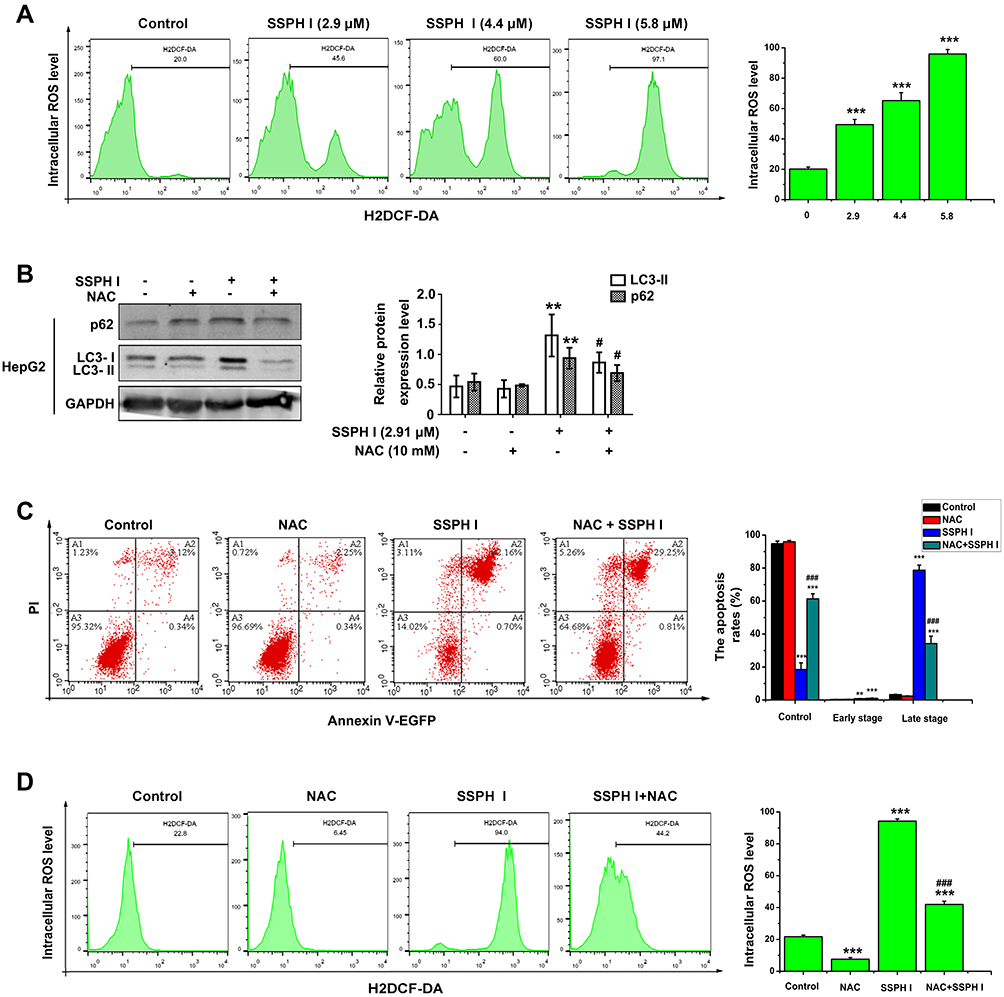 |
Figure 4 SSPH I induced the intracellular ROS of HepG2 cells and the effect of ROS on the autophagy and apoptosis. (A) Effect of SSPH I on the intracellular ROS accumulation in HepG2 cells. (B) Effect of the combination of SSPH I (2.91 μM) and NAC (10 mM) on p62 and LC3-II levels in HepG2 cells. (C) Effect of the combination of SSPH I (5.8 μM) and NAC (10 mM) on apoptosis in HepG2 cells. (D) Effect of the combination of SSPH I (5.8 μM) and NAC (10 mM) on intracellular ROS level of HepG2 cells. Experiments were performed for three independent times. **P< 0.01, ***P< 0.001 vs untreated cells; #P<0.05, ###P < 0.001 vs SSPH I-treated cells. Abbreviations: ROS, reactive oxygen species; H2DCF-DA, 2ʹ7ʹ-dichlorodihydrofluorescein diacetate; NAC, N-acetyl-cysteine; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PI, propidium iodide; EGFP, enhanced green fluorescent protein. |
SSPH I Activated the MAPK/ERK1/2 Signaling Pathway
As shown in Figure 5A, when compared with the control group, the protein expression of ERK1/2 phosphorylation, LC3-II and p62 increased with SSPH I treatment. When the increase of ERK1/2 phosphorylation induced by SSPH I was blocked by U0126, the expression of LC3-II and p62 proteins decreased compared with the cells treated by SSPH I alone, indicating that the inhibition of autophagy was reversed by inhibiting ERK1/2 phosphorylation (Figure 5A). Additionally, when compared with the control group, the protein expression of caspase-3 and caspase-9 were both downregulated (even though there was no significant difference, it showed the downregulated trends) (Figure 5A) and the fluorescence value increased significantly (Figure 5B), when they were blocked by U0126, the protein expression of caspase-3 and caspase-9 were both upregulated (Figure 5A) and the fluorescence value