")
Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 8
Spotlight on taliglucerase alfa in the treatment of pediatric patients with type 1 Gaucher disease
Authors Gupta P, Pastores GM
Received 6 February 2017
Accepted for publication 27 March 2017
Published 16 June 2017 Volume 2017:8 Pages 73—81
DOI https://doi.org/10.2147/PHMT.S93634
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Punita Gupta,1 Gregory M Pastores2
1Division of Genetics, Department of Pediatrics, St. Joseph’s Children’s Hospital, Paterson, New Jersey, USA; 2National Center for Inherited Metabolic Disorders, Mater Misericordiae University Hospital, University College Dublin, Dublin, Ireland
Abstract: Gaucher disease (GD) is a heritable storage disorder caused by functional defects of the lysosomal acid β-glucosidase and the accumulation of glucosylceramide within macrophages, resulting in multiple organ dysfunction. There are three commercially available enzyme replacement therapy (ERT) products for the treatment of GD type 1 (GD1): imiglucerase, velaglucerase alfa, and taliglucerase alfa. Imiglucerase and velaglucerase alfa are produced in different mammalian cell systems; imiglucerase requires postproduction deglycosylation to expose terminal α-mannose residues, which are required for mannose receptor-mediated uptake by target macrophages. These steps are critical to the success of ERT for the treatment of visceral and hematologic manifestations of GD. Taliglucerase alfa is the first US Food and Drug Administration-approved plant-cell-expressed recombinant human protein, using carrot root cell cultures. Furthermore, it does not require postproduction glycosidic modifications. It is indicated for treatment of adults with GD1 in the US, Israel, Australia, Canada, Chile, Brazil, and other countries, and it is additionally approved for the treatment of pediatric patients in the US, Australia, and Canada and for the treatment of hematologic manifestations in pediatric patients with Type 3 GD in Canada and other countries. Our review focuses on the role of taliglucerase alfa in the pediatric population. A literature search through PubMed (from 1995 up till November 2016) of English language articles was performed with the following terms: Gaucher disease, lysosomal storage disease, taliglucerase. Secondary and tertiary references were obtained by reviewing related articles as well as the website www.Clinicaltrials.gov. It has been demonstrated that taliglucerase alfa is efficacious, with a well-established safety profile in pediatric, ERT-naïve patients with symptomatic GD1, as well as for those patients previously treated with imiglucerase.
Keywords: Gaucher disease, enzyme replacement therapy, taliglucerase alfa, pediatrics
Introduction
Gaucher disease (GD) is a lysosomal storage disorder arising from mutations in the gene GBA1 encoding acid β-glucosidase (glucocerebrosidase [EC 3.2.1.45]).1 It has an estimated prevalence of approximately 1 in 111,000 to 1 in 57,000,1,2 with a higher prevalence noted within the Ashkenazi Jewish population of 1 in 855.1,3 Insufficient enzyme activity leads to accumulation of glucosylceramide and other glycosphingolipids within the lysosomes of various cells and tissues, which results in varying degrees of visceral and bone pathology, with neuronal involvement in a subset.1
GD is classified into GD type 1 (non-neuronopathic), GD type 2 (acute neuronopathic), and GD type 3 (chronic neuronopathic) according to the presence of neurological deterioration, age at identification, and rate of disease progression.4,5 Many patients with GD type 1 have signs or symptoms during childhood, which include anemia, thrombocytopenia, hepatosplenomegaly, bone disease, pubertal delay, and growth retardation.6,7 Earlier onset correlates with more severe disease and a high risk of morbidity. Splenomegaly, hepatomegaly, and bone disease are each found in more than 80% of children at diagnosis. Anemia and moderate-to-severe thrombocytopenia, both found in approximately 40% of children, are caused by bone marrow infiltration and hypersplenism.6 Growth and puberty are significantly delayed in ~60% of adolescents with untreated GD and is a significant disease burden,7 which can have a considerable psychological impact.