decreased significantly (Figure 5B), indicating that the SSPH I-enhanced caspases-dependent apoptosis were reversed by inhibiting ERK1/2 phosphorylation. Furthermore, Hoechst 33,258 staining showed that SSPH I could activate HepG2 cell apoptosis and cause chromatin concentration, nuclear atrophy, nuclear fragmentation, enhanced fluorescence intensity (Figure 5B). In the presence of U0126, the apoptotic effect of SSPH I decreased (Figure 5B). Lyso-Tracker Red and Magic Red Cathepsin B confirmed that U0126 could reverse the deduced number of lysosomes and the inhibited activity of cathepsin B by SSPH I (Figure 5C). Together, these data provided additional evidence for the involvement of the ROS and MAPK/ERK signaling pathways, by which SSPH I promotes apoptosis and suppresses the autophagy in HepG2 cells.
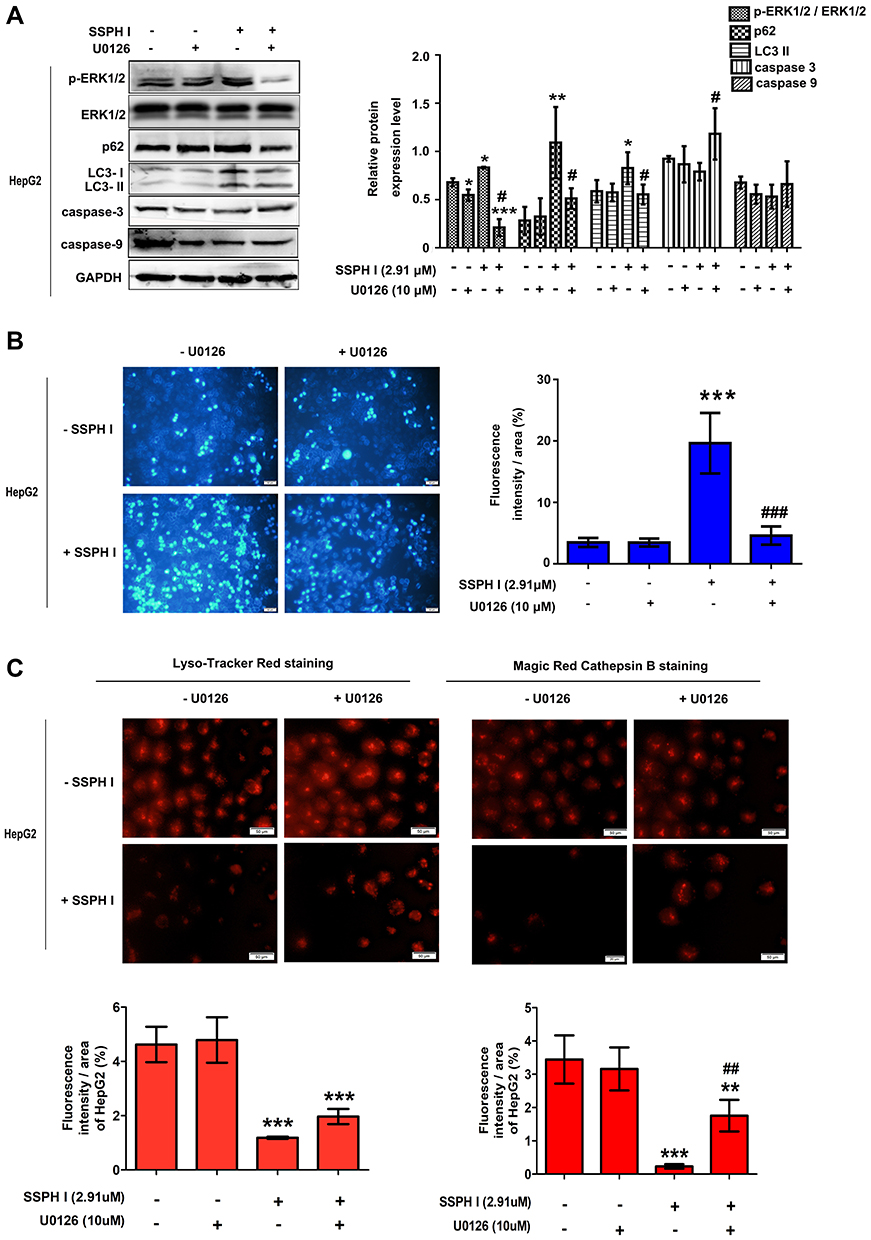 |
Figure 5 Effect of SSPH I on MAPK/ERK1/2 signaling pathway in HepG2 cells. (A) Effect of the combination of SSPH I (2.91 μM) and U0126 (10 μM) on p-ERK1/2, ERK1/2, LC3-II, p62, caspase-3, and caspase-9 levels in HepG2 cells. (B) Effect of the combination of SSPH I (2.91 μM) and U0126 (10 μM) on morphology of apoptosis in HepG2 cells. Scale bar, 50 µm. (C) Effect of the combination of SSPH I (2.91 μM) and U0126 (10 μM) on lysosomes and cathepsin B in HepG2 cells. Scale bar, 50 µm. Experiments were performed for three independent times. *P< 0.05, **P< 0.01, ***P< 0.001 vs untreated cells; #P<0.05, ##P<0.01, ###P < 0.001 vs SSPH I-treated cells. Abbreviations: MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; p-ERK, phosphorylation of extracellular signal-regulated kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. |
Discussion
Cancer cells have the characteristic of infinite proliferation.27 We used MTT assay and clone formation test to prove the effect of SSPH I on the viability of HepG2 and BEL-7402 cells. MTT assay, a method for detecting cell viability, is mostly used to reflect the short-term cell viability due to its characteristics of simplicity and rapid. While clone formation test is mostly used to evaluate the integrity of cell reproduction, particularly in reflecting the long-term therapeutic effect of drugs with the advances of better accuracy and reliability. The results showed that SSPH I possessed similar anti-viability and anti-proliferative effects against HepG2 and BEL-7402 cells in a concentration dependent manner. For this reason, we took HepG2 cell line, which is more sensitive to SSPH I, to explore the mechanism of effective anti-tumor action of SSPH I. We found that SSPH I promoted apoptosis of HepG2 cells, and this conclusion was supported the characteristics of apoptosis by TEM as well.
The WB results indicated that SSPH I induced the changes of LC3. However, the process of autophagy cannot be judged only by the changes of LC3. Autophagic flux, the process of autophagy, starts with the formation and maturation of autophagosome, and then the autophagosome fuses with lysosomes to form an autolysosome, by which the autophagic substrates are ultimately degraded.28 The increased shift of LC3-I to LC3-II only indicates that the accumulation of autophagosomes but does not indicate an actual increase in the autophagy flux.29 Therefore, to confirm autophagy regulatory activity, it is highly recommended to use multiple indicators such as autophagic substrate p62 or RFP-GFP-LC3, and activators and inhibitors of this process, such as CQ or bafilomycin.30,31 Thus, we used p62 and CQ to identified autophagy regulatory activity, and found p62 increased time-dependently and dose-dependently, and SSPH I increased LC3 and p62 as CQ did. These results indicated that autophagy-lysosome fusion was blocked by SSPH I.