In non-neuropathic GD, often described incorrectly as an adult disorder, the majority of patients manifest symptoms in childhood, with diagnosis usually established before age 20.8 Enzyme replacement therapy (ERT) is recommended for patients, including children, with GD who manifest progressive signs and symptoms. Early intervention with ERT in symptomatic children may prevent the development of irreversible pathology. Treatment is also important to improve growth and reduce the impact of the disease on physical and psychosocial development.6,9
Successful treatment of GD requires targeting of the infused enzyme to lysosomes within macrophages. This occurs via uptake of appropriately glycosylated enzyme to permit efficient uptake by mannose receptors on the surface of macrophages.10
Three ERTs are available for the treatment of Type 1 GD: imiglucerase, produced in a Chinese hamster ovary cell culture system;11 velaglucerase alfa, produced in a human fibroblast cell system;12 and taliglucerase alfa, an ERT produced in a plant-cell-based expression system.13 Taliglucerase alfa is the first US Food and Drug Administration-approved plant-cell-expressed recombinant therapeutic protein.13 It is indicated for treatment of adults with Type 1 GD in the US, Israel, Australia, Canada, Chile, Brazil, and other countries, and it is additionally approved for the treatment of pediatric patients in the US, Australia, and Canada and for the treatment of hematologic manifestations in pediatric patients with Type 3 GD in Canada. The plant cell production system allows for generation of appropriately glycosylated glucocerebrosidase without the need for postproduction enzymatic modification. Moreover, it is free from mammalian-derived components in the production process and cannot be contaminated with or propagate mammalian pathogens, and it also has a potentially lower manufacturing cost.14–16
In the taliglucerase alfa clinical development program, a phase 1 study was conducted in six healthy adult volunteers.17 Subsequently, patients with GD1 have been enrolled in several clinical studies, as well as compassionate use programs, to receive treatment with taliglucerase alfa. All of the clinical trials have been completed (Table 1).3,18–23
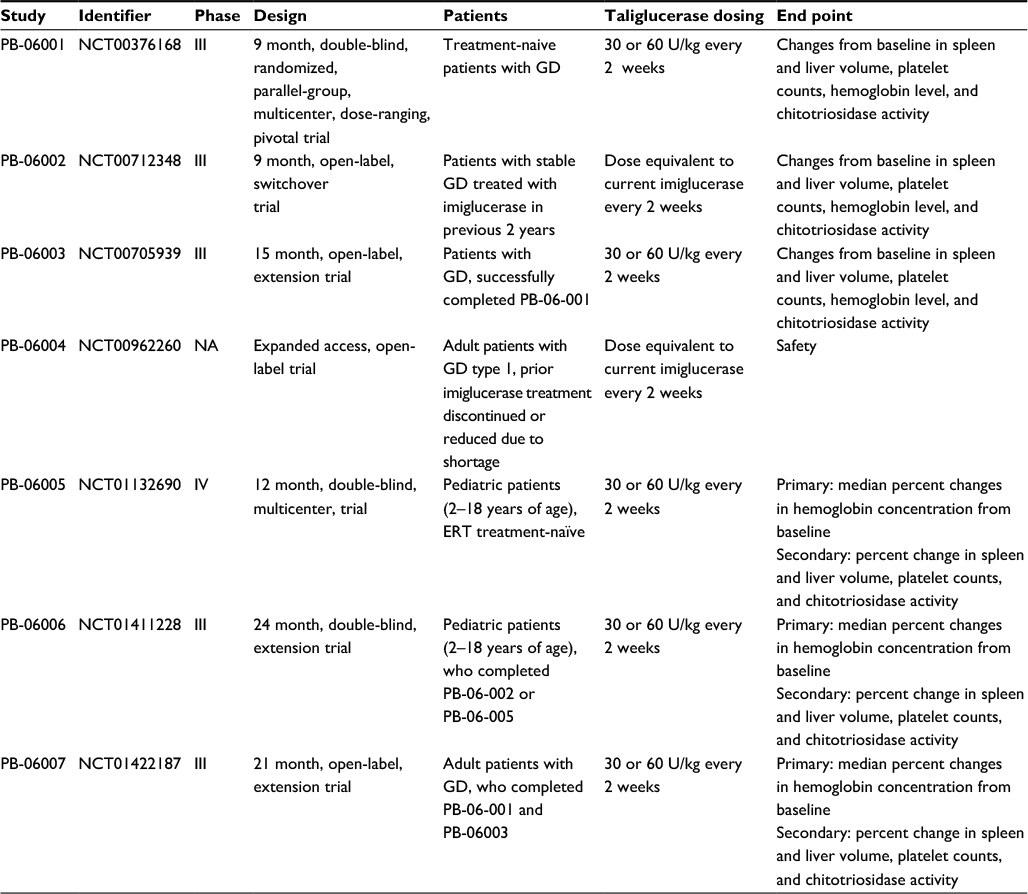 | Table 1 Summary of phase III and IV taliglucerase alfa clinical trials, including an expanded access, open-label trial Note: Reprinted from Molecular Genetics and Metabolism, vol 112. Grabowski GA, Golembo M, Shaaltiel Y. Taliglucerase alfa: an enzyme replacement therapy using plant cell expression technology. Pages: 1–8. Copyright 2014, with permission from Elsevier.18 Abbreviations: ERT, enzyme replacement therapy; GD, Gaucher disease; NA, not applicable. |
The studies were carried out in a number of medical centers worldwide and were designed to assess the safety and efficacy of taliglucerase alfa in both adult and pediatric patients who were ERT-naive or ERT-experienced and switched from imiglucerase to taliglucerase alfa.3,18–24 Treatment was generally well tolerated, and all drug-related adverse events (AEs) were mild/moderate and transient.