Autophagy dependent cell death is a regulated cell death determined by autophagy mechanism, which is mainly used for adaptive cell protection. In some cases of hepatotoxicity, autophagy dependent apoptosis can be induced in hepatocytes through lysosomal mitochondrial axis.32 Lysosomal dependent cell death is a regulated type of cell death caused by lysosomal membrane permeability (LMP), which is related to inflammation, tissue degradation, aging, neurodegeneration and intracellular pathogen response. Due to the process of apoptosis and necrosis, LMP may occur after mitochondrial membrane permeability.8 However, lysosomes can penetrate into mitochondria before Bax raised lysosomal membrane. Oxidative stress and lipid peroxidation of lysosomal membrane may also contribute to LMP. Primary LMP can also be produced by tumor necrosis factor. In autophagy, the lysosome needs the activity of protease to degrade cell macromolecular protein. Cathepsin, the main lysosomal protease, plays an important role in maintaining cell homeostasis and differentiation by recovering cell contents.33 In addition, since the lysosomal associated membrane proteins LAMP1 and LAMP2A are essential for the fusion of autophagy and lysosome,34,35 we detected the proteins levels of LAMP1, LAMP2A, CTSB and CTSD to further elucidate the molecular mechanism of SSPH I inhibiting autophagy in HepG2 cells. The results showed that SSPH I significantly decreased the protein levels of LAMP1 and LAMP2A in HepG2 cells. Taken together, these results indicated that SSPH I could inhibit lysosomal protein hydrolytic activity by changing lysosomal pH and down regulating CTSB and CTSD protein levels, and inhibit autophagy of HepG2 cells by consuming LAMP1 and LAMP2A protein to cause the fusion of autophagosome and lysosome to be blocked.
Mitogen-activated protein kinase (MAPK) signal transduction pathway organizes a huge network to regulate several physiological processes, such as cell growth, differentiation and apoptosis cell death. Because of the importance of this signaling pathway, the maladjustment of MAPK signaling cascade involves the pathogenesis of various human cancer types. Oxidative stress is one of the important factors, which leads to carcinogenesis through the disorder of the signaling pathway. Reactive oxygen species (ROS) are by-products of oxidative energy metabolism and represent important physiological regulators of several intracellular signaling pathways, including the MAPK pathway.14 Although the proliferation of cancer cells can be stimulated by low dose of superoxide or hydrogen peroxide, excessive ROS levels can lead to irreversible damage of cancer cells. Moreover, enhanced mitochondrial oxidative stress results in caspases activation and cell death.36 As a member of the MAPK family, the ERK signaling pathway has been found playing an important role in various aspects of cell biological functions including proliferation, differentiation, autophagy, and death.37,38 It has been previously documented that SSPH I increases ROS levels in HCC in vivo and in vitro, and produces sustained activation of the ERK1/2 signaling pathway, and then results in inhibiting proliferation and inducing apoptosis.39 In our present study, SSPH I inhibited autophagy and induced apoptosis of HepG2 cells by increasing ROS mitochondrial pathway and activating MAPK/ERK1/2 signal pathway. When the inhibition of autophagy was blocked by ERK1/2 inhibitor U0126 (Figure 5) or ROS inhibitor NAC (Figure 4), the induction of apoptosis was partially blocked, suggesting that SSPH I may inhibit autophagy of HepG2 cells by activating MAPK/ERK1/2 signaling pathway and improving ROS mitochondrial pathway to promote apoptosis.
In conclusion, the present study identified the anti-cancer activity of the novel SSPH I steroidal saponin in vitro, and revealed the underlying mechanism of the suppressed autophagy and promoted apoptosis after the treatment for the first time, where the involvement of ROS and MAPK/ERK1/2 signaling pathway is highlighted (Figure 6). Our present study therefore suggests that SSPH I is expected to be a novel chemical drug and provide an option for the treatment of HCC or even other cancer types. Further tests are still required to confirm the efficacy of this drug in animal models and clinical trials.
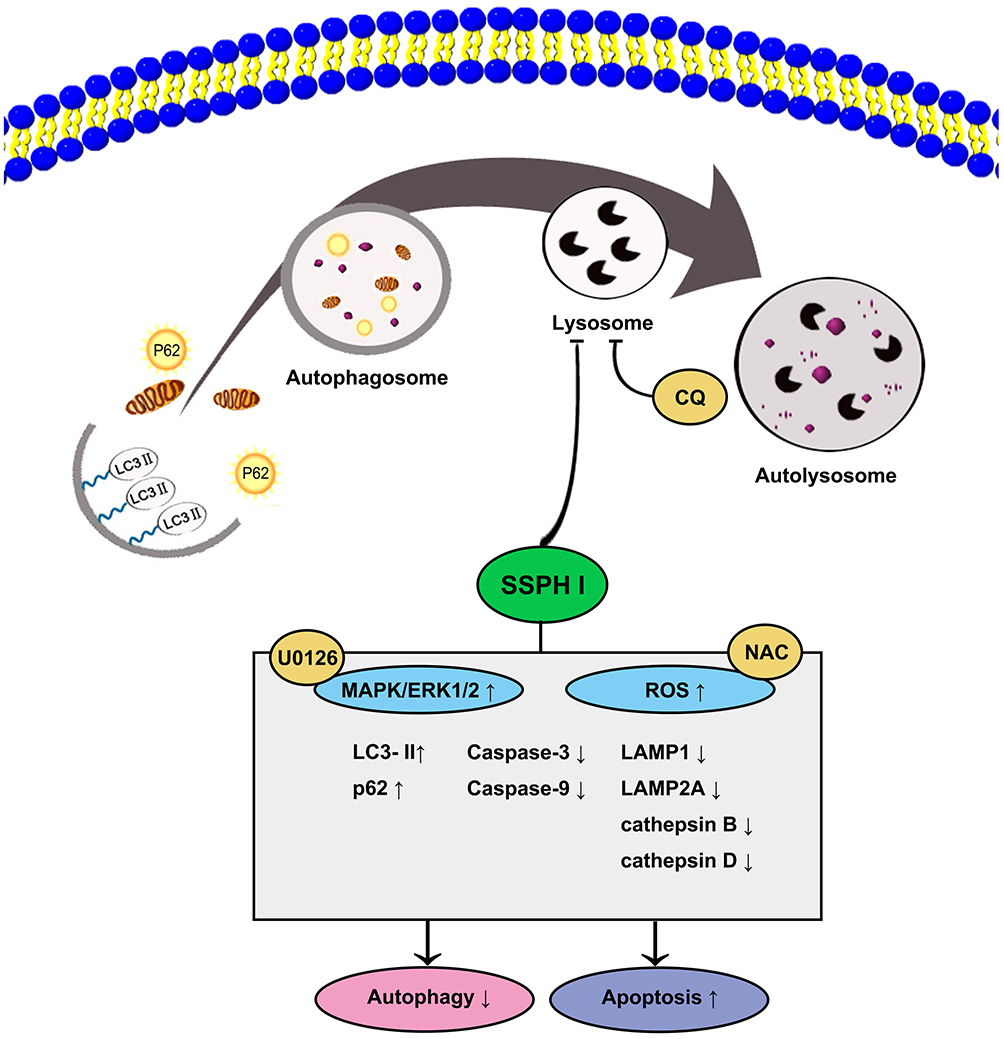 |
Figure 6 The proposed mechanism of the anti-cancer effect of SSPH I in vitro model of HCC. The lysosomal function was impaired by SSPH I, resulting in the autophagosome–lysosome fusion, consequently, exerting antiproliferative action and induces caspase-dependent apoptosis in HepG2 cells. The mechanism was verified using the autophagy inhibitor CQ, ROS inhibitor NAC, and MEK inhibitor U0126. The obtained results indicated that the inhibition of autophagy-dependent apoptosis of HCC cells by SSPH I could be attributed to the increase in ROS and MAPK/ERK1/2 signaling pathway. Abbreviations: HCC, human hepatocellular carcinoma; CQ, chloroquine; NAC, N-acetyl-cysteine; ROS, reactive oxygen species; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; MEK, mitogen-activated protein kinase kinase. |
Acknowledgments
This study was supported by grant no. 81460620 from the National Natural Science Foundation of China.