The objective of this review is to discuss the published data on pediatric population from these studies.
Patients and methods
The safety and efficacy of taliglucerase alfa have been examined in Studies PB-06-002 in patients previously treated with imiglucerase through 9 months, PB-06-005 in treatment-naive pediatric patients through 12 months, and in extension study PB-06-006 for an additional 24 months, for a maximum duration of 33–36 total months of treatment (Figure 1).
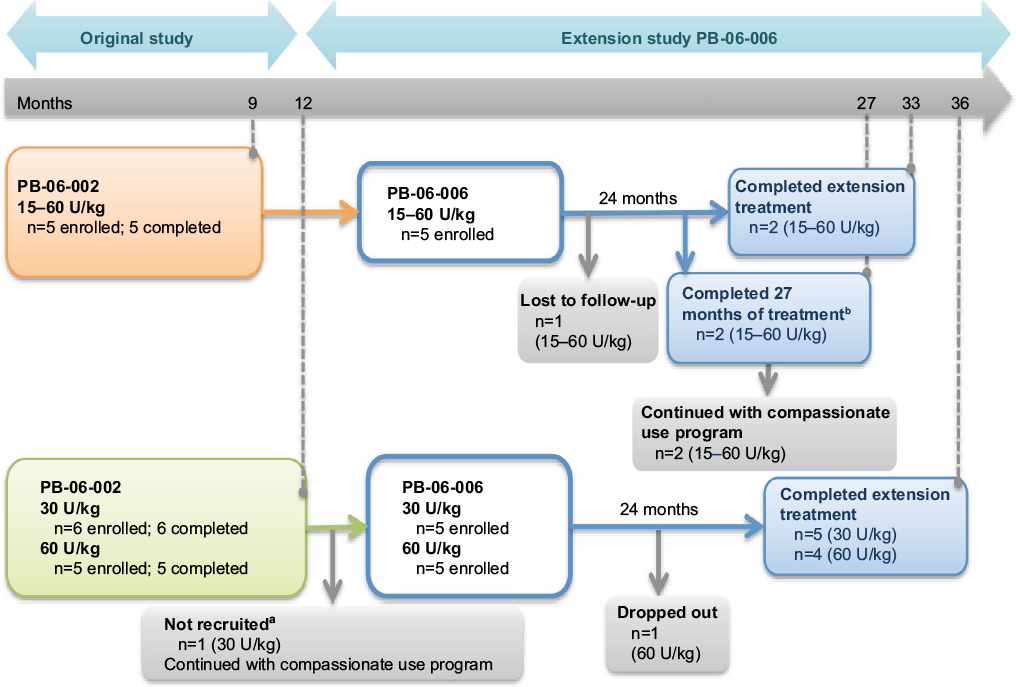 | Figure 1 PB-06-002 was a switchover study from imiglucerase to taliglucerase alfa. Notes: aPatient had GD Type 3c and cardiac involvement requiring intervention due to decompensation. bPatients did not reach 33 total months of treatment due to early closure of study. Patients were either maintained on the same dose as received on imiglucerase, or if they were on <60 U/kg of imiglucerase, dose could be increased based on medical condition and at the investigator’s discretion. The five pediatric patients who completed this trial were enrolled in the PB-06-006 trial. Of these, one was lost to follow-up, two completed the 24 months duration of the extension trial, and two completed only 18 months of the extension trial due to early closure of the study; however, both patients continued to receive therapy through the compassionate use program. PB-06-005 involved the treatment of naïve pediatric patients who received either 30 or 60 U/kg of taliglucerase alfa for 12 months. Out of the 11 patients who completed the PB-06-005 trial, 10 were enrolled in the PB-06-006 extension trial. One patient had Type 3c GD and cardiac involvement requiring intervention due to decompensation and was not recruited into the extension phase, one patient dropped out from the trial, and nine patients completed the PB-06-006 extension trial. Out of the 15 patients enrolled in the PB-06006 extension trial, 11 completed. Reprinted from Blood Cells Mol Dis. Vol S1079–9796(16). Zimran A, Gonzalez-Rodriguez DE, Abrahamov A, et al. Long-term safety and efficacy of taliglucerase alfa in pediatric Gaucher disease patients who were treatment-naïve or previously treated with imiglucerase. Pages 30221–30222. Copyright 2016, with permission from Elsevier.25 Abbreviation: GD, Gaucher disease. |
PB-06-005 was a phase 3B multicenter, randomized, double-blind, 2-dose trial of taliglucerase alfa (30 and 60 U/kg per infusion every other week) in pediatric patients (aged 2 to <18 years) through 12 months (US National Institutes of Health www.clinicaltrials.gov identifier NCT01132690). Male and female patients aged 2 to <18 years were required to have a diagnosis of GD with leukocyte acid β-glucosidase activity level ≤30% of the mean activity of the reference range for healthy individuals. Patients were eligible if they were determined in need of treatment with ERT based on clinical condition and the opinion of the local investigator and had neither received ERT in the past or within the previous 12 months and had a negative anti-glucosidase antibody assay assessment, nor been treated with substrate reduction therapy for GD in the past 12 months. The trial was conducted at 3 centers from August 2010 until May 2012. Eleven patients were randomized to receive taliglucerase alfa 30 U/kg or 60 U/kg per infusion every other week for 12 months and all 11 completed the study.3 PB-06-002 was a Phase 3, multicenter, open-label, switchover trial (US National Institutes of Health www.clinicaltrials.gov identifier NCT00712348) designed to assess the safety and efficacy of the switch to treatment with taliglucerase alfa in adults and pediatric patients through 9 months. Eligible patients included males and females (≥2 years of age) with a diagnosis of stable GD and leukocyte glucocerebrosidase (β-glucosidase) activity level ≤3 nmol/mL/h (≤30% of the mean activity of the reference range).
Stable GD was initially defined as follows:
- stable hemoglobin concentration during the 12-week stability evaluation period, with no value >15% below or above the mean value of the stability evaluation period measurements or the mean historical value for patients affected by the shortage;
- stable platelet count during the stability evaluation period, with no values outside the defined range below or above the mean value of the stability evaluation period measurements or the mean historical value (>40% below or above the mean value if the mean value was >120,000 or >20% below or above the mean value if the mean value was ≤120,000);
- no major surgery in the last year;
- no blood transfusion;
- no major bleeding episode;
- no acute avascular necrosis in the last year; and
- no evidence of worsening spleen or liver enlargement as detected by palpation, ultrasound, or magnetic resonance imaging (MRI) over the last year while being treated with ERT.
The stability definition was amended to allow the use of 6 historical hemoglobin concentration values and platelet counts for patients affected by the 2009 global imiglucerase shortage.23 Patients were required to have received imiglucerase therapy for at least the last 2 years and were currently on a stable maintenance regimen (ie, dose had not been changed except for a situation that arose because of the drug shortage) for at least the last 6 months prior to enrollment. The study was conducted at 11 medical centers worldwide from December 2008 until January 2013.
Taliglucerase alfa was administered by intravenous infusion over 1–2 h at 2-week intervals at the dosage equal to the patient’s imiglucerase dose at screening or prior to the imiglucerase shortage. Of the 31 patients allocated to taliglucerase alfa, 5 were pediatric patients, and all 5 of them completed the study.23
PB-06-006 was a multicenter extension study. Patients who completed Studies PB-06-005 and PB-06-002 were eligible for inclusion in Study PB-06-006 (US National Institutes of Health www.clinicaltrials.gov identifier NCT01411228). The trial ran from September 2011 to July 2014. The patients continued to receive the same dose of taliglucerase alfa as in the original studies for a maximum additional 24 months. Fifteen patients were enrolled in the study, and 11 completed it.22,25
Safety assessments
Safety assessments included the incidence and severity of AEs, the incidence of serious AEs (SAEs), the presence of anti-taliglucerase alfa antibodies, changes upon physical examination, and changes in clinical laboratory values, electrocardiogram, echocardiogram, and pulmonary function tests. AEs were assessed at all study visits. Immunogenicity analysis of patient samples included testing of anti-taliglucerase alfa-specific immunoglobulin G, immunoglobulin E, and neutralizing antibody activity assessment, which was conducted using both an in vitro and a cell-based neutralizing antibody assay. Growth and developmental assessments in patients <18 years of age included height and weight for growth evaluation, growth velocity, bone age by X-ray of left hand and wrist, and Tanner stage for sexual development. Occurrence of bone events, including bone crises, was part of the analysis of AEs.3,22–25
Efficacy assessments
In the PB-06-002 (‘switch’) study, the primary efficacy outcome was clinical stability during treatment with taliglucerase alfa. Clinical deterioration as assessed by platelet counts, hemoglobin concentration, spleen volume, and liver volume were defined as follows: 1) a decrease in platelet count of > 20% from the mean of 6 stability evaluation period for values ≤ 120,000 and a >40% reduction from the mean of 6 stability evaluation period for values of >120,000; or 2) a decrease in hemoglobin concentration >20% from the mean of 6 stability evaluation period values; or 3) an increase in spleen volume >20% by MRI from baseline to month 9; or 4) an increase in liver volume >10% by MRI from baseline to month 9. Secondary efficacy outcomes included assessment of the disease biomarkers chitotriosidase (activity) and CCL18 (concentration).23
In the PB-06-005 (‘naive-pediatric’) and PB-06-006 (‘extension’) studies, the primary end point was the median percent change in hemoglobin concentration from baseline and the interquartile range of median percent change in hemoglobin levels from baseline. Secondary end points included the percent changes from baseline in spleen volume, liver volume, platelet counts, and either chitotriosidase or CCL18 activity.