Disclosure
All authors report grants from The National Natural Science Foundation of China, during the conduct of the study. In addition, Professor Gang Liang has patents “Application of saponins extract from Schizocapsa plantaginea in anti-hepatocellular carcinoma and nasopharyngeal carcinoma” and “The separation method of a saponin compound” issued. The authors report no other conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Hassanipour S, Vali M, Gaffari-Fam S, et al. The survival rate of hepatocellular carcinoma in Asian countries: a systematic review and meta-analysis. EXCLI J. 2020;19:108–130. doi:10.17179/excli2019-1842
3. Ravikumar B, Futter M, Jahreiss L, et al. Mammalian macroautophagy at a glance. J Cell Sci. 2009;122(11):1707–1711. doi:10.1242/jcs.031773
4. Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–892. doi:10.1083/jcb.200911078
5. Wang L, Li H, Zhen Z, et al. CXCL17 promotes cell metastasis and inhibits autophagy via the LKB1-AMPK pathway in hepatocellular carcinoma. Gene. 2019;690:129–136. doi:10.1016/j.gene.2018.12.043
6. Peng WX, Xiong EM, Ge L, et al. Egr-1 promotes hypoxia-induced autophagy to enhance chemo-resistance of hepatocellular carcinoma cells. Exp Cell Res. 2016;340:62–70. doi:10.1016/j.yexcr.2015.12.006
7. Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int J Biochem Cell Biol. 2004;36(12):2405–2419. doi:10.1016/j.biocel.2004.04.011
8. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541.
9. Wang M, Huang C, Su Y, Yang C, Xia Q, Xu DJ. Astragaloside II sensitizes human hepatocellular carcinoma cells to 5-fluorouracil via suppression of autophagy. J Pharm Pharmacol. 2017;69:743–752. doi:10.1111/jphp.12706
10. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14(1):48. doi:10.1186/s12943-015-0321-5
11. Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. doi:10.1038/cdd.2009.33
12. Wang Y, Pang B, Zhang R, Fu Y, Pang Q. Ubenimex induces apoptotic and autophagic cell death in rat GH3 and MMQ cells through the ROS/ERK pathway. Drug Des Devel Ther. 2019;13:3217–3228. doi:10.2147/DDDT.S218371
13. Shi JM, Bai LL, Zhang DM, et al. Saxifragifolin D induces the interplay between apoptosis and autophagy in breast cancer cells through ROS-dependent endoplasmic reticulum stress. Biochem Pharmacol. 2013;85:913–926. doi:10.1016/j.bcp.2013.01.009
14. Song S, Tan J, Miao Y, Li M, Zhang Q. Crosstalk of autophagy and apoptosis: involvement of the dual role of autophagy under ER stress. J Cell Physiol. 2017;232(11):2977–2984. doi:10.1002/jcp.25785
15. Xia X, Wang X, Zhang S, et al. miR-31 shuttled by halofuginone-induced exosomes suppresses MFC-7 cell proliferation by modulating the HDAC2/cell cycle signaling axis. J Cell Physiol. 2019;234(10):18970–18984. doi:10.1002/jcp.28537
16. Zhao L, Nicholson JK, Lu A, et al. Targeting the human genome-microbiome axis for drug discovery: inspirations from global systems biology and traditional Chinese medicine. J Proteome Res. 2012;11(7):3509–3519. doi:10.1021/pr3001628
17. Liu R, Dong HF, Jiang MS. Artemisinin: the gifts from traditional Chinese medicine not only for malaria control but also for schistosomiasis control. Parasitol Res. 2012;110:2071–2074. doi:10.1007/s00436-011-2707-7
18. Al-Hrout A, Chaiboonchoe A, Khraiwesh B, et al. Safranal induces DNA double-strand breakage and ER-stress-mediated cell death in hepatocellular carcinoma cells. Sci Rep. 2018;8(1):16951. doi:10.1038/s41598-018-34855-0
19. Al-Dabbagh B, Elhaty IA, Al Hrout A, et al. Antioxidant and anticancer activities of trigonella foenum-graecum, cassia acutifolia and rhazya stricta. BMC Complement Altern Med. 2018;18(1):240. doi:10.1186/s12906-018-2285-7