Exploratory end points included change in height, weight, puberty, and bone age (based on radiograph of the left hand and wrist); occurrence of bone events including bone crises; and quality of life using Child Health QuestionnaireTM (healthactchq, Inc., Boston, MA, USA; valid for patients aged 5–18 years).3,22
Results
All five pediatric patients, enrolled in the switch study, completed the study. Disease parameters including hemoglobin concentration, platelet count, spleen volume, liver volume, and biomarker activity remained stable through 9 months of treatment.23
In the naive-pediatric study for treatment naive pediatric patients, six were randomized to the 30 U/kg dose arm and five were randomized to the 60 U/kg dose arm. All 11 pediatric patients completed the study. Results through 12 months in these pediatric patients demonstrated that taliglucerase alfa infusions q 2 weeks at 30 and 60 U/kg, respectively, increased median hemoglobin concentration (by 12.2% and 14.2%, respectively) and mean platelet count (by 30.9% and 73.7%, respectively), while also reducing mean spleen volume, liver volume, and chitotriosidase activity.3
Patients who completed the naive-pediatric and switch studies were eligible for inclusion in the pediatric extension study. The patients continued to receive the same dose of taliglucerase alfa as in the original studies, but for a maximum additional period of 24 months. Fifteen patients were enrolled in the study and were equally divided among the different treatment groups: 30 U/kg arm (n–5), 60 U/kg arm (n–5), and the dose-adjusted switchover arm (n–5). Patients’ mean age was 12 years, approximately two-thirds were male, 13 were Caucasian, and one was a Pacific Islander; ethnicity was not provided by one patient.23 Of the 11 patients who completed the naive-pediatric study, 1 patient who had type 3c GD did not enroll in the pediatric extension study because of worsening of cardiac valve involvement requiring intervention; however, this patient continued taliglucerase alfa treatment in a compassionate use program. Of the 10 patients from the naive-pediatric study who enrolled in the pediatric extension study, 9 completed 36 total months of treatment; 1 patient withdrew voluntarily (parents were unhappy with the child’s growth). All five patients who were previously treated with imiglucerase and completed taliglucerase alfa treatment in the switch study were enrolled in the pediatric extension study. Of these, one patient was lost to follow-up. Based on results from previous and current studies confirming the efficacy and safety of taliglucerase alfa and to ease the burden of clinical studies for pediatric patients, the sponsor decided to close the extension study prematurely. At that point, two patients who switched from imiglucerase completed 18 months of treatment in the pediatric extension study (27 total months of treatment) and continued treatment under a compassionate use program.25
Efficacy
Hemoglobin concentration
In the switchover study, the change in mean hemoglobin after a total of 33 months of treatment was from 13.5 to 14.5 g/dL, corresponding to a 3.3% increase. In the treatment- naive group, the change in mean hemoglobin was from 11.2 to 13.2 and 10.6 to 12.7 g/dL in the 30 and 60 U/kg dose arms, respectively, after completing a total of 36 months on taliglucerase alfa.25 These changes correspond to an increase in hemoglobin concentration of 19.7% and 23.3%, respectively.
Platelet count
In the switchover study, the change in mean platelet count after a total of 33 months of treatment was from 164,587 to 210,000/mm3, corresponding to a 2.3% increase. In the treatment- naive arm, the change in mean platelet count was from 182,000 to 220,200/mm3 and from 99,600 to 243,750/mm3 in the 30 and 60 U/kg dose arms, respectively, after completing a total of 36 months on taliglucerase alfa.25 These changes correspond to an increase in platelet count of 23.9% and 156.6%, respectively.
Spleen volume
In the switchover study, the change in mean spleen volume after a total of 33 months of treatment was from 4.1 to 1.7 multiples of normal (MN), corresponding to a 5.3% decrease. In the treatment- naive arm, the change in mean spleen volume was from 23.9 to 9.0 MN and from 29.4 to 6.6 MN in the 30 and 60 U/kg dose arms, respectively, after completing a total of 36 months on taliglucerase alfa.25 These changes correspond to a decrease in spleen volume of 67.8% and 68.9%, respectively.
Liver volume
In the switchover study, the change in mean liver volume after a total of 33 months of treatment was from 1.3 to 0.9 MN, corresponding to an 8.8% decrease. In the treatment- naive arm, the change in mean liver volume was from 1.9 to 1.3 MN and from 2.2 to 1.5 MN in the 30 and 60 U/kg dose arms, respectively, after completing a total of 36 months on taliglucerase alfa.25 These changes correspond to a decrease in liver volume of 37% and 34.3%, respectively.
Chitotriosidase
In the switchover study, the change in chitotriosidase activity after a total of 33 months of treatment was from 10,432 to 370.5 nmol/mL/h, corresponding to a 97.1% decrease. In the treatment- naive arm, the change in mean biomarker activity was from 30,783 to 7,640.6 nmol/mL/h and from 34,968 to 5,204.0 nmol/mL/h in the 30 and 60 U/kg dose arms, respectively, after completing a total of 36 months on taliglucerase alfa.25 These changes correspond to a decrease in chitotriosidase activity of 72.2% and 84.4%, respectively.
Growth
After completion of 9 months of treatment with taliglucerase alfa in the switchover trial, there was an increase in mean height and weight in pediatric patients. Mean growth velocity for the five pediatric patients was estimated to be 4.2 cm per year. Bone age increased age appropriately from baseline to month 9.23
In patients who switched from imiglucerase and completed the pediatric extension study, the height velocity through the end of the study (or last visit if a patient did not complete the study) revealed mean (standard error, SE) rates of 2.5 (0.7) cm/yr (n=5). Two patients who switched from imiglucerase advanced from Tanner stage 3 and 2, respectively, to stage 4 by the end of the study.25
After completion of 12 months of treatment with either 30 U/kg or 60 U/kg of taliglucerase alfa in the naive-pediatric trial, increases in height and weight were seen in both dose groups. Mean growth velocity was estimated to be 5.1 and 8.0 cm/yr in the 30 and 60 U/kg treatment arms, respectively.3 All growth parameters were physiological and increased proportionally with age.
In treatment-naive patients who completed the pediatric extension study, the height velocity through the end of the study (or last visit if a patient did not complete the study) revealed mean (SE) rates of 5.5 (0.8) cm/yr with 30 U/kg (n=5) and 6.7 (0.6) cm/yr with 60 U/kg (n=5).25
The pubertal status did not change during this extension study for most patients. All treatment- naive patients were ≤ 10 years of age at baseline; nine patients were at stage 1 and one patient was at stage 3. Two patients treated with taliglucerase alfa 30 U/kg had pubertal advancement; one patient progressed from Tanner stage 1 at baseline to stage 2 by 12 months and continued to advance to stage 3 by the end of this study. The other patient advanced from Tanner stage 3 to stage 4 by 18 months.25
At baseline, in the treatment-naive patients, bone age for all treated patients was considered to be delayed relative to chronological age, except for one 13-year-old patient, whose bone age was equivalent to chronological age at baseline. After 12 months of treatment with taliglucerase alfa in the nine pediatric patients evaluated, bone age advancement of 1–4 years was noted.3
At the completion of the extension study, bone age increased from baseline by a mean (SE) of 3.6 (0.9) years (range, 1.5–6.5 years) over the 36 months of the combined study for the 30 U/kg cohort (n=5) and by 4.6 (0.6) years (range, 3.0–5.5 years) for the 60 U/kg cohort (n=4) in treatment-naive patients. For three patients who switched from imiglucerase and had baseline bone age measurements, bone age increased by a mean (SE) of 2.3 (0.3) years (range, 2.0–3.0 years) by 24 months (no data available over 33 months).25
Z-scores from bone mineral density analysis by dual energy X-ray absorptiometry showed mean decreases at the lumbar spine of −0.20 and femoral neck of −0.30 in the taliglucerase alfa 30-U/kg dose group, whereas mean increases at the lumbar spine of 0.27 and femoral neck 0.20 in the taliglucerase alfa 60-U/kg dose group were noted.3
Based on the responses to the Child Health Questionnaire for Quality of Life assessment, which was performed for treatment-naive patients compared to baseline, more parents/guardians rated their children’s global health as very good or excellent after 12 months of treatment with taliglucerase alfa.3 Subsequently more parents/guardians rated their children’s global health as “very good” at 36 months (7/9) than at baseline (1/9).25
AEs/safety
Four of the five pediatric patients who received treatment with taliglucerase alfa in the switch study experienced a total of nine AE. The most commonly experienced AEs (ie, occurring in >10% of patients) were nasopharyngitis, infusion-related reaction, arthralgia, and headache. No severe AEs were reported by any pediatric patients.23