20. Hamza AA, Ahmed MM, Elwey HM, Amin A, Ahmad A. Melissa officinalis protects against doxorubicin-induced cardiotoxicity in rats and potentiates its anticancer activity on MCF-7 cells. PLoS One. 2016;11(11):e0167049. doi:10.1371/journal.pone.0167049
21. Tan SM, Li F, Rajendran P, Kumar AP, Hui KM, Sethi G. Identification of beta-escin as a novel inhibitor of signal transducer and activator of transcription 3/Janus-activated kinase 2 signaling pathway that suppresses proliferation and induces apoptosis in human hepatocellular carcinoma cells. J Pharmacol Exp Ther. 2010;334:285–293. doi:10.1124/jpet.110.165498
22. Li B, Wang Z, Xie J-M, et al. TIGAR knockdown enhanced the anticancer effect of aescin via regulating autophagy and apoptosis in colorectal cancer cells. Acta Pharmacol Sin. 2019;40(1):111–121. doi:10.1038/s41401-018-0001-2
23. Cheng CL, Chao WT, Li YH, et al. Escin induces apoptosis in human bladder cancer cells: an in vitro and in vivo study. Eur J Pharmacol. 2018;840:79–88. doi:10.1016/j.ejphar.2018.09.033
24. Cheong D, Arfuso F, Sethi G, et al. Molecular targets and anti-cancer potential of escin. Cancer Lett. 2018;422:1–8. doi:10.1016/j.canlet.2018.02.027
25. Qiu HC, Sun YW, Luo SR, et al. Effects of saponins from Schizocapsa plantaginea Hance on proliferation, migration and apoptosis of human hepatocellular carcinoma cells and its toxicity to normal hepatocytes. Shandong Med J. 2017;57:
26. Sun YW, Qiu HC, Ou MC, Chen RL, Liang G. Saponins isolated from Schizocapsa plantaginea inhibit human hepatocellular carcinoma cell growth in vivo and in vitro via mitogen-activated protein kinase signaling. Chin J Nat Med. 2018;16:29–40. doi:10.1016/S1875-5364(18)30027-X
27. Wang S, Zhou D, Xu Z, et al. Anti-tumor drug targets analysis: current insight and future prospect. Curr Drug Targets. 2019;20:1180–1202.
28. Singh K, Sharma A, Mir MC, et al. Autophagic flux determines cell death and survival in response to Apo2L/TRAIL (dulanermin). Mol Cancer. 2014;13:70. doi:10.1186/1476-4598-13-70
29. Zhou J, Li G, Zheng Y, et al. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy. 2015;11(8):1259–1279. doi:10.1080/15548627.2015.1056970
30. Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1(2):84–91. doi:10.4161/auto.1.2.1697
31. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi:10.1016/j.cell.2010.01.028
32. Wang Y, Liu Y, Liu X, et al. Citreoviridin induces autophagy-dependent apoptosis through lysosomal-mitochondrial axis in human liver HepG2 cells. Toxins (Basel). 2015;7(8):3030–3044. doi:10.3390/toxins7083030
33. Fernández ÁF, López-Otín C. The functional and pathologic relevance of autophagy proteases. J Clin Invest. 2015;125(1):33–41. doi:10.1172/JCI73940
34. Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8(9):528–539. doi:10.1038/nrclinonc.2011.71
35. Kucharewicz K, Dudkowska M, Zawadzka A, et al. Simultaneous induction and blockade of autophagy by a single agent. Cell Death Dis. 2018;9(3):353. doi:10.1038/s41419-018-0383-6
36. Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell. 2014;25(5):563–573. doi:10.1016/j.ccr.2014.03.015
37. Bonjardim CA. Viral exploitation of the MEK/ERK pathway – A tale of vaccinia virus and other viruses. Virology. 2017;507:267–275. doi:10.1016/j.virol.2016.12.011
38. Zhang K, Ding J. In vitro anticancer effects of levopimaric acid in cisplatin-resistant human lung carcinoma are mediated via autophagy, ROS-mediated mitochondrial dysfunction, cell apoptosis and modulation of ERK/MAPK/JNK signaling pathway. J BUON. 2020;25:248–254.
39. Kawai A, Uchiyama H, Takano S, Nakamura N, Ohkuma S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy. 2007;3(2):154–157. doi:10.4161/auto.3634
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.