In the naive-pediatric study, two patients in the 60-U/kg treatment group experienced eight events that were considered to be treatment-related by the investigator. These events were itchy throat, chest discomfort, gastroenteritis, and vomiting. One patient in the taliglucerase alfa 60-U/kg group experienced an SAE during the first infusion visit (gastroenteritis, requiring hospitalization for rehydration) that resolved after 1 day (and recurred as a nonserious AE). No patient withdrew from this trial due to an AE.3
In the pediatric extension study, one patient contracted dengue fever, which was considered an SAE. The other AEs reported were lymphadenopathy in three patients, ear pain in two patients, abdominal pain in three patients, diarrhea in three patients, upper respiratory tract infection in four patients, nasopharyngitis in three patients, pain in extremity in three patients, headache in four patients, and cough in four patients.25,26
Discussion
The combined results from the three studies in pediatric patients demonstrated clinically significant improvements in hemoglobin concentration and platelet count, and a decrease in spleen and liver volumes, as well as in the GD plasma biomarkers chitotriosidase and CCL18. Responses were observed as early as 3 months from the start of therapy and continued through the extension period.3,25
In the switch population, primary efficacy parameters were generally stable over time.23 The efficacy results in the pediatric population from both the switch and naive taliglucerase alfa clinical trials followed by the extension trial reflect those seen in the phase 3 pivotal taliglucerase alfa clinical trial in adults as well as the combined extension study population.3,21–25 The improvements in visceral and hematologic manifestations of GD observed during the 33–36 months of treatment with taliglucerase alfa in the overall pediatric population were generally consistent with findings for pediatric patients receiving imiglucerase and velaglucerase alfa.6,27
Taliglucerase alfa infusions were shown to have a positive safety profile in children and adolescents throughout the studies. The safety results in the pediatric population reflect those seen in the pivotal phase 3 trial in adults, as well as in the extension study, although vomiting was reported more often in the pediatric population.3,21–25 No new safety concerns were identified in the pediatric subgroup. No SAE were deemed related to taliglucerase alfa, and no patients withdrew from the study because of an AE.
Bone involvement is a serious and debilitating aspect of GD that can lead to complications such as growth retardation and skeletal deformity in children.6 Bone involvement begins early in life and low bone mineral density may be observed as early as 5 years of age, and is putatively most prevalent in adolescence.3 Pubertal delay is an additional feature of pediatric GD.6 Achieving optimal peak bone mass and minimizing bone-related disease manifestations are vital in preventing long-term morbidity, which can adversely affect quality of life.3,6
In these studies, assessment of growth and development included measurements of changes in height, weight, bone age, bone mineral density, puberty, occurrence of bone crises, and quality of life. Increases were seen in both the switch and naive taliglucerase alfa patients in mean height, weight, and bone age consistent with a positive benefit of early treatment to enable normal growth.3,25
While the interpretation of the reported findings may be limited by both the small number of patients and overall duration of studies (33–36 months), the positive results are reflective of the collective experience that children respond similarly to adults to ERT for GD at a weight-based dosage.3,6,
Conclusion
Taliglucerase alfa treatment had a positive effect on key efficacy parameters in children with GD, which included improvement in growth, hematological, and visceral manifestations. Treatment was well tolerated, and no unexpected safety concerns were identified. The production process of the plant-cell-expressed enzyme introduces benefits such as the inability to be contaminated with or propagate mammalian pathogens, along with a potentially lower manufacturing cost. The cost to benefit ratio and greater safety may help address rising concerns regarding health care expenditure for orphan diseases. Increased clinical experience will demonstrate the extent to which taliglucerase alfa is incorporated in the therapeutic armamentarium for GD.
Acknowledgments
None of the authors received compensation for their contributions to this manuscript. Editorial support was provided by Julie Collins, CMPP, Peloton Advantage.
Author contributions
Drs Gupta and Pastores contributed to the writing of the manuscript and approved the final manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors did not receive financial support for the preparation of the manuscript. Pfizer provided a courtesy medical accuracy review and offered to pay the very minor permission fee for obtaining copyrighted information for the table and figure on the authors’ behalf. The authors report no other conflicts of interest in this work.
References
Grabowski GA, Petsko GA, Kolodny EH. Gaucher disease. In: Scriver CR, Beudet AL, Sly WS, editors. The Metabolic and Molecular Basis of Inherited Disease. New York, NY: McGraw-Hill; 2010:1–143. | ||
Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. | ||
Zimran A, Gonzalez-Rodriguez DE, Abrahamov A, et al. Safety and efficacy of two dose levels of taliglucerase alfa in pediatric patients with Gaucher disease. Blood Cells Mol Dis. 2015; 54(1):9–16. | ||
Zimran A, Elstein D. Lipid storage diseases. In: Lichtman MA, Kipps T, Seligsohn U, Kaushansky K, Prchal JT, editors. Williams Hematolog. 8th ed. New York, NY: McGraw-Hill; 2010:1065–1071. | ||
Mistry P, Germain DP. Phenotype variations in Gaucher disease. Rev Med Interne. 2006;27:S3–S10. | ||
Andersson H, Kaplan P, Kacena K, Yee J. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics. 2008;122:1182–1190. | ||
Kauli R, Zaizov R, Lazar L, et al. Delayed growth and puberty in patients with Gaucher disease type 1: natural history and effect of splenectomy and/or enzyme replacement therapy. Isr Med Assoc J. 2000;158–163. | ||
Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160:2835–2843. | ||
Charrow J, Andersson HC, Kaplan P, et al. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: con- sensus recommendations. J Pediatr. 2004;144:112–120. | ||
Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency—macrophage-targeted glucocerebrosidase for Gaucher’s disease. N Engl J Med. 1991;324:1464–1470. | ||
Cerezyme [package insert]. Cambridge, MA: Genzyme Corporation; 2009. | ||
VPRIV [package insert]. Lexington, MA: Shire Human Genetic Therapies, Inc.; 2013. | ||
Elelyso [package insert]. New York, NY: Pfizer Labs; 2014. | ||
Shaaltiel Y, Bartfeld D, Hashmueli S, et al. Production of gluco- cerebrosidase with terminal mannose glycans for enzyme replacement therapy of Gaucher’s disease using a plant cell system. Plant Biotechnol J. 2007;5:579–590. | ||
Fox JL. First plant-made biologic approved. Nat Biotechnol. 2012; 30:472. | ||
Opar A. ‘Pharmers’ hope for first plant drug harvest. Nat Rev Drug Discov. 2011;10: 81–82. | ||
Aviezer D, Brill-Almon E, Shaaltiel Y, et al. A plant-derived recombinant human glucocerebrosidase enzyme—a preclinical and phase I investigation. PLoS One. 2009;4: e4792. | ||
Grabowski GA, Golembo M, Shaaltiel Y. Taliglucerase alfa: an enzyme replacement therapy using pla nt cell expression technology. Mol Genet Metab. 2014;112:1–8. | ||
ClinicalTrials.gov, National Institutes of Health. Plant cell expressed recombinant human glucocerebrosidase extension trial. NLM identifier: NCT00705939. | ||
ClinicalTrials.gov, National Institutes of Health. Expanded access trial of plant expressed recombinant glucocerebrosidase (prGCD) in patients with Gaucher disease. NLM identifier: NCT00962260. | ||
Zimran A, Brill-Almon E, Chertkoff R, et al. Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood. 2011;118:5767–5773. | ||
Zimran A, Gonzalez-Rodriguez DE, Abrahamov A, et al. Long-term safety and efficacy of taliglucerase alfa in pediatric patients with Gaucher disease who were treatment-naïve previously treated with immiglucerase. Abstract 293. Mol Genet Metab. 2015;114(2): S129. | ||
Pastores GM, Petakov M, Giraldo P, et al. A Phase 3, multicenter, open-label, switchover trial to assess the safety and efficacy of taliglucerase alfa, a plant cell-expressed recombinant human glucocerebrosidase, in adult and pediatric patients with Gaucher disease previously treated with imiglucerase. Blood Cells Mol Dis. 2014;53(4):253–260. | ||
ClinicalTrials.gov, National Institutes of Health. A multicenter extension study of taliglucerase alfa in adult subjects with Gaucher disease. NLM identifier: NCT01422187. | ||
Zimran A, Gonzalez-Rodriguez DE, Abrahamov A, et al. Long-term safety and efficacy of taliglucerase alfa in pediatric Gaucher disease patients who were treatment-naïve or previously treated with imiglucerase. Blood Cells Mol Dis. 2016;S1079-9796(16):30221–30222. | ||
ClinicalTrials.gov, National Institutes of Health. A multicenter extension study of taliglucerase alfa in pediatric subjects with Gaucher disease. NLM identifier: NCT01411228. | ||
Hughes DA, Gonzalez DE, Lukina EA, et al. Velaglucerase alfa (VPRIV) enzyme replacement therapy in patients with Gaucher disease: long-term data from phase III clinical trials. Am J Hematol. 2015;90(7):584–591. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.